

Abstract
Background
Understanding longitudinal variability of the microbiome in ill patients is critical to moving microbiome-based measurements and therapeutics into clinical practice. However, the vast majority of data regarding microbiome stability are derived from healthy subjects. Herein, we sought to determine intra-patient temporal microbiota variability, the factors driving such variability, and its clinical impact in an extensive longitudinal cohort of hospitalized cancer patients during chemotherapy.
Methods
The stool (n = 365) and oral (n = 483) samples of 59 patients with acute myeloid leukemia (AML) undergoing induction chemotherapy (IC) were sampled from initiation of chemotherapy until neutrophil recovery. Microbiome characterization was performed via analysis of 16S rRNA gene sequencing. Temporal variability was determined using coefficients of variation (CV) of the Shannon diversity index (SDI) and unweighted and weighted UniFrac distances per patient, per site. Measurements of intra-patient temporal variability and patient stability categories were analyzed for their correlations with genera abundances. Groups of patients were analyzed to determine if patients with adverse outcomes had significantly different levels of microbiome temporal variability. Potential clinical drivers of microbiome temporal instability were determined using multivariable regression analyses.
Results
Our cohort evidenced a high degree of intra-patient temporal instability of stool and oral microbial diversity based on SDI CV. We identified statistically significant differences in the relative abundance of multiple taxa amongst individuals with different levels of microbiota temporal stability. Increased intra-patient temporal variability of the oral SDI was correlated with increased risk of infection during IC (P = 0.02), and higher stool SDI CVs were correlated with increased risk of infection 90 days post-IC (P = 0.04). Total days on antibiotics was significantly associated with increased temporal variability of both oral microbial diversity (P = 0.03) and community structure (P = 0.002).
Conclusions
These data quantify the longitudinal variability of the oral and gut microbiota in AML patients, show that increased variability was correlated with adverse clinical outcomes, and offer the possibility of using stabilizing taxa as a method of focused microbiome repletion. Furthermore, these results support the importance of longitudinal microbiome sampling and analyses, rather than one time measurements, in research and future clinical practice.
Electronic supplementary material
The online version of this article (doi:10.1186/s13073-017-0409-1) contains supplementary material, which is available to authorized users.
Keywords: Microbiome, Temporal variability, Leukemia, Chemotherapy, Antibiotics
Background
There is an increasing appreciation for the role the human microbiome plays in many aspects of human physiology, health, and disease. Several studies of healthy human cohorts have found that although each person has a relatively distinct gastrointestinal microbiome signature, a healthy individual’s microbiome remains relatively stable over time [1–4]. Although several factors, such as diet, drive normal levels of day-to-day microbiota variability, it appears that a steady-state equilibrium both ecologically and functionally is required for health. In contrast, acute perturbations of an individual’s microbiome stability within a temporal context can lead to an unhealthy status [5, 6]. Considering that one of the principal aims of the microbiome research community is to use the microbiome as either an indicator for morbidity or to improve human health, an enhanced understanding of the kinetics and taxonomic characterization of microbiome stability in acutely ill patients is of paramount importance [7–11].
Although several studies have been done in healthy subjects, relatively scant data are available as to the stability, resilience, and temporal dynamics of the gastrointestinal microbiome in acutely ill patients [1, 3, 4, 12–16]. Many of the previous investigations examining temporal variability of the microbiome using healthy participants have been limited by small numbers of volunteers [4, 12, 13], short periods of longitudinal sampling [2, 3, 16], or by being focused on only one site of collection [1, 4, 14, 17]. On the other hand, the limited number of temporal variability studies among ill patients have typically been in cohorts with chronic ailments such as atopic dermatitis or colitis [18–20]. A study of stool samples from 14 patients under intensive care described rapid shifts in microbiome composition to ultra-low diversity communities comprised of four or less taxa as a result of aggressive antibiotic treatment and other intensive care medication stresses, such as opioids [11]. Similar dramatic changes in the microbiome were also observed in patients undergoing hematopoietic stem cell transplant, where increased microbial chaos early after transplant is thought to be a potential risk factor for subsequent graft versus host disease [21, 22]. However, quantitative measurements of longitudinal microbial variability among ill patients and an analysis of factors affecting microbiome temporal stability are lacking [21, 23–25]. Moreover, despite many reports associating low microbial diversity with different illnesses, most studies associate only one-time microbiome measurements with subsequent clinical outcomes, which could be potentially problematic in settings of significant temporal variability [24, 26].
Our group previously reported that a single measurement of baseline stool microbial diversity was associated with infectious risk for 34 patients during induction chemotherapy (IC) for acute myelogenous leukemia (AML) [25]. Similar to other studies of ill patients, we observed instances of rapid and profound shifts in the microbiota in our AML cohort [11, 21, 22]. Thus, herein, we sought to quantify the overall intra-patient temporal variability of the oral and stool microbiome of this cohort expanded to 59 patients. In addition, we sought to determine the consequences of microbiome temporal instability on patient outcomes and clinical factors driving intra-patient temporal variability of the microbiome during IC. We chose to study such patients because of the opportunity to characterize the microbiome prior to receipt of chemotherapy and intense antibiotic exposure (i.e., prior to severe perturbations) and the capacity to obtain dense longitudinal sampling over the course of intensive treatment due to the extended inpatient nature of IC. Moreover, AML patients are at high risk for infection during IC and such infections are generally derived from the commensal microflora [21, 23]. We hypothesized that higher microbiome intra-patient temporal variability, driven by prolonged antibiotic exposure, would be associated with poorer clinical outcomes.
Methods
Patient recruitment and specimen collection
Study subjects included 59 newly diagnosed adult AML patients undergoing IC at MD Anderson Cancer Center (MDACC) in Houston, TX from September 2013 to October 2014. AML patients initiating inpatient IC at MDACC were approached for study inclusion unless they had systemic infection. AML patients receiving IC at MDACC are routinely prescribed a prophylactic fluoroquinolone or cephalosporin prior to the initiation of therapy. In this study, 100% of patients received routine prophylaxis, with 64% of baseline stool, and 55% of baseline oral samples taken after the patient had already started prophylactics. AML patients over 50 years receiving IC are treated in a laminar-air flow isolation until neutrophil counts recover to >500 cells/μL or until day 28. Patients aged under 50 years are admitted for the duration of the chemotherapy (approximately 4–5 days) and then followed as an outpatient with clinic visits three times a week until neutrophil recovery or 28 days.
Buccal and fecal specimens were collected from each patient at baseline, continued approximately every 96 h as available, and stopped upon neutrophil recovery. Baseline samples were considered up to 8 days before and 24 h following IC initiation. As per availability of samples, 55 (93%) of the patients had oral samples collected before or at the same time as the initiation of chemotherapy, while 35 (59%) of the patients had stool samples collected before or at the same time as the initiation of chemotherapy. The buccal mucosa of each individual was swabbed three times on each side using a Catch-All™ Sample Collection Swab (Epicentre). Patient stool samples were either collected in a stool hat or using a BBL™ CultureSwab® (BD Diagnostics). All samples were placed in sterile 2-mL cryovials and stored immediately at −80 °C until further processing.
16S rRNA sequencing and data processing
Bacterial genomic DNA was extracted from buccal and stool specimens using the MO BIO PowerSoil DNA Isolation Kit (MO BIO Laboratories). The 16S rRNA V4 region was PCR amplified and sequenced on the Illumina MiSeq platform using a 2 × 250-bp paired-end protocol adapted from the Human Microbiome Project (HMP) methods [16, 27]. All samples from the same patient and site were processed and sequenced together to minimize batching issues. Amplification primers contained adapters for MiSeq sequencing and single-index barcodes resulting in PCR products that were pooled and sequenced directly. Read pairs were de-multiplexed based on barcodes and merged using USEARCH v7.0.100. 16S rRNA gene sequences were allocated to specific operational taxonomic units using a UPARSE pipeline and aligned to the V4 region within the SILVA SSURef_NR99_119 database [28]. Analysis of microbiome communities was performed in R (R Core Team 2015, version 3.2.2, http://www.R-project.org), using phyloseq [29] to calculate α- and β-diversity metrics. The Shannon Diversity Index (SDI) was used for α-diversity calculations, and weighted and unweighted UniFrac for β-diversity distances [30]. The 16S V3–V4 region HMP sequencing reads were obtained from http://hmpdacc.org/HMQCP, trimmed to match the region amplified by this study, and processed identically to AML patient samples.
Microbiome community and statistical analyses
Intra-patient temporal variability of microbial diversity was defined as the coefficient of variation (CV) of a longitudinal collection of α-diversity values, and was calculated for each patient’s set of oral and stool samples. Higher values were indicative of more variable microbial diversity. Temporal variability in community composition, or β-diversity, of each patient was determined for the oral and stool by calculating the CV of the weighted and unweighted UniFrac distances of longitudinal samples collected from each individual per site. Again, higher values were indicative of more variable communities. Pairwise differences in temporal variability across body sites were made using Mann–Whitney U test, whereas pairwise differences among infection or response groups was performed using Student’s t-test with Welch’s correction. Linear correlations between CVs at different body sites were determined using Pearson’s r and P values generated in GraphPad Prism 6.
Heatmaps analyzing genera abundance over time among patients with increasing temporal variability were generated with the publically available pheatmap R package version 1.0.8. (http://CRAN.R-project.org/package=pheatmap), and include correlation metrics calculated with R’s cor and cor.test stats package functions. P values were corrected for multiple comparisons using the Benjamini and Hochberg method.
For each body habitat the population was divided into quartiles based on CV of the weighted UniFrac distance values or SDI where the first quartile was defined as stable, second and third as average, and fourth as variable as previously described [15]. To determine significant differences in genera abundance between stable, average, and variable individuals, we tested for differences between groups using non-parametric Kruskal–Wallis analysis of variance in R for genera across individuals, then corrected for the false discovery rate using the Benjamini and Hochberg method.
Multivariable regression analyses were performed using base R (R Core Team 2015, version 3.2.2, http://www.R-project.org ) and included age, antibiotic type, chemotherapy regimen, and exposure to antibiotics as covariates. Antibiotic types were subdivided into three major broad spectrum β-lactam antibiotics received by this cohort, namely, cefepime, carbapenems (primarily meropenem), and piperacillin-tazobactam. Chemotherapy regimens were subdivided into fludarabine-containing regimens, high intensity non-fludarabine-containing regimens, hypomethylators, or other. Fludarabine-containing regimens included fludarabine in combination with idarubicin and cytarabine [31], or fludarabine/idarubicin/cytarabine with G-CSF (FLAG-Ida). High intensity non-fludarabine-containing regimens were purine analog of clofarabine or cladrabine in combination with idarubicin and cytarabine. Hypomethylator-based combinations included decitabine and azacytidine [32].
Clinical definitions
Infections were defined as microbiologically defined infections (MDIs) or clinically defined infections as described previously [25]. Subsequent infectious episodes were defined as MDIs that occurred within 90 days of cessation of longitudinal sampling. Complete remission (CR) of AML was assessed using standard definitions [33].
Results
AML patients undergoing IC exhibit temporal instability of the stool and oral microbiome diversity
In order to understand the intra-patient temporal variability of the microbiome among hospitalized patients with AML, we performed sequencing of the V4 region of the 16S rRNA gene via the MiSeq platform (Illumina) using the 2 × 250-bp protocol [34] on a total of 901 longitudinal samples collected twice weekly from initiation of chemotherapy until neutrophil recovery for 59 AML patients undergoing IC. Of the samples, 848 (84%, n = 365 stool and 483 oral) passed sequencing quality control measures for further analyses. For these samples, we obtained a total of 24,271,698 reads, for an average of 28,622 reads per sample. Patient demographics and clinical metadata can be found in Table 1.
Table 1.
Clinical features of 59 AML patients
| Characteristic | Number (%) |
|---|---|
| Demographics | |
| Median age in years a | 55 (49–68) |
| Male | 31 (52.5) |
| Female | 28 (47.5) |
| Median days on study | 28 (25–35) |
| Median number of oral samples | 8 (6–9) |
| Median number of stool samples | 6 (4–8) |
| Chemotherapy | |
| Hypomethylatorsb | 14 (23.7) |
| Non-fludarabine high intensityc | 19 (32.2) |
| Fludarabine-containingd | 19 (32.2) |
| Othere | 7 (11.8) |
| Chemotherapeutic response | |
| Complete remission after IC | 20 (33.8) |
| Overall response ratef | 43 (72.8) |
| Infectionsg | |
| Microbiologically documented infection | 15 (25.4) |
| Clinically documented infection | 14 (23.7) |
| No infection | 30 (50.8) |
| Antimicrobial administration | |
| Received treatment antibioticsh | 53 (89.8) |
| Carbapenem >72 h | 39 (66.1) |
| Piperacillin/tazobactam >72 h | 14 (23.7) |
| Cefepime >72 h | 26 (44.1) |
| Received prophylactic antibiotics | 59 (100) |
| Median number of antibiotics administered | 6 (4–7) |
| Median number of days exposed to all antibioticsi | 28 (24–35) |
| Median number of days exposed to treatment antibiotics | 16 (9–24) |
| Median number of days exposed to prophylactic antibiotics | 16 (8–28) |
a All median values in this table have the interquartile range in parentheses
b These chemotherapies included: 1) vasoroxin in combination with decitabine; 2) decitabine alone; 3) azacytidine in combination with pracinostat; 4) azacytidine in combination with quidartinib; and 5) SGI-110
c These chemotherapies included: 1) CIA, 2) CLIA, 3) or CIA + sorafanib
d These chemotherapies included: 1) FLAG-Ida or 2) FIA regimens
eOther chemotherapies included:1) omacetaxine in combination with low-dose cytarabine or 2) Clad + LDAC
f Includes CR (morphologic complete remission), CRi (morphologic complete remission with incomplete bloodcount recovery), and CRp (morphologic complete remission with incomplete platelet recovery)
g Specific information on microbiologically and clinically documented infections can be found in the “Methods”
h Refers to any antibiotic/antimicrobial-based therapy given for suspected or proven infection, that is, not included as prophylaxis (cephalosporins or fluoroquinilones). Denoted are the three most common broad spectrum antibiotics given in the study. Note that numbers of individual antibiotics add up to >100% because some patients received more than one of the listed antimicrobials during IC
i Includes prophylactic antibiotics
Currently, the majority of 16 s rRNA microbiome-based data are summarized using either numerical or index-based measurements of species richness and/or evenness within a habitat (i.e., α-diversity) or characterization of differences in microbial community composition by measuring the distance or dissimilarity between samples (i.e., β-diversity). We first sought to determine intra-patient temporal variability of α-diversity by calculating the CV of the SDI for both the oral and the stool samples for each patient. The coefficient of variation is defined as the ratio of the standard deviation to the mean; thus, a low CV would mean an individual had relatively stable species diversity over time whereas a high CV would reflect more variation. We found considerable heterogeneity in the temporal stability values of both stool (mean SDI CV 0.48 ± 0.25) and oral (mean SDI CV 0.42 ± 0.26) samples among AML patients during IC (Fig. 1a). There was no statistically significant difference in CV values between the two sites (P = 0.16). This finding is in contrast to previous studies performed in healthy individuals where the microbiota of oral samples have been shown to be less variable compared to stool [2, 15]. The intra-patient temporal variability of other α-diversity metrics, specifically the Chao-1 diversity index and Simpson’s diversity index, were also analyzed for the oral (mean Chao CV 0.39 ± 0.18, mean Simpson CV 0.33 ± 0.24) and stool samples (mean Chao CV 0.48 ± 0.22, mean Simpson CV 0.37 ± 0.27) of the AML cohort (Additional file 1: Figure S1a, b). Assessment of the temporal variability of α-diversity also revealed that the SDI CV of oral and stool samples from the same patients were statistically moderately correlated (P = 0.01, r = 0.33; Fig. 1b). The relationship between the two sites leads to the postulation that factors influencing temporal variability of microbial diversity in treated cancer patients may be acting on both sites concurrently. Conversely, it has been reported that the variability of one body site was not associated with the variability of other body habitats in healthy cohorts [15].
Fig. 1.
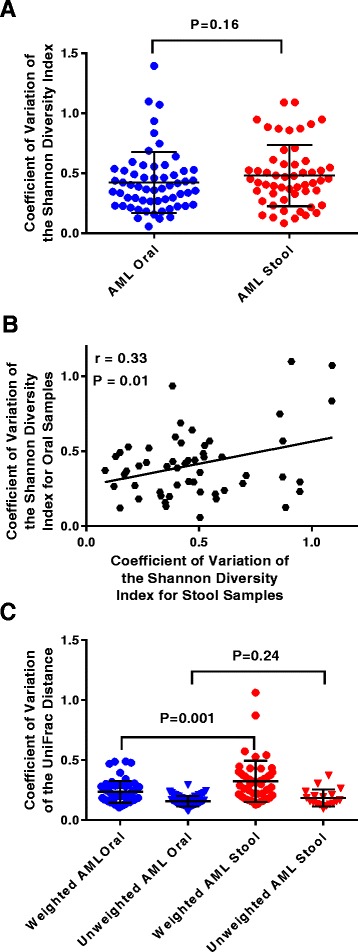
Intra-patient temporal variability in oral and stool microbiomes of hospitalized AML patients undergoing IC. a The oral and stool microbial α-diversity intra-patient temporal variability. Each point represents the coefficient of variation (CV) of the Shannon diversity index (SDI) for samples derived from each patient. b The correlation between the CV of the SDI values originating from oral and stool samples from the same patient. The Pearson’s correlation (r) value and P value from correlation analyses also are indicated. c The oral and stool microbial β-diversity intra-patient temporal variability using either the CV of the weighted or unweighted UniFrac distances for samples derived from each patient. In panels a and c, the bars represent mean ± standard deviation, and P values comparing the different body sites were calculated using a Mann–Whitney U-test
High intra-patient temporal variability of oral and stool microbiome among AML patients is associated with increased pathogenic-associated genera abundance
Next we sought to determine the temporal variability in microbiome community structure and membership as represented by quantitative and qualitative measurements of β-diversity using weighted and unweighted UniFrac distance measurements, respectively. Here, we considered the CV of each patient’s samples per site in order to characterize the dispersion of β-diversity metrics. The mean CVs of weighted and unweighted UniFrac distances for the cohort were 0.24 ± 0.1 and 0.16 ± 0.04 for the oral samples and 0.32 ± 0.2 and 0.20 ± 0.08 for stool samples, respectively (Fig. 1c). Contrary to SDI CVs, the temporal variability of the weighted UniFrac distances between the oral and stool of patients was not significantly correlated (P = 0.10; Additional file 1: Figure S1d). Reports in healthy persons have observed associations between diversity and temporal stability, such as individuals with a more diverse microbiome are likely to have a more stable microbiome over time [4, 14, 15]. However, we did not find any statistically significant correlations between either the baseline or median SDI values of patients and their temporal variability as measured by the CV of the weighted UniFrac distances of their samples, suggesting microbiome structural variability does not appear to be affected by α-diversity in treated AML patients (Additional file 1: Figure S2).
It is well known that patients in the hospital are at risk for colonization and intestinal domination by pathogenic bacteria and that a diverse microbiome provides colonization resistance against many such organisms [7, 11, 21]. Thus, to begin to investigate factors that might influence temporal instability in our cohort, we sought to test the hypothesis that temporal instability was influenced by increasing relative abundance of pathogenic-associated genera, such as Enterococcus and Staphylococcus. In order to visualize this relationship, we ranked patients and their samples by CV of weighted UniFrac from smallest to greatest (low variability to high variability of microbial community structure), and correlated this with the relative abundance of specific genera (Fig. 2). Consistent with our hypothesis, high weighted UniFrac CV values were moderately positively correlated with the relative abundance of pathogenic genera such as Staphylococcus (P < 0.001, r = 0.3), Streptococcus (P = 0.02, r = 0.2), and Stenotrophomonas (P = 0.01, r = 0.2) in the oral samples. Similarly, instability of community structure was statistically positively correlated with the abundance of Staphylococcus (P < 0.001, r = 0.2) and Streptococcus (P < 0.001, r = 0.2) in the stool samples. The same genera were correlated with temporal instability of microbial α-diversity (SDI CV) in oral and stool samples as well (Fig. 3). Also of note, the abundance of non-pathogenic organisms in the stool, such as Akkermansia, were associated with increased temporal stability of both the microbiome community structure and diversity (both stool weighted UniFrac CV and SDI CV P < 0.001, r = −0.2). Thus, our data imply that patients with a relatively high relative abundance of commensal organisms, like Akkermansia, maintain a more stable microbiome over the course of their hospitalization whereas higher relative abundances of pathogenic-associated bacteria, like Staphylococcus and Streptococcus, are associated with temporal instability of both the oral and gastrointestinal microbiome.
Fig. 2.

Temporal instability of microbiome community structure correlates with increasing abundance of pathogenic-associated genera over time. Heatmap of all samples and untransformed relative abundance values of indicated bacterial taxa colored white to red as denoted in the figure. Samples from each patient are clustered together and arranged by timepoint (i.e., consecutive samples) from left to right. Additionally, clusters of patient samples are organized in accordance with temporal variability as determined by the coefficient of variation of the weighted UniFrac distance (cv_w.unifrac) with increasing variability from left to right. Taxa are organized from top to bottom by highest positive correlation of relative abundance of genera with CV of the weighted UniFrac distance, to negative correlation as determined by Pearson’s correlation (r) values depicted in the colored inlaid figure legend. A P value for the correlation’s significance (Corr.SigLvl) was derived from the test statistic based on Pearson’s product moment correlation coefficient, corrected for multiple comparisons with the Benjamini and Hochberg method, and displayed on the plot
Fig. 3.

Temporal instability of microbial α-diversity correlates with increasing abundance of pathogenic-associated genera over time. This figure is arranged in the same way as Fig. 2, except temporal variability is defined using coefficient of variation of the Shannon diversity index (cv_shannon)
Stabilizing and destabilizing taxa can be inferred by categorizing AML patients into stable, average, or variable microbiomes during IC
We next sought to determine if we could infer stabilizing or destabilizing taxa by looking at significant differences in the relative abundance of taxa between patients with stable, average, or variable microbiomes based on their intra-patient microbial temporal stability. Patients were defined as stable, average, or variable according to their weighted UniFrac CV and SDI CV by dividing the population into quartiles with the bottom quartile (lowest CVs) being considered stable, and the top quartile (highest CVs) being considered variable. Figure 4 denotes all genera that had statistically significant abundance differences between stability categories defined by both SDI and weighted UniFrac CVs at the same body site. Pathogenic associated genera such as Streptococcus and Staphylococcus were more abundant in both oral and stool samples of individuals that were categorized as temporally variable in both the α- and β-diversity (Fig. 4). Confirming the analyses performed in Fig. 2, Akkermensia and Subdilogranulum were more abundant in the stool of stable individuals, alluding to a possible stabilizing effect of these genera. Additionally, Pseudobutyrivibrio was also found in greater abundance in the stool of individuals with a stable microbiome, signifying its potential importance in microbiome integrity as well. Other results for relative abundance differences in specific genera (those >1% abundance) between patient stability categories can be found in Additional file 1: Figure S3.
Fig. 4.
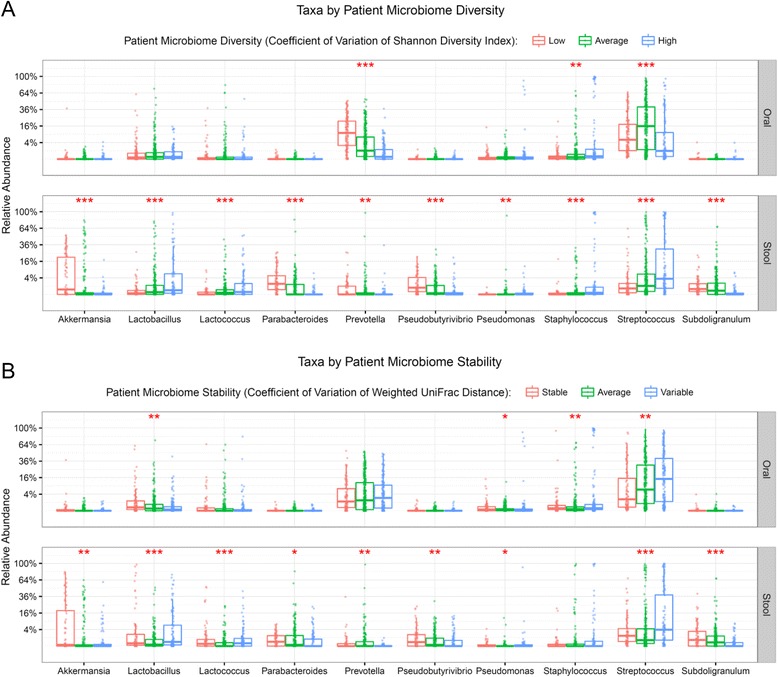
Taxonomic composition differences among different stability categories. The significant differences in relative abundances of genera between different patient stability categories based on either the coefficient of variation (CV) of the Shannon diversity index (SDI; top two panels) or the CV of the weighted UniFrac distances (bottom two panels). For each body habitat the population was divided into quartiles, where the first quartile was defined as low/stable, second and third as average, and fourth as high/variable. Differences in genera abundance across categories were determined using non-parametric Kruskal–Wallis analysis of variance, then corrected for the false discovery rate using the Benjamini and Hochberg method. Asterisks indicate adjusted P values: *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, respectively. a Taxa by patient microbiome diversity. b Taxa by patient microbiome stability
Intra-patient temporal instability of microbial diversity is linked to adverse infectious outcomes during and after IC
To evaluate the clinical consequences of the observed differences in temporal stability in our cohort, we sought to determine if intra-patient temporal variability of the microbiome could be linked with adverse outcomes during and following IC. Specifically, we analyzed if the measures of temporal variability (CVs of the SDI and weighted and unweighted Unifrac distances) were correlated with infection during IC, infection in the 90 days post-IC neutrophil recovery, or response of the leukemia to chemotherapy.
Although temporal variability of neither oral nor stool community membership (unweighted UniFrac CV) and structure (weighted UniFrac CV) was significantly correlated with infection during IC (Additional file 1: Figure S4), patients who had higher levels of oral variability based on α-diversity had significantly higher rates of infection during IC (P = 0.02) (Fig. 5a, b). Conversely, stool microbial α-diversity temporal variability levels, but not oral, were significantly higher during IC in patients who subsequently developed an infection within 90 days post-neutrophil recovery from IC (P = 0.04; Fig. 5c, d). None of the intra-patient microbiome temporal variability outputs were significantly associated with response of the leukemia to chemotherapy (Additional file 1: Figure S5). Moreover, we went on to assess if those genera found to be statistically correlated with microbial instability (e.g., Staphylococcus, Streptococcus, and Stenotrophomonas) were also correlated with infection. Indeed, individuals who contracted an infection during IC exhibited significantly higher relative abundances of Streptococcus (P < 0.05 for oral) and Stenotrophomonas (P < 0.001 for oral, P < 0.05 for stool) compared to those that did not. (Fig. 5e). These data indicate temporal variability and microbiome composition dynamics are likely important markers for infectious risk during and after IC.
Fig. 5.
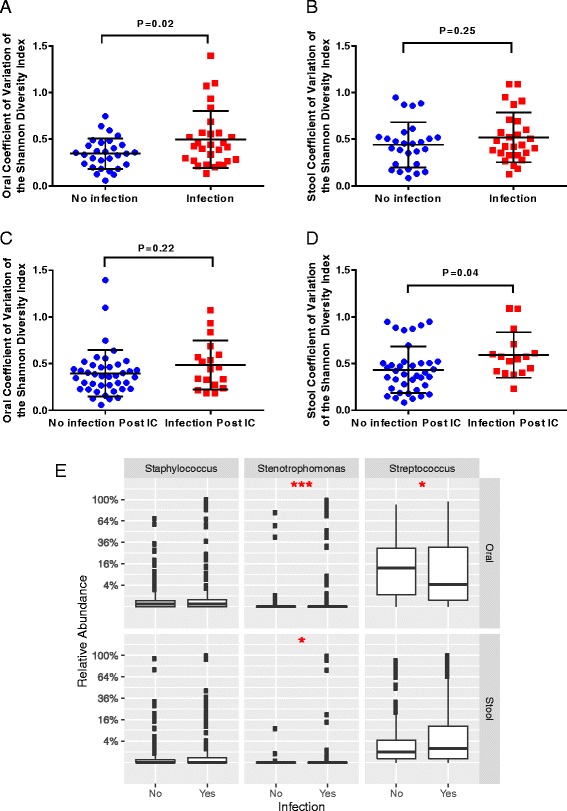
Temporal instability of microbiome α-diversity is associated with infectious outcomes during and after chemotherapy. Shown are the coefficient of variation (CV) of the Shannon diversity index (SDI) for oral (a) and stool (b) samples stratified by patients who did or did not contract an infection during the induction phase of chemotherapy before neutrophil recovery. c, d The CVs of the SDI for oral and stool samples, respectively, stratified by patients who did or did not contract an infection in the 90 days following neutrophil recovery. In all panels the bars represent mean ± standard deviation, and P values comparing SDI CV values among infectious outcomes were calculated using a two-sample t-test with Welch’s correction. e Summarized genera abundance differences between patients who did or did not contract an infection during IC. Significance was determined by individual Mann–Whitney tests for the three different genera (*P < 0.05, **P < 0.01, ***P < 0.001)
Duration of antibiotic treatment is associated with temporal instability of the oral microbiome during IC in AML patients
Due to the fact that temporal instability of the oral and gastrointestinal microbiome is associated with infectious risk among AML patients, we sought to determine clinical factors associated with microbiome temporal instability. Multivariable regression analysis was performed using clinical variables previously implicated in microbiome stability, including age, antibiotic type and duration, and chemotherapy regimen [2, 7, 35–37]. Only total days on antibiotics was statistically correlated with temporal variability of the oral microbial diversity (SDI CV P = 0.031), community membership (unweighted UniFrac CV P < 0.001), and population structure (weighted UniFrac CV P = 0.002) measurements (Table 2; Additional file 1: Tables S1–S3). Interestingly, none of the clinical factors analyzed were significantly correlated with intra-patient temporal instability of the stool microbiome by multivariable regression analyses (Table 2; Additional file 1: Tables S4–6). Thus, our data suggest a site-specific effect of antimicrobials on temporal variability in the AML cohort.
Table 2.
Multivariable regression analyses of potential clinical factors associated with the intra-patient temporal instability of the oral and stool microbiomes of AML patients
| P value | ||||||
|---|---|---|---|---|---|---|
| Variables | Oral SDI CV | Oral UUCV | Oral WUCV | Stool SDI CV | Stool UUCV | Stool WUCV |
| Age | 0.287 | 0.864 | 0.529 | 0.425 | 0.779 | 0.885 |
| Received piperacillin/tazobactam >72 h | 0.475 | 0.208 | 0.175 | 0.507 | 0.215 | 0.973 |
| Received cefepime >72 h | 0.108 | 0.557 | 0.943 | 0.508 | 0.669 | 0.639 |
| Received carbapenem >72 h | 0.748 | 0.762 | 0.832 | 0.360 | 0.482 | 0.681 |
| Days on all antibioticsa | 0.031b | 0.0001b | 0.002b | 0.392 | 0.858 | 0.580 |
| Days on treatment antibiotics | 0.205 | 0.060 | 0.163 | 0.917 | 0.633 | 0.451 |
| Number of antibiotics received | 0.089 | 0.380 | 0.874 | 0.357 | 0.724 | 0.167 |
| Non-fludarabine high intensity chemotherapy | 0.723 | 0.581 | 0.935 | 0.415 | 0.605 | 0.456 |
| Hypomethylator-based chemotherapy | 0.734 | 0.712 | 0.181 | 0.244 | 0.900 | 0.612 |
a Includes prophylactic antibiotics
b Significant P values
UUCV unweighted UniFrac coefficient of variation, WUCV weighted UniFrac coefficient of variation
Prolonged antibiotic exposure is associated with long-term infectious outcomes among AML patients undergoing IC
To this point we had found that total antibiotic exposure was significantly associated with microbiome temporal instability in AML patients undergoing IC, and that microbiome instability could be associated with adverse infectious outcomes. Thus, we next sought to determine if increased antibiotic exposure was associated with long-term infectious outcomes in this cohort. Indeed, individuals that developed an infection in the 90 days post-neutrophil recovery had significantly lengthier exposure to treatment antibiotics (P = 0.02; Fig. 6a). When exposure to all antibiotics, to include prophylactic antimicrobials, was analyzed between infection and non-infection groups post-IC, there was a trend towards increasing infection rates among patients who had received longer duration of antimicrobials, although it was not statistically significant (P = 0.07; Fig. 6b).
Fig. 6.

Prolonged antibiotic exposure is associated with long-term infectious complications. Illustrated are patients who did or did not contract a microbiologically determined infection in the 90 days post-neutrophil recovery and their a days on treatment antibiotics or b days on all antibiotics to include prophylaxis. In both panels the bars represent mean ± standard deviation, and P values comparing antibiotic exposure among infectious outcomes were calculated using a two-sample t-test with Welch’s correction
Discussion
There is tremendous enthusiasm for using measurements and manipulation of the microbiome as a means to improve different aspects of human health. Indeed, multiple studies have shown that the microbiome can significantly impact a broad variety of pathophysiologic processes, from carcinogenesis to autoimmunity to serious infections [7, 9, 18, 24, 25, 38, 39]. As almost all of the existing datasets ascertaining longitudinal variability exist in healthy controls, the limited understanding of the variability in microbiota composition for sick patients impedes the capacity to readily translate microbiome measurements into the clinical setting. The urgent need to monitor and understand microbiome dysbiosis during critical illness has been expressed with recent initiatives such as the ICU Microbiome Project (http://americangut.org/the-icu-microbiome-project-is-there-a-better-way-to-treat-infections-than-antibiotics/), the National Microbiome Initiative (https://www.whitehouse.gov/the-press-office/2016/05/12/fact-sheet-announcing-national-microbiome-initiative), and the Center for Disease Control’s recent Broad Agency Announcement for Advanced and Innovative Solutions to Improve Public Health, which includes the request for microbiome assessment and intervention to address antibiotic resistance in both healthy individuals and in healthcare settings. Herein, we contribute to addressing this knowledge gap by analyzing the inter-patient variability of both the oral and stool microbiome for 59 AML patients using >800 samples collected over a median of 28 days, the factors driving differential variability in this cohort, and the association of inter-patient variability with clinical outcomes.
When considering how our cohort of leukemia patients compares to healthy individuals, we obtained, trimmed, and processed data from the Human Microbiome Project (HMP) [27] to match the V4 region of the 16S rRNA gene amplified by this study’s primers and processing protocols. Although a direct statistical comparison is not suitable due to differences between study methodologies, our mean SDI CV values for both oral and stool were two- to fourfold higher than both the HMP as well as a separate cohort of healthy subjects studied by Flores et al. [15, 16] (HMP mean SDI CV values were 0.2 for oral and 0.09 for stool, and mean SDI CV for stool and tongue samples for the Flores et al. data set was approximately 0.17 and 0.1, respectively), indicating that our cohort of treated AML patients had high intra-patient temporal variability of α-diversity compared to healthy subjects (Additional file 1: Figure S1c). A high level of intra-patient temporal variability found within our cohort is in concordance with observations of rapid fluctuations in microbiota composition reported in previous cohorts of intensive care unit and stem cell transplant patients [7, 11, 21].
Although the overall cohort had high levels of variability, there was a wide range, showing that many patients maintained a relatively stable microbiome despite the significant stress of AML therapy and a prolonged hospital stay. It has been previously demonstrated that specific enterotypes and the diversity of the microbiome influence the potential adverse impact of antibiotics on microbial communities [40]. In our cohort, variability was associated with the predominance of potentially pathogenic genera such as Staphylococcus whereas more stable microbiomes were characterized by high levels of commensals such as Akkermansia, Subdilogranulum, and Pseudobutyrivibrio (Figs. 2, 3, and 5). Akkermansia spp. have been described to be important in host metabolic homeostasis, anti-inflammatory functions, such as interleukin secretion, and promoting intestinal epithelial integrity in murine models and human colonic cell lines [38, 41]. Moreover, butyrate-producing commensal microorganisms, like Akkermansia and Pseudobutyrivibrio, are important in maintaining the health of the intestinal epithelium, which in turn provides nutrients necessary for microbiota stability [9, 38, 42]. However, Akkermansia spp. have also been recently associated with loss of the colonic mucus layer and compromised intestinal barrier function in mice with graft versus host disease [43]. More perplexing was the increased abundance of Lactobacillus and Lactococcus in the stool of variable individuals, as these organisms have often been associated with microbiome maintenance and mucosal integrity [39, 44–47]. These results suggest the importance of performing metagenomic shotgun sequencing in order to determine the specific species among these complex genera that are associated with maintenance and variability. Therefore, in combination with the aforementioned studies, our data appear to suggest that stable microbiomes with high levels of specific commensal microorganisms might be implicated in mechanisms protecting patients from intestinal domination and subsequent infection by pathogenic bacteria. These observations raise the question of whether fecal microbiota transplantation or targeted species repletion of patients with a microbiota dominated by pathogenic bacteria could restore intestinal homeostasis [8].
Interestingly, although antibiotic exposure is often associated with reduced stool microbial diversity [7, 35, 36], our results show total days on antibiotics was significantly correlated with temporal variability of the oral microbial diversity, but not the stool. These findings are in agreement with recent findings using generalized linear models which identified antibiotic use as a significant predictor of temporal variability of the tongue, but not the gut, in healthy subjects [15]. Given previous literature associating other clinical factors, such as the age and chemotherapy, with microbiome variability and dysbiosis, it was surprising that total antibiotic exposure to all antibiotics, including prophylaxis, was the only clinical variable tested to be statistically correlated with any measure of temporal variability [2, 37]. This suggests the very treatment applied to protect the patient appears to predispose the patient to recurrent infectious-related issues. Thus, greater efforts need to be taken towards antibiotic stewardship as well as tailoring antimicrobial treatments within the context of the microbiome.
Several limitations of our study bear mentioning. First, all of our patients had a single disease and were recruited from a single center, which means we do not know how these data represent the broader array of hospitalized patients. However, the relative homogeneity of the cohort was chosen in order to facilitate comparative analyses of the complex data generated in microbiome studies and to be able to characterize the microbiota prior to the patient becoming seriously ill. With this in mind, while the majority of samples were true baselines, a number of baseline samples, particularly fecal samples, were taken within the 24 h after initiation of chemotherapy due to sample availability. To our knowledge, there are no time-wise studies showing the effects of chemotherapy on the microbiome as the studies published to date comparing the microbiome before and after chemotherapy consider a wide range of days post-chemotherapy analyzed together [37]. Moreover, it is also known that the impact of chemotherapy on host factors is not typically observed until 7–10 days following chemotherapy initiation (e.g., neutropenia, mucositis, etc.). So, although we believe 24 h provides a reasonable window to collect baseline samples from patients urgently receiving chemotherapy, we cannot exclude the concern that chemotherapy could affect the microbiome within 24 h as this is currently unknown. Second, we used 16S rRNA analyses to determine microbial composition, limiting our classification of bacteria to the genus level. A species level analysis could be performed via shotgun metagenomic sequencing, which would also permit functional metabolomics studies that could help to elucidate mechanistic bases for our observations. However, applying such methodology to the very large number of samples in our study is currently cost prohibitive. Finally, although we had >800 microbiome measurements in our cohort, the complexity of the clinical course of our patients meant that we were limited in our ability to detect certain associations between clinical factors and microbiota variability. For example, most of our patients received various durations and combinations of antimicrobial administration that were problematic to reduce to discrete categories that could be incorporated into multivariable regression analyses. However, clinical studies of the microbiota in the acute care setting will face this same challenge which mandates large sample sizes and careful collection and analyses of both clinical and microbiome data.
Conclusions
We have provided the largest dataset to date quantifying the longitudinal variability of the oral and stool microbiome in ill, hospitalized patients. We have identified particular bacterial taxa that are positively and negatively correlated with microbiome instability and demonstrated that high temporal variability is associated with increased rates of infectious outcomes. The characterization of microbiome temporal fluctuations described herein contribute to the first steps towards advancing microbiome-based diagnostic and therapeutic interventions that can be applicable in a wide range of ailments such as cancer, critical illness, and other immune-compromised individuals. Our previous data revealed low baseline stool α-diversity was associated with infectious risk during IC while decreases in both the oral and stool α-diversity between baseline and last samples were associated with infection post-IC. [25]. These previous findings, combined with the data herein, indicate that both microbial diversity as well as intra-patient temporal variability have infectious implications when studying acutely ill patients. Our finding of high intra-patient variability having clinical implications in these patients signifies that making conclusions based on single microbiome measurements in ill patients is likely to be problematic and suggests that statistical mechanisms that capture both the composition and the overall trajectory of a patient’s microbiota during illness will likely be required to fully integrate microbiome measurements into the clinical arena.
Acknowledgements
We thank Lisa Marsh and Lily Carlin for assistance with sample collection, and any other clinical or nursing staff that assisted with patient enrollment or sample collection. We thank members of the Center for Metagenomics and Microbiome Research for processing and sequencing of samples, specifically Tulin Ayvaz.
Funding
This work was supported by The University of Texas MD Anderson Cancer Center AML MoonShot Knowledge Gap Project (to Dimitrios P. Kontoyiannis), and the Odyssey Program and CFP Foundation at The University of Texas MD Anderson Cancer Center (to Jessica Galloway-Peña). W. Duncan Wadsworth is supported by NIH grant NCI T32 CA096520 at Rice University. Supported by the NIH/NCI under award number P30CA016672 and used the Bioinformatics Shared Resource.
Availability of data and materials
The dataset supporting the conclusions of this article is available in the NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra). The datasets generated during the current study are available in the NCBI Sequence Read Archive under BioProject ID PRJNA352060. BioSample accession numbers for the data generated are listed in Additional file 1: Table S7.
Authors’ contributions
JGP: conceptualization, methodology, validation, formal analysis, investigation, data curation, writing (original draft), writing (review and editing), visualization, project administration, and funding acquisition. DPS: methodology, software, validation, formal analysis, resources, data curation, writing (review and editing), visualization, and project administration. PS: methodology, investigation, resources, and writing (review) and editing. WDW: methodology, software, validation, formal analysis, data curation, writing (review and editing), and visualization. BF: methodology, software, validation, formal analysis, data curation, writing (review and editing), and visualization. NJA: methodology, software, validation, formal analysis, resources, data curation, writing (review and editing), visualization, and project administration. EJS: writing (review and editing) and funding acquisition. NGD: investigation, resources, writing (review and editing), and visualization. MG: methodology, software, validation, formal analysis, data curation, writing (original draft), writing (review and editing), and visualization. JFP: software, validation, formal analysis, resources, data curation, writing (review and editing), visualization, and project administration. DPK: conceptualization, methodology, resources, writing (original draft), writing (review and editing), and funding acquisition. SAS: conceptualization, methodology, formal analysis, investigation, data curation, writing (review and editing), visualization, supervision, project administration, and funding acquisition. All authors read and approved the final manuscript.
Competing interests
Nadim J. Ajami and Joseph F. Petrosino are the Project Director and Founder/Chief Science Officer, respectively, of Diversgen. Dimitrios P. Kontoyiannis reports research support from Merck, Pfizer, and Astellas; service on the Merck Advisory Board; acting as a consultant for Astellas and F2G; and honoraria from Gilead, T2 Biosystems, and Mylan, Inc., during the course of the study. The other authors declare that they have no competing interests.
Consent for publication
All patient information was anonymized at source and unique ID codes were used to identify cases. Publication of de-identified results from all consenting participants was approved in the protocol below (MDACC IRB PA13-0339).
Ethics approval and consent to participate
The study protocol was approved by the MDACC Institutional Review Board (PA13-0339) and was conducted in compliance with the Declaration of Helsinki. Written informed consent was obtained from all participants before enrollment.
Abbreviations
- AML
Acute myelogenous leukemia
- CR
Complete remission
- CV
Coefficient of variation
- HMP
Human Microbiome Project
- IC
Induction chemotherapy
- MDACC
MD Anderson Cancer Center
- MDI
Microbiologically defined infection
- SDI
Shannon diversity index.
Additional file
Figure S1. The temporal variability of the oral and stool microbial α-diversity of the AML and HMP cohorts. Figure S2. No correlation between temporal stability of community structure and bacterial diversity found among treated acute myeloid leukemia patients. Figure S3. Taxonomic composition differences among different stability categories. Figure S4. Temporal instability of microbiome community structure is not associated with infection during chemotherapy. Figure S5. Temporal variability of the microbiome is not associated with chemotherapeutic response. Table S1. Multivariable regression analyses of potential clinical factors associated with the intra-patient temporal instability of the stool as measured by the coefficient of variation of the Shannon diversity index. Table S2. Multivariable regression analyses of potential clinical factors associated with the intra-patient temporal instability of the stool as measured by the coefficient of variation of the unweighted Unifrac distance. Table S3. Multivariable regression analyses of potential clinical factors associated with the intra-patient temporal instability of the stool as measured by the coefficient of variation of the weighted Unifrac distance. Table S4. Multivariable regression analyses of potential clinical factors associated with the intra-patient temporal instability of oral samples as measured by the coefficient of variation of the Shannon diversity index. Table S5. Multivariable regression analyses of potential clinical factors associated with the intra-patient temporal instability of oral samples as measured by the coefficient of variation of the unweighted Unifrac distance. Table S6. Multivariable regression analyses of potential clinical factors associated with the intra-patient temporal instability of oral samples as measured by the coefficient of variation of the weighted Unifrac distance. Table S7. Biosample accession numbers. (PDF 1329 kb)
References
- 1.Cameron SJ, Huws SA, Hegarty MJ, Smith DP, Mur LA. The human salivary microbiome exhibits temporal stability in bacterial diversity. FEMS Microbiol Ecol. 2015;91(9):fiv091. [DOI] [PubMed]
- 2.Claesson MJ, Cusack S, O’Sullivan O, Greene-Diniz R, de Weerd H, Flannery E, et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4586–4591. doi: 10.1073/pnas.1000097107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science. 2009;326(5960):1694–1697. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oh J, Byrd AL, Park M, Program NCS, Kong HH, Segre JA. Temporal stability of the human skin microbiome. Cell. 2016;165(4):854–866. doi: 10.1016/j.cell.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moya A, Ferrer M. Functional redundancy-induced stability of gut microbiota subjected to disturbance. Trends Microbiol. 2016;24(5):402–413. doi: 10.1016/j.tim.2016.02.002. [DOI] [PubMed] [Google Scholar]
- 6.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489(7415):220–230. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Halpin AL, de Man TJ, Kraft CS, Perry KA, Chan AW, Lieu S, et al. Intestinal microbiome disruption in patients in a long-term acute care hospital: a case for development of microbiome disruption indices to improve infection prevention. Am J Infect Control. 2016;44(7):830–836. doi: 10.1016/j.ajic.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laufer Halpin A, McDonald LC. The dawning of microbiome remediation for addressing antibiotic resistance. Clin Infect Dis. 2016;62(12):1487–1488. doi: 10.1093/cid/ciw187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lei YM, Nair L, Alegre ML. The interplay between the intestinal microbiota and the immune system. Clin Res Hepatol Gastroenterol. 2015;39(1):9–19. doi: 10.1016/j.clinre.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parfrey LW, Knight R. Spatial and temporal variability of the human microbiota. Clin Microbiol Infect. 2012;18(Suppl 4):8–11. doi: 10.1111/j.1469-0691.2012.03861.x. [DOI] [PubMed] [Google Scholar]
- 11.Zaborin A, Smith D, Garfield K, Quensen J, Shakhsheer B, Kade M, et al. Membership and behavior of ultra-low-diversity pathogen communities present in the gut of humans during prolonged critical illness. MBio. 2014;5(5):e01361–01314. doi: 10.1128/mBio.01361-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belstrom D, Holmstrup P, Bardow A, Kokaras A, Fiehn NE, Paster BJ. Temporal stability of the salivary microbiota in oral health. PLoS One. 2016;11(1):e0147472. doi: 10.1371/journal.pone.0147472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caporaso JG, Lauber CL, Costello EK, Berg-Lyons D, Gonzalez A, Stombaugh J, et al. Moving pictures of the human microbiome. Genome Biol. 2011;12(5):R50. doi: 10.1186/gb-2011-12-5-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Meij TG, Budding AE, de Groot EF, Jansen FM, Frank Kneepkens CM, Benninga MA, et al. Composition and stability of intestinal microbiota of healthy children within a Dutch population. FASEB J. 2016;30(4):1512–1522. doi: 10.1096/fj.15-278622. [DOI] [PubMed] [Google Scholar]
- 15.Flores GE, Caporaso JG, Henley JB, Rideout JR, Domogala D, Chase J, et al. Temporal variability is a personalized feature of the human microbiome. Genome Biol. 2014;15(12):531. doi: 10.1186/s13059-014-0531-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Human Microbiome Project Consortium Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Falony G, Joossens M, Vieira-Silva S, Wang J, Darzi Y, Faust K, et al. Population-level analysis of gut microbiome variation. Science. 2016;352(6285):560–564. doi: 10.1126/science.aad3503. [DOI] [PubMed] [Google Scholar]
- 18.Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. 2012;22(5):850–859. doi: 10.1101/gr.131029.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Angelberger S, Reinisch W, Makristathis A, Lichtenberger C, Dejaco C, Papay P, et al. Temporal bacterial community dynamics vary among ulcerative colitis patients after fecal microbiota transplantation. Am J Gastroenterol. 2013;108(10):1620–1630. doi: 10.1038/ajg.2013.257. [DOI] [PubMed] [Google Scholar]
- 20.Martinez C, Antolin M, Santos J, Torrejon A, Casellas F, Borruel N, et al. Unstable composition of the fecal microbiota in ulcerative colitis during clinical remission. Am J Gastroenterol. 2008;103(3):643–648. doi: 10.1111/j.1572-0241.2007.01592.x. [DOI] [PubMed] [Google Scholar]
- 21.Taur Y, Xavier JB, Lipuma L, Ubeda C, Goldberg J, Gobourne A, et al. Intestinal domination and the risk of bacteremia in patients undergoing allogeneic hematopoietic stem cell transplantation. Clin Infect Dis. 2012;55(7):905–914. doi: 10.1093/cid/cis580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jenq RR, Ubeda C, Taur Y, Menezes CC, Khanin R, Dudakov JA, et al. Regulation of intestinal inflammation by microbiota following allogeneic bone marrow transplantation. J Exp Med. 2012;209(5):903–911. doi: 10.1084/jem.20112408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Montassier E, Al-Ghalith GA, Ward T, Corvec S, Gastinne T, Potel G, et al. Pretreatment gut microbiome predicts chemotherapy-related bloodstream infection. Genome Med. 2016;8(1):49. doi: 10.1186/s13073-016-0301-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taur Y, Jenq RR, Perales MA, Littmann ER, Morjaria S, Ling L, et al. The effects of intestinal tract bacterial diversity on mortality following allogeneic hematopoietic stem cell transplantation. Blood. 2014;124(7):1174–1182. doi: 10.1182/blood-2014-02-554725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galloway-Pena JR, Smith DP, Sahasrabhojane P, Ajami NJ, Wadsworth WD, Daver NG, et al. The role of the gastrointestinal microbiome in infectious complications during induction chemotherapy for acute myeloid leukemia. Cancer. 2016;122(14):2186–2196. doi: 10.1002/cncr.30039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang JY, Antonopoulos DA, Kalra A, Tonelli A, Khalife WT, Schmidt TM, et al. Decreased diversity of the fecal microbiome in recurrent Clostridium difficile-associated diarrhea. J Infect Dis. 2008;197(3):435–438. doi: 10.1086/525047. [DOI] [PubMed] [Google Scholar]
- 27.Human Microbiome Project Consortium A framework for human microbiome research. Nature. 2012;486(7402):215–221. doi: 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41(Database issue):D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8(4):e61217. [DOI] [PMC free article] [PubMed]
- 30.Morgan XC, Huttenhower C. Human microbiome analysis. PLoS Comput Biol. 2012;8(12):e1002808. doi: 10.1371/journal.pcbi.1002808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nazha A, Kantarjian H, Ravandi F, Huang X, Choi S, Garcia-Manero G, et al. Clofarabine, idarubicin, and cytarabine (CIA) as frontline therapy for patients </=60 years with newly diagnosed acute myeloid leukemia. Am J Hematol. 2013;88(11):961–966. doi: 10.1002/ajh.23544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Montalban-Bravo G, Garcia-Manero G. Novel drugs for older patients with acute myeloid leukemia. Leukemia. 2015;29(4):760–769. doi: 10.1038/leu.2014.244. [DOI] [PubMed] [Google Scholar]
- 33.Cheson BD, Bennett JM, Kopecky KJ, Buchner T, Willman CL, Estey EH, et al. Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol. 2003;21(24):4642–4649. doi: 10.1200/JCO.2003.04.036. [DOI] [PubMed] [Google Scholar]
- 34.The Integrative Human Microbiome Project Dynamic analysis of microbiome-host omics profiles during periods of human health and disease. Cell Host Microbe. 2014;16(3):276–289. doi: 10.1016/j.chom.2014.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4554–4561. doi: 10.1073/pnas.1000087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Modi SR, Collins JJ, Relman DA. Antibiotics and the gut microbiota. J Clin Invest. 2014;124(10):4212–4218. doi: 10.1172/JCI72333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Montassier E, Gastinne T, Vangay P, Al-Ghalith GA, Bruley des Varannes S, Massart S, et al. Chemotherapy-driven dysbiosis in the intestinal microbiome. Aliment Pharmacol Ther. 2015;42(5):515–528. doi: 10.1111/apt.13302. [DOI] [PubMed] [Google Scholar]
- 38.Derrien M, Belzer C, de Vos WM. Akkermansia muciniphila and its role in regulating host functions. Microb Pathog. 2016. doi: 10.1016/j.micpath.2016.02.005. http://www.sciencedirect.com/science/article/pii/S0882401015301789. [Epub ahead of print]. [DOI] [PubMed]
- 39.Kozakova H, Schwarzer M, Tuckova L, Srutkova D, Czarnowska E, Rosiak I, et al. Colonization of germ-free mice with a mixture of three Lactobacillus strains enhances the integrity of gut mucosa and ameliorates allergic sensitization. Cell Mol Immunol. 2016;13(2):251–262. doi: 10.1038/cmi.2015.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raymond F, Ouameur AA, Deraspe M, Iqbal N, Gingras H, Dridi B, et al. The initial state of the human gut microbiome determines its reshaping by antibiotics. ISME J. 2016;10(3):707–720. doi: 10.1038/ismej.2015.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reunanen J, Kainulainen V, Huuskonen L, Ottman N, Belzer C, Huhtinen H, et al. Akkermansia muciniphila adheres to enterocytes and strengthens the integrity of the epithelial cell layer. Appl Environ Microbiol. 2015;81(11):3655–3662. doi: 10.1128/AEM.04050-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mathewson ND, Jenq R, Mathew AV, Koenigsknecht M, Hanash A, Toubai T, et al. Gut microbiome-derived metabolites modulate intestinal epithelial cell damage and mitigate graft-versus-host disease. Nat Immunol. 2016;17(5):505–513. doi: 10.1038/ni.3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shono Y, Docampo MD, Peled JU, Perobelli SM, Velardi E, Tsai JJ, et al. Increased GVHD-related mortality with broad-spectrum antibiotic use after allogeneic hematopoietic stem cell transplantation in human patients and mice. Sci Transl Med. 2016;8(339):339ra371. [DOI] [PMC free article] [PubMed]
- 44.Lam EK, Tai EK, Koo MW, Wong HP, Wu WK, Yu L, et al. Enhancement of gastric mucosal integrity by Lactobacillus rhamnosus GG. Life Sci. 2007;80(23):2128–2136. doi: 10.1016/j.lfs.2007.03.018. [DOI] [PubMed] [Google Scholar]
- 45.Yu Q, Yuan L, Deng J, Yang Q. Lactobacillus protects the integrity of intestinal epithelial barrier damaged by pathogenic bacteria. Front Cell Infect Microbiol. 2015;5:26. doi: 10.3389/fcimb.2015.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bernbom N, Licht TR, Brogren CH, Jelle B, Johansen AH, Badiola I, et al. Effects of Lactococcus lactis on composition of intestinal microbiota: role of nisin. Appl Environ Microbiol. 2006;72(1):239–244. doi: 10.1128/AEM.72.1.239-244.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Araos R, Tai AK, Snyder GM, Blaser MJ, D’Agata EM. Predominance of Lactobacillus spp. among patients who do not acquire multidrug-resistant organisms. Clin Infect Dis. 2016;63(7):937–943. doi: 10.1093/cid/ciw426. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The dataset supporting the conclusions of this article is available in the NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra). The datasets generated during the current study are available in the NCBI Sequence Read Archive under BioProject ID PRJNA352060. BioSample accession numbers for the data generated are listed in Additional file 1: Table S7.