

ABSTRACT
Recently, Rhizobium etli, in addition to Agrobacterium spp., has emerged as a prokaryotic species whose genome encodes a functional machinery for DNA transfer to plant cells. To understand this R. etli-mediated genetic transformation, it would be useful to define how its vir genes respond to the host plants. Here, we explored the transcriptional activation of the vir genes contained on the R. etli p42a plasmid. Using a reporter construct harboring lacZ under the control of the R. etli virE promoter, we show that the signal phenolic molecule acetosyringone (AS) induces R. etli vir gene expression both in an R. etli background and in an Agrobacterium tumefaciens background. Furthermore, in both bacterial backgrounds, the p42a plasmid also promoted plant genetic transformation with a reporter transfer DNA (T-DNA). Importantly, the R. etli vir genes were transcriptionally activated by AS in a bacterial species-specific fashion in regard to the VirA/VirG signal sensor system, and this activation was induced by signals from the natural host species of this bacterium but not from nonhost plants. The early kinetics of transcriptional activation of the major vir genes of R. etli also revealed several features distinct from those known for A. tumefaciens: the expression of the virG gene reached saturation relatively quickly, and virB2, which in R. etli is located outside the virB operon, was expressed only at low levels and did not respond to AS. These differences in vir gene transcription may contribute to the lower efficiency of T-DNA transfer of R. etli p42a than of T-DNA transfer of pTiC58 of A. tumefaciens.
IMPORTANCE The region encoding homologs of Agrobacterium tumefaciens virulence genes in the Rhizobium etli CE3 p42a plasmid was the first endogenous virulence system encoded by the genome of a non-Agrobacterium species demonstrated to be functional in DNA transfer and stable integration into the plant cell genome. In this study, we explored the transcriptional regulation and induction of virulence genes in R. etli and show similarities to and differences from those of their A. tumefaciens counterparts, contributing to an understanding and a comparison of these two systems. Whereas most vir genes in R. etli follow an induction pattern similar to that of A. tumefaciens vir genes, a few significant differences may at least in part explain the variations in T-DNA transfer efficiency.
KEYWORDS: Rhizobium etli, plant genetic transformation, virulence genes
INTRODUCTION
Agrobacterium tumefaciens genetically transforms the host plants by transferring and integrating a segment of its own DNA into the genome of its host cells (1, 2). This virulence is conferred by a large plasmid (Ti plasmid) that harbors both the genes required for the DNA transfer, i.e., the virulence (vir) genes, and the DNA sequence to be transferred (transfer DNA [T-DNA]). Whereas in nature A. tumefaciens transfers specific genes that cause plant cell proliferation and the production of opines, i.e., small amino acid derivatives used as sources of carbon and nitrogen by the bacteria (3), the DNA transfer mechanism per se is not sequence specific, making A. tumefaciens the most widely used vector for plant genetic transformation (4). Traditionally, the ability of A. tumefaciens to mediate the bacterium-to-eukaryote DNA transfer has been considered unique in the living world. Indeed, other bacterial species within the Rhizobiales could acquire this ability only if they are provided with the vir region from a virulent strain of A. tumefaciens. For example, several species of Rhizobium, Sinorhizobium, and Mesorhizobium (5–7) and, later, Ensifer adhaerens (Ensifer and Sinorhizobium, in fact, represent the same genus) (8, 9) have been shown to become capable of plant genetic transformation when provided with the A. tumefaciens Ti plasmid functions. Only very recently, we demonstrated that another bacterial species, Rhizobium etli strain CE3, known to be a symbiotic plant-associated bacterium able to fix nitrogen like many other members of the Rhizobiales family, has evolved its own functional bacterium-to-plant DNA transfer machinery (10). Although R. etli can potentially be associated with different host plant species, this bacterium represents the predominant species nodulating common beans (Phaseolus vulgaris), particularly in its region of origin, Latin America (11). Specifically, a region of R. etli plasmid p42a contains genes with extensive homology to all the essential vir genes of A. tumefaciens. Complete genome sequencing of R. etli CFN42 revealed that it harbors six plasmids, designated p42a to p42f, with different functions. Among these plasmids, p42a is a 194-kb self-conjugative plasmid containing a segment of 30 kb that carries genes involved in conjugative transfer (12), similar to the A. tumefaciens Ti plasmid. The similarity between these two plasmids, however, is restricted to the vir region, and no T-DNA-like sequences have been detected in R. etli (10, 12). The activity of the p42a vir region does not appear to affect symbiosis directly, as normal nodulation was observed with mutants with mutations in the virG and virE2 genes (13); however, p42a is involved in the conjugative transfer of the symbiotic plasmid p42d/pSym (14). When supplied with a binary plasmid that contains a T-DNA sequence but not the vir region, R. etli was able to genetically transform plant cells (10). In A. tumefaciens, vir genes are transcriptionally activated in response to signaling molecules, such as the phenolic compound acetosyringone (AS) or small phenolics contained in plant exudates (15, 16). Here, we investigated whether the vir genes of R. etli also are induced by similar signals and examined potential similarities and differences in transcriptional activation of the virulence systems encoded by R. etli and A. tumefaciens.
RESULTS
Sequence analysis of vir promoter regions in R. etli p42a.
We analyzed the sequences of the vir genes and operons contained within the R. etli p42a plasmid (Table 1) for the presence of vir box motifs in the promoter regions. These motifs, first identified in both octopine and nopaline strains A. tumefaciens (17, 18), comprise a short sequence of 10 to 12 nucleotides with a loose consensus sequence of 5′-dPu(T/A)TDCAATTGHAAPy (where dPu is a deoxypurine; H is A, C, or T; D is A, G, or T; and Py is a pyrimidine) (19) and are found in promoters of vir operons, usually between 50 and 200 bp upstream of the translation initiation codon of the first gene of each vir operon. The vir boxes are directly recognized by the transcriptional activator VirG, which binds to their sequences (19, 20). We performed a manual search for conserved motifs in the promoter regions of R. etli vir operons (Table 1). Putative vir boxes were found for all vir operons of R. etli on the basis of the comparison with their closest counterparts identified in A. tumefaciens (17). Similar to the vir box sequences in different Agrobacterium strains, the identified vir boxes of R. etli are not strongly conserved, but each vir operon in the p42a plasmid contains at least one vir box homolog.
TABLE 1.
Sequence homologs of the A. tumefaciens vir box in vir promoter regions of R. etli/p42a
|
R. etli CFN42 |
A. tumefaciens |
No. of nucleotide hits/total no. of nucleotides | ||
|---|---|---|---|---|
| Operon | Sequence (position)a | Operon | Sequence (position)b | |
| virB | AAAAATCGAAAA (−102) | virB | AGCAATTGAAAA* (−110) | 9/12 |
| virG | TAAAATTGAAAT (−78) | 9/12 | ||
| virD | CGAATTCAAAAT (−415) | virC | CGAATTTGAAAT* (−116) | 10/12 |
| TTAATTTGCAAG (−188) | virD | TTAAATTGCAAT (−151) | 10/12 | |
| AGCAGTTCAATG (−178) | virB | AGCAATTGAAAA* (−110) | 9/12 | |
| AATGATTGCGAT (−143) | virG | AACGATTGAGAA (−99) | 9/12 | |
| TGCGATTGTAAT (−137) | virD | TTCAATTGTAAT (−141) | 10/12 | |
| ATAAACTGAAAT (−122) | virB | TTCAATTGAAAT* (−130) | 9/12 | |
| virB | TTCAAATGAAAT (−132) | 9/12 | ||
| ATAAACTGAAAT (−123) | virC | TA-AAATTGAAAT (−117) | 11/12 | |
| virE | TACATA-TGAAAC (−126) | 10/12 | ||
| TAAACTGAAATT (−125) | virC | TACAATAAAATT (−122) | 9/12 | |
| TATAATATTGAT (−102) | virD | TATAATTTCAAT (−147) | 9/12 | |
| virE | TTCAGATGAAGC (−436) | virB | TTCAAATGAAAT (−132) | 9/12 |
| TTCACTTCAATT (−376) | virA | TTCACTTGAAAC (−117) | 9/12 | |
| virD | TTCAATTTTATT (−163) | 10/12 | ||
| virB2 | TTCGCTTCAAAT (−537) | virA | TTCACTTGAAAC (−117) | 10/12 |
| virB | TTCAATTGAAAT* (−130) | 10/12 | ||
| TGAGATTGAAAT (−526) | virC | CGAATTTGAAAT* (−116) | 9/12 | |
| virC | TAAAATTGAAAT (−117) | 10/12 | ||
| AGAAATCGAAAA (−512) | virB | AGCAATTGAAAA* (−110) | 10/12 | |
| virF1 | TGTGATTGAAAG (−33) | virE | TTCAATTGAAAT* (−130) | 10/12 |
| virF2 | ACCATTTGAAAG (−210) | virB | AGCAATTGAAAA* (−110) | 9/12 |
| virA | TTCATTTGAAAC (−117) | 9/12 | ||
| virC | TTCAAAGGGATT (−742) | virB | TTCAAATGAAAT (−132) | 9/12 |
| AACAAGTGCTAC (−613) | virC | TATAATTGCTAC* (−147) | 10/12 | |
| CCATCTTGAAAT (−460) | virC | CGAATTTGAAAT* (−116) | 11/12 | |
| AAATATAAAATT (−109) | virC | TACAATAAAATT (−122) | 10/12 | |
| TTCGATTAATAG (−99) | virB | TTCAATTGAAAT* (−130) | 9/12 | |
| virG | TATGTTTGTAAC (−59) | virD | TTTTATTGTAAT (−158) | 10/12 |
| virE | TACATATGAAAC (−126) | 10/12 | ||
Consensus bases of the vir box sequences (19) are indicated by boldface. Position coordinates are relative to the position of the annotated translation initiation codons.
Sequences that have been validated experimentally are indicated by asterisks.
Activation of the R. etli p42a virE promoter and T-DNA transfer capability in R. etli and A. tumefaciens backgrounds.
To assess and compare the activation of a vir promoter from the R. etli p42a origin in the R. etli and A. tumefaciens backgrounds, we designed a construct, designated pREp-lacZ (Table 2), that harbors the β-galactosidase reporter gene under the control of the promoter region of the virE operon of R. etli p42a, which includes the sequence ca. 500 bp upstream of the translation initiation codon of the virE1 gene. R. etli and A. tumefaciens strains transformed with this reporter plasmid were then used to measure promoter activity in response to challenge with AS (Fig. 1) under the conditions known to favor AS induction in A. tumefaciens, i.e., a low pH, a low phosphate concentration, and the presence of glucose (21). In R. etli harboring the wild-type p42a plasmid, the β-galactosidase activity increased significantly upon treatment with 50 and 100 μM AS, whereas the basal expression level was observed in the absence of AS. In contrast, the expression levels remained very low in the R. etli ΔvirG mutant strain, showing that activation of the virE promoter depends on the VirA/VirG two-component system. Furthermore, in the absence of AS, the R. etli ΔvirG mutant exhibited lower reporter expression levels than the wild-type R. etli strain (Fig. 1), suggesting that the basal, uninduced level of vir gene expression in R. etli also requires VirG. As expected, when the pREp-lacZ reporter construct was introduced into the A. tumefaciens C58 strain cured of its Ti plasmid (C58NT1), only residual β-galactosidase activity was observed (Fig. 1). However, when C58NT1 was cotransformed with p42a and pREp-lacZ, AS induced reporter expression to levels similar to those observed in the wild-type R. etli background (Fig. 1). Thus, the vir gene induction machinery encoded by the p42a plasmid remains functional in the A. tumefaciens background.
TABLE 2.
Plasmids and bacterial strains used in this study
| Plasmid or bacterial strain | Description | Source and/or reference |
|---|---|---|
| Plasmids | ||
| pBp | For gene expression in A. tumefaciens | This study |
| pE2431 | For gene expression in A. tumefaciens from the A. tumefaciens virB promoter | S. B. Gelvin (Purdue University) |
| pREp-lacZ | For lacZ expression in A. tumefaciens and R. etli from the R. etli virE promoter | This study |
| pAEp-lacZ | For lacZ expression in A. tumefaciens and R. etli from the A. tumefaciens C58 virE promoter | This study |
| pREp-virB2 | For R. etli virB2 expression in R. etli from the R. etli virE promoter | This study |
| pE2431-RevirG | For R. etli virG expression in A. tumefaciens and R. etli from the R. etli virG promoter | This study |
| p302T-GFP | Binary plasmid with a GFP expression cassette in its T-DNA | 41 |
| p42a Specr | R. etli CFN42 p42a plasmid with a spectinomycin resistance cassette inserted in the RHE_PA00165 locus | S. Brom (University of Mexico) (35) |
| Strains | ||
| CE3 | R. etli with wild-type p42a plasmid | R. Carlson (University of Georgia) |
| CFN42 ΔvirG | R. etli ΔvirG mutant | J. Handelsman (Yale University) (13) |
| C58 | A. tumefaciens strain with wild-type pTiC58 plasmid | Lab collection |
| C58NT1 | A. tumefaciens strain C58 with no Ti plasmid | Lab collection |
FIG 1.
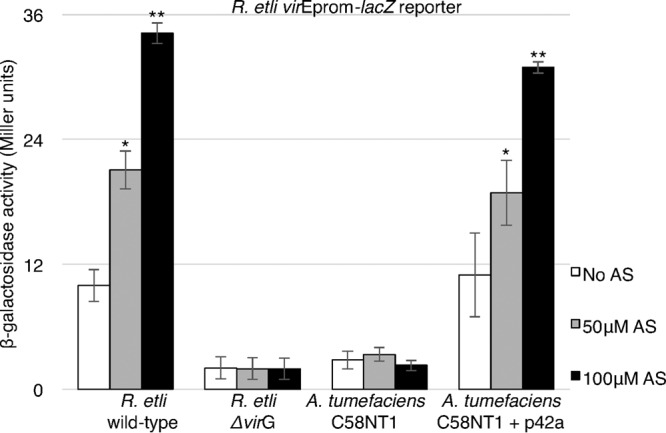
AS-dependent induction of p42a virE promoter activity in the R. etli and A. tumefaciens backgrounds. Wild-type R. etli carrying p42a, R. etli ΔvirG, A. tumefaciens C58NT1, and A. tumefaciens C58NT1 carrying the p42a-Specr plasmid were transformed with the pREp-lacZ reporter plasmid (R. etli virEprom-lacZ). An assay measuring β-galactosidase activity was used to measure virE promoter activity upon induction by the indicated concentrations of AS. All data represent average values from three independent experiments with the indicated standard deviations. Asterisks indicate the statistical significance (*, P < 0.05; **, P < 0.01) of the difference between data from noninduced control and AS-induced cells within each experiment, as determined by the two-tailed Student's t test.
Next, we examined whether the vir region of the p42a plasmid is fully functional, i.e., able to mediate T-DNA transfer to host cells, in A. tumefaciens. To this end, we introduced both p42a and p302T-GFP, a binary reporter construct with a green fluorescent protein (GFP) expression cassette in its T-DNA, into the disarmed C58NT1 strain (Table 2). When the resulting cultures were infiltrated into Nicotiana benthamiana leaves, multiple GFP-expressing plant cells were observed (Fig. 2), indicating transient genetic transformation and, consequently, the ability of the R. etli p42a vir region to mediate T-DNA transfer from the A. tumefaciens background. In positive-control experiments, infiltration of N. benthamiana leaves with A. tumefaciens strain C58, carrying the wild-type Ti plasmid pTiC58 and the p302T-GFP reporter construct, resulted in high levels of transformation, whereas in negative-control experiments, no GFP expression was observed after infiltration with C58NT1 carrying p302T-GFP but no Ti plasmid (Fig. 2A). Quantification of the transient transformation efficiency indicated that A. tumefaciens carrying its native, wild-type Ti plasmid was approximately 10 times more efficient than A. tumefaciens carrying the R. etli p42a plasmid (Fig. 2B).
FIG 2.
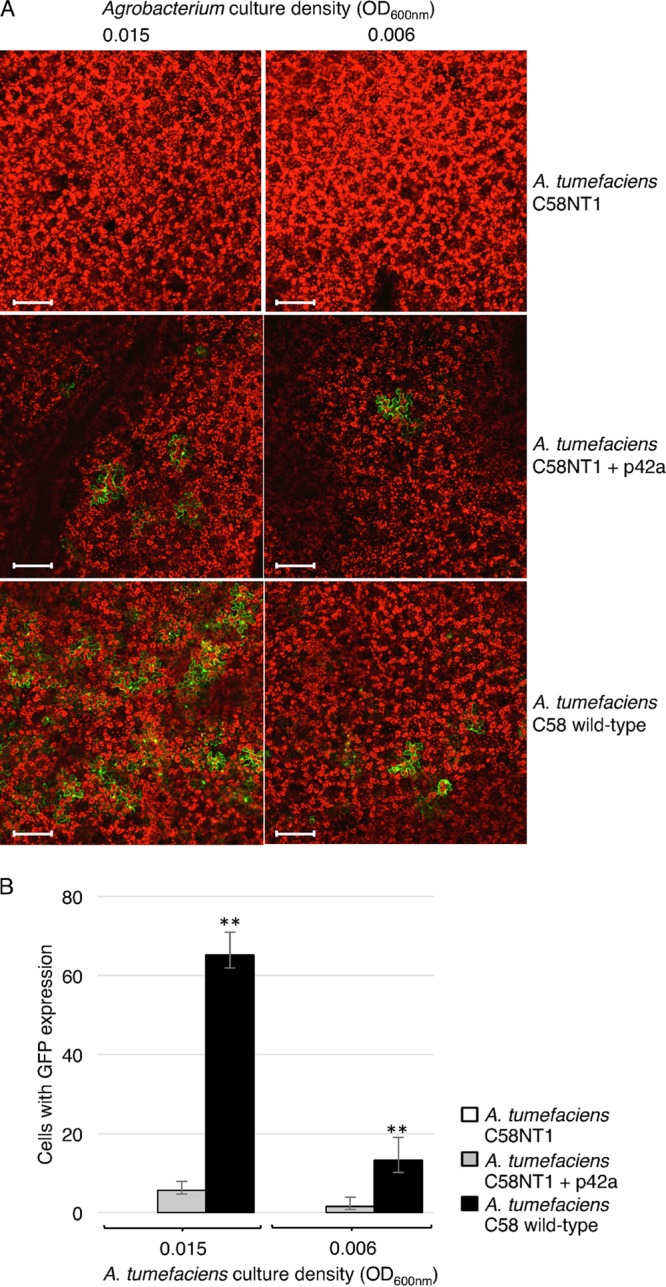
Comparison between T-DNA transfer efficiencies mediated by vir genes from R. etli/p42a or A. tumefaciens C58/pTiC58 in the A. tumefaciens background. N. benthamiana leaves were infiltrated with A. tumefaciens C58NT1, A. tumefaciens C58NT1 carrying R. etli p42a, and wild-type A. tumefaciens strain C58 carrying pTiC58. All bacterial strains also harbored the pCB302T-GFP reporter plasmid. (A) Representative confocal images of the infiltrated leaf areas. Green, GFP; red, plastid autofluorescence. All images are single confocal sections. Bars = 100 μm. (B) Efficiency of T-DNA transfer estimated by scoring the number of GFP-expressing cells per leaf surface area. All data represent average values from three independent experiments with the indicated standard deviations. Asterisks indicate the statistical significance (**, P < 0.01) of the difference between experimental and control data, as determined by the two-tailed Student's t test.
R. etli VirG is required for R. etli virE promoter activation.
To explore further the compatibility between induction of the R. etli virE promoter and the A. tumefaciens VirA/VirG vir gene induction system, we utilized the pREp-lacZ construct, which expresses the lacZ reporter from the R. etli virE promoter (Table 2). Unexpectedly, when the pREp-lacZ reporter construct was introduced into the A. tumefaciens C58 strain, only a very weak expression of the β-galactosidase reporter activity, which was not inducible by AS, was observed (Fig. 3A). These data suggest that the R. etli virE promoter cannot be induced by the A. tumefaciens VirG protein and that the induction of the R. etli vir genes most likely is species specific in regard to the VirA/VirG system. As a positive control, substantial and AS-dependent induction of the lacZ reporter gene was observed in wild-type R. etli cells containing pREp-lacZ (Fig. 3A). Because VirG functions as a transcriptional activator that directly binds vir promoters, we reasoned that it is the native VirG protein of R. etli that the R. etli virE promoter requires for activation. To test this hypothesis, we generated a plasmid, designated pE2431-RevirG (Table 2), which expresses the R. etli virG gene from its own promoter, and introduced it into wild-type A. tumefaciens strain C58 together with the pREp-lacZ reporter construct. Figure 3A shows that the AS-inducible expression of the R. etli virE promoter activity was restored to levels similar to those observed with the p42a plasmid.
FIG 3.
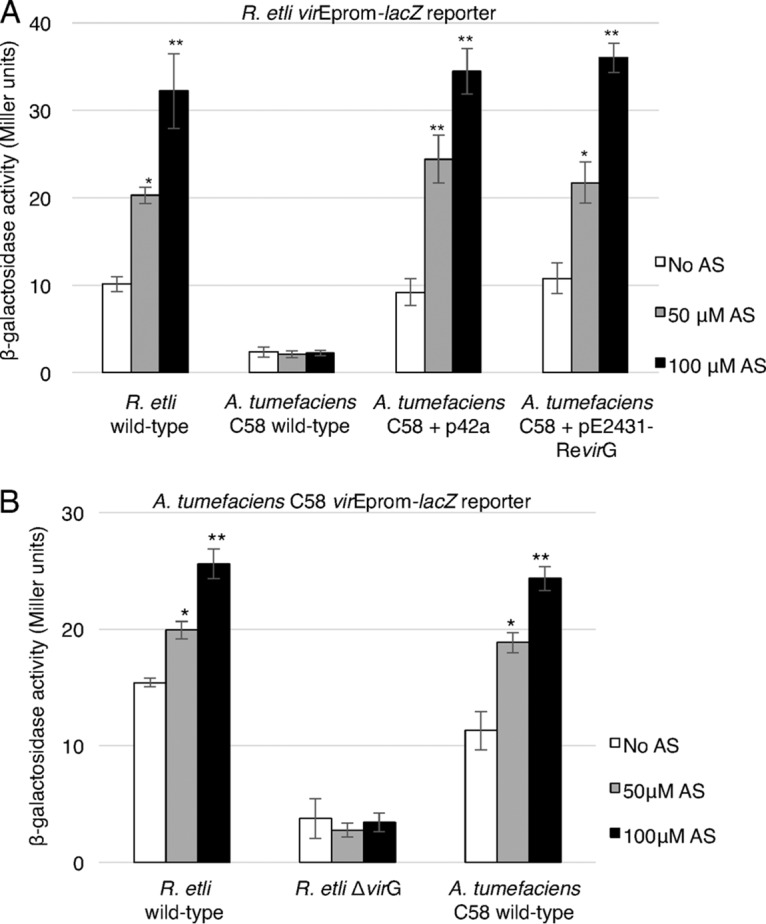
R. etli VirG is required for activation of the R. etli virE promoter. (A) Expression of the lacZ reporter under the control of the R. etli virE promoter (R. etli virEprom-lacZ) in wild-type R. etli carrying p42a, wild-type A. tumefaciens strain C58 carrying pTiC58, A. tumefaciens C58 carrying pTiC58 and the R. etli p42a plasmid, and A. tumefaciens C58 carrying pTiC58 and the R. etli virG expression construct pE2431-RevirG. (B) Expression of the lacZ reporter under the control of the A. tumefaciens C58 virE promoter (A. tumefaciens C58 virEprom-lacZ reporter) in wild-type R. etli, R. etli ΔvirG, and A. tumefaciens C58 carrying wild-type pTiC58. Reporter expression was induced by the indicated concentrations of AS, and the level of expression was determined by measuring β-galactosidase activity. All data represent average values from three independent experiments with the indicated standard deviations. Asterisks indicate the statistical significance (*, P < 0.05; **, P < 0.01) of the difference between the experimental data for noninduced control and AS-induced cells, as determined by the two-tailed Student's t test.
Next, we examined a reciprocal scenario, i.e., whether R. etli VirG can activate the A. tumefaciens virE promoter. Figure 3B shows that when the pAEp-lacZ construct that expresses the lacZ reporter from the A. tumefaciens C58 virE promoter (Table 2) was introduced into the wild-type R. etli strain, significant levels of AS-dependent induction of the lacZ reporter were observed. This induction depended on the presence of the R. etli virG gene because it did not occur in the R. etli ΔvirG mutant cells (Fig. 3B). Similar levels of induction by increasing concentrations of AS were achieved by the native A. tumefaciens VirG protein when pAEp-lacZ was introduced into the wild-type A. tumefaciens C58 strain (Fig. 3B).
Activation of the R. etli virE promoter by extracts from host and nonhost plant species.
Besides AS, plants secrete numerous other phenolic compounds that can activate the VirA/VirG two-component system and, thus, vir gene expression (22, 23). Because A. tumefaciens and R. etli differ in their host range, it was interesting to investigate how the virulence induction system of R. etli responded to vir gene-inducing phenolics contained in extracts from host and nonhost plant species. For the host species, we selected Phaseolus vulgaris (the common bean), and for the nonhost species, we utilized Glycine max (soybean) and Nicotiana tabacum (tobacco). R. etli cells containing p42a and the pREp-lacZ reporter construct were challenged with concentrated ethanolic extracts of the roots and shoots of these plants. Among these plant species, only extracts from P. vulgaris displayed a significant ability to induce the virE promoter; the highest levels of this induction were obtained with the extract of plant shoots, and they were comparable to the induction levels achieved with AS. Roots of P. vulgaris had modest but significant vir gene-inducing activity, whereas both the roots and shoots of nonhost plants had little or no vir gene-inducing activity (Fig. 4).
FIG 4.
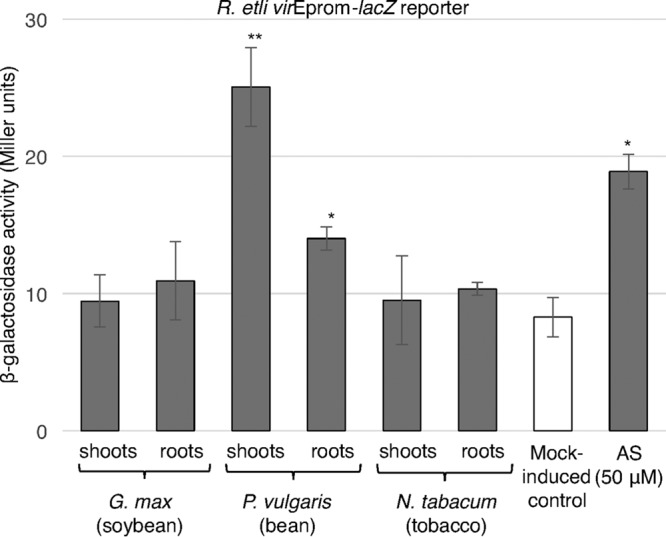
Induction of R. etli virE promoter activity by extracts from host and nonhost plant species. The level of expression of the lacZ reporter under the control of the R. etli virE promoter (R. etli virEprom-lacZ) was determined by measuring the β-galactosidase activity in the cell culture of wild-type R. etli carrying p42a supplemented with identical volumes of ethanol (mock-induced control), AS, and ethanolic extracts of the roots and shoots of plantlets from the indicated species. All data represent average values from three independent experiments with the indicated standard deviations. Asterisks indicate the statistical significance (*, P < 0.05; **, P < 0.01) of the difference between the experimental data for noninduced control and AS-induced cells, as determined by the two-tailed Student's t test.
Kinetics of early transcriptional activation of different vir genes of R. etli.
Next, we expanded our analysis of transcriptional activation to additional R. etli vir genes representing four major vir operons. Specifically, we measured the kinetics of vir gene expression directly by reverse transcription (RT)-quantitative real-time PCR (qPCR) amplification of the specific transcripts of the virB4, virB5, virD2, virE2, virG, and virB2 genes (see Table S1 in the supplemental material) at 6, 16, and 24 h following the beginning of treatment with AS. The virB4, virB5, virD2, and virE2 genes exhibited similar patterns of transcriptional activation by AS, ranging from a 2-fold to a 4-fold increase at 6 h of AS treatment to a 8-fold to 12-fold increase after 24 h of AS treatment (Fig. 5A to D). This is unlike the transcription of virG, which, having increased ca. 4-fold after 6 h of AS treatment, remained relatively stable thereafter, still displaying a 5-fold increase after 24 h of treatment (Fig. 5E). Thus, the temporal pattern of transcriptional regulation of the R. etli AS-sensing VirA/VirG two-component system most likely differs from that of other major vir operons.
FIG 5.
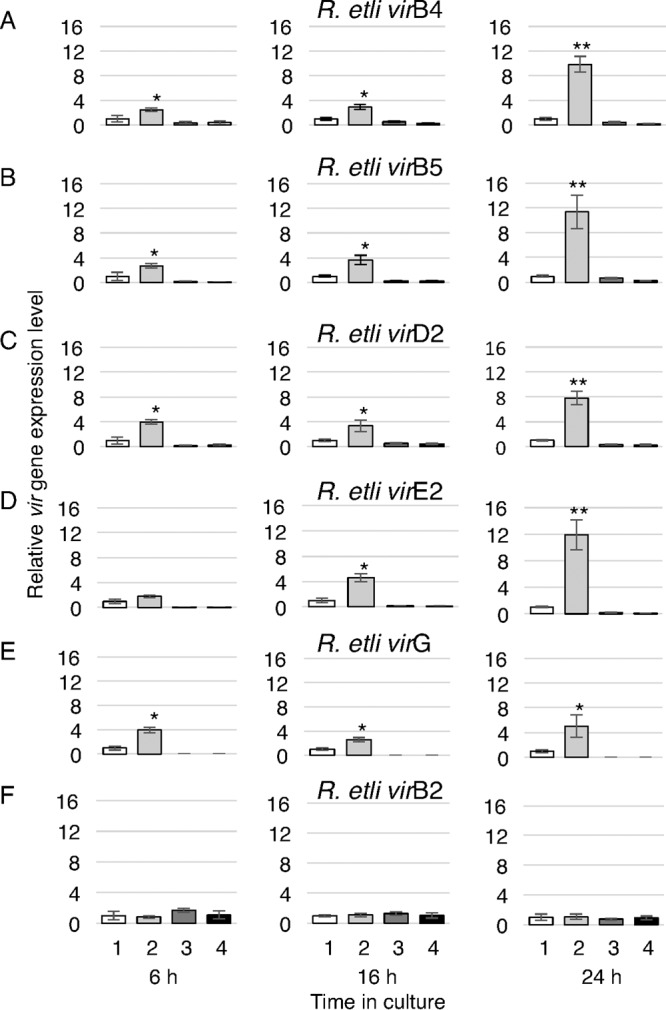
RT-qPCR analysis of early activation of R. etli vir gene transcription. Relative transcript levels were determined for the R. etli virB4 (A), virB5 (B), virD2 (C), virE2 (D), virG (E), and virB2 genes (F) after 6, 16, and 24 h of growth of the following bacterial cultures: wild-type R. etli without AS induction (bars labeled 1), wild-type R. etli with 50 μM AS induction (bars labeled 2), R. etli ΔvirG without AS induction (bars labeled 3), and R. etli ΔvirG with 50 μM AS induction (bars labeled 4). All data represent average values from two independent biological replicates and three technical replicates with the indicated standard deviations. Asterisks indicate the statistical significance (*, P < 0.05; **, P < 0.01) of the difference between the experimental data for noninduced control and AS-induced cells, as determined by the two-tailed Student's t test.
One of the R. etli vir genes, virB2 is located separately from the other virB genes in a discrete locus on the p42a plasmid, unlike its A. tumefaciens homolog, which is a part of the virB operon. Consistent with this positional uniqueness, R. etli virB2 did not respond to the presence of AS, showing no transcriptional activation even 24 h of treatment with AS (Fig. 5F).
Next, we examined whether this weak and uninducible expression of virB2 might contribute to the lower efficiency of transient genetic transformation of plants mediated by the R. etli p42a plasmid (Fig. 2) (10). We produced a pREp-virB2 plasmid that expresses R. etli virB2 from the R. etli virE promoter (Table 2), which is inducible by AS, and introduced it into the wild-type R. etli cells carrying p42a together with the p302T-GFP reporter plasmid. Quantification of the transient expression of GFP detected no differences in this transient transformation efficiency between wild-type R. etli and the R. etli strain that expresses the inducible virB2 (Fig. 6).
FIG 6.
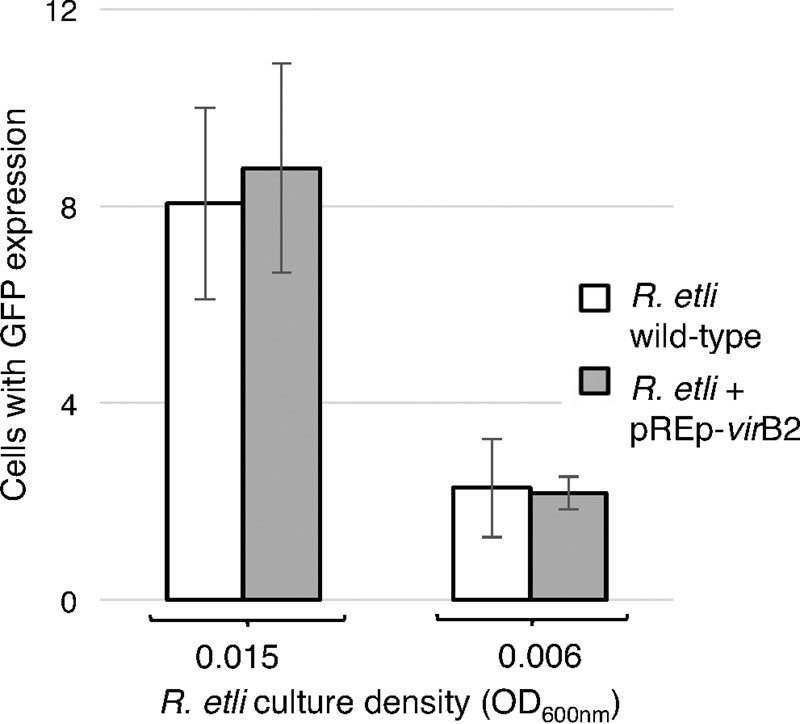
Effect of enhanced expression of virB2 on efficiencies of T-DNA transfer from R. etli CFN42. N. benthamiana leaves were infiltrated with wild-type R. etli carrying p42a and R. etli carrying the pREp-virB2 construct, which expresses R. etli virB2 under the control of the R. etli virE promoter. Both bacterial strains also harbored the pCB302T-GFP reporter plasmid. The efficiency of T-DNA transfer was estimated by scoring the number of GFP-expressing cells per leaf surface area. All data represent average values from three independent experiments with the indicated standard deviations.
DISCUSSION
Recently, R. etli has emerged as only the second known prokaryote, in addition to Agrobacterium spp., that encodes a functional protein machinery for prokaryote-to-eukaryote DNA transfer. Here, we demonstrate that this virulence machinery, the vir genes, is transcriptionally activated by AS, a paradigm of plant phenolic signals that activate the vir genes of A. tumefaciens. Furthermore, also similar to the A. tumefaciens vir genes, the AS-induced expression of the R. etli vir genes required the presence of the VirG constituent of the VirA/VirG two-component system.
One interesting aspect of R. etli vir gene induction is its requirement for the native vir gene transcriptional activator VirG, such that the R. etli VirG but not the A. tumefaciens VirG can activate the R. etli vir promoters in the heterologous background of A. tumefaciens cells. Previously, similar compatible interactions between VirA/VirG and the regulatory sequences of other vir genes have been reported for different A. tumefaciens strains (24). On the other hand, the vir region of the R. etli p42a plasmid can function autonomously in a heterologous cellular background of disarmed A. tumefaciens C58 that lacks its own Ti plasmid-based virulence machinery. Importantly, this functionality of p42a vir genes was essentially complete, as it supported both vir gene induction and the transfer of T-DNA into the plant cells, followed by its transient expression. Quantitatively, however, the level of induction of the R. etli vir promoter observed here using the β-galactosidase reporter was 3 to 25 times lower than the level of induction of the same reporter gene measured in earlier studies of the vir genes of A. tumefaciens (24, 25). However, our reporter system utilizes the specific 527-bp sequence of the R. etli virE promoter fused to lacZ, whereas most induction assays with A. tumefaciens have been done using lacZ insertion into the Ti plasmid or a larger segment of the vir region (19, 25). Thus, it is possible that other untranslated sequences of the vir region contribute to the induction efficiency. For example, these regions may encode regulatory small RNA molecules that have been shown to activate A. tumefaciens virulence (26–28). This notion is lent support by our observations that AS induction of the defined, 1,384-bp-long A. tumefaciens virE promoter by the A. tumefaciens VirG was comparable to that of the R. etli virE promoter by the R. etli VirG. Furthermore, the R. etli VirG activated the virE promoter from both A. tumefaciens and R. etli, suggesting the broader interspecies recognition of vir promoter sequences by the R. etli VirG than by the A. tumefaciens VirG, which was species specific.
RT-qPCR analysis of the expression kinetics of major vir operons, which represents the first use of this approach to quantify vir gene expression, revealed two variations in AS-induced transcriptional activation of the tested vir genes. First, whereas the three main vir operons, virB, virD, and virE, exhibited a continuous increase in expression over the period of at least 24 h of AS treatment, expression of virG already reached apparent saturation after 6 h of treatment. In A. tumefaciens, on the other hand, induction of expression of virG was observed to increase for at least 24 h of AS treatment (29), although its differences with the induction patterns of other vir genes have not been examined. Second, in R. etli, the virB2 gene is located outside the virB operon, is expressed constitutively at relatively low levels, and shows no detectable inducibility by AS. Although the virB2 promoter region contains several potential vir boxes, it may be lacking nonconserved sequences downstream of the vir box, known to be required for efficient VirG binding and transcriptional activation (30). In contrast, A. tumefaciens virB2 is a part of the virB operon and is induced by AS. A. tumefaciens virB2 is considered essential for virulence (31), but A. tumefaciens mutants unable to form VirB2 pili still can transfer DNA (32). Thus, in R. etli, the weak expression of virB2 most likely is sufficient for the native bacterial virulence.
Another interesting aspect of the transcriptional activation of the R. etli vir genes is its differential response to signal molecules originated from different plant species. Generally, besides AS, vir genes respond to numerous other plant phenolic signals, and the nature of the inducer molecules produced by a given plant species might affect the host specificity for different bacterial strains. Our data suggest that the R. etli vir genes respond differently to cell extracts from host and nonhost species. Specifically, cell extracts from the common bean, which is nodulated by R. etli in nature, elicited significant levels of vir gene expression comparable to those elicited by synthetic AS, while extracts from the nonhosts tobacco and soybean had no effect. Whereas the induction was particularly strong with the shoot extract of the common bean, it was lower, but still significant, with the root extract, which may simply reflect differences in the concentrations of the vir gene-inducing compounds between the root and shoot extracts. Collectively, our data demonstrate that (i) the vir genes of R. etli—which, together with their A. tumefaciens homologs, encode the only known functional bacterial machineries for DNA transfer to eukaryotic cells—are transcriptionally induced by plant phenolic signals, (ii) the R. etli vir genes are induced by signals from the natural host species of this bacterium, and (iii) the induction pattern and extent exhibit small but most likely functionally relevant differences between R. etli and A. tumefaciens.
The cumulative differences between R. etli and A. tumefaciens in the transcriptional activation efficiency of one or more of their vir operons may also underlie the significantly lower efficiency of T-DNA transfer by the chimeric A. tumefaciens/p42a system than by the conventional A. tumefaciens/pTiC58 system. This is because the only difference between these two systems is the plasmid carrying the vir region, i.e., p42a and pTiC58, rather than other functions of the bacterial cell, such as attachment to the host cell surface, which relies on chromosomal factors (33, 34). Obviously, the involvement of factors other than vir gene expression, e.g., functional differences in specific vir gene products, such as the ability of VirE2 to bind single-stranded DNA, cannot be ruled out.
MATERIALS AND METHODS
R. etli and A. tumefaciens strains.
Three different strains of R. etli were used in this study. The wild-type strain R. etli CE3, a streptomycin-resistant isolate of strain CFN42, was kindly provided by Russell Carlson, University of Georgia, Athens, GA. The virG mutant of R. etli CFN42 (13) was a gift from Jo Handelsman, Yale University, New Haven, CT. The modified p42a plasmid carrying the spectinomycin resistance gene (35) located in the RHE_PA00165 locus was extracted from an R. etli strain provided by Susana Brom, National Autonomous University of Mexico, Cuernavaca, Mexico. These R. etli strains were grown in TY medium, composed of 5 g · liter−1 tryptone, 3 g · liter−1 yeast extract, and 10 mM CaCl2. A. tumefaciens strains C58, a nopaline strain carrying the wild-type pTiC58 plasmid, and its derivative, C58NT1, i.e., the C58 strain cured of its TiC58 plasmid, were grown as described previously (36). The different plasmids used in this study and summarized in Table 2 were introduced into the R. etli and A. tumefaciens strains using the classical CaCl2 protocol, with minor modifications being used in the case of R. etli (37).
Plasmid construction.
The pREp-lacZ plasmid, which expresses lacZ from the R. etli virE promoter, was produced in three steps. First, the virB promoter from the A. tumefaciens pTiA6 plasmid was amplified with the primer pair 5′-ATGCCATGGCGATCCGTCTTGCGCTGACAG-3′/5′-CCGCTCGAGGTCGACACTAGTAGATCTAAGCCTACCTTATCTCCTTAGCTCGCAAC-3′ and introduced into the NcoI-SalI sites of the pCB302 (38) backbone amplified with the primer pair 5′-CGCGTCGACATCGATGGTACCGGATCCGAATTCCGCTCACCGGGCTGGTTG-3′/5′-ATGCCATGGAGTAAAGCGCTGGCTGAACCC-3′, resulting in pBp. Then, the R. etli virE promoter region, comprising 527 bp upstream of the translation initiation codon of the virE1 gene, was amplified from p42a with the primer pair 5′-GGACCATGGTCCACCTCTCCGCTGTGGA-3′/5′-GGAAAGCTTGTCGTTGTTTCTCCTGCAAAACTTGC-3′ and inserted into the NcoI-HindIII sites of pBp, replacing the virB promoter and resulting in pREp. Finally, the lacZ gene, encoding β-galactosidase, was amplified from the pET30b:β-gal plasmid (Novagen) with the primer pair 5′-GGAAAGCTTATGACCATGATTACGGATTCACTGG-3′/5′-GGAGGATCCTTATTTTTGACACCAGACCAACTGGTAATG-3′ and inserted into the HindIII-BamHI sites of pREp, resulting in pREp-lacZ. To produce the pAEp-lacZ plasmid, which expresses lacZ from the A. tumefaciens virE promoter, the promoter region of 1,384 bp upstream of the translation initiation codon of virE1 was amplified from pTi58 with the primer pair 5′-CATGTCATGAGTCGACCGCTGAGGTTGAATATAC-3′/5′-CCCAAGCTTATCATTGTTTCTCCTACAGA-3′ and inserted into the NotI-HindIII sites of pREp-lacZ, replacing the R. etli virE promoter. For expression of the R. etli virG gene from its own promoter, the segment containing virG and its promoter region, comprising the sequence 98 bp upstream of the translation initiation codon, was amplified from the p42a plasmid with the primer pair 5′-GGCGAGCTCATGTTGCTCAACTTTCAAGCAGGC-3′/5′-CGGGGTACCTCAGGCAGCCATCACTCCT-3′ and inserted into the SacI-KpnI sites of the pE2431 plasmid (kindly provided by Stanton B. Gelvin, Purdue University, West Lafayette, IN), replacing the virB promoter with the R. etli virG expression cassette and resulting in pE2431-RevirG. For expression of the R. etli virB2 gene from the R. etli virE promoter, the virB2 coding sequence was amplified from the p42a plasmid with the primer pair 5′-CCCAAGCTTATGATGCGATGCTTTGAGAGATAC-3′/5′-CGCGGATCCTCAACCACCTCCAGTCAGCG-3′ and inserted into the HindIII-BamHI sites of pREp-lacZ, replacing lacZ and producing pREp-virB2.
Ethanolic plant extracts.
The shoots and roots of 3- to 4-week-old plants grown in vitro on Murashige and Skoog basal medium (Sigma) supplemented with 30 g · liter−1 sucrose and 4 g · liter−1 agar were flash-frozen in liquid nitrogen and ground with a mortar and pestle, resuspended in 100% ethanol, and extracted for 5 min at 4°C. The extract was then concentrated 100 times in a SpeedVac concentrator (50 rpm at room temperature) and stored at −20°C before use.
β-Galactosidase assay.
The β-galactosidase assay was performed as described previously (25, 39, 40) with modifications. Bacteria were grown for 24 h at 28°C and 240 rpm in induction medium [1× AB salts, 2 mM phosphate buffer (pH 5.6), 50 mM 2-(4-morpholino)ethanesulfonic acid (MES), 0.5% glucose] (21) without inducer, with AS (50 or 100 μM), or with 10 μl of ethanolic plant extract, and the cell density of the culture (optical density at 600 nm [OD600]) was determined. Then, 0.2 ml of the bacterial suspension was added to 1.8 ml of Z buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4, 50 mM β-mercaptoethanol), combined with 100 μl chloroform and 50 μl 0.1% SDS, and vigorously vortexed, after which 0.4 ml of medium A [60 mM K2HPO4, 33 mM KH2PO4, 7.5 mM (NH4)2SO4, 2.0 mM Na citrate] containing 4 g · liter−1 ortho-nitrophenyl-β-galactoside (ONPG) was added, and the mixture was incubated at 28°C and 240 rpm for approximately 40 min. When a light yellow color appeared upon visual examination, the reaction was stopped by the addition of 1 ml of 1 M Na2CO3. After centrifugation (10 min, 20,000 × g) to pellet the cell debris, the OD420 of the supernatant was determined. The resulting measurements of the β-galactosidase activity, expressed in Miller units, were calculated using the following equation: (1,000 × OD420)/(OD600 × v × t), where v is the volume of the bacterial suspension (in milliliters) and t is the reaction time (in minutes).
Transient GFP expression in plant cells.
A. tumefaciens or R. etli strains carrying pCB302T-GFP (10) were grown for 24 h at 28°C and infiltrated into intact N. benthamiana leaves as described previously (41). The bacterial suspension was first adjusted to an OD600 of 0.6 and then diluted 40 or 100 times, i.e., to OD600s of 0.015 and 0.006, respectively, in MES buffer before infiltration. Three days after infiltration, the expression of the GFP reporter was analyzed under a Zeiss LSM 5-Pa confocal microscope at low magnification with a 10× objective; the number of GFP-expressing cells per square centimeter of infiltrated leaf surface was scored as described previously (41).
RNA extraction and RT-qPCR.
Different bacterial strains were grown for 6, 16, or 24 h, as described above for the β-galactosidase assays, without AS or with 50 μM AS. Two milliliters of bacterial suspension was first mixed with 4 ml of RNAprotect Bacteria reagent (Qiagen), and the mixture was incubated for 5 min at room temperature to stabilize the RNA. After centrifugation, total RNA was extracted from the pellet using an RNeasy minikit (Qiagen) and treated with DNase I (New England BioLabs) to eliminate potential DNA contamination. The resulting total RNA preparation (1.0 μg) was used for cDNA synthesis with a RevertAid RT kit (Thermo) with random hexamer primers. The cDNA preparation was diluted 1:100 for the reference 16S RNA gene or 1:10 for the tested vir genes, and 0.5 μl of the resulting cDNA solution was analyzed by qPCR with the Maxima SYBR green–carboxy-X-rhodamine qPCR master mix (Thermo-Fisher) in a Mini Opticon real-time PCR system (catalog number PTC1148; Bio-Rad). The primer sequences for the different vir and control genes are summarized in Table S1 in the supplemental material. Data were analyzed with the CFX Manager software program (Bio-Rad), using 16S RNA as an internal control, and are presented as relative gene expression levels, with the values obtained for uninduced wild-type R. etli being set equal to 1.
Supplementary Material
ACKNOWLEDGMENTS
We thank Russell Carlson for the R. etli CE3 strain, Jo Handelsman for the R. etli CFN42 ΔvirG strain, Susana Brom for the R. etli CFN42 strain carrying plasmid p42a expressing spectinomycin resistance, and Stanton B. Gelvin for the pE2431 plasmid.
The work in the V.C. laboratory is supported by grants from USDA/NIFA, NIH, NSF, BARD, and BSF to V.C. L.W. is supported by the National Natural Science Foundation of China (grant no. 31471812 and 31672075) and the China Scholarship Council (no. 201506850021).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00841-16.
REFERENCES
- 1.Gelvin SB. 2003. Agrobacterium-mediated plant transformation: the biology behind the “gene-jockeying” tool. Microbiol Mol Biol Rev 67:16–37. doi: 10.1128/MMBR.67.1.16-37.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lacroix B, Citovsky V. 2013. The roles of bacterial and host plant factors in Agrobacterium-mediated genetic transformation. Int J Dev Biol 57:467–481. doi: 10.1387/ijdb.130199bl. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Escobar MA, Dandekar AM. 2003. Agrobacterium tumefaciens as an agent of disease. Trends Plant Sci 8:380–386. doi: 10.1016/S1360-1385(03)00162-6. [DOI] [PubMed] [Google Scholar]
- 4.Banta L, Montenegro M. 2008. Agrobacterium and plant biotechnology, p 72–147. In Tzfira T, Citovsky V (ed), Agrobacterium: from biology to biotechnology. Springer, New York, NY. [Google Scholar]
- 5.Broothaerts W, Mitchell HJ, Weir B, Kaines S, Smith LM, Yang W, Mayer JE, Roa-Rodriguez C, Jefferson RA. 2005. Gene transfer to plants by diverse species of bacteria. Nature 433:629–633. doi: 10.1038/nature03309. [DOI] [PubMed] [Google Scholar]
- 6.Hooykaas PJJ, Klapwijk PM, Nuti MP, Schilperoort RA, Rorsch A. 1977. Transfer of the Agrobacterium tumefaciens Ti plasmid to avirulent agrobacteria and to Rhizobium ex planta. J Gen Microbiol 98:477–484. doi: 10.1099/00221287-98-2-477. [DOI] [Google Scholar]
- 7.Wendt T, Doohan F, Winckelmann D, Mullins E. 2011. Gene transfer into Solanum tuberosum via Rhizobium spp. Transgenic Res 20:377–386. doi: 10.1007/s11248-010-9423-4. [DOI] [PubMed] [Google Scholar]
- 8.Wendt T, Doohan F, Mullins E. 2012. Production of Phytophthora infestans-resistant potato (Solanum tuberosum) utilising Ensifer adhaerens OV14. Transgenic Res 21:567–578. doi: 10.1007/s11248-011-9553-3. [DOI] [PubMed] [Google Scholar]
- 9.Zuniga-Soto E, Mullins E, Dedicova B. 2015. Ensifer-mediated transformation: an efficient non-Agrobacterium protocol for the genetic modification of rice. Springerplus 4:600. doi: 10.1186/s40064-015-1369-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lacroix B, Citovsky V. 2016. A functional bacterium-to-plant DNA transfer machinery of Rhizobium etli. PLoS Pathog 12:e1005502. doi: 10.1371/journal.ppat.1005502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Segovia L, Young JP, Martínez-Romero E. 1993. Reclassification of American Rhizobium leguminosarum biovar phaseoli type I strains as Rhizobium etli sp. nov. Int J Syst Bacteriol 43:374–377. doi: 10.1099/00207713-43-2-374. [DOI] [PubMed] [Google Scholar]
- 12.González V, Santamaría RI, Bustos P, Hernández-González I, Medrano-Soto A, Moreno-Hagelsieb G, Janga SC, Ramírez MA, Jiménez-Jacinto V, Collado-Vides J, Dávila G. 2006. The partitioned Rhizobium etli genome: genetic and metabolic redundancy in seven interacting replicons. Proc Natl Acad Sci U S A 103:3834–3839. doi: 10.1073/pnas.0508502103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bittinger MA, Gross JA, Widom J, Clardy J, Handelsman J. 2000. Rhizobium etli CE3 carries vir gene homologs on a self-transmissible plasmid. Mol Plant Microbe Interact 13:1019–1021. doi: 10.1094/MPMI.2000.13.9.1019. [DOI] [PubMed] [Google Scholar]
- 14.Brom S, Girard L, Tun-Garrido C, García-de los Santos A, Bustos P, González V, Romero D. 2004. Transfer of the symbiotic plasmid of Rhizobium etli CFN42 requires cointegration with p42a, which may be mediated by site-specific recombination. J Bacteriol 186:7538–7548. doi: 10.1128/JB.186.22.7538-7548.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brencic A, Winans SC. 2005. Detection of and response to signals involved in host-microbe interactions by plant-associated bacteria. Microbiol Mol Biol Rev 69:155–194. doi: 10.1128/MMBR.69.1.155-194.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stachel SE, Messens E, Van Montagu M, Zambryski PC. 1985. Identification of the signal molecules produced by wounded plant cell that activate T-DNA transfer in Agrobacterium tumefaciens. Nature 318:624–629. doi: 10.1038/318624a0. [DOI] [Google Scholar]
- 17.Steck TR, Morel P, Kado CI. 1988. Vir box sequences in Agrobacterium tumefaciens pTiC58 and A6. Nucleic Acids Res 16:8736. doi: 10.1093/nar/16.17.8736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Winans SC, Allenza P, Stachel SE, McBride KE, Nester EW. 1987. Characterization of the virE operon of the Agrobacterium Ti plasmid pTiA6. Nucleic Acids Res 15:825–837. doi: 10.1093/nar/15.2.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pazour GJ, Das A. 1990. Characterization of the VirG binding site of Agrobacterium tumefaciens. Nucleic Acids Res 18:6909–6913. doi: 10.1093/nar/18.23.6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin SG, Roitsch T, Christie PJ, Nester EW. 1990. The regulatory VirG protein specifically binds to a cis-acting regulatory sequence involved in transcriptional activation of Agrobacterium tumefaciens virulence genes. J Bacteriol 172:531–537. doi: 10.1128/jb.172.2.531-537.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gelvin SB. 2006. Agrobacterium virulence gene induction. Methods Mol Biol 343:77–84. doi: 10.1385/1-59745-130-4:77. [DOI] [PubMed] [Google Scholar]
- 22.Joubert P, Beaupère D, Wadouachi A, Chateau S, Sangwan RS, Sangwan-Norreel BS. 2004. Effect of phenolic glycosides on Agrobacterium tumefaciens virH gene induction and plant transformation. J Nat Prod 67:348–351. doi: 10.1021/np030281z. [DOI] [PubMed] [Google Scholar]
- 23.Melchers LS, Regensburg-Tuink AJ, Schilperoort RA, Hooykaas PJJ. 1989. Specificity of signal molecules in the activation of Agrobacterium virulence gene expression. Mol Microbiol 3:969–977. doi: 10.1111/j.1365-2958.1989.tb00246.x. [DOI] [PubMed] [Google Scholar]
- 24.Krishnamohan A, Balaji V, Veluthambi K. 2001. Efficient vir gene induction in Agrobacterium tumefaciens requires virA, virG, and vir box from the same Ti plasmid. J Bacteriol 183:4079–4089. doi: 10.1128/JB.183.13.4079-4089.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stachel SE, An G, Flores C, Nester EW. 1985. A Tn3 lacZ transposon for the random generation of beta-galactosidase gene fusions: application to the analysis of gene expression in Agrobacterium. EMBO J 4:891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Becker A, Overlöper A, Schlüter JP, Reinkensmeier J, Robledo M, Giegerich R, Narberhaus F, Evguenieva-Hackenberg E. 2014. Riboregulation in plant-associated alpha-proteobacteria. RNA Biol 11:550–562. doi: 10.4161/rna.29625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dequivre M, Diel B, Villard C, Sismeiro O, Durot M, Coppée JY, Nesme X, Vial L, Hommais F. 2015. Small RNA deep-sequencing analyses reveal a new regulator of virulence in Agrobacterium fabrum C58. Mol Plant Microbe Interact 28:580–589. doi: 10.1094/MPMI-12-14-0380-FI. [DOI] [PubMed] [Google Scholar]
- 28.Lee K, Huang X, Yang C, Lee D, Ho V, Nobuta K, Fan JB, Wang K. 2013. A genome-wide survey of highly expressed non-coding RNAs and biological validation of selected candidates in Agrobacterium tumefaciens. PLoS One 8:e70720. doi: 10.1371/journal.pone.0070720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winans SC, Kerstetter RA, Nester EW. 1988. Transcriptional regulation of the virA and virG genes of Agrobacterium tumefaciens. J Bacteriol 170:4047–4054. doi: 10.1128/jb.170.9.4047-4054.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roitsch T, Jin S, Nester EW. 1994. The binding site of the transcriptional activator VirG from Agrobacterium comprises both conserved and specific nonconserved sequences. FEBS Lett 338:127–132. doi: 10.1016/0014-5793(94)80349-8. [DOI] [PubMed] [Google Scholar]
- 31.Berger BR, Christie PJ. 1994. Genetic complementation analysis of the Agrobacterium tumefaciens virB operon: virB2 through virB11 are essential virulence genes. J Bacteriol 176:3646–3660. doi: 10.1128/jb.176.12.3646-3660.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sagulenko V, Sagulenko E, Jakubowski S, Spudich E, Christie PJ. 2001. VirB7 lipoprotein is exocellular and associates with the Agrobacterium tumefaciens T pilus. J Bacteriol 183:3642–3651. doi: 10.1128/JB.183.12.3642-3651.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Douglas CJ, Staneloni RJ, Rubin RA, Nester EW. 1985. Identification and genetic analysis of an Agrobacterium tumefaciens chromosomal virulence region. J Bacteriol 161:850–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu X, Zhao J, Degrado WF, Binns AN. 2013. Agrobacterium tumefaciens recognizes its host environment using ChvE to bind diverse plant sugars as virulence signals. Proc Natl Acad Sci U S A 110:678–683. doi: 10.1073/pnas.1215033110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.López-Fuentes E, Torres-Tejerizo G, Cervantes L, Brom S. 2014. Genes encoding conserved hypothetical proteins localized in the conjugative transfer region of plasmid pRet42a from Rhizobium etli CFN42 participate in modulating transfer and affect conjugation from different donors. Front Microbiol 5:793. doi: 10.3389/fmicb.2014.00793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wise AA, Liu Z, Binns AN. 2006. Culture and maintenance of Agrobacterium strains. Methods Mol Biol 343:3–13. [DOI] [PubMed] [Google Scholar]
- 37.Vincze E, Bowra S. 2006. Transformation of rhizobia with broad-host-range plasmids by using a freeze-thaw method. Appl Environ Microbiol 72:2290–2293. doi: 10.1128/AEM.72.3.2290-2293.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiang C, Han P, Lutziger I, Wang K, Oliver DJ. 1999. A mini binary vector series for plant transformation. Plant Mol Biol 40:711–717. doi: 10.1023/A:1006201910593. [DOI] [PubMed] [Google Scholar]
- 39.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 40.Stachel SE, Nester EW. 1986. The genetic and transcriptional organization of the vir region of the A6 Ti plasmid of Agrobacterium. EMBO J 5:1445–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lacroix B, Citovsky V. 2011. Extracellular VirB5 enhances T-DNA transfer from Agrobacterium to the host plant. PLoS One 6:e25578. doi: 10.1371/journal.pone.0025578. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.