

Abstract
Background
While NGS allows rapid global detection of transcripts, it remains difficult to distinguish ncRNAs from short mRNAs. To detect potentially translated RNAs, we developed an improved protocol for bacterial ribosomal footprinting (RIBOseq). This allowed distinguishing ncRNA from mRNA in EHEC. A high ratio of ribosomal footprints per transcript (ribosomal coverage value, RCV) is expected to indicate a translated RNA, while a low RCV should point to a non-translated RNA.
Results
Based on their low RCV, 150 novel non-translated EHEC transcripts were identified as putative ncRNAs, representing both antisense and intergenic transcripts, 74 of which had expressed homologs in E. coli MG1655. Bioinformatics analysis predicted statistically significant target regulons for 15 of the intergenic transcripts; experimental analysis revealed 4-fold or higher differential expression of 46 novel ncRNA in different growth media. Out of 329 annotated EHEC ncRNAs, 52 showed an RCV similar to protein-coding genes, of those, 16 had RIBOseq patterns matching annotated genes in other enterobacteriaceae, and 11 seem to possess a Shine-Dalgarno sequence, suggesting that such ncRNAs may encode small proteins instead of being solely non-coding. To support that the RIBOseq signals are reflecting translation, we tested the ribosomal-footprint covered ORF of ryhB and found a phenotype for the encoded peptide in iron-limiting condition.
Conclusion
Determination of the RCV is a useful approach for a rapid first-step differentiation between bacterial ncRNAs and small mRNAs. Further, many known ncRNAs may encode proteins as well.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3586-9) contains supplementary material, which is available to authorized users.
Background
Bacterial RNA molecules consist of non-coding RNAs (ncRNAs including rRNAs and tRNAs), and protein-coding mRNAs. ncRNAs are encoded either in cis or in trans of coding genes and their size ranges from 50–500 nt [1, 2]. Cis-encoded ncRNA templates are localized opposite to the gene to be regulated and, accordingly, have full complementarity to the mRNA. Their expression leads to a negative or positive impact on the expression of the regulated gene [3–5]. This type of gene regulation has been exploited in applied molecular biology [6]. However, only few experimentally verified cis-encoded ncRNAs exist, in contrast to trans-encoded ncRNAs. Trans-encoded ncRNAs are usually found in intergenic regions and have a limited complementarity to the regulated gene. Recent research has led to the view that trans-encoded ncRNAs are involved in the regulation of almost all bacterial metabolic pathways (see [7], and references therein).
The number of annotated ncRNAs known from different bacterial species is rapidly increasing. For instance, 329 ncRNAs are annotated for E. coli O157:H7 str. EDL933 [2]. Around 80 of them have been experimentally verified in E. coli [8]. Numerous bioinformatic studies on E. coli K12 and other bacterial species predicted the number of ncRNAs to range between 100 and 1000 (e.g. [9–11]). As E. coli O157:H7 strain EDL933 (EHEC) contains a core genome of 4.1 Mb which is well conserved among all E. coli strains [12], many similar or identical ncRNAs are assumed to exist in EHEC.
In the past, ncRNAs have been predicted by different bioinformatics methods (see [13] for a review about ncRNA detection in bacteria). A commonly used tool in ncRNA-prediction is RNAz, which has been used to predict ncRNAs in Bordetella pertussis [14], Streptomyces coelicolor [15] and others. However, any such studies require experimental verification [13] of which next-generation sequencing is of prime interest for this task.
While experimental large scale screenings for ncRNAs, especially strand-specific transcriptome sequencing using NGS, are becoming more and more important (e.g. [16–18]), it is not possible to determine whether a transcript is translated, based solely on RNAseq (see, e.g. [19]). In order to distinguish “true” ncRNAs from translated short mRNAs, we modified the ribosomal profiling approach developed by Ingolia et al. for yeast [20] and applied this technique to E. coli O157:H7 strain EDL933. Ribosomal profiling, which is also termed ribosomal footprinting or RIBOseq, detects RNAs which are covered by ribosomes and which are, therefore, assumed to be involved in the process of translation. The RNA population which is covered by ribosomes is termed “translatome” [21] and bioinformatics tools are now available to analyze these novel data [22]. Combined with strand-specific RNA-sequencing, we suggest that this approach provides additional evidence to distinguish between non-coding RNAs and RNAs covered by ribosomes.
In the past, RNAs have been found which function as ncRNA (i.e. having a function as RNA molecule not based on encoding a peptide chain) and, at the same time, as mRNA (i.e. encoding a peptide chain). Therefore, those RNAs were either termed dual-functioning RNAs (dfRNAs [23]) or coding non-coding RNAs (cncRNAs [24]). The former name is now used for RNAs with any two different functions (e.g., base-pairing and protein binding [25]), the latter describes the fact that the DNA-encoded entity functions on the level of RNA (hence, non-coding) and additionally on the level of an peptide (i.e. coding). Less than ten examples of cncRNAs are known from prokaryotes, e.g., RNAIII, SgrS, SR1, PhrS, gdpS, irvA, and others [23, 24, 26, 27].
Methods
Microbial strain
Strain E. coli O157:H7 EDL933 was obtained from the Collection l’Institute de Pasteur (Paris) under the collection number CIP 106327 (= WS4202, Weihenstephan Microbial Strain Collection) and was used in all experiments. The strain was originally isolated from raw hamburger meat, first described in 1983 [28], originally sequenced in 2001 [12] and its sequence improved recently [29]. The genome of WS4202 was re-sequenced by us to check for laboratory derived changes (GenBank accession CP012802).
RIBOseq
Ribosomal footprinting was conducted according to Ingolia et al. [20], but was adapted to sequence bacterial footprints using strand-specific libraries obtained with the TruSeq Small RNA Sample Preparation Kit (Illumina, USA). Cells were grown in ten-fold diluted lysogeny broth (LB; 10 g/L peptone, 5 g/L yeast extract, 10 g/L NaCl) with shaking at 180 rpm. At the transition from late exponential to early stationary phase the cultures were supplemented with 170 μg/mL chloramphenicol to stall the ribosomes (about 6-times above the concentration at which trans-translation occurs [30]). After two minutes, cells were harvested by centrifugation at 6000 × g for 3 min at 4 °C. Pellets were resuspended in lysis buffer (20 mM Tris-Cl at pH8, 140 mM KCl, 1.5 mM MgCl2, 170 μg/mL chloramphenicol, 1% v/v NP40; 1.5 mL per initial liter of culture) and the suspension was dripped into liquid nitrogen and stored at −80 °C. The cells were ground with pestle and mortar in liquid nitrogen and 2 g sterile sand for about 20 min. The powder was thawed on ice and centrifuged twice, first at 3000 × g at 4 °C for 5 min and next at 20,000 × g at 4 °C for 10 min. The supernatant was saved and A260nm determined. After dilution to an A260nm of 200, RNase I (Ambion AM2294) was added to the sample to a final concentration of 3 U/μL and the sample was gently rotated at room temperature (RT) for 1 h. Remaining intact ribosomes with protected mRNA-fragments (footprints) were enriched by gradient centrifugation. A sucrose gradient was prepared in gradient buffer (20 mM Tris-Cl at pH 8, 140 mM KCl, 5 mM MgCl2, 170 μg/mL chloramphenicol, 0.5 mM DTT, 0.013% SYBR Gold). Nine different sucrose concentrations were prepared in 5% (w/v) steps ranging from 10 to 50% and 1.5 mL of each concentration was loaded to a centrifuge tube. Five hundred μL of the crude ribosome sample were loaded onto each gradient tube and centrifuged at 104,000 × g at 4 °C for 3 h. The layer containing the ribosomes was visualized using UV-light and the tube was pierced at the bottom to slowly release the gradient and the band containing intact 70S ribosomes was collected. To ensure that RNA which is not protected by ribosomes is fully digested, and to get a highly enriched ribosomal fraction, the procedure of RNase-digestion and gradient centrifugation was repeated: The ribosomal fraction was diluted 1:1 with gradient buffer (without SYBR Gold and sucrose) and was loaded on a sucrose gradient without the 10% sucrose layer. After centrifugation, complete 70S ribosomes were collected by slowly releasing the gradient as described above and frozen in liquid nitrogen. To obtain the protected ribosomal footprints, 1 mL Trizol was added to 200 μL of the ribosome suspension following the manual for Trizol extraction of RNA (life technologies, USA). The final footprint-RNA pellet was dissolved in RNase free water. To ensure no carry-over of genomic DNA fragments, DNase treatment was performed using the TURBO DNA-free Kit (Applied Biosystems, USA) according to the manual. For footprint size-selection, the crude RNA-preparation was loaded to a 15% denaturing polyacrylamide gel. An oligonucleotide of 28 bp was used as a marker which is about the size of a ribosomal footprint [31, 32]. After staining with SYBR Gold, the region of about 28 nt was excised from the gel. The RNA was extracted from the gel slice as described [20]. Results of pilot experiments showed that RNase I cuts the 5′ ends of the 16S rRNA producing a fragment of about the size expected for the footprints, contributing about 50% to the size-selected RNA fragments after sequencing. For this reason, these fragments were removed with oligonucleotides complementary to the 5′-end of the 16S rRNA using the MICROBExpress bacterial mRNA enrichment kit (life technologies, USA) following the manual. Furthermore, true footprints were found to be shorter than expected (see Results). Enriched footprint-RNAs were dephosphorylated using Antarctic phosphatase (10 units per 300 ng RNA, supplemented with 10 units Superase, 37 °C for 30 min). Footprints were recovered using the miRNeasy Mini Kit (Qiagen, Germany). Subsequent phosphorylation was carried out using T4 polynucleotide kinase (20 units supplemented with 10 units Superase, 37 °C for 60 min) and cleaned using the miRNeasy Mini Kit as before. Finally, the entire sample was processed with the TruSeq Small RNA Sample Preparation Kit (Illumina) according to the manual, using 11 PCR cycles, and was sequenced on an Illumina MiSeq.
Transcriptome sequencing
The same cultures used for ribosomal footprinting were also used for transcriptome sequencing (i.e., strand specific RNAseq). Fifty μL of the diluted cell extract with an A260nm of 200 units (see above) were added to one 1 mL of Trizol and total RNA was isolated. Since 90–95% of the total RNA consists of ribosomal RNA [33], the Ribominus Transcriptome Isolation Kit (Yeast and Bacteria, Invitrogen, USA) was applied according to the manual and the RNA was precipitated with the help of glycogen and two volumes 100% ethanol. DNase treatment was performed as described above. One μg RNA was fragmented as described [34] and the RNA-fragments were precipitated with glycogen and 2.5 volumes 100% ethanol. For sequencing on an Illumina MiSeq, the fragments were resuspended in 25 μL RNase free water and further processed like the cleaned footprint-RNAs (see above).
Northern blots
RNA was isolated in the same manner and under the same conditions as for the NGS experiments. Northern blots were performed using the DIG Northern Starter kit (Roche, Switzerland). Primers to generate DIG (digoxygenin) labeled probes are listed in Additional file 1: Table S1. For preparation of the probes, electroblotting, crosslinking, hybridization and detection, the manufacturer’s protocol was followed, except that electroblotting was performed using polyacrylamide gels and that for crosslinking EDC (1-ethyl-3-(3-dimethylaminopropyl) carbodiimide) was used [35]. After exposure to CDP-Star (included in the DIG Northern Starter kit), luminescence activity of the hybridized probes was measured using an In-Vivo Imaging System (PerkinElmer, USA).
Competitive growth assays for the overexpression phenotype of RyhP
For the production of the peptide RyhP encoded in RyhB, two versions of the corresponding ORF (named P1 and P2) were cloned onto pBAD/Myc-His C (Invitrogen). Similarly, two versions of this ORF with either the second or the third codon changed into stop codons to terminate translation were used as negative controls (named T2 and T3). For cloning, primer pairs (for primer see Additional file 1: Table S1) were hybridized forming RyhP-coding dsDNA fragments. The pBAD was opened by NcoI and BglII in restriction buffer NEB3.1 (NEB) and was subsequently column cleaned (Genelute PCR Clean-Up Kit, Sigma-Aldrich). RyhP-DNA fragments and pBAD were ligated (T4 ligase, NEB) and transformed in E. coli TOP10. After sequencing (eurofins), verified plasmids were transformed in E. coli O157:H7 EDL933. EHEC strains (containing either P1, P2, T2 or T3) were grown overnight in LB medium with a final concentration of 120 μg/ml ampicillin. The cell was density measured and both strains were mixed 50:50. Minimal Medium (MM) M9 without any iron added [36], but supplemented with a final concentration of 120 μg/ml ampicillin and 0.2% arabinose (for induction), was inoculated 1:1000 using the mixture and incubated 24 h at 37 °C with shaking at 150 rpm. Of both, the initial mixture and of the MM-culture, the plasmids were isolated and Sanger sequenced using the primer pBAD-C-R. The peak heights of the two nucleotides changed to form the stop codon in T2 or T3 were measured in comparison to the P variants, and the mean CI was calculated according to CI = (T(out) · P(in))/(P(out) · T(in)) [37] of P1 against, T2, P1 against T3 and P2 against T3. Given are mean and the standard deviations of three biological independent experiments.
Bioinformatics procedures
NGS mapping and evaluation
Raw data were deposited at the Gene Expression Omnibus [GEO: GSE94984]. Illumina output files (FASTQ files in Illumina format) were converted to plain FASTQ using FastQ Groomer [38] in Galaxy [38, 39]. The FASTQ files were mapped to the reference genome (NC_002655) using Bowtie2 [40] with default settings, except for a changed seed length of 19 nt and zero mismatches permitted within the seed in the Illumina data due to the short length of the footprints. Visualization of the data was carried out using our own NGS-Viewer [41] or BamView [42] implemented in Artemis 15.0.0 [43].
The number of reads was normalized to reads per kilobase per million mapped reads (RPKM) [44]. Using this method, the number of reads is normalized both with respect to the sequencing depth and the length of a given transcript. For determination of counts and RPKM values, BAM files were imported into R (R Development Team [45]) using Rsamtools [46]. For further processing, the Bioconductor [47] packages GenomicRanges [48] and IRanges were used [49]. The locations of the 16S rRNA and 23S rRNA are given by the RNT file from RefSeq [50]. findOverlaps of IRanges [49] was used to determine the remaining reads overlapping a 16S or 23S rRNA gene on the same strand. Reads from these rRNA-genes were excluded from further analysis as most rRNA had been removed using the Ribominus kit, as described above. countOverlaps can also determine the number of reads overlapping a gene on the same strand (counts). Using these counts, RPKM values were generated. For the value “million mapped reads”, the number of reads mapped to the genome, less the remaining reads overlapping a 16S or 23S rRNA gene, were used. Pearson correlation was calculated using Excel and Spearman rank correlation according to Wessa [51].
RCV thresholds
To distinguish between translated and non-translated for a given RNA, the ribosomal coverage value (i.e., reads of ribosomal footprints per reads of mRNA) was examined [52]. A negative control set contains the RCVs of tRNAs (“untranslated”). Sixteen phage encoded tRNAs, one tRNA annotated as a pseudogene, and one tRNA containing less than 20 reads in the combined transcriptome data set were disregarded since phage tRNAs sometimes have unusual properties [53, 54]. The RCVs of the tRNAs were transformed to ln(RCV), abbreviated LRCV. A density function LRCV-tRNA(x), with x = LRCV, was estimated by a kernel density estimation with Gaussian kernels and bandwidth selection according to Scott’s rule [55], furthermore a normal distribution was fitted as well for comparison. This was also conducted for the annotated genes (i.e., “translated” set), excluding zero RCVs (261 genes). To test the hypothesis “the RCV of the RNA belongs to the tRNA distribution”, we used the estimated tRNA LRCV distribution to compute a P value for an observed ncRNA with LRCV x as
where we numerically evaluate the density function. For example, the hypothesis will be rejected for α = 0.05 for any x ≥ −1.816817 which corresponds to an RCV of 0.162542. Similar for α = 0.01 we obtain an RCV of 0.354859. For α = 0.05 we reject 52 of 115 annotated ncRNAs to be not translated, and for α = 0.01 we reject 63.
Since the interpretation of the results depends on the assumed distribution, we also used, at least for tRNAs, a fit of the normal distribution. The tails of the normal distribution tend to zero faster than before, which results in different P values. For example, for α = 0.05 a corresponding RCV of 0.646079 is obtained and for α = 0.01 the bound for the RCV is 0.928702. However, the normal distribution has no good fit (not shown) and is henceforth excluded.
In a similar way as for the tRNAs, we can use the gene distribution to test the hypothesis “the RCV of the RNA belongs to the mRNA distribution” by using the RCV of all annotated genes (aORFs) as a negative control set. In this case, the P value is computed by
For the latter function, we obtained the bounds 0.532837 and 0.197320 for α = 0.05 and α = 0.01, respectively. Thus, all RNAs above those values might be considered mRNAs.
Examination of known and novel ncRNAs
Escherichia coli O157:H7 EDL933 (genbank accession AE005174) contains 329 known ncRNAs (Rfam database, April, 30th 2014 [56]). All ncRNAs which should naturally have ribosomal footprints (e.g., are leader peptides, riboswitches (several contain a translatable ORF [57]), occur within genes on the same strand, or tmRNA) were excluded from the analysis, as well as rRNAs and tRNAs. Thus, the excluded RNAs are 5S_rRNA (8x), ALIL (19x), Alpha_RBS, C4, Cobalamin, cspA (4x), DnaX, FMN, greA, His_leader, IS009 (3x), IS102 (2x), iscRS, isrC (2x), isrK (2x), JUMPstart (3x), Lambda_thermo (2x), Leu_leader, Lysine, Mg_sensor, mini-ykkC, MOCO_RNA_motif, nuoG, Phe_leader (2x), PK-G12rRNA (7x), QUAD_2, rimP, rncO, rnk_leader, rne5, ROSE_2, S15, SECIS (3x), SgrS, ssrA (tmRNA), sok (10x), SSU_rRNA_archaea (14x), STnc40, STnc50, STnc370, t44/ttf, Thr_leader, TPP (3x), tRNAs (99x), tRNA-Sec, Trp_leader, and yybP-ykoY. The remaining 116 RNAs were grouped in translated, non-translated and undecided according to their RCV. Translated ncRNAs were three-frame translated and proteins sequences were searched against the non-redundant database “nr” of genbank using blastp [58]. Cases in which the ORFs of the ncRNA generated a single hit to the database were excluded since a false annotation of the hit is likely for those.
In order to provide an initial in silico characterization of the putative function for the novel intergenically-encoded ncRNAs, we used CopraRNA [59, 60] and examined the functional enrichments returned for the predictions. CopraRNA was called with default parameters for each set of putative ncRNA homologs. To find ncRNA homologs for the CopraRNA prediction, GotohScan (v1.3 stable) [61] was run with an e value threshold of 10−2 against the set of genomes listed in the Additional file 2: Table S2. The highest scoring homolog (i.e. having the lowest e value) for each organism was retained, if more than one GotohScan hit was present.
Ka/Ks ratio
The most likely ORF encoding a peptide was chosen according to the RIBOseq data. Homologs were searched using NCBI Web BLAST in the database nr using blastn. Hits with the highest e value but still achieving 100% coverage and displaying no gaps in the alignment were chosen (Additional file 3: Table S3). Gene pairs were examined using the KaKs_Calculator 2.0 [62] providing a number of algorithms which are compared and evaluated.
Shine-Dalgarno prediction
For any novel ncRNA with a significant blastp hit (e value ≤ 10−3, see above), a start codon (ATG, GTG, TTG) of the respective frame was searched closest to the start position of the ncRNA (except sgrS for which the start codon position is known, but ATG in E. coli K12 corresponds to ATT in EHEC, a rare but possible start codon; see Discussion). The maximum distance allowed between the ncRNA start coordinate and proposed start codon was ±30 bp. The region upstream of the putative start codon was examined for the presence of a Shine-Dalgarno sequence (optimum taAGGAGGt) according to [63] and [64]. A Shine-Dalgarno motif was assumed to be present at a ΔG° threshold of ≤ −2.9 kcal/mol (according to [63]) to allow weak Shine-Dalgarno sequences to be reported since even leaderless mRNAs exist [65].
For global examinations, we used PRODIGAL bins of the Shine-Dalgarno sequence and their distance to the start codon (Additional file 4: File S1) according to Hyatt et al. [66]. Bins without genes were omitted, and bins containing less than 100 genes were combined to superbins: S0, S2-3-4, S6, S7-8-9-12, S13, S14-15, S16, S18-19-20, S22, and S23-24-26-27 containing 629, 115, 116, 133, 1095, 664, 1191, 145, 687, and 327 genes, respectively.
Results and discussion
Sequencing statistics and footprint size
Two biologically independent replicates were used to assay reproducibility (Additional file 5: Figure S1). The numbers of footprint reads per gene of both RIBOseq replicates have a Pearson correlation of 0.86 and a Spearman rank correlation of 0.92, which was found to be slightly less compared to other NGS experiments [17, 67]. Nevertheless, the data sets were combined to increase the overall sequencing depth. In summary, 32.0 million transcriptome reads and 20.6 million translatome reads could be mapped to the EHEC genome (NC_002655; see Additional file 6: Table S4). Interestingly, the percentage of tRNA, an RNA species not translated, in both experiments was quite different. In the transcriptome, tRNAs contributed 31% of the library, whereas in the footprint libraries, tRNAs contributed only 0.3%. Such a difference is expected, since in the transcriptome sequencing, the tRNAs are processed together with the total RNA isolated. In contrast, in translatome sequencing, only translated RNAs are sequenced since the RNase digestion will destroy any RNA outside the ribosomes, including most tRNAs. However, some tRNAs might be trapped in the ribosomes and are recorded despite the RNase treatment. Thus, we reasoned that tRNAs would represent the best maximum background value for any carry-over of a non-translated RNA in the translatome sequencing.
The number of nucleotides which are protected by the ribosomes, i.e., the size of the footprints, was reported to be 28 nt in prokaryotes as well as in eukaryotes [20, 31, 32, 34, 68, 69]. Additionally, other studies using ribosome profiling in eukaryotes were able to determine the ribosome position of the footprints at sub-codon resolution (e.g. [70, 71]). The situation is quite different in bacteria: In one of the first studies in bacteria, Li et al. [72] determined the footprint size to range between 25 and 40 nt. Based on these results, O’Connor et al. [73] suggested that the footprint size may vary due to different progression rates of the ribosome. However, the enzyme used to obtain the bacterial ribosomal footprints in these studies was micrococcal nuclease which is known to prefer sites rich in adenylate, deoxyadenylate or thymidylate, which explains the varying length of the footprints [72]. In our study, after sequencing E. coli ribosomal footprints, the major peak of fragment sizes was observed at 23 nt, even despite the size-selection targeting 28 nt. We believe that RNase I, which we used, is a better choice [74, 75]. We also tested a number of commercially available RNases and mixtures of endo- and exo-cutting enzymes and received a consistent footprint size of about 23 nt and not 28 nt (unpublished data). The observed value of 23 nt may be explained by the different size of prokaryotic and eukaryotic ribosomes. Klinge et al. [76] estimated the mass of ribosomes to be 3.3 MDa for the eukaryotic and 2.5 MDa for prokaryotic, respectively. Assuming a roughly proportional scaling between the mass of the ribosome and its diameter suggest a bacterial footprint size of about 23 nt.
Putative novel ncRNAs with low ribosomal coverage
The ribosome coverage value (RCV) gives the ratio of RPKM footprints over RPKM transcriptome. ncRNAs should have low RCVs. The RCV is similar to the “translational efficiency” applied for eukaryotes [77] to determine the translatability of a given mRNA. The RCV varied between zero (for 261 annotated genes) and a maximum value of nearly 39 for an annotated gene. Low or zero RCVs for annotated genes can be explained by the internal status of the cells controlling translation independent of transcription. For instance, some mRNAs are blocked by riboswitches or bound by ncRNA (e.g. [78]). We examined the genes with zero reads in some detail. This group contains about 3-times more phage associated genes compared to all genes (36% versus 13%). The genes are shorter compared to all (about half the size) and a larger fraction is annotated as hypothetical (50% compared to 30% in the annotation NC_002655). We looked for transcription under any of 11 different growth conditions [17] and found transcription for less than 20% of those genes under any condition. However, the other genes might be activated in specific circumstances not tested yet. This is corroborated by our findings that some genes were induced when EHEC was grown in co-culture with amoeba (unpublished results), but are not activated in any other condition of the published data set [17].
To analyze the data for novel ncRNAs, the transcriptome data was analyzed for contiguous transcription patterns (no gaps allowed) containing at least 20 transcriptome reads which do not correspond to an annotated gene (i.e., in a distance of more than 100 nt to a same-strand annotated ORF of a gene). Start and end of the novel ncRNAs were defined as the first and last nt of the contiguous read pattern. The chosen value of 20 reads was applied independently of any length restriction. For a 100-bp transcript in our dataset this approximately corresponds to an RPKM of 20, which is about 200-times above background level for transcriptome sequencing [17].
Each novel transcript was analyzed for its RCV to determine whether it is potentially translated. As a negative control, we chose tRNAs which have RCVs in a range between 0.000173 and 0.094843. While the RCVs are small for tRNAs, the ratio between the highest and lowest RCV of the tRNAs is about 500-fold. We surmised that tRNA abundance might correlate either to the RCV or to the codon usage of EHEC (which correlates with tRNA abundance). However, no relationship was found (not shown) and the reasons for the difference in RCV remain unknown. For convenience, the RCV is shown as ln(RCV) (=LRCV) in Fig. 1. Figure 1a shows a histogram of the LRCV of tRNAs together with an estimated density function LRCV (x) obtained by a kernel density estimation (blue line). Next, the LRCV distribution of the annotated genes is shown in Fig. 1b (green line). Finally, Fig. 1c shows the LRCV of all annotated ncRNAs (red line; less those known to be translated; see Table 1). To determine, whether the RCV of a given RNA belongs either to the tRNA distribution group or the gene distribution group, we determined the lower and upper limit of the RCV corresponding to a probability of error of 99% (α = 0.01), respectively (see Methods). Below the RCV threshold 0.197 a transcript is considered to be untranslated and above 0.355 it is considered to be a candidate for translation. Thus, a transcript is qualified as a putative novel ncRNA only, if its RCV was below the lower threshold.
Fig. 1.
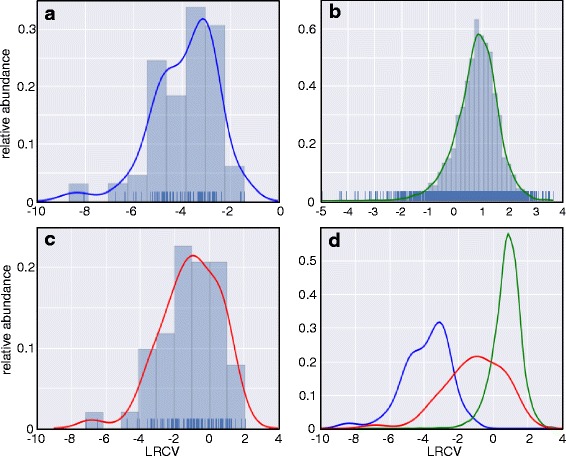
Logarithmic (ln) ribosomal coverage (LRCV) of tRNAs, annotated genes, annotated ncRNAs and a merger of the former. a Histogram of the LRCVs (X-axis) of the tRNAs together with either the estimated density function (blue curve). The density of the individual tRNAs is shown as little blue bars on top of the X-axis. b LRCV histogram as before, but of the annotated genes and their estimated density function (green). c LRCV histogram as before, but of the known ncRNAs (see Table 1) together with their estimated density function (red). d A combination of the estimated density functions for the tRNAs (blue), the annotated genes (green) and the ncRNAs (red) of the former panels, shown a substantial overlap between the annotated genes and the ncRNAs supposedly non-coding
Table 1.
Transcriptome and translatome profiles of 115 ncRNAs known from E. coli O157:H7 EDL933
| Name | Start position in the genome | Length | Strand | Number of transcriptome reads | Number of footprint reads | RPKM transcriptome | RPKM footprints | RCV | P value* | Northern Blot/Shine Dalgarno |
|---|---|---|---|---|---|---|---|---|---|---|
| DicF_1/Z1327 | 1255006 | 52 | - | 2 | 7 | 2 | 16 | 8.00 | 1.55E-11 | |
| STnc70 | 719959 | 94 | + | 47 | 141 | 28 | 182 | 6.50 | 4.83E-11 | |
| RyhB | 4367464 | 65 | - | 92 | 192 | 80 | 359 | 4.49 | 1.77E-09 | |
| OmrA-B_2 | 3766084 | 82 | - | 504 | 844 | 348 | 1251 | 3.59 | 1.47E-08 | |
| OrzO-P_2 | 2954314 | 76 | + | 5057 | 8198 | 3764 | 13114 | 3.48 | 1.97E-08 | taaagtggt |
| STnc100_10 | 2995675 | 210 | - | 496 | 742 | 134 | 430 | 3.21 | 4.12E-08 | tatgggata |
| STnc550 | 2412748 | 391 | - | 533 | 779 | 77 | 242 | 3.14 | 4.96E-08 | caaatagtg |
| RtT_3 | 1824178 | 132 | - | 22 | 28 | 9 | 26 | 2.89 | 1.03E-07 | |
| RprA | 2445280 | 108 | + | 568 | 745 | 297 | 839 | 2.82 | 1.25E-07 | |
| STnc180 | 2250970 | 203 | - | 1225 | 1534 | 341 | 919 | 2.70 | 1.86E-07 | caagcgggg |
| GadY | 4474223 | 114 | + | 213 | 248 | 106 | 264 | 2.49 | 3.55E-07 | |
| STnc630 | 5216481 | 166 | + | 502 | 572 | 171 | 419 | 2.45 | 4.05E-07 | aacggagga |
| STnc100_1 | 902843 | 159 | + | 1046 | 1049 | 372 | 802 | 2.16 | 1.11E-06 | |
| CyaR_RyeE | 2912765 | 86 | + | 16620 | 16668 | 10932 | 23563 | 2.16 | 1.11E-06 | |
| sroE | 3426663 | 92 | - | 64 | 63 | 39 | 83 | 2.13 | 1.22E-06 | |
| Z6077/DicF_4 | 2325956 | 52 | + | 118 | 112 | 128 | 262 | 2.05 | 1.64E-06 | |
| C0299 | 1763522 | 79 | + | 1 | 1 | 1 | 2 | 2.00 | 1.96E-06 | |
| RtT_2 | 1824000 | 132 | - | 3 | 2 | 1 | 2 | 2.00 | 1.96E-06 | gaccaaggt |
| QUAD_7 | 4002118 | 150 | - | 859 | 791 | 324 | 641 | 1.98 | 2.12E-06 | |
| tpke11 | 14107 | 78 | + | 59 | 51 | 43 | 79 | 1.84 | 3.64E-06 | |
| STnc100_5 | 1866224 | 209 | + | 5038 | 4068 | 1364 | 2366 | 1.73 | 5.48E-06 | |
| MicA | 3606250 | 72 | + | 1500 | 1180 | 1178 | 1992 | 1.69 | 6.54E-06 | |
| STnc100_3 | 1353605 | 206 | + | 2403 | 1688 | 660 | 996 | 1.51 | 1.41E-05 | |
| sroD | 2565135 | 86 | - | 94 | 65 | 62 | 92 | 1.48 | 1.58E-05 | |
| MicC | 2113860 | 122 | - | 43 | 29 | 20 | 29 | 1.45 | 1.83E-05 | |
| frnS | 2168565 | 118 | - | 175 | 106 | 84 | 109 | 1.30 | 3.70E-05 | tcagggcaa |
| OmrA-B_1 | 3765887 | 88 | - | 696 | 380 | 447 | 525 | 1.17 | 6.73E-05 | |
| ArcZ | 4160147 | 108 | + | 3234 | 1708 | 1694 | 1923 | 1.14 | 8.20E-05 | |
| STnc130 | 1161203 | 135 | - | 2 | 1 | 1 | 1 | 1.00 | 1.66E-04 | |
| STnc560 | 1939628 | 214 | + | 132 | 58 | 35 | 33 | 0.94 | 2.27E-04 | |
| sraL | 5161197 | 141 | - | 627 | 265 | 252 | 228 | 0.90 | 2.81E-04 | |
| RydB | 2439675 | 61 | - | 280 | 102 | 260 | 203 | 0.78 | 5.76E-04 | |
| RtT_4 | 1824474 | 131 | - | 30 | 10 | 13 | 9 | 0.69 | 9.91E-04 | |
| sroC | 767984 | 163 | - | 3945 | 1269 | 1369 | 946 | 0.69 | 9.99E-04 | |
| CRISPR-DR4_2 | 1058550 | 28 | + | 3 | 1 | 6 | 4 | 0.67 | 1.16E-03 | |
| STnc100_2 | 1267542 | 167 | + | 3718 | 1129 | 1259 | 822 | 0.65 | 1.27E-03 | |
| sok_15/sokX | 3674872 | 152 | - | 93 | 28 | 35 | 22 | 0.63 | 1.49E-03 | tcaggtata |
| STnc100_4 | 1641323 | 191 | + | 4486 | 1215 | 1329 | 773 | 0.58 | 2.02E-03 | positive |
| GcvB | 3732394 | 206 | + | 13532 | 3307 | 3716 | 1952 | 0.53 | 2.96E-03 | negative/tgagccgga |
| Spot_42/spf | 4914606 | 119 | + | 323 | 77 | 154 | 79 | 0.51 | 3.22E-03 | gtagggtac |
| STnc450 | 5326800 | 58 | - | 20 | 5 | 20 | 10 | 0.50 | 3.52E-03 | |
| CRISPR-DR4_1 | 1058490 | 28 | + | 4 | 1 | 8 | 4 | 0.50 | 3.52E-03 | |
| STAXI_4 | 1482887 | 131 | + | 4 | 1 | 2 | 1 | 0.50 | 3.52E-03 | |
| RybB | 1014999 | 79 | - | 1953 | 439 | 1398 | 676 | 0.48 | 3.95E-03 | gcagggcat |
| sroB | 572997 | 84 | + | 704 | 151 | 474 | 219 | 0.46 | 4.59E-03 | |
| P26 | 5058572 | 62 | + | 261 | 52 | 238 | 102 | 0.43 | 5.83E-03 | |
| sok_14 | 2777459 | 175 | - | 1539 | 298 | 497 | 207 | 0.42 | 6.35E-03 | tgaggccca |
| sroH | 5068058 | 161 | - | 606 | 114 | 213 | 86 | 0.40 | 6.97E-03 | |
| DicF_2 | 1881271 | 52 | - | 5 | 1 | 5 | 2 | 0.40 | 7.16E-03 | |
| rdlD_3 | 1807675 | 60 | + | 58 | 10 | 55 | 20 | 0.36 | 9.36E-03 | |
| OrzO-P_1 | 2953705 | 74 | + | 7227 | 1195 | 5524 | 1963 | 0.36 | 9.96E-03 | |
| sok_10 | 1888482 | 175 | + | 3663 | 598 | 1184 | 415 | 0.35 | 1.03E-02 | tgaggctca |
| ryfA | 3444344 | 305 | + | 16 | 3 | 3 | 1 | 0.33 | 1.18E-02 | |
| IS061 | 2172064 | 180 | - | 10 | 1 | 3 | 1 | 0.33 | 1.18E-02 | |
| rdlD_4 | 4509509 | 66 | + | 78 | 11 | 67 | 20 | 0.30 | 1.49E-02 | |
| rdlD_2 | 1807146 | 60 | + | 59 | 8 | 56 | 16 | 0.30 | 0.02 | |
| sok_7 | 1480784 | 158 | + | 2602 | 366 | 932 | 282 | 0.29 | 0.02 | |
| RyeB | 2600241 | 100 | - | 2380 | 314 | 1346 | 382 | 0.28 | 0.02 | |
| QUAD_1 | 2898598 | 149 | + | 358 | 47 | 136 | 38 | 0.28 | 0.02 | |
| MicF | 3117339 | 94 | + | 1059 | 132 | 637 | 171 | 0.27 | 0.02 | |
| STnc100_6 | 1893978 | 190 | + | 6373 | 703 | 1897 | 450 | 0.24 | 0.03 | |
| OxyS | 5033797 | 110 | - | 106 | 11 | 55 | 12 | 0.22 | 0.03 | |
| arrS | 4467416 | 69 | - | 266 | 22 | 201 | 36 | 0.18 | 0.04 | |
| istR | 4712705 | 130 | - | 99 | 8 | 43 | 7 | 0.16 | 0.05 | |
| SraB | 1590770 | 169 | + | 511 | 38 | 171 | 27 | 0.16 | 0.05 | |
| QUAD_6 | 4001742 | 150 | - | 771 | 54 | 291 | 44 | 0.15 | 0.06 | |
| DsrA | 2725072 | 87 | - | 82 | 6 | 53 | 8 | 0.15 | 0.06 | |
| StyR-44_7 | 5087479 | 109 | + | 1784 | 125 | 926 | 139 | 0.15 | 0.06 | |
| QUAD_5 | 3861645 | 151 | + | 1621 | 113 | 607 | 91 | 0.15 | 0.06 | |
| StyR-44_5 | 4902290 | 109 | + | 1846 | 127 | 958 | 142 | 0.15 | 0.06 | |
| QUAD_4 | 3861252 | 151 | + | 2395 | 153 | 897 | 123 | 0.14 | 0.07 | |
| StyR-44_4 | 4806012 | 109 | + | 1761 | 112 | 914 | 125 | 0.14 | 0.07 | |
| StyR-44_1 | 228975 | 109 | + | 1908 | 111 | 990 | 124 | 0.13 | 0.08 | |
| STnc240 | 2830003 | 75 | - | 112 | 6 | 84 | 10 | 0.12 | 0.08 | |
| Bacteria_small_SRP /ffs | 542524 | 97 | + | 230378 | 12741 | 134343 | 15969 | 0.12 | 0.08 | positive |
| STnc100_9 | 2773346 | 167 | - | 3475 | 184 | 1177 | 134 | 0.11 | 0.09 | |
| GlmZ_SraJ_2 | 4848834 | 207 | + | 7351 | 364 | 2009 | 214 | 0.11 | 0.10 | positive |
| SraC_RyeA | 2600138 | 145 | + | 2011 | 91 | 784 | 76 | 0.10 | 0.12 | |
| GlmY_tke1_2 | 4848836 | 149 | + | 7310 | 323 | 2775 | 264 | 0.10 | 0.12 | |
| StyR-44_6 | 5046470 | 109 | + | 4004 | 161 | 2078 | 180 | 0.09 | 0.14 | |
| STnc100_8 | 2314989 | 167 | - | 706 | 23 | 239 | 17 | 0.07 | 0.19 | |
| RtT_1 | 867059 | 143 | + | 3357 | 102 | 1328 | 87 | 0.07 | 0.21 | |
| C4_2 | 2673794 | 88 | + | 108363 | 3042 | 69654 | 4203 | 0.06 | 0.23 | |
| sok_6 | 1389612 | 175 | - | 934 | 22 | 302 | 15 | 0.05 | 0.29 | |
| STnc100_7 | 2145571 | 190 | - | 327 | 7 | 97 | 4 | 0.04 | 0.35 | |
| CsrB | 3714213 | 360 | - | 43044 | 748 | 6763 | 253 | 0.04 | 0.38 | |
| CsrC | 4915753 | 254 | + | 25764 | 425 | 5738 | 203 | 0.04 | 0.40 | |
| RydC | 2079463 | 64 | + | 1636 | 27 | 1446 | 51 | 0.04 | 0.40 | |
| RNaseP_bact_a /rnpB | 4077043 | 377 | - | 39359 | 640 | 5905 | 206 | 0.03 | 0.40 | |
| GlmZ_SraJ_1 | 3481543 | 185 | - | 7668 | 122 | 2345 | 80 | 0.03 | 0.41 | |
| GlmY_tke1_1 | 3481544 | 148 | - | 7634 | 119 | 2918 | 98 | 0.03 | 0.42 | |
| 6S/ssrS | 3860420 | 184 | + | 470148 | 7239 | 144532 | 4783 | 0.03 | 0.42 | |
| QUAD_3 | 2899260 | 144 | + | 3436 | 44 | 1350 | 37 | 0.03 | 0.47 | |
| symR | 5467620 | 77 | + | 726 | 6 | 533 | 9 | 0.02 | 0.60 | |
| sRNA-Xcc1 | 1392052 | 89 | - | 40293 | 290 | 25609 | 396 | 0.02 | 0.62 | |
| rdlD_1 | 1806611 | 66 | + | 2090 | 8 | 1791 | 15 | 0.01 | 0.76 | |
| StyR-44_3 | 4229125 | 109 | - | 2523 | 2 | 1309 | 2 | 0.00 | 0.96 | |
| StyR-44_2 | 3519339 | 109 | - | 2499 | 1 | 1297 | 1 | 0.00 | 0.97 | |
| HPnc0260 | 2421623 | 163 | - | 1 | 0 | 0 | 0 | N/A | N/A | |
| rseX | 2733408 | 90 | + | 4 | 0 | 3 | 0 | N/A | N/A | |
| sok_12 | 2152486 | 125 | - | 13 | 0 | 6 | 0 | N/A | N/A | |
| SraG | 4120940 | 172 | + | 1 | 0 | 0 | 0 | N/A | N/A | |
| STAXI_1 | 1087216 | 64 | + | 6 | 0 | 5 | 0 | N/A | N/A | |
| STAXI_2 | 1087280 | 131 | + | 2 | 0 | 1 | 0 | N/A | N/A | |
| STAXI_3 | 1482823 | 64 | + | 3 | 0 | 3 | 0 | N/A | N/A | |
| STnc100_11 | 3553828 | 189 | - | 387 | 0 | 116 | 0 | N/A | N/A | |
| STnc410 | 4777710 | 158 | + | 3 | 0 | 1 | 0 | N/A | N/A | |
| tp2 | 127504 | 114 | - | 1 | 0 | 0 | 0 | N/A | N/A | |
| sraA | 524870 | 96 | - | 0 | 0 | 0 | 0 | N/A | N/A | |
| STnc480 | 635390 | 67 | + | 0 | 0 | 0 | 0 | N/A | N/A | |
| sar | 1661162 | 67 | - | 0 | 0 | 0 | 0 | N/A | N/A | |
| group-II-D1D4-2 | 2037712 | 118 | - | 0 | 0 | 0 | 0 | N/A | N/A | |
| DicF_3 | 2159230 | 56 | + | 0 | 0 | 0 | 0 | N/A | N/A | |
| C0465 | 2649880 | 76 | + | 0 | 0 | 0 | 0 | N/A | N/A | |
| STnc430 | 5118969 | 150 | - | 0 | 0 | 0 | 0 | N/A | N/A |
*; The P values give the probability that the RCV of the given RNA is similar to / results from the RCV distribution of the tRNAs. Thus, RNAs with high P values are probably not translated and vice versa
Annotated ncRNAs which are not independent of translation (e.g. leader peptides or ribosomal RNAs, etc.) are not shown (see text). The genome position (start) of each ncRNA is indicated, the ncRNAs are sorted according to their RCV. Transcripts examined via Northern blots are indicated and putative Shine-Dalgarno sequences are shown. An overview of all data for the 115 known ncRNAs is found in Additional file 8: Table S6
Using the RCV limits mentioned in the methods section (i.e., RCV <0.197), 150 putative ncRNAs were discovered of which three examples are shown in Fig. 2. All novel ncRNA candidates are listed in Table 2, including the read counts, RPKM values and RCV values for each transcript. The putative novel ncRNAs range between 27 and 268 nt with an average size of 77 nt. One (ncR3609372) had a match in the Rfam database [56] as being a tRNA. We analyzed these transcripts to see whether they contained a potentially protein coding ORF. Of the 150 identified transcripts, 44 do not contain any ORF at all and only a minority of 6 candidates contains a putative ORF coding for more than 30 amino acids, indicating that most transcripts identified are truly non-coding. This agrees with the fact that all RCVs are below the threshold for translation. The RPKM-transcriptome values of the novel ncRNA transcripts range between 8 and 8857, the average being 198 (Table 2).
Fig. 2.
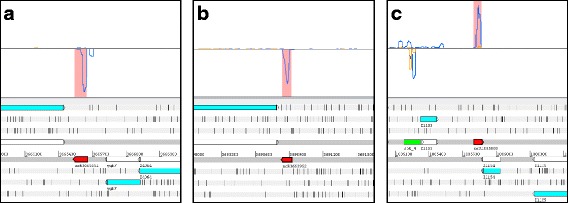
Three examples of novel ncRNAs detected using transcriptome and translatome analysis. A genomic area is visualized in Artemis 15.0.0 [43]. In the lower part of the panels, the genome (shown as grey lines) is visualized in a six-frame translation mode. Numbers given between the grey lines indicate the genome coordinates. On top of the forward strand are three reading frames and on the reverse DNA strand are three further reading frames. Each reading frame represented is visible by the indicated stop codons (vertical black bars). Annotated genes are shown in their respective reading frame (turquoise arrows) and also on the DNA strand itself (white arrows). The gene name is written below each arrow. Any protein-coding ORF must be at least located between two black bars, with the downstream stop codon being the translational stop. In the upper part of the panels, the DNA is indicated by a thin black line and the sequencing reads matching to the forward or reverse strand are shown above or below this line. The sequencing reads from the footprint (yellow line) and transcriptome (blue line) sequencing are shown as coverage plot, respectively. The pink shaded area in the coverage plot corresponds to the novel ncRNAs, which are drawn in by red arrows. Novel ncRNAs were identified by their very low RCV, thus, hardly any footprint reads (in yellow) but a number of transcriptome reads (in blue; see Table 2). Known ncRNAs are indicated on the DNA by a bright green arrow. Since ncRNAs supposedly do not contain a protein-coding ORF, these genes are only shown on the DNA. a ncR3665651. b ncR3690952. c ncR1085800
Table 2.
Novel non-coding RNA (ncRNA) candidates (150 in total) based on transcriptome sequencing and ribosomal profiling. ncRNAs are identified by their start position on the genome given in the name (abbreviated as ncR#)
| Name | Length | Max. ORF (nt) | Strand | Number of transcriptome reads | Number of footprint reads | RPKM transcriptome | RPKM footprints | RCV | Northern Blot | CopraRNA | CopraRNA term |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ncR1085800 | 72 | - | + | 11274 | 532 | 8857 | 898 | 0.1 | positive | ||
| ncR1481381 | 99 | 9 | + | 11208 | 516 | 6404 | 634 | 0.1 | positive | ||
| ncR3690952 | 145 | 54 | - | 4898 | 26 | 1911 | 22 | 0.01 | negative | 1.18 | ecc00620:Pyruvate metabolism |
| ncR1636218 | 78 | 27 | - | 1034 | 22 | 750 | 34 | 0.05 | negative | 1.31a | GO:0042364~water-soluble vitamin biosynthetic process |
| ncR3545929 | 41 | - | + | 542 | 2 | 748 | 6 | 0.01 | |||
| ncR3665651 | 109 | 45 | - | 1442 | 2 | 748 | 2 | 0 | negative | 1.24 | GO:0045426~quinone cofactor biosynthetic process |
| ncR3860554 | 73 | 24 | - | 637 | 3 | 494 | 5 | 0.01 | 0.98 | 2Fe-2S | |
| ncR1088953 | 37 | - | + | 295 | 4 | 451 | 13 | 0.03 | |||
| ncR3066135 | 48 | - | + | 354 | 0 | 417 | 0 | 0 | |||
| ncR1484560 | 42 | - | + | 294 | 0 | 396 | 0 | 0 | |||
| ncR5355946 | 47 | 21 | - | 246 | 4 | 296 | 10 | 0.04 | |||
| ncR16920 | 36 | - | - | 164 | 4 | 258 | 14 | 0.05 | |||
| ncR165975 | 49 | 9 | + | 216 | 4 | 249 | 10 | 0.04 | negative | ||
| ncR5223290 | 66 | - | + | 265 | 3 | 227 | 6 | 0.02 | |||
| ncR1641710 | 114 | 105 | - | 430 | 28 | 213 | 30 | 0.14 | |||
| ncR1765944 | 114 | 54 | + | 406 | 33 | 201 | 35 | 0.18 | 5.48a | membrane | |
| ncR2358348 | 45 | - | - | 151 | 1 | 190 | 3 | 0.01 | |||
| ncR1888606 | 46 | 18 | - | 147 | 4 | 181 | 11 | 0.06 | |||
| ncR2530362 | 64 | 6 | - | 196 | 0 | 173 | 0 | 0 | |||
| ncR5133665 | 29 | - | - | 86 | 0 | 168 | 0 | 0 | |||
| ncR2638864 | 38 | - | - | 100 | 2 | 149 | 6 | 0.04 | |||
| ncR326492 | 42 | - | + | 110 | 9 | 148 | 26 | 0.18 | |||
| ncR622277 | 95 | 63 | + | 248 | 1 | 148 | 1 | 0.01 | negative | ||
| ncR2549762 | 59 | 45 | - | 150 | 2 | 144 | 4 | 0.03 | |||
| ncR2287 | 90 | 45 | - | 218 | 8 | 137 | 11 | 0.08 | |||
| ncR1019437 | 57 | - | - | 130 | 2 | 129 | 4 | 0.03 | |||
| ncR1694864 | 51 | 18 | + | 116 | 1 | 129 | 2 | 0.02 | |||
| ncR1864748 | 174 | 105 | - | 370 | 32 | 120 | 22 | 0.19 | 4.00a | cell membrane | |
| ncR3526958 | 96 | 45 | - | 193 | 9 | 114 | 11 | 0.1 | 2.92a | ecd00190:Oxidative phosphorylation | |
| ncR867065 | 45 | 21 | - | 76 | 1 | 96 | 3 | 0.03 | |||
| ncR1079732 | 27 | - | - | 41 | 2 | 86 | 9 | 0.11 | |||
| ncR3020266 | 116 | 57 | + | 168 | 9 | 82 | 9 | 0.12 | |||
| ncR1328373 | 40 | 21 | - | 55 | 1 | 78 | 3 | 0.04 | |||
| ncR3094200 | 36 | 9 | + | 47 | 3 | 74 | 10 | 0.14 | |||
| ncR774709 | 58 | 39 | + | 74 | 1 | 72 | 2 | 0.03 | |||
| ncR3725111 | 44 | - | - | 52 | 1 | 67 | 3 | 0.04 | |||
| ncR451977 | 43 | 21 | - | 50 | 3 | 66 | 8 | 0.13 | |||
| ncR4881271 | 105 | 75 | + | 123 | 7 | 66 | 8 | 0.12 | 1.70a | GO:0022900~electron transport chain | |
| ncR4922734 | 44 | 27 | + | 49 | 2 | 63 | 6 | 0.09 | |||
| ncR1748457 | 38 | 9 | - | 40 | 0 | 60 | 0 | 0 | |||
| ncR4393950 | 74 | - | - | 78 | 5 | 60 | 8 | 0.14 | |||
| ncR5324582 | 92 | 24 | + | 94 | 4 | 58 | 5 | 0.09 | |||
| ncR1847082 | 38 | - | + | 38 | 1 | 57 | 3 | 0.06 | |||
| ncR2820623 | 107 | 66 | - | 105 | 9 | 56 | 10 | 0.18 | 1.89a | lipoprotein | |
| ncR1114186 | 94 | 60 | - | 91 | 3 | 55 | 4 | 0.07 | |||
| ncR3583650 | 35 | - | - | 34 | 1 | 55 | 3 | 0.06 | |||
| ncR1509794 | 96 | 60 | - | 91 | 5 | 54 | 6 | 0.12 | |||
| ncR4391372 | 28 | - | - | 26 | 2 | 53 | 9 | 0.17 | |||
| ncR4546182 | 36 | 12 | - | 34 | 2 | 53 | 7 | 0.13 | |||
| ncR612919 | 36 | - | + | 32 | 0 | 50 | 0 | 0 | |||
| ncR426804 | 47 | - | + | 41 | 3 | 49 | 8 | 0.16 | |||
| ncR3164662 | 66 | 9 | + | 56 | 3 | 48 | 6 | 0.12 | |||
| ncR2585184 | 44 | - | + | 36 | 1 | 46 | 3 | 0.06 | |||
| ncR2930972 | 38 | 6 | - | 30 | 2 | 45 | 6 | 0.14 | |||
| ncR3527530 | 119 | 51 | - | 95 | 4 | 45 | 4 | 0.09 | 1.97a | GO:0046395~carboxylic acid catabolic process | |
| ncR4520884 | 50 | 24 | + | 40 | 3 | 45 | 7 | 0.16 | |||
| ncR2498369 | 53 | 33 | - | 41 | 1 | 44 | 2 | 0.05 | |||
| ncR4161484 | 42 | 39 | + | 33 | 1 | 44 | 3 | 0.07 | |||
| ncR2699447 | 35 | 18 | + | 26 | 1 | 42 | 3 | 0.08 | |||
| ncR5210782 | 28 | 18 | + | 21 | 0 | 42 | 0 | 0 | |||
| ncR205409 | 74 | 45 | - | 53 | 0 | 41 | 0 | 0 | 1.02 | IPR014021:Helicase, superfamily 1 and 2, ATP-binding | |
| ncR1868696 | 103 | 30 | - | 72 | 6 | 40 | 7 | 0.18 | |||
| ncR3915561 | 37 | - | - | 26 | 2 | 40 | 7 | 0.17 | |||
| ncR1462015 | 40 | 21 | - | 27 | 0 | 38 | 0 | 0 | |||
| ncR1475353 | 73 | - | - | 48 | 4 | 37 | 7 | 0.18 | |||
| ncR397399 | 68 | - | - | 44 | 0 | 37 | 0 | 0 | |||
| ncR4645569 | 57 | 15 | - | 36 | 0 | 36 | 0 | 0 | |||
| ncR1239030 | 54 | - | + | 33 | 1 | 35 | 2 | 0.07 | |||
| ncR1645154 | 42 | - | - | 26 | 1 | 35 | 3 | 0.08 | |||
| ncR3553461 | 57 | 33 | + | 35 | 1 | 35 | 2 | 0.06 | |||
| ncR4853400 | 65 | 15 | + | 40 | 3 | 35 | 6 | 0.16 | |||
| ncR1143400 | 43 | 18 | + | 25 | 2 | 33 | 6 | 0.17 | |||
| ncR2693045 | 49 | 6 | - | 29 | 2 | 33 | 5 | 0.15 | |||
| ncR3735643 | 69 | 45 | + | 40 | 2 | 33 | 4 | 0.11 | |||
| ncR3991822 | 44 | 9 | - | 26 | 0 | 33 | 0 | 0 | |||
| ncR4714439 | 67 | 6 | - | 39 | 2 | 33 | 4 | 0.11 | |||
| ncR1960332 | 35 | - | + | 20 | 1 | 32 | 3 | 0.11 | 1.96a | GO:0034660~ncRNA metabolic process | |
| ncR2885483 | 44 | - | - | 25 | 1 | 32 | 3 | 0.09 | |||
| ncR4463425 | 63 | 12 | - | 36 | 1 | 32 | 2 | 0.06 | |||
| ncR501481 | 70 | 21 | + | 39 | 0 | 32 | 0 | 0 | |||
| ncR963596 | 50 | 9 | - | 28 | 1 | 32 | 2 | 0.08 | |||
| ncR1152534 | 90 | 51 | + | 50 | 4 | 31 | 5 | 0.17 | 0.84 | ecq00052:Galactose metabolism | |
| ncR2602372 | 62 | 12 | - | 34 | 2 | 31 | 4 | 0.13 | |||
| ncR2062548 | 38 | - | + | 20 | 0 | 30 | 0 | 0 | |||
| ncR4770438 | 40 | 12 | + | 21 | 1 | 30 | 3 | 0.1 | |||
| ncR865067 | 103 | 51 | - | 54 | 1 | 30 | 1 | 0.04 | |||
| ncR11537 | 73 | 48 | + | 37 | 2 | 29 | 3 | 0.12 | |||
| ncR3040352 | 77 | 18 | - | 40 | 3 | 29 | 5 | 0.16 | |||
| ncR4249267 | 64 | 9 | + | 33 | 2 | 29 | 4 | 0.13 | |||
| ncR1592436 | 48 | 9 | - | 24 | 1 | 28 | 3 | 0.09 | |||
| ncR3583545 | 40 | - | - | 20 | 1 | 28 | 3 | 0.11 | |||
| ncR725615 | 61 | - | + | 30 | 2 | 28 | 4 | 0.14 | |||
| ncR1066434 | 66 | 21 | - | 32 | 1 | 27 | 2 | 0.07 | |||
| ncR4163613 | 125 | 45 | + | 59 | 5 | 27 | 5 | 0.18 | 1.29 | antibiotic resistance | |
| ncR15950 | 44 | 18 | + | 20 | 0 | 26 | 0 | 0 | |||
| ncR2452385 | 45 | 18 | - | 21 | 0 | 26 | 0 | 0 | |||
| ncR2841773 | 79 | 18 | + | 37 | 0 | 26 | 0 | 0 | |||
| ncR3320428 | 75 | 51 | + | 34 | 1 | 26 | 2 | 0.06 | |||
| ncR2903620 | 140 | 33 | - | 61 | 3 | 25 | 3 | 0.11 | |||
| ncR3042903 | 164 | 57 | - | 69 | 3 | 24 | 2 | 0.09 | |||
| ncR543583 | 67 | 51 | + | 29 | 0 | 24 | 0 | 0 | |||
| ncR1585703 | 88 | 48 | + | 36 | 2 | 23 | 3 | 0.12 | |||
| ncR1999946 | 51 | 15 | + | 21 | 0 | 23 | 0 | 0 | |||
| ncR5077759 | 50 | 6 | - | 20 | 1 | 23 | 2 | 0.11 | |||
| ncR1640190 | 56 | - | - | 22 | 2 | 22 | 4 | 0.2 | |||
| ncR1752346 | 64 | - | + | 25 | 0 | 22 | 0 | 0 | |||
| ncR3074598 | 54 | 36 | - | 21 | 0 | 22 | 0 | 0 | |||
| ncR3320341 | 85 | 18 | + | 33 | 0 | 22 | 0 | 0 | |||
| ncR3330508 | 57 | - | - | 22 | 0 | 22 | 0 | 0 | |||
| ncR4096595 | 60 | - | - | 23 | 2 | 22 | 4 | 0.19 | |||
| ncR4137844 | 268 | 195 | - | 102 | 7 | 22 | 3 | 0.15 | |||
| ncR4414172 | 53 | - | - | 21 | 0 | 22 | 0 | 0 | |||
| ncR492696 | 51 | - | - | 20 | 0 | 22 | 0 | 0 | |||
| ncR1215540 | 74 | 21 | + | 28 | 2 | 21 | 3 | 0.15 | |||
| ncR2254917 | 76 | - | + | 28 | 2 | 21 | 3 | 0.15 | |||
| ncR2902855 | 104 | - | - | 39 | 3 | 21 | 4 | 0.17 | |||
| ncR752395 | 172 | 66 | + | 64 | 2 | 21 | 1 | 0.07 | |||
| ncR283226 | 71 | 27 | + | 25 | 0 | 20 | 0 | 0 | |||
| ncR1049002 | 77 | 48 | - | 26 | 1 | 19 | 2 | 0.08 | 1.84a | GO:0050890~cognition | |
| ncR1216838 | 100 | 12 | - | 33 | 1 | 19 | 1 | 0.07 | |||
| ncR4829752 | 78 | - | - | 26 | 1 | 19 | 2 | 0.08 | |||
| ncR1483108 | 77 | 63 | + | 23 | 1 | 17 | 2 | 0.09 | |||
| ncR155024 | 78 | 18 | + | 23 | 0 | 17 | 0 | 0 | |||
| ncR4156147 | 78 | - | - | 23 | 1 | 17 | 2 | 0.09 | 1.75a | GO:0032196~transposition | |
| ncR4788281 | 97 | 78 | - | 30 | 0 | 17 | 0 | 0 | 0.99 | topological domain:Periplasmic | |
| ncR1854285 | 91 | 48 | - | 25 | 1 | 16 | 1 | 0.09 | |||
| ncR1942672 | 85 | - | + | 24 | 1 | 16 | 1 | 0.09 | 2.09a | GO:0015031~protein transport | |
| ncR2614043 | 86 | 57 | - | 25 | 0 | 16 | 0 | 0 | |||
| ncR4741832 | 75 | 30 | - | 21 | 0 | 16 | 0 | 0 | |||
| ncR484751 | 94 | 51 | - | 27 | 2 | 16 | 3 | 0.16 | |||
| ncR5283975 | 98 | 84 | - | 28 | 0 | 16 | 0 | 0 | 2.14a | GO:0009386~translational attenuation | |
| ncR1314229 | 101 | 48 | + | 26 | 2 | 15 | 2 | 0.17 | |||
| ncR1893573 | 187 | 87 | - | 48 | 4 | 15 | 3 | 0.18 | |||
| ncR3609575 | 74 | - | + | 20 | 0 | 15 | 0 | 0 | 1.72a | eum00660:C5-Branched dibasic acid metabolism | |
| ncR3724967 | 105 | 42 | - | 28 | 0 | 15 | 0 | 0 | |||
| ncR4245110 | 118 | 48 | + | 30 | 1 | 14 | 1 | 0.07 | |||
| ncR866957 | 79 | 42 | - | 20 | 0 | 14 | 0 | 0 | |||
| ncR336953 | 96 | 27 | + | 22 | 2 | 13 | 3 | 0.2 | |||
| ncR3997954 | 104 | 81 | + | 23 | 2 | 13 | 2 | 0.19 | |||
| ncR411208 | 99 | 6 | - | 22 | 2 | 13 | 2 | 0.2 | 2.64a | GO:0015980~energy derivation by oxidation of organic compounds | |
| ncR1736783 | 100 | 57 | - | 22 | 1 | 12 | 1 | 0.1 | |||
| ncR745748 | 107 | 54 | + | 23 | 1 | 12 | 1 | 0.09 | |||
| ncR3609372 | 201 | - | + | 38 | 1 | 11 | 1 | 0.06 | Rfam match: tRNA RF00005b | ||
| ncR3890479 | 104 | 51 | - | 20 | 1 | 11 | 1 | 0.11 | |||
| ncR1087411 | 224 | 135 | + | 41 | 2 | 10 | 1 | 0.11 | |||
| ncR3874837 | 127 | 48 | - | 22 | 2 | 10 | 2 | 0.2 | |||
| ncR4187491 | 122 | 9 | - | 22 | 0 | 10 | 0 | 0 | |||
| ncR769665 | 198 | 102 | + | 34 | 2 | 10 | 1 | 0.13 | |||
| ncR5157894 | 218 | 114 | + | 33 | 3 | 9 | 2 | 0.2 | |||
| ncR196880 | 135 | 81 | - | 20 | 1 | 8 | 1 | 0.11 | 1.23 | ecc00330:Arginine and proline metabolism |
aDAVID enrich. score; signif. ≥ 1.3. btRNA
The longest potential ORF is indicated for each ncRNA. RPKM values of transcriptome and translatome are shown, as well as the ribosome coverage value (RCV). A transcript is considered non-coding if it has at least 20 reads in the transcriptome data and the RCV is below 0.197 (α = 0.01). Transcripts examined via Northern blots are indicated and CopraRNA functional enrichments are shown
Presence of novel ncRNAs in E. coli K12
In E. coli O157:H7 EDL933, 329 ncRNAs have been annotated [2], but various bioinformatic studies suggest the existence of up to 1000 ncRNAs in E. coli (e.g. [8–11]) and probably in other bacteria as well (e.g. [19, 79]). Our current study presents even under a single growth condition 150 new ncRNA candidates. For comparison, we determined the presence of corresponding regions in the E. coli K12 strain MG1655. We found 102 of 150 novel ncRNAs regions present in MG1655. Next, we searched data of prokaryotes having both, transcriptome and translatome data of the same experiment. Only a single study was published by the Weissman group of MG1655 grown in MOPS glucose medium [80]. In addition, the ArrayExpress database contains a further dataset of MG1655 grown in LB (E-MTAB-2903). In MOPS medium with glucose at OD 0.3 and in LB medium at an OD of about 0.5, 43 and 66 of the 102 putative ncRNAs were found to be transcribed in MG1655, respectively. Combining both datasets confirmed transcription (without translation) of 74 of the 102 ncRNAs under either condition in E. coli MG1655 (Additional file 7: Table S5).
Detection of ncRNAs by Northern blots
To verify the existence of at least some annotated ncRNAs, Northern blot analysis was conducted for five of the annotated ncRNAs of different length and strength. Three were verified, namely ffs, sraJ, and STnc100_4 (Table 1 and Fig. 3). We then chose seven exemplary novel ncRNAs to be confirmed using Northern blots. However, of the novel ncRNAs only the two transcripts with the highest RPKM in the transcriptome of 8857 and 6404 could be verified as sum signal since they are indistinguishable on the basis of Northern blots (Fig. 3). Obviously, Northern blots have a certain detection limit. Under the conditions applied in this study, any RNA required an RPKM value of about 2000 to be detectable. RNAs transcribed at lower levels were not detected via hybridization. A sufficiently high number of RNA molecules are needed to generate a signal passing the detection threshold, a problem also common to microarrays [81, 82].
Fig. 3.
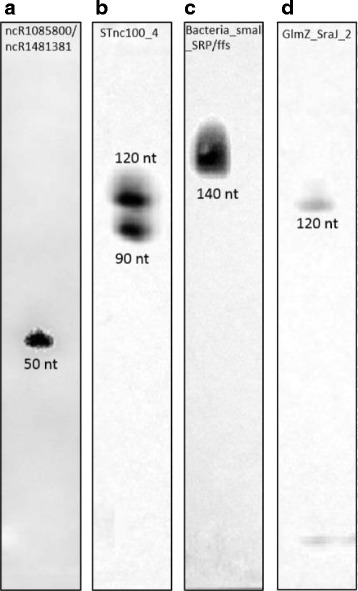
Detection of novel and annotated ncRNAs by Northern blots. Since ncRNAs do not have defined ends like, e.g., ORFs which have start and stop codons, their actual length may differ somewhat from the expected length (compare to Table 1). The contrast of the bands has been adjusted by gamma correction using digital image processing for better visibility. a ncR1085800 and ncR1481381. Both ncRNAs are indistinguishable by their sequence. b STnc100_4. c Bacteria_small SRP/ffs. d GlmZ_SraJ_2
Putative functions and differential expression of the novel ncRNAs
To examine putative functions of the novel ncRNA candidates, we used the sRNA target prediction tool CopraRNA [59, 60]. For 14 of the 150 novel ncRNAs, a significant functional enrichment was found (Table 2). The targets include a diversity of metabolic and regulatory functions within the cell, e.g., synthesis pathways of amino acids and vitamins; but also respiratory functions and oxidation of components etc.
Interestingly, 121 of the novel ncRNAs were found to be expressed (i.e., ≥ 10 RPKM, which is ≥ 100-fold above background) in the data of a former study [17], when grown in eleven different growth conditions for at least one condition. Forty-six novel ncRNAs revealed 4-fold differential expression in at least one other condition when compared to plain LB. Example data are given in Table 3, the full data set can be found in the Additional file 7: Table S5. Combining these findings of CopraRNA predictions (14), Rfam match (1), expression (121), and regulation (46) suggests that at least 126 out of 150 putative ncRNAs are not just a random by-product of pervasive transcriptional activity, but might fulfill specific functions in the cell.
Table 3.
Expression of exemplary novel ncRNAs under 11 different growth conditions (MM, minimal medium)
| Name | Length [nt] | LB plain | MM | LB + nitrite | LB pH9 | Radish sprouts | Spinach juice | LB 15°C | LB + antibiotics | Cow dung | LB solid medium | LB pH4 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ncR1085800 | 72 | 4177 | 4563 | 4990 | 345 | 25504 | 3228 | 1940 | 655 | 11683 | 1410 | 9815 |
| ncR1114186 | 94 | 641 | 0 | 416 | 684 | 102 | 29 | 1382 | 0 | 65 | 246 | 26 |
| ncR1481381 | 99 | 2774 | 3227 | 3740 | 291 | 19298 | 2504 | 1266 | 411 | 8632 | 1108 | 6997 |
| ncR1483108 | 77 | 128 | 188 | 223 | 46 | 31 | 79 | 33 | 42 | 0 | 48 | 32 |
| ncR1509794 | 96 | 628 | 0 | 529 | 628 | 50 | 7 | 1407 | 0 | 51 | 288 | 43 |
| ncR1641710 | 114 | 168 | 356 | 662 | 533 | 504 | 183 | 215 | 14 | 21 | 65 | 7 |
| ncR1854285 | 91 | 153 | 30 | 45 | 10 | 92 | 30 | 34 | 232 | 27 | 8 | 109 |
| ncR1864748 | 174 | 23 | 26 | 24 | 3 | 28 | 4 | 24 | 262 | 28 | 13 | 19 |
| ncR1868696 | 103 | 293 | 210 | 353 | 185 | 267 | 59 | 387 | 16 | 106 | 43 | 48 |
| ncR1999946 | 51 | 34 | 18 | 215 | 43 | 117 | 0 | 251 | 0 | 95 | 0 | 0 |
| ncR2585184 | 44 | 119 | 41 | 109 | 20 | 27 | 76 | 372 | 0 | 0 | 51 | 0 |
| ncR348122 | 91 | 382 | 30 | 324 | 136 | 355 | 1440 | 1152 | 36 | 67 | 172 | 244 |
| ncR3526958 | 96 | 18 | 28 | 21 | 5 | 212 | 56 | 112 | 0 | 101 | 8 | 69 |
| ncR4137844 | 268 | 551 | 249 | 558 | 821 | 304 | 283 | 1113 | 55 | 77 | 265 | 55 |
| ncR4546182 | 36 | 129 | 75 | 343 | 135 | 0 | 0 | 227 | 0 | 34 | 207 | 0 |
| ncR4853400 | 65 | 134 | 139 | 369 | 341 | 331 | 341 | 102 | 0 | 75 | 149 | 0 |
| ncR612919 | 36 | 80 | 50 | 38 | 49 | 0 | 224 | 0 | 0 | 34 | 21 | 0 |
Evidence for translation of annotated ncRNAs
To our own surprise, a significant number of annotated ncRNAs had high RCVs indicating translation, which we examined further. Table 1 shows the known ncRNAs which i) are independent from protein coding genes (i.e., are not leader peptides or riboswitches, etc.), ii) are not ribosomal RNA or iii) do not encode tRNAs. The remaining 115 annotated ncRNAs were categorized according to their RCV (Fig. 1c; Additional file 8: Table S6). As expected for ncRNAs, 52 of these ncRNAs are not translated and have a low RCV (RCV ≤ 0.16). This indicates transcription but no translation. Surprisingly, we identified 52 ncRNAs with an RCV higher than 0.355 (α = 0.01) which we used as lower limit for considering a transcript to be translated (Additional file 9: Figure S2). For both cases, an ncRNA example with low (csrB) and high (arcZ) RCV is shown in Fig. 4. Eleven ncRNAs fall in an RCV range above the upper limit for untranslated and below the lower limit for translated RNAs and, thus, their translation status (i.e., either untranslated or weakly translated) is difficult to assess. In summary, the ncRNAs were divided into three groups with different ribosome coverage: low RCV similar to untranslated RNAs (52 or 45.5%), such of ambiguous nature (11 or 9%), and those with high RCV similar to translated genes (52 or 45.5%). Clearly, the RCV threshold at which an RNA is considered to be translated depends on the assumed distribution fitted to the tRNA values (see Methods). In any case, different thresholds only alter the region of uncertainty, but do not invalidate our principal finding that quite a number of annotated ncRNAs appear to be associated with ribosomes. Normally, translation is considered the main cause for ribosome binding of an RNA in RIBOseq experiments [83].
Fig. 4.
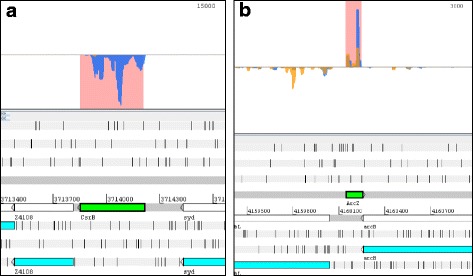
Visualization of ribosomal footprints and transcript reads mapping to annotated ncRNAs as coverage plots. A genomic area is visualized in Artemis 15.0.0 [43]. In the lower part of the panels, the genome (shown as grey lines) is visualized in a six-frame translation mode. Numbers given between the grey lines indicate the genome coordinates. On top of the forward strand are three reading frames and on the reverse DNA strand are three further reading frames. Each reading frame represented is visible by the indicated stop codons (vertical black bars). Annotated genes are shown in their respective reading frame (turquoise arrows) and also on the DNA strand itself (white arrows). The gene name is written below each arrow. Any protein-coding ORF must be at least located between two black bars, with the downstream stop codon being the translational stop. In the upper part of the panels, the DNA is indicated by a thin black line and the sequencing reads matching to the forward or reverse strand are shown above or below this line. The sequencing reads from the footprint (yellow) and transcriptome (blue) sequencing are shown as filled coverage plots, respectively. The known ncRNAs are indicated on the DNA by a bright green arrow. Since ncRNAs supposedly do not contain a protein-coding ORF, these genes are only shown on the DNA. a csrB: Very few footprint reads are seen for CsrB, indicating that this ncRNA is not translated. b arcZ: In contrast, ArcZ is covered with many footprints and a number of transcript reads are found. All further examples are shown in Additional file 9: Figure S2
We analyzed the potential ORFs of the 52 ncRNAs covered by ribosomes for their annotation status in other organisms using blastp [58]. Twenty were found to contain ORFs which achieve blastp-hits to multiple genes annotated in other enterobacteria (e value 10−3 or lower), mainly in other Escherichia coli strains. From these, 15 are annotated as hypothetical proteins, two belong to toxin-antitoxin systems, one encodes a conserved domain of phage origin and the remaining two are membrane proteins (Additional file 8: Table S6).
Correlation of translation with Shine-Dalgarno sequences
The presence or absence of a Shine-Dalgarno sequence in proper distance to the start codon can be an indicator for a translational start [66]. A strong Shine-Dalgarno sequence should correspond to a high RCV. On a global scale, i.e. taking average values of all genes with comparable Shine-Dalgarno sequences, such a correlation was found (Additional file 4: File S1). However, predictions are unreliable for single genes. Since several genes exist which either have no Shine-Dalgarno or are completely leaderless [65], a missing Shine-Dalgarno is not necessarily an indication for absent translation. We then searched for the presence of a Shine-Dalgarno sequence for those 20 ncRNAs which have a blastp hit. A start codon in reasonable distance to the start coordinate of the ncRNA was selected (see Methods) and a possible Shine-Dalgarno sequence was determined according to Ma et al. [63], also including weak Shine-Dalgarno sequences. In 11 of 20 cases, a putative Shine-Dalgarno sequence was found (Table 1, Additional file 8: Table S6). The Shine-Dalgarno sequences were also determined as above according to Hyatt et al. [66] (see Additional file 4: File S1), but this method is more stringent and misses some of the weaker sequences (Additional file 8: Table S6). The observation that 11 out of 20 translated ncRNAs with blastP hit (i.e., 55%) have Shine-Dalgarno sequences compares well to about 57% annotated genes possessing such a sequence in E. coli K12 [64].
Why are ncRNAs covered with ribosomes?
Translational profiling showed that 52 annotated ncRNAs have high RCVs. High RCVs may occur due to incomplete digestion of free RNA. Therefore, we had performed two rounds of RNase I digestion and sucrose density gradient centrifugation for ribosomal enrichment, which makes this assumption very unlikely. Most ncRNAs are reported in the Rfam database to bind Hfq and regulate via antisense pairing to their target genes; some ncRNAs are of completely unknown function, and few are involved in toxin-antitoxin interactions. We consider it unlikely that the high numbers of footprints are false-positives in all cases. While the phenomenon of “translated ncRNAs” is highly discussed for eukaryotes [70, 71, 84–89], this observation has, to our knowledge, only rarely been reported for bacteria, i.e. SgrS/SgrT or the “ncRNA” C0343 ([90]; see below, [91]).
In any case, the ribosomal “coverage” of tRNAs (median RCV 0.03), taken as background in this study, is far below the high ribosomal coverage of some ncRNAs. Finally, another explanation for ribosomal coverage of ncRNAs is regulatory functions performed by interaction of the ncRNA with the ribosomes and, thereby, causing accidental carry-over. However, ribosome-interacting ncRNAs are a minority according to Guttman et al. [86].
RNAs functioning as both ncRNA and mRNA?
A few ncRNAs which are also translated have been suggested to exist in bacteria and are termed coding non-coding RNAs (cncRNAs) [24]. sgrS/sgrT is the only known example for E. coli K12 [90]. In EHEC EDL933, the ATG start codon used by E. coli K12 is mutated to ATT. In addition, the Shine-Dalgarno sequence has changed from AAGGGGGT in K12 to AAGGAGGT in EDL933, the very best category S27 of Hyatt et al. [66]. Since a strong SD sequence compensates a weak start codon [63], and sgrS has an RCV of 1.55 (Table 1), and ATT is known to be a (very rare) start codon in E. coli [92–94], we hypothesize that EHEC synthesizes SgrT using the uncommon start codon ATT. Interestingly, the ORF encoding for SgrT gave a Ka/Ks ratio below 1, i.e. 0.15 with a P value of about 0.002. Unfortunately, most ORFs found covered with footprints proved to be too short for any meaningful Ka/Ks analysis (data not shown). Only one other footprint-covered ORF of the ncRNA MicA gave significant results. This ORF had a Ka/Ks ratio of about 0.35 with a P value of about 0.018 (Additional file S3: Table S3).
Not all former entities named as ncRNA in the past, however, are cncRNAs. For instance, C0343 had formerly been described as ncRNA, but contains an ORF and yields an RCV of 2.49 in our study (not shown). This validates Washietl et al. [91] who shows that C0343 encodes a short 57-aa protein. Consequently, this entity was possibly falsely labelled as ncRNA and it had been removed from the Rfam database. However, a former study described 72 novel intergenic small protein-coding genes of EHEC [83]. We found six instances in which the locus of a novel protein-coding gene overlaps fully or partly with the locus of one of the ncRNAs (Additional file 8: Table S6), which also hints towards cncRNAs.
In any case, we suggest being cautious in labeling any ribosome covered “ncRNA” of E. coli found in this study as cncRNA since further experimental evidence is needed. Based on our current results, we conclude that ribosome covered ncRNAs may represent a mixture of misannotated short mRNAs, ncRNAs with a regulatory function including potential ribosomal binding, and cncRNAs translated indeed. To corroborate this hypothesis about additional cncRNAs and to confirm the existence of novel peptides from so called “non-coding” RNAs as indicated by ribosomal footprints, we tested the footprint-covered ORF of ryhB for a phenotype (see below).
ryhB supposedly is a novel cncRNA, encoding the RNA RyhB and a phenotype-causing peptide, RyhP
Closer examination of footprint signals for several ncRNAs revealed possible ORFs which encode novel peptides. We chose ryhB for further examination, since the encoded RNA-molecule RyhB has a well-known function in iron homeostasis for many bacteria [95–97]. Accordingly, we expected iron-limiting to be the most likely condition in which a phenotype for this novel peptide might be found. Thus, we picked the best matching ORF according to the RIBOseq data, coding for the nona-peptide MAHIASSIT (Fig. 5; start codon ATT) and named it ryh B-encoded peptide, RyhP, in the following. This ORF was introduced on a high-copy arabinose-inducible plasmid in EHEC wild type. In cloning, we omitted all non-coding parts of ryhB, to limit any effect the expressed (m)RNA-fragment might have (sequence P1). To even further reduce the possibility that the expressed RNA and not the peptide itself causes the phenotype, we changed all codons of the ORF such that the same peptide is produced, but the underlying RNA sequence differs maximally from the wild type sequence (P2). This strategy prevents the RNA made hybridizing with any natural target RNAs [e.g., 99]. Two negative controls were created, either with the second (T2) or third codon (T3) changed into a stop codon, terminating RyhP translation prematurely. Competitive indices (CI) under RyhB-inducing condition (i.e. low-iron) showed a significant advantage of the strain possessing the RyhP-producing plasmid over those strains containing a plasmid with stop codons in the RyhP-ORF (Table 4).
Fig. 5.
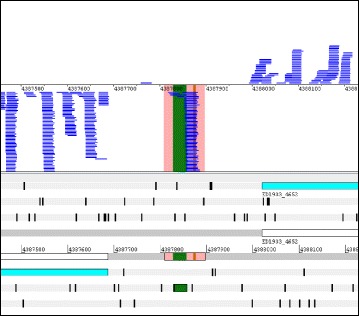
Visualization of individual ribosomal footprints mapping to rhyB. The genomic area is visualized in Artemis 15.0.0 [43]. In the lower part of the panels, the genome (shown as grey lines) is visualized in a six-frame translation mode. Numbers given between the grey lines indicate the genome coordinates. On top of the forward strand are three reading frames and on the reverse DNA strand are three further reading frames. Each reading frame represented is visible by the indicated stop codons (vertical black bars). Annotated genes are shown in their respective reading frame (turquoise arrows) and also on the DNA strand itself (white arrows). The gene name is written below each arrow. In the upper part of the panels, the DNA is indicated by a thin black line and the footprint reads (blue) matching to the forward or reverse strand are shown above or below this line. The shaded areas indicate ryhB (pink), the coding ORF RyhP (green) and a putative weak Shine-Dalgarno sequence (brown; ggagaa)
Table 4.
Competitive index values (CI) for EHEC strains possessing a wild-type like ORF encoding RyhP (P1 or P2) or an ORF with a premature stop codon (T2 or T3) plusminus their standard deviations (Std)
| Wild-type like RyhP-ORF | Terminated RyhP-ORF | CI | ±Std |
|---|---|---|---|
| P1 | T2 | 0.79 | 0.08 |
| P1 | T3 | 0.19 | 0.08 |
| P2 | T3 | 0.38 | 0.06 |
Strains are competitively grown in minimal medium M9 with no iron added for 24 h. The RyhP-encoding ORF was transcriptionally induced with 0.2% arabinose
RyhB folds when not bound to its regulated target RNA (Fig. 6) and this, assumedly, makes the coding ORF unavailable for translation. However, ribosomes are able to resolve secondary structures of mRNAs [98]. Furthermore, RyhP has a weak putative Shine-Dalgarno motif (i.e., ggagaa) upstream. Upon binding a target mRNA like sodA [99], the RNA structure opens and the Shine-Dalgarno sequence is set free (Fig. 6). If this opening facilitates ribosomal binding for translation initiation of the RyhB RNA, and subsequent progression of ribosomes along the RNA, must remain open.
Fig. 6.
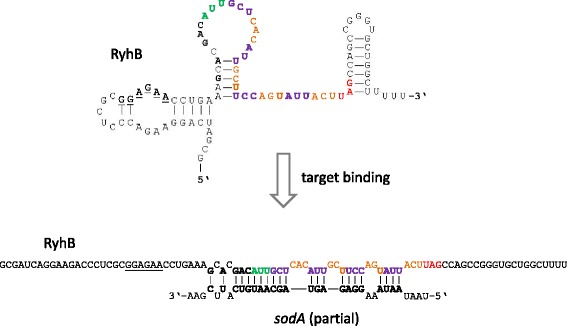
Overview of the secondary structures formed by RyhB for the molecule on its own (top) and after binding to a target RNA, like sodA (bottom). Structures are taken from [99]. Individual bases have been highlighted. Underlined, putative Shine-Dalgarno sequence; green, start codon; violet/orange, individual codons along the frame; red, stop codon, bold; bases involved in hybridization to the sodA-target
Conclusion
In the past, very short proteins or peptides were excluded from annotation and believed to be unlikely. Some short mRNAs could have been labeled as ncRNA solely on this presumption. However, more and more small proteins are being discovered. For instance, a number of small genes have been described for E. coli in recent years. These genes were hard to detect because they appear to be membrane proteins and are induced under stress conditions only [100, 101]. In another study, we confirmed the existence of 72 novel and short protein-coding genes in the EHEC genome, some which were verified by proteome data [83]. Similar findings have been made by other groups (see, e.g., [102–104]), and future research could confirm the existence of more of these proteins similar to studies conducted in eukaryotic ribosomal profiling [70, 105–107].
Acknowledgements
None.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft DFG (SCHE316/3-2, KE740/13-2, BO867/23-2, SCHO 1576/1, and BA2168/4-3 within SPP 1395 InKoMBio); and by the German Federal Ministry of Education and Research BMBF (grant 031 6165A within e:Bio RNAsys). The funding bodies had no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
All additional files supporting the results of this article are available in the repository labarchives.com (http://www.labarchives.com/) using the link http://dx.doi.org/10.6070/H4G15XX9. All the supporting data are included as Additional files.
Authors’ contributions
RL and KN conceived the project. RL conducted transcriptome and translatome experiments, and analyzed the data with help of SSi. RCV thresholds were provided by StSch; Shine-Dalgarno predictions by KN. Putative ncRNA functions were examined by PRW, CS and RB. RW conducted the competitive growth assays. RL and KN wrote the manuscript with the help of all other authors. KN, DAK, and SSche supervised the study. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
No ethics approval was required for this study since it involved bacteria only.
Abbreviations
- aa
Amino acids
- blast
Basic local alignment search tool
- bp
Base pairs
- cncRNA
coding non-coding RNA
- dfRNA
dual-functioning RNA
- DTT
Dithiothreitol
- EHEC
Enterohemorrhagic E. coli
- LB
Luria-Bertani medium
- NEB
New England Biolabs
- NGS
Next Generation Sequencing
- nt
Nucleotides
- OD
Optical density
- ORF
Open reading frame
- RCV
Ribosome coverage value
- RIBOseq
Ribosomal footprinting
- rP
Pearson correlation
- RPKM
Reads per kilobase per million mapped reads
- rpm
Revolutions per minute
- rS
Spearman rank correlation
Additional files
Oligos used in this study to prepare ncRNA-specific probes. (DOC 51 kb)
Bacterial reference genomes used for CopraRNA. (DOCX 15 kb)
Gene pairs for Ka/Ks analysis and results. (XLSX 16 kb)
Coverage with ribosomal footprints correlates globally with the conservation of the Shine-Dalgarno sequence. (DOCX 78 kb)
Correlation of the RPKM translatome between the replicate footprint experiments 1 and 2. (PPTX 142 kb)
Sequencing statistics. The number of mapped reads is listed for the transcriptome and the ribosomal profiling experiments. Additionally, the numbers of reads mapping to rRNA and tRNA genes are shown, as well as the number of remaining reads. (DOCX 14 kb)
Full data set of the 150 novel ncRNAs and their properties in EHEC. Expressed homologs in E. coli K12 are indicated, CopraRNA predictions are shown, as well as regulation in eleven diverse growth conditions. (XLSX 81 kb)
Data set of the 115 annotated ncRNAs and their properties, including blastp hits and proposed Shine-Dalgarno sequences. (XLSX 44 kb)
Overview of 52 known ncRNAs with a translation, i.e. an RCV above the threshold. Panels were drawn using Artemis [43]. (PPTX 5604 kb)
References
- 1.Griffiths-Jones S, Moxon S, Marshall M, Khanna A, Eddy SR, Bateman A. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005;33(Database issue):D121–4. doi: 10.1093/nar/gki081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burge SW, Daub J, Eberhardt R, Tate J, Barquist L, Nawrocki EP, Eddy SR, Gardner PP, Bateman A. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 2013;41(Database issue):D226–32. doi: 10.1093/nar/gks1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gottesman S. Micros for microbes: non-coding regulatory RNAs in bacteria. Trends Genet. 2005;21(7):399–404. doi: 10.1016/j.tig.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 4.Li W, Ying X, Lu Q, Chen L. Predicting sRNAs and their targets in bacteria. Genomics Proteomics Bioinformatics. 2012;10(5):276–84. doi: 10.1016/j.gpb.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Georg J, Hess WR. cis-antisense RNA, another level of gene regulation in bacteria. Microbiol. Mol. Biol. Rev. 2011;75(2):286–300. doi: 10.1128/MMBR.00032-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakashima N, Tamura T. Gene silencing in Escherichia coli using antisense RNAs expressed from doxycycline-inducible vectors. Lett Appl Microbiol. 2013;56(6):436–42. doi: 10.1111/lam.12066. [DOI] [PubMed] [Google Scholar]
- 7.Gelderman G, Contreras LM. Discovery of posttranscriptional regulatory RNAs using next generation sequencing technologies. Methods Mol Biol. 2013;985:269–95. doi: 10.1007/978-1-62703-299-5_14. [DOI] [PubMed] [Google Scholar]
- 8.Raghavan R, Groisman EA, Ochman H. Genome-wide detection of novel regulatory RNAs in E. coli. Genome Res. 2011;21(9):1487–97. doi: 10.1101/gr.119370.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Argaman L, Hershberg R, Vogel J, Bejerano G, Wagner EG, Margalit H, Altuvia S. Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr Biol. 2001;11(12):941–50. doi: 10.1016/S0960-9822(01)00270-6. [DOI] [PubMed] [Google Scholar]
- 10.Chen S, Lesnik EA, Hall TA, Sampath R, Griffey RH, Ecker DJ, Blyn LB. A bioinformatics based approach to discover small RNA genes in the Escherichia coli genome. BioSyst. 2002;65(2–3):157–77. [DOI] [PubMed]
- 11.Rivas E, Klein RJ, Jones TA, Eddy SR. Computational identification of noncoding RNAs in E. coli by comparative genomics. Curr Biol. 2001;11(17):1369–73. [DOI] [PubMed]
- 12.Perna NT, Plunkett G, 3rd, Burland V, Mau B, Glasner JD, Rose DJ, Mayhew GF, Evans PS, Gregor J, Kirkpatrick HA, et al. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature. 2001;409(6819):529–33. doi: 10.1038/35054089. [DOI] [PubMed] [Google Scholar]
- 13.Backofen R, Hess WR. Computational prediction of sRNAs and their targets in bacteria. RNA Biol. 2010;7(1):33–42. doi: 10.4161/rna.7.1.10655. [DOI] [PubMed] [Google Scholar]
- 14.Hot D, Slupek S, Wulbrecht B, D’Hondt A, Hubans C, Antoine R, Locht C, Lemoine Y. Detection of small RNAs in Bordetella pertussis and identification of a novel repeated genetic element. BMC Genomics. 2011;12(1):1. doi: 10.1186/1471-2164-12-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herbig A, Nieselt K. nocoRNAc: characterization of non-coding RNAs in prokaryotes. BMC Bioinformatics. 2011;12(1):1. doi: 10.1186/1471-2105-12-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Solomon KV, Haitjema CH, Thompson DA, O’Malley MA. Extracting data from the muck: deriving biological insight from complex microbial communities and non-model organisms with next generation sequencing. Curr Opin Biotechnol. 2014;28C:103–10. doi: 10.1016/j.copbio.2014.01.007. [DOI] [PubMed] [Google Scholar]
- 17.Landstorfer R, Simon S, Schober S, Keim D, Scherer S, Neuhaus K. Comparison of strand-specific transcriptomes of enterohemorrhagic Escherichia coli O157:H7 EDL933 (EHEC) under eleven different environmental conditions including radish sprouts and cattle feces. BMC Genomics. 2014;15:353. doi: 10.1186/1471-2164-15-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mutz K-O, Heilkenbrinker A, Lönne M, Walter J-G, Stahl F. Transcriptome analysis using next-generation sequencing. Curr Opin Biotechnol. 2013;24(1):22–30. doi: 10.1016/j.copbio.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 19.Kröger C, Dillon SC, Cameron AD, Papenfort K, Sivasankaran SK, Hokamp K, Chao Y, Sittka A, Hebrard M, Handler K, et al. The transcriptional landscape and small RNAs of Salmonella enterica serovar Typhimurium. Proc Natl Acad Sci U S A. 2012;109(20):E1277–86. [DOI] [PMC free article] [PubMed]
- 20.Ingolia NT. Genome-wide translational profiling by ribosome footprinting. Methods Enzymol. 2010;470:119–42. doi: 10.1016/S0076-6879(10)70006-9. [DOI] [PubMed] [Google Scholar]
- 21.Berghoff BA, Konzer A, Mank NN, Looso M, Rische T, Forstner KU, Kruger M, Klug G. Integrative “omics”-approach discovers dynamic and regulatory features of bacterial stress responses. PLoS Genet. 2013;9(6):e1003576. doi: 10.1371/journal.pgen.1003576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Legendre R, Baudin-Baillieu A, Hatin I, Namy O. RiboTools: a Galaxy toolbox for qualitative ribosome profiling analysis. Bioinformatics. 2015;31(15):2586–8. doi: 10.1093/bioinformatics/btv174. [DOI] [PubMed] [Google Scholar]
- 23.Vanderpool CK, Balasubramanian D, Lloyd CR. Dual-function RNA regulators in bacteria. Biochimie. 2011;93(11):1943–9. doi: 10.1016/j.biochi.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumari P, Sampath K. cncRNAs: Bi-functional RNAs with protein coding and non-coding functions. Semin Cell Dev Biol. 2015;47–48:40–51. doi: 10.1016/j.semcdb.2015.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jørgensen MG, Thomason MK, Havelund J, Valentin-Hansen P, Storz G. Dual function of the McaS small RNA in controlling biofilm formation. Genes Dev. 2013;27(10):1132–45. doi: 10.1101/gad.214734.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen C, Zhang X, Shang F, Sun H, Sun B, Xue T. The Staphylococcus aureus protein-coding gene gdpS modulates sarS expression via mRNA-mRNA interaction. Infect Immun. 2015;83(8):3302–10. [DOI] [PMC free article] [PubMed]
- 27.Liu N, Niu G, Xie Z, Chen Z, Itzek A, Kreth J, Gillaspy A, Zeng L, Burne R, Qi F, et al. The Streptococcus mutans irvA gene encodes a trans-acting riboregulatory mRNA. Mol Cell. 2015;57(1):179–90. doi: 10.1016/j.molcel.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wells JG, Davis BR, Wachsmuth IK, Riley LW, Remis RS, Sokolow R, Morris GK. Laboratory investigation of hemorrhagic colitis outbreaks associated with a rare Escherichia coli serotype. J Clin Microbiol. 1983;18(3):512–20. [DOI] [PMC free article] [PubMed]
- 29.Latif H, Li HJ, Charusanti P, Palsson BØ, Aziz RK. A gapless, unambiguous genome sequence of the enterohemorrhagic Escherichia coli O157: H7 strain EDL933. Genome Announc. 2014;2(4):e00821–00814. doi: 10.1128/genomeA.00821-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sunohara T, Jojima K, Tagami H, Inada T, Aiba H. Ribosome stalling during translation elongation induces cleavage of mRNA being translated in Escherichia coli. J Biol Chem. 2004;279(15):15368–75. doi: 10.1074/jbc.M312805200. [DOI] [PubMed] [Google Scholar]
- 31.Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324(5924):218–23. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steitz JA. Polypeptide chain initiation: nucleotide sequences of the three ribosomal binding sites in bacteriophage R17 RNA. Nature. 1969;224(5223):957–64. doi: 10.1038/224957a0. [DOI] [PubMed] [Google Scholar]
- 33.Aigner A, Jansohn M. Gentechnische Methoden: Eine Sammlung von Arbeitsanleitungen für das molekularbiologische Labor. Heidelberg: Elsevier-Spektrum Akademischer Verl.; 2007.
- 34.Flaherty BL, Van Nieuwerburgh F, Head SR, Golden JW. Directional RNA deep sequencing sheds new light on the transcriptional response of Anabaena sp. strain PCC 7120 to combined-nitrogen deprivation. BMC Genomics. 2011;12:332. doi: 10.1186/1471-2164-12-332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pall GS, Hamilton AJ. Improved Northern blot method for enhanced detection of small RNA. Nat Protoc. 2008;3(6):1077–84. doi: 10.1038/nprot.2008.67. [DOI] [PubMed] [Google Scholar]
- 36.Sambrook J, Russell DW. Molecular cloning. A laboratory manual, 3 edn. New York: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- 37.Macho AP, Zumaquero A, Ortiz-Martin I, Beuzon CR. Competitive index in mixed infections: a sensitive and accurate assay for the genetic analysis of Pseudomonas syringae-plant interactions. Mol Plant Pathol. 2007;8(4):437–50. [DOI] [PubMed]
- 38.Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J. Galaxy: a web-based genome analysis tool for experimentalists. Curr. Protoc. Mol. Biol. 2010;Chapter 19:Unit 19 10 11–21. doi: 10.1002/0471142727.mb1910s89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goecks J, Nekrutenko A, Taylor J. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11(8):R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simon S, Oelke D, Landstorfer R, Neuhaus K, Keim D. Visual analysis of next-generation sequencing data to detect overlapping genes. IEEE Symp Biol Data Vis. 2011;1:47–54. doi: 10.1109/BioVis.2011.6094047. [DOI] [Google Scholar]
- 42.Carver T, Bohme U, Otto TD, Parkhill J, Berriman M. BamView: viewing mapped read alignment data in the context of the reference sequence. Bioinformatics. 2010;26(5):676–7. doi: 10.1093/bioinformatics/btq010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16(10):944–5. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 44.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–8. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 45.R_Development_Core_Team . R: a language and environment for statistical computing. 2011. [Google Scholar]
- 46.Morgan M. Rsamtools: Binary alignment (BAM), variant call (BCF), or tabix file import. R package version 1.8.6. 2013. [Google Scholar]
- 47.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5(10):R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aboyoun P, Pages H, Lawrence M. GenomicRanges: Representation and manipulation of genomic intervals. [https://bioconductor.org/packages/release/bioc/html/GenomicRanges.html].
- 49.Pages H, Aboyoun P, Lawrence M. IRanges: Infrastructure for manipulating intervals on sequences. [https://www.bioconductor.org/packages/release/bioc/html/IRanges.html].
- 50.Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35(Database issue):D61–5. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Free Statistics Software version 1.1.23-r7. [http://www.wessa.net/].
- 52.Nakahigashi K, Takai Y, Shiwa Y, Wada M, Honma M, Yoshikawa H, Tomita M, Kanai A, Mori H. Effect of codon adaptation on codon-level and gene-level translation efficiency in vivo. BMC Genomics. 2014;15:1115. doi: 10.1186/1471-2164-15-1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dreher TW. Viral tRNAs and tRNA-like structures. Wiley Interdiscipl Rev RNA. 2010;1(3):402–14. doi: 10.1002/wrna.42. [DOI] [PubMed] [Google Scholar]
- 54.Bailly-Bechet M, Vergassola M, Rocha E. Causes for the intriguing presence of tRNAs in phages. Genome Res. 2007;17(10):1486–95. doi: 10.1101/gr.6649807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scott DW. Multivariate density estimation: theory, practice, and visualization. New York, Chicester: Wiley; 1992. [Google Scholar]
- 56.Nawrocki EP, Burge SW, Bateman A, Daub J, Eberhardt RY, Eddy SR, Floden EW, Gardner PP, Jones TA, Tate J, et al. Rfam 12.0: updates to the RNA families database. Nucleic Acids Res. 2015;43(Database issue):D130–7. doi: 10.1093/nar/gku1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Caron MP, Bastet L, Lussier A, Simoneau-Roy M, Masse E, Lafontaine DA. Dual-acting riboswitch control of translation initiation and mRNA decay. Proc Natl Acad Sci U S A. 2012;109(50):E3444–53. doi: 10.1073/pnas.1214024109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 59.Wright PR, Richter AS, Papenfort K, Mann M, Vogel J, Hess WR, Backofen R, Georg J. Comparative genomics boosts target prediction for bacterial small RNAs. Proc Natl Acad Sci U S A. 2013;110(37):E3487–96. doi: 10.1073/pnas.1303248110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wright PR, Georg J, Mann M, Sorescu DA, Richter AS, Lott S, Kleinkauf R, Hess WR, Backofen R. CopraRNA and IntaRNA: predicting small RNA targets, networks and interaction domains. Nucleic Acids Res. 2014;42(Web Server issue):W119–23. doi: 10.1093/nar/gku359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hertel J, de Jong D, Marz M, Rose D, Tafer H, Tanzer A, Schierwater B, Stadler PF. Non-coding RNA annotation of the genome of Trichoplax adhaerens. Nucleic Acids Res. 2009;37(5):1602–15. doi: 10.1093/nar/gkn1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang D, Zhang Y, Zhang Z, Zhu J, Yu J. KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinformatics. 2010;8(1):77–80. doi: 10.1016/S1672-0229(10)60008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ma J, Campbell A, Karlin S. Correlations between Shine-Dalgarno sequences and gene features such as predicted expression levels and operon structures. J Bacteriol. 2002;184(20):5733–45. doi: 10.1128/JB.184.20.5733-5745.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Starmer J, Stomp A, Vouk M, Bitzer D. Predicting Shine-Dalgarno sequence locations exposes genome annotation errors. PLoS Comput Biol. 2006;2(5):e57. doi: 10.1371/journal.pcbi.0020057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zheng X, Hu G-Q, She Z-S, Zhu H. Leaderless genes in bacteria: clue to the evolution of translation initiation mechanisms in prokaryotes. BMC Genomics. 2011;12(1):361. doi: 10.1186/1471-2164-12-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Haas BJ, Chin M, Nusbaum C, Birren BW, Livny J. How deep is deep enough for RNA-Seq profiling of bacterial transcriptomes? BMC Genomics. 2012;13:734. doi: 10.1186/1471-2164-13-734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vasquez JJ, Hon CC, Vanselow JT, Schlosser A, Siegel TN. Comparative ribosome profiling reveals extensive translational complexity in different Trypanosoma brucei life cycle stages. Nucleic Acids Res. 2014;42(6):3623–37. [DOI] [PMC free article] [PubMed]
- 69.Lareau LF, Hite DH, Hogan GJ, Brown PO. Distinct stages of the translation elongation cycle revealed by sequencing ribosome-protected mRNA fragments. Elife. 2014;3:e01257. doi: 10.7554/eLife.01257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Smith JE, Alvarez-Dominguez JR, Kline N, Huynh NJ, Geisler S, Hu W, Coller J, Baker KE. Translation of small open reading frames within unannotated RNA transcripts in Saccharomyces cerevisiae. Cell Rep. 2014;7(6):1858–66. doi: 10.1016/j.celrep.2014.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chew GL, Pauli A, Rinn JL, Regev A, Schier AF, Valen E. Ribosome profiling reveals resemblance between long non-coding RNAs and 5′ leaders of coding RNAs. Development. 2013;140(13):2828–34. doi: 10.1242/dev.098343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li GW, Oh E, Weissman JS. The anti-Shine-Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature. 2012;484(7395):538–41. doi: 10.1038/nature10965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.O’Connor PB, Li GW, Weissman JS, Atkins JF, Baranov PV. rRNA:mRNA pairing alters the length and the symmetry of mRNA-protected fragments in ribosome profiling experiments. Bioinformatics. 2013;29(12):1488–91. doi: 10.1093/bioinformatics/btt184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shen V, Schlessinger D. 16 RNases, I, II, and IV of Escherichia coli. The enzymes. 1982;15:501–15. doi: 10.1016/S1874-6047(08)60288-7. [DOI] [Google Scholar]
- 75.Delcardayre SB, Raines RT. The extent to which ribonucleases cleave ribonucleic acid. Anal Biochem. 1995;225(1):176–8. doi: 10.1006/abio.1995.1132. [DOI] [PubMed] [Google Scholar]
- 76.Klinge S, Voigts-Hoffmann F, Leibundgut M, Ban N. Atomic structures of the eukaryotic ribosome. Trends Biochem Sci. 2012;37(5):189–98. doi: 10.1016/j.tibs.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 77.Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147(4):789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Coornaert A, Chiaruttini C, Springer M, Guillier M. Post-transcriptional control of the Escherichia coli PhoQ-PhoP two-component system by multiple sRNAs involves a novel pairing region of GcvB. PLoS Genet. 2013;9(1):e1003156. [DOI] [PMC free article] [PubMed]
- 79.Kopf M, Klahn S, Scholz I, Matthiessen JK, Hess WR, Voss B. Comparative analysis of the primary transcriptome of Synechocystis sp. PCC 6803. DNA Res. 2014;21(5):527–39. doi: 10.1093/dnares/dsu018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li GW, Burkhardt D, Gross C, Weissman JS. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell. 2014;157(3):624–35. doi: 10.1016/j.cell.2014.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sirbu A, Kerr G, Crane M, Ruskin HJ. RNA-Seq vs dual- and single-channel microarray data: sensitivity analysis for differential expression and clustering. PLoS One. 2012;7(12):e50986. doi: 10.1371/journal.pone.0050986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kane MD, Jatkoe TA, Stumpf CR, Lu J, Thomas JD, Madore SJ. Assessment of the sensitivity and specificity of oligonucleotide (50mer) microarrays. Nucleic Acids Res. 2000;28(22):4552–7. doi: 10.1093/nar/28.22.4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Neuhaus K, Landstorfer R, Fellner L, Simon S, Marx H, Ozoline O, Schafferhans A, Goldberg T, Rost B, Küster B, et al. Translatomics combined with transcriptomics and proteomics reveals novel functional, recently evolved orphan genes in Escherichia coli O157:H7 (EHEC) BMC Genomics. 2016;7:133. doi: 10.1186/s12864-016-2456-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Brar GA, Yassour M, Friedman N, Regev A, Ingolia NT, Weissman JS. High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science. 2012;335(6068):552–7. doi: 10.1126/science.1215110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.van Heesch S, van Iterson M, Jacobi J, Boymans S, Essers PB, de Bruijn E, Hao W, Macinnes AW, Cuppen E, Simonis M. Extensive localization of long noncoding RNAs to the cytosol and mono- and polyribosomal complexes. Genome Biol. 2014;15(1):R6. doi: 10.1186/gb-2014-15-1-r6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Guttman M, Russell P, Ingolia NT, Weissman JS, Lander ES. Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins. Cell. 2013;154(1):240–51. doi: 10.1016/j.cell.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ruiz-Orera J, Messeguer X, Subirana JA, Alba MM. Long non-coding RNAs as a source of new peptides. Elife. 2014;3:e03523. doi: 10.7554/eLife.03523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ingolia NT. Ribosome profiling: new views of translation, from single codons to genome scale. Nat Rev Genet. 2014;15(3):205–13. doi: 10.1038/nrg3645. [DOI] [PubMed] [Google Scholar]
- 89.Ingolia NT, Brar GA, Stern-Ginossar N, Harris MS, Talhouarne GJ, Jackson SE, Wills MR, Weissman JS. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Rep. 2014;8(5):1365–79. doi: 10.1016/j.celrep.2014.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ulveling D, Francastel C, Hube F. When one is better than two: RNA with dual functions. Biochimie. 2010;93(4):633–44. doi: 10.1016/j.biochi.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 91.Washietl S, Findeiss S, Muller SA, Kalkhof S, von Bergen M, Hofacker IL, Stadler PF, Goldman N. RNAcode: robust discrimination of coding and noncoding regions in comparative sequence data. RNA. 2011;17(4):578–94. doi: 10.1261/rna.2536111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Binns N, Masters M. Expression of the Escherichia coli pcnB gene is translationally limited using an inefficient start codon: a second chromosomal example of translation initiated at AUU. Mol Microbiol. 2002;44(5):1287–98. [DOI] [PubMed]
- 93.Prère MF, Canal I, Wills NM, Atkins JF, Fayet O. The interplay of mRNA stimulatory signals required for AUU-mediated initiation and programmed −1 ribosomal frameshifting in decoding of transposable element IS911. J Bacteriol. 2011;193(11):2735–44. doi: 10.1128/JB.00115-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sussman JK, Simons EL, Simons RW. Escherichia coli translation initiation factor 3 discriminates the initiation codon in vivo. Mol Microbiol. 1996;21(2):347–60. doi: 10.1046/j.1365-2958.1996.6371354.x. [DOI] [PubMed] [Google Scholar]
- 95.Masse E, Salvail H, Desnoyers G, Arguin M. Small RNAs controlling iron metabolism. Curr Opin Microbiol. 2007;10(2):140–5. doi: 10.1016/j.mib.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 96.Oglesby-Sherrouse AG, Murphy ER. Iron-responsive bacterial small RNAs: variations on a theme. Metallomics. 2013;5(4):276–86. doi: 10.1039/c3mt20224k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Salvail H, Massé E. Regulating iron storage and metabolism with RNA: an overview of posttranscriptional controls of intracellular iron homeostasis. Wiley Interdiscip Rev. 2012;3(1):26–36. doi: 10.1002/wrna.102. [DOI] [PubMed] [Google Scholar]
- 98.Qu X, Wen J-D, Lancaster L, Noller HF, Bustamante C, Tinoco I. The ribosome uses two active mechanisms to unwind messenger RNA during translation. Nature. 2011;475(7354):118–21. doi: 10.1038/nature10126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tjaden B, Goodwin SS, Opdyke JA, Guillier M, Fu DX, Gottesman S, Storz G. Target prediction for small, noncoding RNAs in bacteria. Nucleic Acids Res. 2006;34(9):2791–802. doi: 10.1093/nar/gkl356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hemm MR, Paul BJ, Miranda-Rios J, Zhang A, Soltanzad N, Storz G. Small stress response proteins in Escherichia coli: proteins missed by classical proteomic studies. J Bacteriol. 2010;192(1):46–58. doi: 10.1128/JB.00872-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hemm MR, Paul BJ, Schneider TD, Storz G, Rudd KE. Small membrane proteins found by comparative genomics and ribosome binding site models. Mol Microbiol. 2008;70(6):1487–501. doi: 10.1111/j.1365-2958.2008.06495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Boekhorst J, Wilson G, Siezen RJ. Searching in microbial genomes for encoded small proteins. J Microbial Biotechnol. 2011;4(3):308–13. doi: 10.1111/j.1751-7915.2011.00261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hobbs EC, Fontaine F, Yin X, Storz G. An expanding universe of small proteins. Curr Opin Microbiol. 2011;14(2):167–73. doi: 10.1016/j.mib.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Storz G, Wolf YI, Ramamurthi KS. Small proteins can no longer be ignored. Annu Rev Biochem. 2014;83:753–77. doi: 10.1146/annurev-biochem-070611-102400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Banfai B, Jia H, Khatun J, Wood E, Risk B, Gundling WE, Jr, Kundaje A, Gunawardena HP, Yu Y, Xie L, et al. Long noncoding RNAs are rarely translated in two human cell lines. Genome Res. 2012;22(9):1646–57. doi: 10.1101/gr.134767.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Slavoff SA, Mitchell AJ, Schwaid AG, Cabili MN, Ma J, Levin JZ, Karger AD, Budnik BA, Rinn JL, Saghatelian A. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nat Chem Biol. 2013;9(1):59–64. doi: 10.1038/nchembio.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Stern-Ginossar N, Weisburd B, Michalski A, Le VT, Hein MY, Huang SX, Ma M, Shen B, Qian SB, Hengel H, et al. Decoding human cytomegalovirus. Science. 2012;338(6110):1088–93. doi: 10.1126/science.1227919. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All additional files supporting the results of this article are available in the repository labarchives.com (http://www.labarchives.com/) using the link http://dx.doi.org/10.6070/H4G15XX9. All the supporting data are included as Additional files.