

Abstract
DNA repair by homologous recombination is under stringent cell cycle control. This includes the last step of the reaction, disentanglement of DNA joint molecules (JMs). Previous work has established that JM resolving nucleases are activated specifically at the onset of mitosis. In case of budding yeast Mus81‐Mms4, this cell cycle stage‐specific activation is known to depend on phosphorylation by CDK and Cdc5 kinases. Here, we show that a third cell cycle kinase, Cdc7‐Dbf4 (DDK), targets Mus81‐Mms4 in conjunction with Cdc5—both kinases bind to as well as phosphorylate Mus81‐Mms4 in an interdependent manner. Moreover, DDK‐mediated phosphorylation of Mms4 is strictly required for Mus81 activation in mitosis, establishing DDK as a novel regulator of homologous recombination. The scaffold protein Rtt107, which binds the Mus81‐Mms4 complex, interacts with Cdc7 and thereby targets DDK and Cdc5 to the complex enabling full Mus81 activation. Therefore, Mus81 activation in mitosis involves at least three cell cycle kinases, CDK, Cdc5 and DDK. Furthermore, tethering of the kinases in a stable complex with Mus81 is critical for efficient JM resolution.
Keywords: cell cycle, genome stability, homologous recombination, joint molecule resolution, post‐translational modification
Subject Categories: Cell Cycle; DNA Replication, Repair & Recombination
Introduction
Many DNA transactions are under cell cycle control to adjust them to cell cycle phase‐specific features of chromosomes (Branzei & Foiani, 2008). Homologous recombination (HR) is cell cycle‐regulated at several steps including the first, DNA end resection, and the last, JM removal (Heyer et al, 2010; Ferretti et al, 2013; Mathiasen & Lisby, 2014; Matos & West, 2014). Given that JMs provide stable linkages between sister chromatids, they will interfere with chromosome segregation and therefore need to be disentangled before sister chromatid separation during mitosis. Accordingly, JM resolvases, such as budding yeast Mus81‐Mms4 (Interthal & Heyer, 2000; Schwartz et al, 2012) or Yen1 (Ip et al, 2008), become activated during mitosis (Matos et al, 2011, 2013; Gallo‐Fernández et al, 2012; Szakal & Branzei, 2013; Blanco et al, 2014; Eissler et al, 2014). In contrast, the alternative JM removal pathway, JM dissolution by the Sgs1‐Top3‐Rmi1 complex, is thought to be constantly active throughout the cell cycle (Mankouri et al, 2013; Bizard & Hickson, 2014). The activation of JM resolvases in mitosis therefore leads to a shift in the balance between JM removal pathways, with dissolution being preferred outside of mitosis, but JM resolution becoming increasingly important in mitosis (Matos et al, 2011, 2013; Gallo‐Fernández et al, 2012; Dehé et al, 2013; Saugar et al, 2013; Szakal & Branzei, 2013; Wyatt et al, 2013). It has been hypothesized that JM resolvases are downregulated at cell cycle stages other than mitosis in order to counteract crossover‐induced loss of heterozygosity or to prevent over‐active resolvases from interfering with S phase by, for example, cleaving stalled replication forks (Gallo‐Fernández et al, 2012; Szakal & Branzei, 2013; Blanco et al, 2014).
Budding yeast Mus81‐Mms4 has previously been shown to be targeted by two cell cycle kinases, cyclin‐dependent kinase Cdc28 (CDK) and the yeast polo‐kinase Cdc5 (Matos et al, 2011, 2013; Gallo‐Fernández et al, 2012; Szakal & Branzei, 2013). The corresponding Mms4 phosphorylation events were shown to correlate with and to be required for activation of Mus81‐Mms4 in mitosis. In 2014, we showed that in mitosis Mus81‐Mms4 also forms a complex with Slx4‐Slx1 and the scaffold proteins Dpb11 and Rtt107 (Gritenaite et al, 2014). Interestingly, mass spectrometric analysis of this complex (Gritenaite et al, 2014) revealed that Cdc5 and a third cell cycle kinase Dbf4‐Cdc7 (Dbf4‐dependent kinase, DDK) are also a stable part of this protein assembly (see Appendix Fig S1A). Here, we investigate the role of DDK in Mus81‐Mms4 regulation and find that DDK can phosphorylate Mms4 and that DDK and Cdc5 target Mus81‐Mms4 in an interdependent manner. Moreover, we show that Rtt107 promotes the association of both kinases with the Mus81‐Mms4 complex. The DDK‐dependent regulation of Mus81‐Mms4 is critical for Mus81 activity thus revealing DDK as a novel regulator of homologous recombination.
Results
Mus81‐Mms4 is a DDK phosphorylation target
The cell cycle regulation of JM resolution by Mus81‐Mms4 is intricate and involves phosphorylation by the cell cycle kinases CDK and Cdc5 (Matos et al, 2011, 2013; Gallo‐Fernández et al, 2012; Szakal & Branzei, 2013) as well as complex formation with the scaffold proteins Dpb11, Slx4 and Rtt107 (Gritenaite et al, 2014). To study this protein complex, we performed an analysis of Mms43FLAG interactors in mitosis by SILAC‐based quantitative mass spectrometry (Gritenaite et al, 2014) and found in addition to Dpb11, Slx4, Rtt107 and Cdc5, also Cdc7 and Dbf4 as specific interactors of Mms4 (Appendix Fig S1A). We verified that Cdc7 binds to Mus81‐Mms4 in an Mms43FLAG pull down from mitotic cells analysed by Western blots (Fig 1A). The fact that Mus81‐Mms4 binds to DDK suggested that it might be involved in the phosphorylation cascade that occurs on Mms4 and controls Mus81 activity in mitosis. Accordingly, we found that purified DDK was able to phosphorylate both subunits of purified Mus81‐Mms4 in vitro (Fig 1B, lane 3). When we furthermore compared the DDK‐dependent phosphorylation signal to Mms4 phosphorylation by CDK and Cdc5 (Fig 1B, lanes 2–4), we observed different degrees of phosphorylation shifts indicating that the three kinases phosphorylate Mms4 at distinct sites and/or to different degrees. DDK target sites on other proteins have been studied in detail, and in several cases, DDK was found to target (S/T)(S/T) motifs, where phosphorylation was stimulated by a priming phosphorylation usually on the second (S/T) (Masai et al, 2006; Montagnoli et al, 2006; Randell et al, 2010; Lyons et al, 2013). Intriguingly, Mms4 contains 15 of these motifs and we therefore tested whether these could be targeted by DDK and would depend on priming phosphorylation. We therefore turned to a peptide‐based assay where Mms4 phosphorylation states are precisely defined. To this end, we synthesized peptides corresponding to two (S/T)(S/T) motifs of Mms4. We chose two representative motifs: S222, as it harbours a minimal CDK consensus motif (S/T)P, and S134, as it contains a non‐(S/T)P consensus for CDK [(S/T)X(K/R)(K/R) (Suzuki et al, 2015)]. For each of these motifs, we generated peptides in three different phosphorylation states: non‐phosphorylated, phosphorylated at the second serine and doubly phosphorylated (Fig 1C and Appendix Fig S1B). When using such peptides as substrates in in vitro kinase reactions, we saw that CDK targeted specifically only the second serine in each peptide, although much stronger for S222 than for S134, consistent with these residues matching CDK consensus motifs (Fig 1C). In contrast, DDK showed only little activity towards the non‐phosphorylated peptides, but was strongly stimulated when the second residue in the (S/T)(S/T) motif was in a phosphorylated state (Fig 1C). DDK may thus be stimulated by priming phosphorylation in order to efficiently phosphorylate Mms4 on (S/T)(S/T) sites. However, using the full‐length protein as a phosphorylation substrate, we did not obtain evidence for a stimulatory effect on DDK by prior CDK phosphorylation (Fig 1B and Appendix Fig S1C), perhaps because over the whole 15 (S/T)(S/T) motifs CDK phosphorylation plays a minor role. We also did not reveal any priming activity of either CDK or DDK for Mms4 phosphorylation by Cdc5 (Fig 1B and Appendix Fig S1D). Overall, the data in Fig 1 thus identify Mus81‐Mms4 as an interaction partner and potential substrate of DDK.
Figure 1. Dbf4‐dependent kinase (DDK) binds to the Mus81‐Mms4 complex in mitosis and can phosphorylate Mms4 at (S/T)(S/T) motifs.
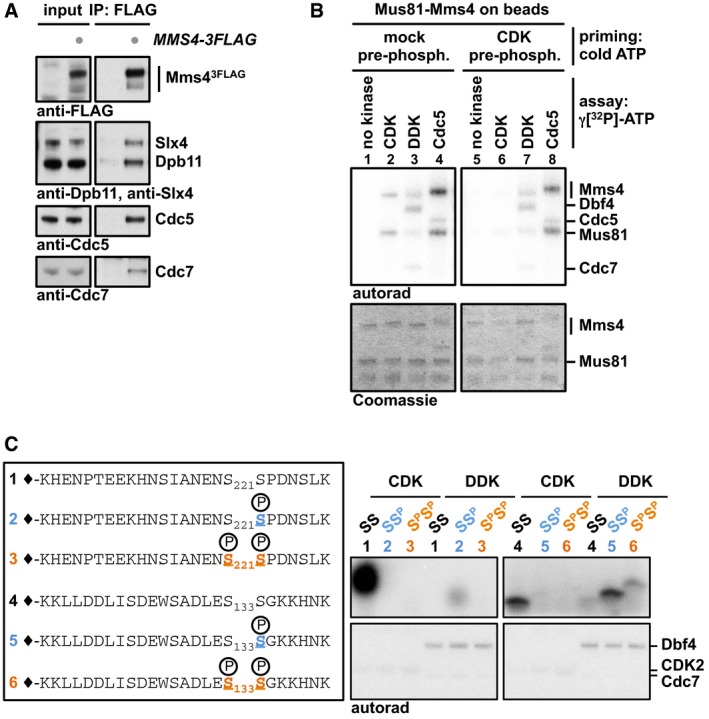
- Cdc7 and Cdc5 are specifically enriched in Mms43FLAG co‐IPs from cells arrested in mitosis (with nocodazole). Under the same conditions, Mus81‐Mms4 associates with scaffold proteins such as Dpb11 and Slx4 (Appendix Fig S1A and Gritenaite et al, 2014).
- DDK can phosphorylate Mus81‐Mms4 in vitro. Purified, immobilized Mus81‐Mms4 is incubated in an in vitro kinase assay with purified CDK2/cycAN170 (a model CDK), DDK or Cdc5 (lanes 1–4). Additionally, Mus81‐Mms4 is incubated with respective kinases after a non‐radioactive priming step with CDK (lanes 5–8).
- DDK phosphorylates Mms4 peptides at (S/T)(S/T) motifs and is enhanced by priming phosphorylation. Mms4 peptides including (S/T)(S/T) motifs (221/222; 133/134) were synthesized in different phosphorylation states (depicted in left panel) and incubated in an in vitro kinase assay with either CDK or DDK. CDK targets unphosphorylated Mms4 peptides 1 and (to a weaker extent) 4 consistent with its substrate specificity (Mok et al, 2010), while DDK primarily targets Mms4 peptides 2 and 5, which harbour a priming phosphorylation at the C‐terminal (S/T) site (see Appendix Fig S1B for in‐gel running behaviour of peptides).
Mus81‐Mms4 is phosphorylated by a mitotic Cdc5‐DDK complex
DDK is present and active throughout S phase and mitosis until anaphase when the Dbf4 subunit is degraded by APC/CCdc20 (Cheng et al, 1999; Weinreich & Stillman, 1999; Ferreira et al, 2000). We therefore tested at which cell cycle stage DDK would associate with Mus81‐Mms4 using cells synchronously progressing through the cell cycle. Figure 2A shows that DDK did not associate with Mus81‐Mms4 in S phase, but only once cells had reached mitosis. Strikingly, DDK binding therefore coincided with binding of Cdc5, Slx4 and Dpb11 and most notably the appearance of the hyperphosphorylated form of Mms43FLAG (Fig 2A).
Figure 2. DDK and Cdc5 target Mus81‐Mms4 in an interdependent manner.
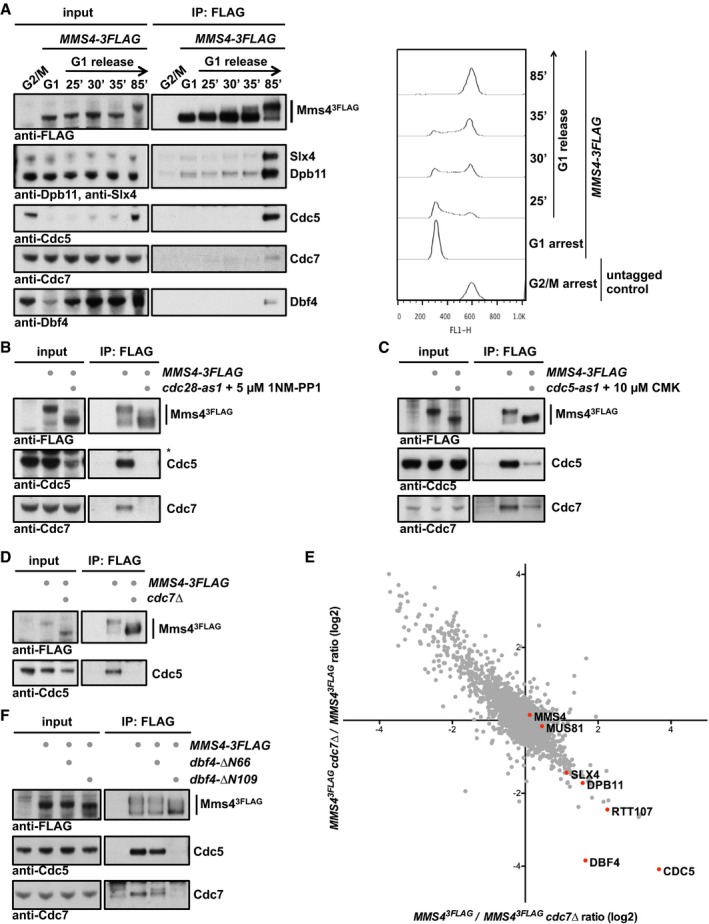
-
ADDK stably associates with Mus81‐Mms4 in mitosis, but not in S phase or G1. Mms43FLAG pull down experiment (left panel, as in Fig 1A) from cells arrested in G1 (with alpha‐factor) or in cells progressing synchronously through S phase until mitosis (arrest with nocodazole) reveals that DDK binds specifically in mitosis concomitant with the raise in Cdc5 levels and Cdc5 binding to Mus81‐Mms4. A nocodazole‐arrested untagged strain was used as a control. Right panel shows measurements of DNA content by FACS from the respective samples.
-
BCDK activity is required for DDK and Cdc5 association with Mus81‐Mms4. Mms43FLAG pull down as in (A), but in mitotic WT or cdc28‐as1 mutant cells treated with 5 μM 1NM‐PP1 for 1 h. Additional Western blots of this experiment are shown in Appendix Fig S5B, including as a control the identical anti‐FLAG Western blot.
-
CCdc5 activity is required for DDK association with Mus81‐Mms4. Mms43FLAG pull down as in (A), but with mitotically arrested WT or cdc5‐as1 mutant cells treated with 10 μM CMK for 1 h.
-
D, EDDK is required for Cdc5 binding to Mus81‐Mms4 in mitosis and the mitotic Mms4 phospho‐shift. (D) Mms43FLAG pull down using mitotically arrested cells as in (A), but using a bob1‐1 background (all samples), where the DDK subunit Cdc7 could be deleted. (E) SILAC‐based quantification of Mms43FLAG pull downs in mitotically arrested bob1‐1 vs. bob1‐1 cdc7Δ cells. Plotted are the H/L ratios of two independent experiments including label switch.
-
FThe Cdc5 binding region on Dbf4 is required for interaction of DDK and Cdc5 with Mus81‐Mms4 and for efficient Mms4 phosphorylation. Mms43FLAG pull down as in (A), but using mitotically arrested cells expressing N‐terminal truncation mutants of Dbf4 lacking aa2–66 (including a D‐box motif) or 2–109 [additionally including the Cdc5 binding site (Miller et al, 2009)].
Given this late timing of the association, we tested in co‐immunoprecipitation (co‐IP) experiments whether DDK binding to Mus81‐Mms4 would depend on CDK or Cdc5 activity. Using analog‐sensitive mutant yeast strains for CDK [cdc28‐as1 (Bishop et al, 2000)] and for Cdc5 [cdc5‐as1 (Snead et al, 2007)], we observed that inhibition of these kinases in mitotically arrested cells strongly reduced the hyperphosphorylation shift of Mms4 (see also Matos et al, 2013) and compromised the association with DDK (Fig 2B and C, and Appendix Fig S2A–C). Notably, both conditions also interfered with Cdc5 binding (Fig 2B and C, and Appendix Fig S2A), suggesting that the association of DDK may follow a similar regulation as Cdc5.
Next, we tested whether conversely DDK is involved in Mms4 phosphorylation. To bypass the essential function of DDK in DNA replication, we used the mcm5 bob1‐1 allele (Hardy et al, 1997), which allowed us to test a cdc7Δ mutant. Using Western blot and SILAC‐based mass spectrometry as a read‐out of Mms43FLAG co‐IPs from cells arrested in mitosis, we found that Cdc5 association with Mus81‐Mms4 was strongly reduced in the cdc7Δ mutant strain (Fig 2D and E). Moreover, we observed that Mms43FLAG phosphorylation as indicated by mobility shift was decreased in the absence of DDK, although not to the same extent as upon CDK or Cdc5 inhibition (Fig 2D and Appendix Fig S2C). Additionally, as an alternative way to deregulate DDK, we used the cdc7‐1 temperature‐sensitive mutant. Even with WT cells, we observed that elevated temperature (38°C) leads to a slight reduction in Cdc5 binding to Mus81‐Mms4. However, in cdc7‐1 mutant cells, incubation at 38°C leads to the complete disappearance of Cdc5 binding to Mus81‐Mms4 (Appendix Fig S2D). Therefore, we conclude from these data that DDK and Cdc5 bind to Mus81‐Mms4 in an interdependent fashion.
Interestingly, Cdc5 was previously shown to interact with DDK via a non‐consensus polo‐box binding site within Dbf4 (Miller et al, 2009; Chen & Weinreich, 2010). The proposed model based on genetic experiments suggested that DDK binding antagonizes mitotic functions of Cdc5. However, the catalytic activity of Cdc5 was not inhibited in this complex (Miller et al, 2009) and we reason that DDK may simply target Cdc5 to a specific set of substrates. Since the Cdc5 binding site was mapped to the N‐terminal portion of Dbf4 (Miller et al, 2009), we tested whether N‐terminal truncations of Dbf4 would affect DDK or Cdc5 association with Mus81‐Mms4. While the dbf4‐ΔN66 truncation lacking the first 66 amino acids (including a D‐box motif) did not influence DDK or Cdc5 binding to Mms43FLAG, the dbf4‐ΔN109 truncation, which additionally lacks the Cdc5 binding motif (Miller et al, 2009), showed strongly decreased DDK and Cdc5 binding to Mus81‐Mms4 (Fig 2F). Additionally, also mitotic hyperphosphorylation of Mms4 was diminished when DDK and Cdc5 could not interact with each other (Fig 2F). Overall, these data strongly suggest that Cdc5 and DDK interact with and target Mus81‐Mms4 in an interdependent manner. Furthermore, it is currently unclear whether collaboration of DDK and Cdc5 is a widespread phenomenon that may affect other Cdc5 substrates as well, given that mitotic phosphorylation of two candidate Cdc5 substrates, Ulp2 and Scc1 (Alexandru et al, 2001), was affected to varying degree by the cdc7Δ mutation (Appendix Fig S2E).
Given the known cell cycle regulation of Cdc5 and DDK (Shirayama et al, 1998; Cheng et al, 1999; Weinreich & Stillman, 1999; Ferreira et al, 2000; Mortensen et al, 2005), the limiting factor for the temporal regulation of this complex and its restriction to mitosis is expected to be Cdc5 and not DDK, which is present already throughout S phase. Consistently, we observed that forced expression of Cdc5 (using the galactose‐inducible GAL promoter) in cells that were arrested in S phase by hydroxyurea (HU) led to the premature occurrence of Mms4 hyperphosphorylation (Fig EV1A; Matos et al, 2013), suggesting that S‐phase DDK is in principle competent for Cdc5 binding and joint substrate phosphorylation.
Figure EV1. Cdc5 restricts Mms4 hyperphosphorylation to mitosis.
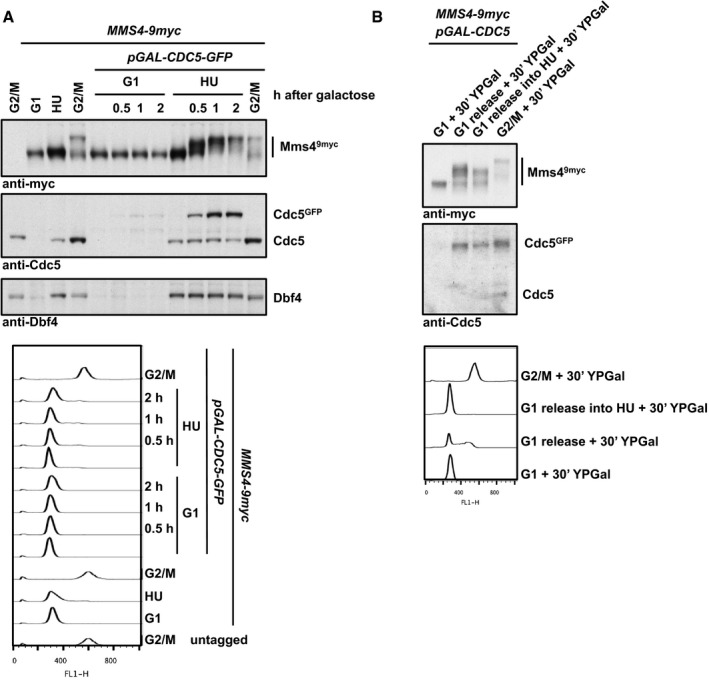
- Overexpression of CDC5 in S phase results in premature Mms4 hyperphosphorylation. Western blot analysis of Mms49myc, Cdc5 and Dbf4 from whole‐cell extracts (upper panel) and FACS data (lower panel). Cells were arrested in G1 (with alpha‐factor), S phase (with HU) or G2/M phase (with nocodazole). After arrest, CDC5 GFP overexpression was induced by addition of 2% galactose for the indicated time to cells harbouring an additional copy of GFP‐tagged CDC5 under the GAL1 promoter. Samples were run in 7% Tris‐acetate gels.
- Mms4 hyperphosphorylation by CDC5 overexpression in S phase is reduced in HU‐treated cells. Western blot analysis of Mms49myc and Cdc5 from precipitated whole‐cell extracts (upper panel) and FACS data (lower panel) of cells arrested in G1 (with alpha factor) or G2/M phase (with nocodazole), or released to S phase (with or without HU). CDC5 GFP overexpression was induced for 30 min by addition of 2% galactose to cells harbouring an additional copy of GFP‐tagged CDC5 under the GAL1 promoter. Note that upon CDC5 overexpression cells are partially defective in bulk replication. Samples were run in 7% Tris‐acetate gels.
Furthermore, we performed additional experiments that addressed the regulation of Mus81‐Mms4 by the DNA damage response. In M‐phase‐arrested cells, association of DDK and Cdc5 with Mus81‐Mms4 was reduced after induction of DNA damage with phleomycin (Appendix Fig S2F), but this treatment was not sufficient to induce a significant reduction in the Mms4 phosphorylation shift. Interestingly, when we forced Cdc5 expression in S‐phase cells and compared normal S‐phase cells to cells treated with hydroxyurea (HU), we observed that the Mms4 phosphorylation shift was less pronounced in the presence of hydroxyurea (HU) (Fig EV1B). These data are therefore consistent with the current view that DNA damage, specifically the DNA damage checkpoint, negatively influences Mus81 resolution activity (Szakal & Branzei, 2013; Gritenaite et al, 2014). Since DDK is known to be targeted and inhibited by the DNA damage checkpoint (Weinreich & Stillman, 1999; Lopez‐Mosqueda et al, 2010; Zegerman & Diffley, 2010), it could become particularly critical to regulate Mms4 phosphorylation after DNA damage.
Even though DDK and Cdc5 seem to target Mus81‐Mms4 in unison, we tested whether it was possible to resolve differences on the level of individual phosphorylation sites. Therefore, we analysed Mms4 phosphorylation sites in M‐phase cells after Cdc5 inhibition (Fig 3A and C) or CDC7 deletion (Fig 3B and D) by SILAC‐based mass spectrometry. We also applied two different experimental set‐ups that used either endogenously expressed Mus81‐Mms4 (Fig 3A and B) or overexpressed Mus81‐Mms4 (Fig 3C and D), as the latter set‐up allowed much better coverage of Mms4 phosphopeptides in higher order phosphorylation states (peptides harbouring > 1 phosphorylated site). Cdc5 inhibition or lack of DDK led to overlapping, but distinct changes in Mms4 phosphorylation sites, suggesting that each kinase phosphorylates specific sites on Mms4. After Cdc5 inhibition, phosphorylation of many sites was reduced and among those were sites that match to a putative Cdc5 consensus [(D/E/N)X(S/T), blue, Fig 3A and C; Mok et al, 2010]. Overall, CDC7 affected Mms4 phosphorylation less than Cdc5 inhibition, but nonetheless, we found widespread changes in the phosphorylation of (S/T)(S/T) motifs (Fig 3B and D). (S/T)(S/T) motifs were found less abundantly in the doubly phosphorylated state (Fig 3D, red), while conversely these motifs were found more abundantly in the state where only the second (S/T) was singly phosphorylated (Fig 3B and D, yellow), as expected for a substrate–product relation. These data are thus consistent with phosphorylation of the second (S/T) priming for phosphorylation at the preceding (S/T) (Appendix Table S1 and Appendix Fig S3).
Figure 3. Analysis of Mms4 phosphorylation sites reveals Cdc5 and DDK target sites, as well as the interdependence between the two.
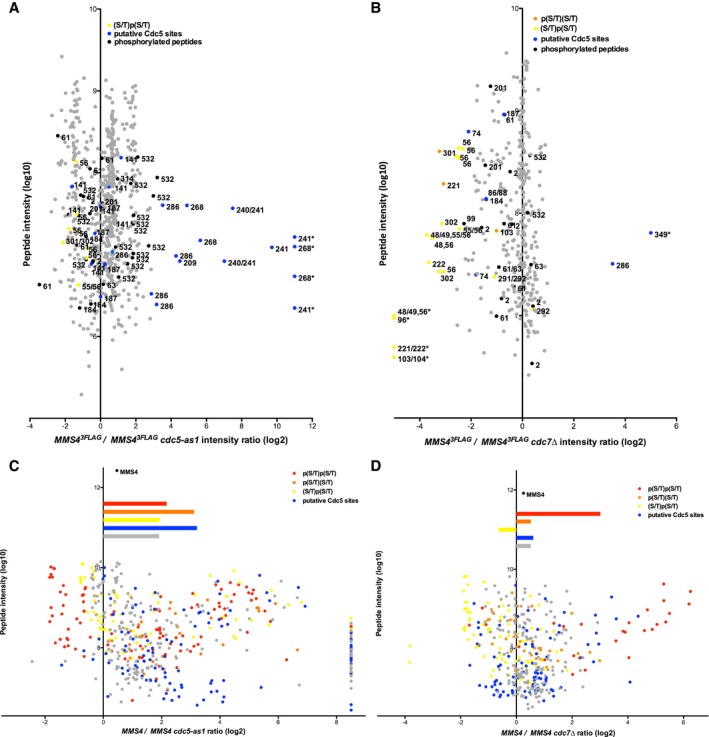
-
A, BDepicted are SILAC‐based intensity ratios of individual MS evidences for peptides of endogenously expressed Mms4. Evidences of non‐phosphorylated Mms4 peptides are shown in grey; evidences of phosphorylated peptides are shown in black, yellow, orange or blue. Blue colour indicates putative Cdc5 phosphorylation as defined by the (D/E/N)X(S/T) consensus (and additionally S268, which was also very strongly deregulated upon Cdc5 inhibition). Yellow or orange colours mark singly phosphorylated (S/T)(S/T) motifs, with orange marking p(S/T)(S/T) and yellow marking (S/T)p(S/T). Numbers indicate the phosphorylated residue in the depicted peptide. An asterisk marks peptide evidences that contained measured intensity values exclusively in the heavy or light sample. For doubly phosphorylated peptides, the two phospho‐sites are separated by a comma. For singly phosphorylated (S/T)(S/T) motifs, peptide ion fragmentation was in some cases unable to unambiguously identify the phosphorylated residue. In these cases, possible phosphorylation sites are indicated as “a/b”. Note that doubly phosphorylated (S/T)(S/T) sites were not reproducibly identified under conditions of endogenous Mus81‐Mms4 expression.
-
C, DAs in panels (A, B) but using Mus81‐Mms4 expressed from a high‐copy promoter. Depicted are SILAC‐based H/L ratios of individual MS evidences for phosphorylated peptides only. Peptides were sorted into categories according to their phosphorylation status: putative DDK target sites ((S/T)(S/T) motifs) were differentiated into the categories p(S/T)p(S/T) (red), p(S/T)(S/T) (orange) or (S/T)p(S/T) (yellow). Phosphorylated peptides matching the Cdc5 consensus site are coloured in blue. All other phosphorylated peptides are marked in grey. Bars depict the mean of the ratios of the respective category. Overall, Mms4 H/L ratio is shown on top.
DDK phosphorylation is required for activation of Mus81‐Mms4 during mitosis
Phosphorylation of Mms4 by CDK and Cdc5 has previously been shown to be required for the upregulation of Mus81‐Mms4 activity during mitosis (Matos et al, 2011, 2013; Gallo‐Fernández et al, 2012; Szakal & Branzei, 2013). Based on our finding that hyperphosphorylation of Mms4 was impaired in the absence of DDK (Fig 2D and Appendix Fig S2C), we predicted that also Mus81‐Mms4 activity would be influenced. Therefore, we tested the activity of endogenous Mus819myc‐Mms43FLAG immunopurified from G2/M arrested cells (approx. 5 fmol) on a nicked Holliday junction (nHJ) substrate (500 fmol) using an assay related to those in (Matos et al, 2011, 2013; Gritenaite et al, 2014). Notably, the activity of the endogenous purified Mus81‐Mms4 from G2/M cells exceeded the activity of recombinant Mus81‐Mms4 (subjected to a dephosphorylation step during the purification), indicating that it is the mitotically activated form (Appendix Fig S4A). Moreover, the activity of endogenous purified Mus81‐Mms4 was not influenced by 350 mM NaCl salt washes. This indicates that the presence of accessory, salt‐labile factors such as Rtt107 or Cdc5 in the reaction is unlikely to contribute to Mus81 activity (Appendix Fig S4B and C).
Importantly, when we used this assay to test immunopurified Mus819myc‐Mms43FLAG from mitotic cells lacking DDK (cdc7Δ or dbf4Δ), we observed a reduced activity compared to Mus819myc‐Mms43FLAG from WT cells (Fig 4A and Appendix Fig S4D; also observed with an RF substrate, Appendix Fig S4E). In order to exclude that indirect effects of the CDC7 deletion may cause the reduction in Mus81 activity, we furthermore created an Mms4 mutant that specifically lacks candidate DDK phosphorylation sites. We chose to mutate (S/T)(S/T) motifs (SS motifs in particular) and created an mms4‐8A mutant that harboured eight S to A exchanges at the N‐terminal (S/T) of the motifs (see Appendix Fig S3A). This mutant appeared less phosphorylated in mitosis as judged by a less pronounced phosphorylation shift (Fig 4B). Furthermore, we observed a reduction in the association of DDK and Cdc5 with the Mus81‐Mms4‐8A complex in pull‐down experiments (Fig 4B), suggesting that phosphorylation of Mms4 also plays a role in tethering these kinases. Notably, Mus819myc‐Mms43FLAG‐8A from mitotic cells showed a moderate but reproducible reduction in resolution activity on nHJ substrates compared to WT Mus819myc‐Mms43FLAG (Fig 4C and Appendix Fig S4F). These data thus indicate that DDK targets Mus81‐Mms4 and that (S/T)(S/T) phosphorylation events are essential for full activation of Mus81 in mitosis.
Figure 4. DDK phosphorylation controls activation of Mus81‐Mms4 resolvase activity in mitosis.
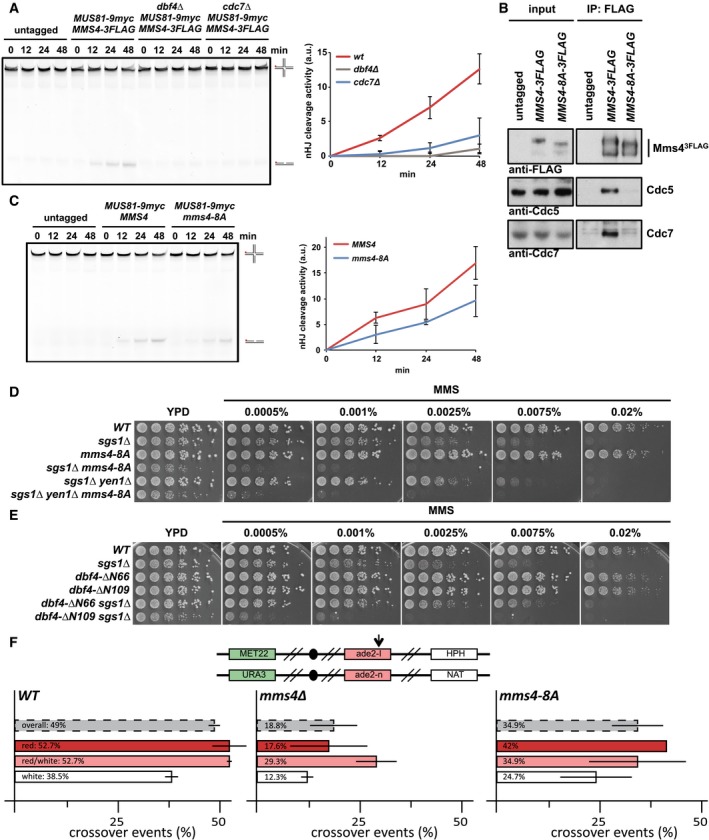
-
ADDK is required for mitotic activation of Mus81‐Mms4. Resolution assay using a nicked Holliday junction (nHJ) substrate and Mus819myc‐Mms43FLAG purified from mitotically arrested bob1‐1 (DDK‐WT+), bob1‐1 dbf4Δ and bob1‐1 cdc7Δ strains or untagged control cells. Right panel: quantification of cleavage products. See Appendix Fig S4D for Western blots samples of anti‐myc IPs. Left panel: representative gel image.
-
BA defect in the phosphorylation of Mms4 (S/T)(S/T) sites causes reduced association of Cdc5 and DDK with Mus81‐Mms4 and reduced phosphorylation of Mms4. Mms43FLAG pull down as in Fig 1A, but using mitotically arrested WT and mms4‐8A mutant cells, which harbour 8 serine to alanine exchanges at (S/T)(S/T) motifs (detailed in Appendix Fig S3).
-
CReduced (S/T)(S/T) phosphorylation of Mms4 generates a defect in Mus81‐Mms4 activity. Resolution assay as in (A), but comparing mitotic Mus81‐Mms4 from untagged, WT and mms4‐8A strains (see Appendix Fig S4F for Western blot samples of anti‐myc IPs).
-
D, EThe mms4‐8A mutation and lack of the Cdc5‐DDK interaction (dbf4‐ΔN109) lead to hypersensitivity towards MMS specifically in the sgs1Δ background. Shown is the growth of indicated strains in fivefold serial dilution on plates containing MMS at indicated concentrations after 2 days at 30°C.
-
FThe mms4‐8A mutant leads to a reduction in crossover formation. Recombination assay between heterologous ade2 alleles in diploid cells as described in Ho et al (2010). The top panel indicates markers on both copies of chromosome XV that are used to determine genetic outcomes of DSB repair. Arrow indicates the I‐SceI cut site. Bottom panel indicates rates of crossover events (%) overall (grey) and in the individual classes (red, red/white, white) that differ in gene conversion tract length. Error bars indicate standard deviation of two independent experiments, each scoring 400–600 colonies per strain.
Additionally, we investigated the relevance of the mms4‐8A mutation in vivo. In comparison with mus81Δ or mms4Δ mutants, the mms4‐8A mutant showed a hypomorphic phenotype. For example, it did neither significantly increase the MMS hypersensitivity of a yen1Δ mutant, nor did it confer synthetic lethality with mutants defective in STR function, such as sgs1Δ, even though the mms4‐8A sgs1Δ double mutant displayed a slow growth phenotype (Figs 4D and EV2A). Importantly, however, we did observe a strongly increased hypersensitivity towards MMS, when we tested an mms4‐8A sgs1Δ double mutant and compared it to an sgs1Δ single mutant (Fig 4D). The mms4‐8A mutation thus leads to a phenotype that is very similar to other activation‐deficient MMS4 mutants in budding and fission yeast (Gallo‐Fernández et al, 2012; Dehé et al, 2013; Matos et al, 2013). Remarkably, the MMS hypersensitivity phenotype of the mms4‐8A mutant was highly similar to that of the Cdc5 binding‐deficient dbf4‐ΔN109 mutant (Figs 4E and EV2B), which also showed reduced survival when combined with sgs1Δ (Fig 4E). These data are therefore consistent with DDK functioning to stimulate JM resolution via Mms4 hyperphosphorylation.
Figure EV2. Phenotypic analysis of Mms4 variants deficient in (S/T)(S/T) phosphorylation sites.
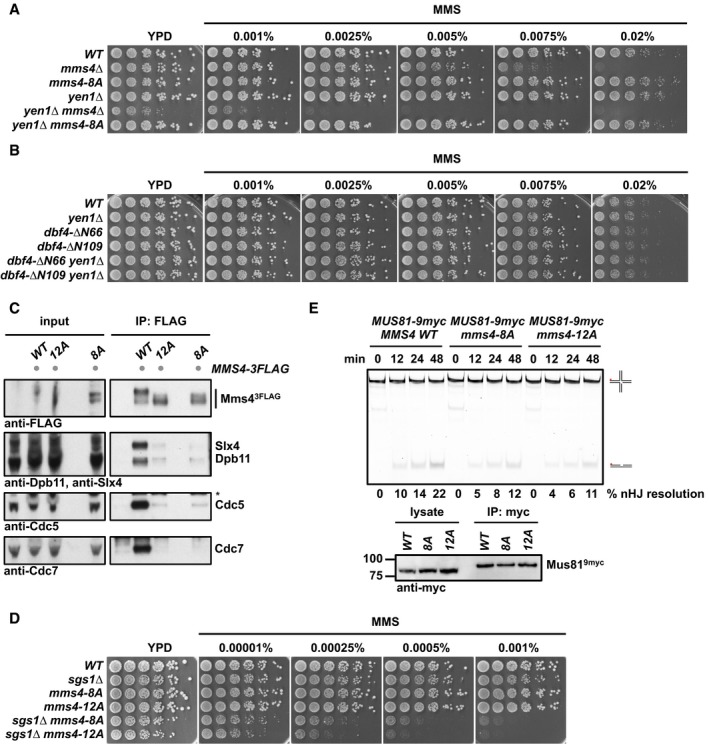
-
A, BThe mms4‐8A mutation or lack of Cdc5‐DDK interaction does not lead to a synthetic hypersensitivity towards MMS in the yen1Δ background. Spotting assay as in Fig 4D and E.
-
C–EAdditional mutation of 4 additional (S/T)(S/T) motifs in the background of the mms4‐8A mutant (mms4‐12A) leads to a reduction in the Mms4 phosphorylation shift (C), increases the hypersensitivity to MMS in the sgs1∆ background (D) and shows a slightly but not significantly decreased activity of Mus81‐Mms4 (E). (C) Mms43FLAG pull down as in Fig 1A, but in G2/M‐arrested cells in untagged, WT, mms4‐12A and mms4‐8A backgrounds. Asterisk marks a cross‐reactive band. (D) Spotting assay as in Fig 4D and E. (E) Resolution assay using a nHJ substrate and Mus819myc‐Mms43FLAG purified from mitotically arrested WT, mms4‐8A or mms4‐12A cells. Lower panel: Western blot samples of anti‐myc IPs.
It is likely that the mms4‐8A mutant is only partially deficient in DDK phosphorylation, since Mms4 contains overall 15 (S/T)(S/T) sites and DDK may phosphorylate the protein on non‐(S/T)(S/T) sites as well. We therefore note that an mms4‐12A mutant, harbouring four additional S to A exchanges on (S/T)(S/T) motifs, showed further increased MMS sensitivity in the mms4‐12A sgs1Δ mutant, when compared to the mms4‐8A sgs1Δ mutant, even though there were only minor additional effects on either the Mms4 mitotic phosphorylation shift or JM resolution activity (Fig EV2C–E).
In order to directly assess whether DDK phosphorylation was required for Mus81 function during JM resolution, we tested the influence of the mms4‐8A mutant in a genetic crossover assay (Ho et al, 2010). In this system, a site‐specific DSB is induced in diploid cells and repair products can be measured by the arrangement of markers and colony sectoring (Fig 4F, upper panel). In this assay, mus81Δ and mms4Δ mutants show a reduction in CO products and a proportional increase in NCO products (Fig 4F; Ho et al, 2010), as would be expected from a defect in JM resolution and the accompanying shift of repair pathways towards JM dissolution. The mms4‐8A mutant shows a similar, albeit weaker defect in the formation of CO products (Fig 4F), suggesting that the defect in Mus81 activation in mitosis results in an overall defect in JM resolution. We therefore conclude that DDK—in conjunction with Cdc5—acts directly on Mms4 and that these phosphorylation events are required for efficient Mus81‐dependent JM resolution in mitosis.
The Dpb11‐Mms4 interaction is not required for DDK‐Cdc5‐dependent activation of Mus81‐Mms4
It is noteworthy that the association of DDK and Cdc5 with Mus81‐Mms4 coincides with the formation of the Mus81‐Mms4 complex with scaffold proteins such as Slx4, Dpb11 and Rtt107, which come together in mitosis (Fig 2A). Therefore, we asked whether the scaffold proteins Dpb11, Slx4 or Rtt107 would be required to target DDK and Cdc5 to Mus81‐Mms4. In order to investigate the influence of Dpb11, we searched for an MMS4 mutant that was deficient in the interaction with Dpb11. When we used a two‐hybrid approach to map the Dpb11 interaction site on Mms4, we found that Mms4 constructs comprising aa 1–212 or 101–230 interacted with Dpb11, while constructs comprising aa 1–195 or 176–230 showed no or reduced interaction (Appendix Fig S5A). This suggested that the Dpb11 binding site may be located between aa 101–212 of Mms4. Consistently, we observed that the Mms4‐S201A mutation abolished binding to Dpb11 in yeast two‐hybrid and co‐IP (Fig 5A and B), while the Mms4‐S184A mutation reduced it (Fig 5A). Serine 201 and 184 are therefore likely candidates for phospho‐sites bound and read by Dpb11. Serine 201 matches the full CDK consensus motif (S/T)PxK, while serine 184 matches the minimal CDK consensus motif (S/T)P. Indeed, we find that CDK inhibition reduced the Dpb11 interaction with Mus81‐Mms4 (Appendix Fig S5B) consistent with a requirement of CDK phosphorylation for a robust interaction between Dpb11 and Mms4.
Figure 5. The interaction between Mms4 and Dpb11 is dispensable for binding of Cdc5 and DDK and mitotic Mus81‐Mms4 activation.

-
A, BSerine 201 of Mms4 is required for Dpb11 binding, but not for interaction with DDK and Cdc5. (A) Two‐hybrid interaction analysis using Gal4‐BD‐Dpb11 with Gal4‐AD‐Mms4, Gal4‐AD‐Mms4‐S184A and Gal4‐AD‐Mms4‐S201A constructs. (B) Mms43FLAG pull downs from mitotically arrested cells as in Fig 1A, but using WT or S201A variants of Mms43FLAG. Asterisks mark cross‐reactive bands.
-
CThe Dpb11 binding‐deficient allele mms4‐SS184,201AA leads to a MMS hypersensitivity specifically in the sgs1Δ background. Spotting assay as in Fig 4D.
When we investigated the phenotype of the mms4‐SS184,201AA mutant, we found that it showed enhanced hypersensitivity to MMS specifically in the sgs1Δ mutant background, consistent with a role of Dpb11 in JM resolution after MMS damage (Fig 5C). We also noted that the phenotype of this MMS4 variant differed from that induced by Dpb11 binding‐deficient version of Slx4 [slx4‐S486A (Gritenaite et al, 2014; Ohouo et al, 2012)]. This could suggest that these mutants are able to separate different Dpb11 functions such as a mitotic function in conjunction with Mus81‐Mms4 and an S‐phase function, which Slx4 and Dpb11 might have independently of Mus81‐Mms4 (Ohouo et al, 2012; Gritenaite et al, 2014; Cussiol et al, 2015; Princz et al, 2015). However, it also needs to be considered that Slx4 and Mus81‐Mms4 may be connected by more than one scaffold protein (see below).
Importantly, however, we did not observe a defect in the association of DDK or Cdc5 with Mus81‐Mms4, when we performed Mms4‐S201A3FLAG pull downs and compared them to a WT Mms43FLAG pull down (Fig 5B). Furthermore, we only observed a very minor defect in the in vitro resolution of nHJ substrates, when we purified Mus81‐Mms4 from mitotically arrested mms4‐S201A cells (Appendix Fig S5C). We therefore reason that Dpb11 is most likely not involved in promoting Mms4 phosphorylation or DDK‐Cdc5‐dependent activation of Mus81‐Mms4.
The Rtt107 scaffold recruits DDK and Cdc5 to Mus81‐Mms4
Having excluded a role of Dpb11 in the recruitment of DDK and Cdc5, we next tested a possible involvement of the Rtt107 scaffold protein. Indeed, when we used an rtt107Δ mutant in IP and SILAC‐based IP‐MS experiments, we observed that DDK and Cdc5 binding to Mus81‐Mms4 was strongly reduced (Fig 6A and Appendix Fig S6A). Interestingly, Rtt107 bound to DDK and Cdc5 even under conditions where Rtt107 binding to Mus81‐Mms4 was abolished (mus81Δ, Appendix Fig S6B). This suggests that Rtt107 may form a subcomplex with DDK and Cdc5. Consistently, we found that Rtt107 bound to Cdc7 in a two‐hybrid assay (Fig 6B). These data therefore suggest that Rtt107 mediates binding of DDK and Cdc5 to the Mus81‐Mms4 complex, most likely via a Cdc7 interaction site on Rtt107.
Figure 6. The Rtt107 scaffold tethers DDK and Cdc5 to Mus81‐Mms4 independently of Slx4 and Dpb11.
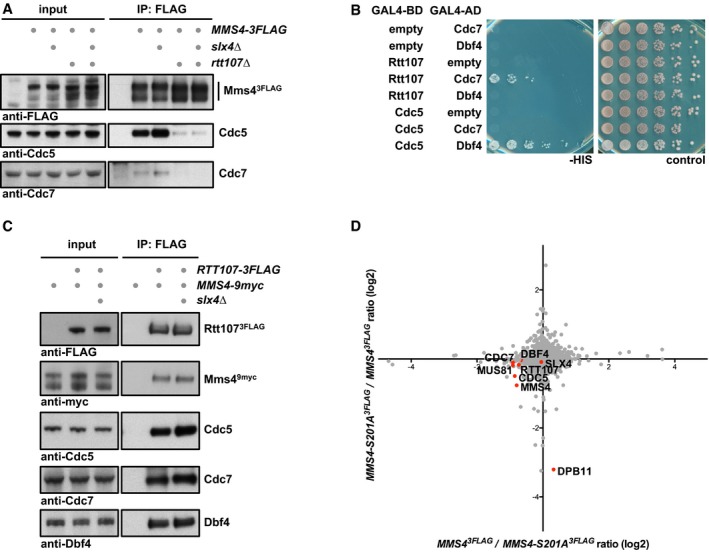
- Rtt107, but not Slx4, is required for DDK and Cdc5 interaction with Mus81‐Mms4. Mms43FLAG pull downs from mitotically arrested cells as in Fig 1A, but specifically comparing interactions of Mus81‐Mms4 in WT, slx4Δ, rtt107Δ and slx4Δ rtt107Δ mutant backgrounds.
- Rtt107 interacts with Cdc7. Two‐hybrid interaction was tested using Gal4‐BD‐Rtt107 constructs and Gal4‐AD‐Cdc7 or Gal4‐AD‐Dbf4 constructs. Interaction between Gal4‐BD‐Cdc5 and Gal4‐AD‐Dbf4 serves as positive control.
- Rtt107 interacts with Mus81‐Mms4, DDK and Cdc5 independently of Slx4. Rtt1073FLAG co‐IPs from untagged control, WT or slx4Δ cells arrested in mitosis were probed for indicated proteins.
- Rtt107 interacts with Mus81‐Mms4 independently of the Mms4‐Dpb11 interaction. SILAC‐based Mms43FLAG pull down in WT and mms4‐S201A cells reveals changes in the Dpb11 association, but not in Rtt107, Slx4, Cdc5 or DDK binding. Plotted are the H/L ratios of two experiments including label switch.
During our co‐IP studies, we furthermore found that the location of Rtt107 in the mitotic Mus81‐Mms4 complex was different than expected. Given that Slx4 was required to bridge between Rtt107 and Dpb11 (Ohouo et al, 2010) and that Mms4 and Dpb11 seemingly interact directly (Gritenaite et al, 2014 and Fig 5A and B), we initially expected that Slx4 and Dpb11 would be required to mediate the interaction between Rtt107 and Mus81‐Mms4. Surprisingly, we found that an slx4Δ mutant did not influence DDK or Cdc5 binding to Mus81‐Mms4 and thereby differed from rtt107Δ (Fig 6A). Therefore, we tested if Rtt107 could bind to Mus81‐Mms4 independently of Slx4 or Dpb11. Indeed, we found that the Mus81‐Mms4 interaction to Rtt107 was not influenced by the slx4Δ mutant (Fig 6C) or the Dpb11 binding‐deficient mms4‐S201A allele (Fig 6D), indicating that Rtt107 binding to the Mus81‐Mms4 complex occurs independently of the other scaffold proteins. In contrast, our data also show that its binding is strongly dependent on kinases and Mms4 phosphorylation, since Rtt107 binding was strongly reduced in the absence of DDK (Fig 2E), after Cdc5 inhibition (Appendix Fig S2A) or in the mms4‐8A phosphorylation site mutant (Fig EV3).
Figure EV3. A defect in the phosphorylation of Mms4 (S/T)(S/T) sites (mms4‐8A) causes reduced association of Cdc5, DDK and Rtt107 with Mus81‐Mms4.
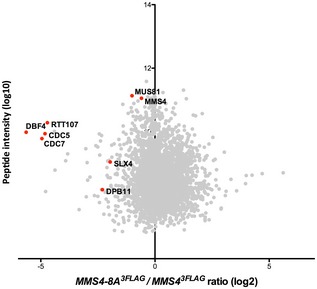
SILAC‐based quantification of Mms43FLAG pull downs in WT vs. mms4‐8A cells. Plotted are the H/L ratios against peptide intensity
Therefore, these data provide novel insight into the role of Rtt107 in Mus81‐Mms4 regulation. First, it shows that Rtt107 mediates the association of DDK and Cdc5 kinases with Mus81‐Mms4. Second, it also suggests that Rtt107 may bind directly to Mus81‐Mms4 and that this binding is dependent on Mms4 phosphorylation and the cell cycle kinases DDK and Cdc5, although an alternative model whereby Rtt107 indirectly promotes DDK and Cdc5 to tightly associate with Mus81‐Mms4 cannot be ruled out entirely. The fact that Rtt107 promotes the interaction of Mus81‐Mms4 with the kinases, yet in turn requires the kinases and Mms4 phosphorylation for interaction, suggests that Rtt107 may be acting after initial Mms4 phosphorylation has occurred and at this late stage tethers the kinases, thus promoting phosphorylation of otherwise inefficiently phosphorylated sites.
Rtt107 stimulates Mms4 hyperphosphorylation in order to enhance Mus81‐Mms4 activity in mitosis
Given Rtt107's involvement in tethering DDK and Cdc5 to the Mus81‐Mms4 complex, we asked whether Rtt107 would mediate mitotic hyperphosphorylation of Mms4 and concomitant activation of the Mus81 nuclease. We observed only a minor effect on the mitotic phospho‐shift of Mms4 when using rtt107Δ mutants (Fig 6A and Appendix Fig S2C). However, as it is still unclear which phosphorylation sites contribute to the Mms4 phospho‐shift, we investigated the effect of rtt107Δ on individual phosphorylation sites in our mass spectrometry data. Appendix Fig S7A and B shows SILAC‐based comparisons of Mms4 phosphorylation sites in WT and rtt107Δ cells, expressing Mus81‐Mms4 from endogenous (Appendix Fig S7A) or high‐copy promoters (Appendix Fig S7B). The overexpression set‐up allowed us to quantify phosphorylation at (S/T)(S/T) motifs, and we found that double phosphorylation of several of these sites was reduced (Appendix Fig S7B), although the change was much smaller compared to cells lacking DDK. On the other hand, while we could not detect higher order phosphorylated Mms4 peptides using endogenous Mus81‐Mms4, we could detect an effect of Rtt107 on several other sites (T209, S241 and S268, and to a lesser extent S286; Appendix Fig S7A), which were also deregulated after Cdc5 inhibition (Fig 3A and C). These data are thus consistent with Rtt107 promoting efficient DDK and Cdc5 phosphorylation of Mms4.
Therefore, we tested whether Rtt107 would affect the mitotic activation of Mus81‐Mms4. We immunopurified Mus819myc‐Mms43FLAG from WT and rtt107Δ cells that were arrested in mitosis and found that Mus81‐Mms4 activity on a nHJ substrate was reduced in the rtt107Δ background (Fig 7A and Appendix Fig S7C). Furthermore, in the background of deficient DDK (cdc7Δ bob1‐1), additional mutation of rtt107Δ did not lead to a further defect in Mus81‐mediated cleavage (Appendix Fig S7D). Therefore, we conclude that Rtt107 is required for full mitotic activation of Mus81‐Mms4 and that it works at least in part through cell cycle kinases such as DDK.
Figure 7. Rtt107 is required for efficient Mus81‐Mms4 activation in mitosis.
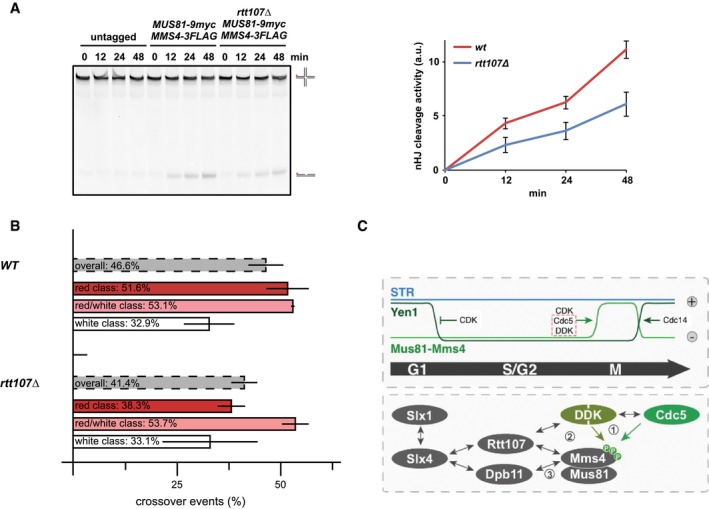
- Mus81‐Mms4 purified from mitotic rtt107Δ cells is less active compared to Mus81‐Mms4 from WT cells. In vitro resolution activity of Mus819myc‐Mms43FLAG purified from WT or rtt107Δ cells is tested on a nHJ substrate (see Appendix Fig S7C for control Western blot). Right panel: quantification of cleavage products from three independent experiments (mean ± SD). Left panel: representative gel picture.
- The rtt107Δ mutant leads to a reduction in crossover formation. Recombination assay as in Fig 4F. Note that the rtt107Δ mutant particularly affects crossover formation in the red class (long conversion tracts), while no significant defect could be observed in the red/white and white class (mean ± SD).
- Hypothetical model of Mus81‐based JM resolution. Upper panel: cell cycle regulation of JM removal pathways, indicating Mus81 activation in mitosis. Lower panel: physical interactions of Mus81‐Mms4 and its regulatory complex in mitotic cells. Grey arrows indicate physical interactions; green arrows specifically indicate kinase–substrate interactions. Genetic data indicate a hierarchy of molecular events leading to Mus81 activation. (1) DDK, Cdc5 and CDK (not shown) phosphorylate Mms4. (2) Rtt107 binds to DDK and Cdc5 and—in a phosphorylation‐dependent manner—associates with Mus81‐Mms4. This interaction is either direct or could potentially depend on bridging effects by DDK and Cdc5. Rtt107 promotes the stable interaction of DDK and Cdc5 with Mus81‐Mms4 and thus full phosphorylation of Mms4 and Mus81 activation. (3) Upon Mms4 phosphorylation, two scaffold proteins, Rtt107 and Dpb11, bind independently to Mus81‐Mms4. Both proteins can also bind to Slx4 enabling two alternative connections of Slx4 with Mus81‐Mms4.
In order to test whether such a defect in Mus81‐Mms4 activation would translate into a shifted balance of JM removal pathways, we measured rates of crossover and non‐crossover formation in the absence of Rtt107. We observed a reduction in crossover rates in the rtt107Δ mutant indicating a shift in the balance of JM removal pathways (Fig 7B). The decrease was mostly visible in one class of recombinants (Fig 7B, “red”) and is smaller compared to the phenotype of a mus81Δ or a mms4‐8A mutant (Ho et al, 2010; Fig 4F), consistent with a stimulatory but non‐essential role of the Rtt107 scaffold in Mus81‐Mms4 function. These data thus provide the first mechanistic insight of how the interaction of the mitotic Mus81‐Mms4 complex with the scaffold proteins influences Mus81 function, as Rtt107 facilitates DDK and Cdc5 tethering, full mitotic phosphorylation of Mms4 and activation of Mus81‐Mms4.
Discussion
Activation of Mus81‐Mms4 during mitosis is critical for the response to DNA damage, in particular to process repair intermediates that may arise from DSBs and stalled replication forks (Matos et al, 2011, 2013; Gallo‐Fernández et al, 2012; Saugar et al, 2013; Szakal & Branzei, 2013). Previously, this regulation was shown to critically depend on phosphorylation by the cell cycle kinases CDK and Cdc5 (Matos et al, 2011, 2013; Gallo‐Fernández et al, 2012; Saugar et al, 2013; Szakal & Branzei, 2013), but also involve the formation of a multi‐protein complex comprising several scaffold proteins (Gritenaite et al, 2014). Here, we not only identify a new cell cycle kinase to be crucial for this regulation—DDK—but moreover show that the two regulatory pathways—cell cycle kinase phosphorylation and scaffold complex formation—are connected by Rtt107 (see Fig 7C for a hypothetical model). Rtt107 association depends on active cell cycle kinases and Mms4 phosphorylation, but in turn Rtt107 is required for stable DDK and Cdc5 association with the Mus81‐Mms4 complex, as well as full phosphorylation of Mms4 and mitotic activation of Mus81. This study thus extends our mechanistic understanding of the regulatory framework that controls cell cycle‐regulated JM resolution.
Interestingly, our work shows that for its function as a regulator of Mus81‐Mms4 DDK must act interdependently and as a complex with Cdc5. DDK and Cdc5 have been shown to interact physically (Miller et al, 2009; Chen & Weinreich, 2010), but until now DDK was viewed to antagonize mitotic functions of Cdc5 (Miller et al, 2009). In contrast, in meiosis I DDK and Cdc5 are known to cooperate in order to promote chromosome segregation and jointly phosphorylate the monopolin and cohesin subunits Lrs4 and Rec8, respectively, as well as the meiotic regulator Spo13 (Matos et al, 2008). We now provide the first example for a joint DDK and Cdc5 substrate in the mitotic cell cycle, suggesting that cooperation between DDK and Cdc5 could be a more widespread phenomenon than previously anticipated. The apparent antagonism between DDK and Cdc5 in the regulation of mitotic exit (Miller et al, 2009), a canonical Cdc5 function, could be explained if DDK targeted Cdc5 to a specialized subset of substrates rather than to substrates involved in mitotic exit. It is also interesting to note that we could detect significant DDK binding to Mus81‐Mms4 only after cells finished S phase (Fig 2A). Therefore, the role of DDK in Mms4 phosphorylation is clearly post‐replicative and further challenges a simplified view of DDK as an S‐phase kinase (Matos et al, 2008). It will therefore be interesting to see whether additional DDK substrates during mitosis can be identified and whether DDK collaborates with Cdc5 for their phosphorylation as well.
Mus81‐Mms4 has previously been shown to be cell cycle‐regulated and Mms4 to be a critical CDK and Cdc5 phosphorylation target (Matos et al, 2011; Gallo‐Fernández et al, 2012). We add DDK to this already complex regulation. Our data clearly show that phosphorylation of (S/T)(S/T) motifs is critical for Mus81‐Mms4 function. The hypomorphic phenotype of the mms4‐8A mutant (Fig 4C, D and F) is likely due to additional DDK phosphorylation sites either on Mms4 or perhaps even on Mus81. Importantly, DDK does not appear to establish the timing of Mms4 phosphorylation in mitosis, as Cdc5 still seems to be the limiting factor for this temporal control in undisturbed cell cycles (Fig EV1B). However, the fact that activation of Mus81‐Mms4 depends on the activity of several kinases makes it a coincidence detector that integrates the activity of several cell cycle regulators. Therefore, it can be envisioned that there are specific cellular conditions under which DDK activity becomes limiting for Mus81‐Mms4 activation. Notably, DNA damage checkpoint kinases are known to phosphorylate DDK and counteract its function during S phase (Weinreich & Stillman, 1999; Lopez‐Mosqueda et al, 2010; Zegerman & Diffley, 2010). Therefore, it can be speculated that the checkpoint acts as a negative regulator of Mus81‐Mms4 activation via inhibition of DDK. Such regulation could therefore explain how the presence of DNA damage restricts Mus81 activity towards replication intermediates (Matos et al, 2011, 2013; Saugar et al, 2013; Szakal & Branzei, 2013; Gritenaite et al, 2014), suggesting that cell cycle and checkpoint pathways converge in the regulation of Mus81.
A second layer of Mus81 regulation relies on the formation of a multi‐protein complex, which assembles specifically in mitosis and contains Mus81‐Mms4, DDK, Cdc5 and Slx4 as well as the scaffold proteins Dpb11 and Rtt107 (Gritenaite et al, 2014). We are only beginning to understand the mechanism whereby this scaffold complex influences Mus81 function. Here, we show that Rtt107, but not Dpb11 or Slx4, promotes the stable association of DDK and Cdc5 with Mus81‐Mms4 (Fig 6), suggesting that one function of the multi‐protein complex is to promote efficient Mus81‐Mms4 phosphorylation. Conversely, our new data as well as our previous work (Gritenaite et al, 2014) show that phosphorylation by cell cycle kinases also regulates the formation of the multi‐protein complex. In particular, Rtt107 association with Mus81‐Mms4 depends strongly on DDK and Cdc5 (Fig 2E and Appendix Fig S2A). A direct interaction of Rtt107 with Mus81‐Mms4 seems the most plausible interpretation of our data, although we currently cannot exclude that Rtt107 may facilitate the interaction of DDK and Cdc5 with Mus81‐Mms4 without a direct interaction. A possible phosphorylation dependence of Rtt107 binding to the complex could thus originate from Mms4 phosphorylation generating a binding site for Rtt107 [e.g. for Rtt107 BRCT domains (Li et al, 2012)].
Importantly, Rtt107 is in turn required for stable binding of DDK and Cdc5 (Fig 6A and Appendix Fig S6A). Via tethering the kinases, Rtt107 regulates the phosphorylation of specific Mms4 sites and is required for full Mus81 activation (Fig 7A and Appendix Fig S7A and B). The interdependence between Rtt107 and Cdc5/DDK phosphorylation therefore suggests that Rtt107 may be part of a signal amplification mechanism, which ensures efficient Mus81‐Mms4 phosphorylation and activation. Mechanistically, Rtt107‐dependent stimulation of Mms4 phosphorylation thus resembles a kinase priming mechanism. It is entirely possible that other kinase priming mechanisms for either Cdc5 or DDK are at work in the Mms4 phosphorylation cascade, although the in vitro kinase assays with full‐length proteins did not provide support for such a mechanism (Fig 1B, and Appendix Fig S1C and D). Altogether, it seems plausible to speculate that Rtt107‐dependent and Rtt107‐independent amplification mechanisms are involved in generating a switch‐like activation of Mus81 in mitosis.
Furthermore, Rtt107 can also bind to Slx4 (Ohouo et al, 2010). There are thus two BRCT‐containing scaffold proteins—Dpb11 (Gritenaite et al, 2014) and Rtt107—that could bridge between Mus81‐Mms4 and Slx4. Interestingly, our data with different mms4 mutants suggest that either one of these BRCT scaffold proteins is sufficient to connect Slx4 and Mus81‐Mms4 [Figs 6D and EV3; note that the rtt107Δ mutant (Appendix Fig S6A) is difficult to interpret in this regard as it also leads to defects in Slx4 phosphorylation and the Slx4‐Dpb11 interaction (Ohouo et al, 2010)]. This redundancy may thus explain the modest phenotype of the mms4‐S201A mutant that is deficient in the Mms4‐Dpb11 interaction (Fig 5C).
Several aspects of Mus81‐Mms4 regulation are conserved throughout eukaryotic evolution. The HJ resolution activity of Mus81‐Eme1 in mammalian cells is cell cycle‐regulated (Matos et al, 2011; Wyatt et al, 2013). Mus81‐Eme1 furthermore binds to Slx4 and forms multi‐protein complexes (Fekairi et al, 2009; Muñoz et al, 2009; Svendsen et al, 2009; Castor et al, 2013; Wyatt et al, 2013), albeit these complexes may have a different organization to that in yeast. Therefore, it will be interesting to explore in the future if in human cells DDK is also required for activation of Mus81‐Eme1 and if this mechanism may contribute to the anti‐tumorigenic activity of DDK inhibitors (Montagnoli et al, 2008).
Materials and Methods
All yeast strains are based on W303 and were constructed using standard methods. Plasmids were constructed using the In‐Fusion HD cloning kit (Clontech Laboratories), and mutations were introduced by site‐directed mutagenesis. A summary of all yeast strains used in this study can be found in the Appendix Table S2.
Cell cycle synchronization was achieved using alpha‐factor (G1), hydroxyurea (S), or nocodazole (mitosis). DNA content was measured by flow cytometry with a BD FACSCalibur system using SYTOX green to stain DNA.
Co‐immunoprecipitations of yeast extracts were performed on anti‐FLAG agarose resin (Sigma) for 2 h with head‐over‐tail rotation at 4°C as previously described (Gritenaite et al, 2014). After bead washing, proteins were eluted by 3X FLAG‐peptide (Sigma), precipitated and separated on 4–12% Bis‐Tris gels. For SILAC‐based mass spectrometry, cells were labelled with heavy‐isotope‐labelled lysine (Lys6 or Lys8), and proteins were digested with Lys‐C. Mass spectrometry data were analysed using MaxQuant (Cox & Mann, 2008).
Yeast two‐hybrid assays, genetic interaction assays, in vitro kinase assays and peptide binding assays were performed as described previously (Pfander & Diffley, 2011; Gritenaite et al, 2014).
Nuclease assays were done as described (Matos et al, 2011, 2013). Briefly, Mus819myc was immunopurified from mitotically arrested cells and mixed with 5′‐Cy3‐end‐labelled nicked Holliday junctions. After incubation at 30°C for the indicated times, the reaction was stopped by proteinase K and SDS for 1 h at 37°C. Products were separated by 10% PAGE, and cleavage efficiency was normalized to the level of immunoprecipitated Mus819myc. Unspecific nHJ cleavage in untagged controls was subtracted in the quantifications.
DSB‐induced recombination assays were performed as described (Ho et al, 2010). Diploids harbouring I‐SceI under the control of the GAL promoter were grown in adenine‐rich raffinose medium and arrested in mitosis. Nuclease expression was induced by addition of galactose for 2.5 h. Cells were plated on YPAD and replica plated on YPAD + Hyg + Nat, YPAD + Hyg, YPAD + Nat, SC‐Met, SC‐Ura and SCR‐ADE + Gal media after 3–4 days to classify recombination events.
Detailed experimental procedures are available in the Appendix.
Data availability
Mass spectrometric datasets are available at EBI PRIDE. DDK and the Rtt107 scaffold promote Mus81‐Mms4 resolvase activation during mitosis (2015). PXD005356.
Author contributions
PW and JM performed in vitro resolution assays of Figs 4A and C, 7A, and EV2E, and Appendix Figs S4A, C, E, and S7D and analysed the data. FJA and MGB provided recombinant purified Mus81‐Mms4 used in Fig 1B, and Appendix Figs S1C and D, and S4A. All other experiments were performed and analysed by LNP, JB and BP. BP wrote the paper and all authors commented on the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank U. Kagerer for technical assistance, D. Gritenaite for early contributions to the project, N. Nagaraj and the proteomics laboratory of the MPIB core facility for proteomics analysis, the MPIB core facility for peptide synthesis and sequencing, J. Diffley and L. Symington for antibodies, plasmid constructs and strains, S. Jentsch, Z. Storchova, C. Biertümpfel and members of the Jentsch and Pfander labs for stimulating discussion and critical reading of the manuscript. Work in the Pfander laboratory is supported by the Max‐Planck Society and the German Research Council (DFG). Work in the Matos laboratory is supported by ETH Zürich and the Swiss National Science Foundation (Grants 31003A_153058 and 155823). Work in the Blanco laboratory is co‐financed by Ministerio de Economía y Competitividad and FEDER (RYC‐2012‐10835 and BFU2013‐41554‐P). Philipp Wild is supported by an EMBO long‐term fellowship (ALTF 475‐2015). F. Javier Aguado is supported by a PhD fellowship from the I2C Program of Xunta de Galicia (ED481A‐2015/011).
The EMBO Journal (2017) 36: 664–678
References
- Alexandru G, Uhlmann F, Mechtler K, Poupart MA, Nasmyth K (2001) Phosphorylation of the cohesin subunit Scc1 by Polo/Cdc5 kinase regulates sister chromatid separation in yeast. Cell 105: 459–472 [DOI] [PubMed] [Google Scholar]
- Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, Wood JL, Morgan DO, Shokat KM (2000) A chemical switch for inhibitor‐sensitive alleles of any protein kinase. Nature 407: 395–401 [DOI] [PubMed] [Google Scholar]
- Bizard AH, Hickson ID (2014) The dissolution of double Holliday junctions. Cold Spring Harb Perspect Biol 6: a016477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco MG, Matos J, West SC (2014) Dual control of Yen1 nuclease activity and cellular localization by Cdk and Cdc14 prevents genome instability. Mol Cell 54: 94–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D, Foiani M (2008) Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol 9: 297–308 [DOI] [PubMed] [Google Scholar]
- Castor D, Nair N, Déclais A‐C, Lachaud C, Toth R, Macartney TJ, Lilley DMJ, Arthur JSC, Rouse J (2013) Cooperative control of Holliday junction resolution and DNA repair by the SLX1 and MUS81‐EME1 nucleases. Mol Cell 52: 221–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YC, Weinreich M (2010) Dbf4 regulates the Cdc5 polo‐like kinase through a distinct non‐canonical binding interaction. J Biol Chem 285: 41244–41254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Collyer T, Hardy CF (1999) Cell cycle regulation of DNA replication initiator factor Dbf4p. Mol Cell Biol 19: 4270–4278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- Cussiol JR, Jablonowski CM, Yimit A, Brown GW, Smolka MB (2015) Dampening DNA damage checkpoint signalling via coordinated BRCT domain interactions. EMBO J 34: 1704–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehé P‐M, Coulon S, Scaglione S, Shanahan P, Takedachi A, Wohlschlegel JA, Yates JR, Llorente B, Russell P, Gaillard P‐HL (2013) Regulation of Mus81–Eme1 Holliday junction resolvase in response to DNA damage. Nat Struct Mol Biol 20: 598–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eissler CL, Mazón G, Powers BL, Savinov SN, Symington LS, Hall MC (2014) The Cdk/Cdc14 module controls activation of the Yen1 Holliday junction resolvase to promote genome stability. Mol Cell 54: 80–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fekairi S, Scaglione S, Chahwan C, Taylor ER, Tissier A, Coulon S, Dong M‐Q, Ruse C, Yates JR, Russell P, Fuchs RP, McGowan CH, Gaillard P‐HL (2009) Human SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell 138: 78–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira MF, Santocanale C, Drury LS, Diffley JF (2000) Dbf4p, an essential S phase‐promoting factor, is targeted for degradation by the anaphase‐promoting complex. Mol Cell Biol 20: 242–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferretti LP, Lafranchi L, Sartori AA (2013) Controlling DNA‐end resection: a new task for CDKs. Front Genet 4: 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo‐Fernández M, Saugar I, Ortiz‐Bazán MÁ, Vázquez MV, Tercero JA (2012) Cell cycle‐dependent regulation of the nuclease activity of Mus81‐Eme1/Mms4. Nucleic Acids Res 40: 8325–8335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritenaite D, Princz LN, Szakal B, Bantele SCS, Wendeler L, Schilbach S, Habermann BH, Matos J, Lisby M, Branzei D, Pfander B (2014) A cell cycle‐regulated Slx4‐Dpb11 complex promotes the resolution of DNA repair intermediates linked to stalled replication. Genes Dev 28: 1604–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy CF, Dryga O, Seematter S, Pahl PM, Sclafani RA (1997) mcm5/cdc46‐bob1 bypasses the requirement for the S phase activator Cdc7p. Proc Natl Acad Sci USA 94: 3151–3155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyer W‐D, Ehmsen KT, Liu J (2010) Regulation of homologous recombination in eukaryotes. Annu Rev Genet 44: 113–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho CK, Mazón G, Lam AF, Symington LS (2010) Mus81 and Yen1 promote reciprocal exchange during mitotic recombination to maintain genome integrity in budding yeast. Mol Cell 40: 988–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Interthal H, Heyer WD (2000) MUS81 encodes a novel helix‐hairpin‐helix protein involved in the response to UV‐ and methylation‐induced DNA damage in Saccharomyces cerevisiae . Mol Gen Genet 263: 812–827 [DOI] [PubMed] [Google Scholar]
- Ip SCY, Rass U, Blanco MG, Flynn HR, Skehel JM, West SC (2008) Identification of Holliday junction resolvases from humans and yeast. Nature 456: 357–361 [DOI] [PubMed] [Google Scholar]
- Li X, Liu K, Li F, Wang J, Huang H, Wu J, Shi Y (2012) Structure of C‐terminal tandem BRCT repeats of Rtt107 protein reveals critical role in interaction with phosphorylated histone H2A during DNA damage repair. J Biol Chem 287: 9137–9146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Mosqueda J, Maas NL, Jonsson ZO, DeFazio‐Eli LG, Wohlschlegel J, Toczyski DP (2010) Damage‐induced phosphorylation of Sld3 is important to block late origin firing. Nature 467: 479–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons NA, Fonslow BR, Diedrich JK, Yates JR, Morgan DO (2013) Sequential primed kinases create a damage‐responsive phosphodegron on Eco1. Nat Struct Mol Biol 20: 194–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankouri HW, Huttner D, Hickson ID (2013) How unfinished business from S‐phase affects mitosis and beyond. EMBO J 32: 2661–2671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masai H, Taniyama C, Ogino K, Matsui E, Kakusho N, Matsumoto S, Kim JM, Ishii A, Tanaka T, Kobayashi T, Tamai K, Ohtani K, Arai K‐I (2006) Phosphorylation of MCM4 by Cdc7 kinase facilitates its interaction with Cdc45 on the chromatin. J Biol Chem 281: 39249–39261 [DOI] [PubMed] [Google Scholar]
- Mathiasen DP, Lisby M (2014) Cell cycle regulation of homologous recombination in Saccharomyces cerevisiae . FEMS Microbiol Rev 38: 172–184 [DOI] [PubMed] [Google Scholar]
- Matos J, Lipp JJ, Bogdanova A, Guillot S, Okaz E, Junqueira M, Shevchenko A, Zachariae W (2008) Dbf4‐dependent CDC7 kinase links DNA replication to the segregation of homologous chromosomes in meiosis I. Cell 135: 662–678 [DOI] [PubMed] [Google Scholar]
- Matos J, Blanco MG, Maslen S, Skehel JM, West SC (2011) Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell 147: 158–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos J, Blanco MG, West SC (2013) Cell‐cycle kinases coordinate the resolution of recombination intermediates with chromosome segregation. Cell Rep 4: 76–86 [DOI] [PubMed] [Google Scholar]
- Matos J, West SC (2014) Holliday junction resolution: regulation in space and time. DNA Repair (Amst) 19: 176–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CT, Gabrielse C, Chen Y‐C, Weinreich M (2009) Cdc7p‐Dbf4p regulates mitotic exit by inhibiting polo kinase. PLoS Genet 5: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok J, Kim PM, Lam HYK, Piccirillo S, Zhou X, Jeschke GR, Sheridan DL, Parker SA, Desai V, Jwa M, Cameroni E, Niu H, Good M, Remenyi A, Ma J‐LN, Sheu Y‐J, Sassi HE, Sopko R, Chan CSM, De Virgilio C et al (2010) Deciphering protein kinase specificity through large‐scale analysis of yeast phosphorylation site motifs. Sci Signal 3: ra12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagnoli A, Valsasina B, Brotherton D, Troiani S, Rainoldi S, Tenca P, Molinari A, Santocanale C (2006) Identification of Mcm2 phosphorylation sites by S‐phase‐regulating kinases. J Biol Chem 281: 10281–10290 [DOI] [PubMed] [Google Scholar]
- Montagnoli A, Valsasina B, Croci V, Menichincheri M, Rainoldi S, Marchesi V, Tibolla M, Tenca P, Brotherton D, Albanese C, Patton V, Alzani R, Ciavolella A, Sola F, Molinari A, Volpi D, Avanzi N, Fiorentini F, Cattoni M, Healy S et al (2008) A Cdc7 kinase inhibitor restricts initiation of DNA replication and has antitumor activity. Nat Chem Biol 4: 357–365 [DOI] [PubMed] [Google Scholar]
- Mortensen EM, Haas W, Gygi M, Gygi SP, Kellogg DR (2005) Cdc28‐dependent regulation of the Cdc5/Polo kinase. Curr Biol 15: 2033–2037 [DOI] [PubMed] [Google Scholar]
- Muñoz IM, Hain K, Déclais A‐C, Gardiner M, Toh GW, Sanchez‐Pulido L, Heuckmann JM, Toth R, Macartney T, Eppink B, Kanaar R, Ponting CP, Lilley DMJ, Rouse J (2009) Coordination of structure‐specific nucleases by human SLX4/BTBD12 Is required for DNA repair. Mol Cell 35: 116–127 [DOI] [PubMed] [Google Scholar]
- Ohouo PY, de Oliveira FMB, Almeida BS, Smolka MB (2010) DNA damage signaling recruits the Rtt107‐Slx4 scaffolds via Dpb11 to mediate replication stress response. Mol Cell 39: 300–306 [DOI] [PubMed] [Google Scholar]
- Ohouo PY, de Oliveira FMB, Liu Y, Ma CJ, Smolka MB (2012) DNA‐repair scaffolds dampen checkpoint signalling by counteracting the adaptor Rad9. Nature 493: 120–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfander B, Diffley JFX (2011) Dpb11 coordinates Mec1 kinase activation with cell cycle‐regulated Rad9 recruitment. EMBO J 30: 4897–4907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Princz LN, Gritenaite D, Pfander B (2015) The Slx4‐Dpb11 scaffold complex: coordinating the response to replication fork stalling in S‐phase and the subsequent mitosis. Cell Cycle 14: 488–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randell JCW, Fan A, Chan C, Francis LI, Heller RC, Galani K, Bell SP (2010) Mec1 is one of multiple kinases that prime the Mcm2‐7 helicase for phosphorylation by Cdc7. Mol Cell 40: 353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saugar I, Vazquez MV, Gallo‐Fernandez M, Ortiz‐Bazan MA, Segurado M, Calzada A, Tercero JA (2013) Temporal regulation of the Mus81‐Mms4 endonuclease ensures cell survival under conditions of DNA damage. Nucleic Acids Res 41: 8943–8958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz EK, Wright WD, Ehmsen KT, Evans JE, Stahlberg H, Heyer WD (2012) Mus81‐Mms4 functions as a single heterodimer to cleave nicked intermediates in recombinational DNA repair. Mol Cell Biol 32: 3065–3080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirayama M, Zachariae W, Ciosk R, Nasmyth K (1998) The Polo‐like kinase Cdc5p and the WD‐repeat protein Cdc20p/fizzy are regulators and substrates of the anaphase promoting complex in Saccharomyces cerevisiae . EMBO J 17: 1336–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snead JL, Sullivan M, Lowery DM, Cohen MS, Zhang C, Randle DH, Taunton J, Yaffe MB, Morgan DO, Shokat KM (2007) A coupled chemical‐genetic and bioinformatic approach to polo‐like kinase pathway exploration. Chem Biol 14: 1261–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Sako K, Akiyama K, Isoda M, Senoo C, Nakajo N, Sagata N (2015) Identification of non‐Ser/Thr‐Pro consensus motifs for Cdk1 and their roles in mitotic regulation of C2H2 zinc finger proteins and Ect2. Sci Rep 5: 7929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svendsen JM, Smogorzewska A, Sowa ME, O'Connell BC, Gygi SP, Elledge SJ, Harper JW (2009) Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell 138: 63–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szakal B, Branzei D (2013) Premature Cdk1/Cdc5/Mus81 pathway activation induces aberrant replication and deleterious crossover. EMBO J 32: 1155–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreich M, Stillman B (1999) Cdc7p‐Dbf4p kinase binds to chromatin during S phase and is regulated by both the APC and the RAD53 checkpoint pathway. EMBO J 18: 5334–5346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt HDM, Sarbajna S, Matos J, West SC (2013) Coordinated actions of SLX1‐SLX4 and MUS81‐EME1 for Holliday junction resolution in human cells. Mol Cell 52: 234–247 [DOI] [PubMed] [Google Scholar]
- Zegerman P, Diffley JFX (2010) Checkpoint‐dependent inhibition of DNA replication initiation by Sld3 and Dbf4 phosphorylation. Nature 467: 474–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Data Availability Statement
Mass spectrometric datasets are available at EBI PRIDE. DDK and the Rtt107 scaffold promote Mus81‐Mms4 resolvase activation during mitosis (2015). PXD005356.