

Abstract
Alzheimer′s disease (AD) is characterized by deposition of amyloid plaques, neurofibrillary tangles, and neuroinflammation. In order to study microglial contribution to amyloid plaque phagocytosis, we developed a novel ex vivo model by co‐culturing organotypic brain slices from up to 20‐month‐old, amyloid‐bearing AD mouse model (APPPS1) and young, neonatal wild‐type (WT) mice. Surprisingly, co‐culturing resulted in proliferation, recruitment, and clustering of old microglial cells around amyloid plaques and clearance of the plaque halo. Depletion of either old or young microglial cells prevented amyloid plaque clearance, indicating a synergistic effect of both populations. Exposing old microglial cells to conditioned media of young microglia or addition of granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) was sufficient to induce microglial proliferation and reduce amyloid plaque size. Our data suggest that microglial dysfunction in AD may be reversible and their phagocytic ability can be modulated to limit amyloid accumulation. This novel ex vivo model provides a valuable system for identification, screening, and testing of compounds aimed to therapeutically reinforce microglial phagocytosis.
Keywords: Alzheimer's disease, amyloid plaque clearance, microglia, neurodegeneration
Subject Categories: Molecular Biology of Disease, Neuroscience
Introduction
Alzheimer′s disease (AD) is the most prevalent neurodegenerative disorder and is pathologically defined by extracellular amyloid β‐peptide (Aβ) deposition, neurofibrillary tangles, and neuroinflammation (Holtzman et al, 2011). Amyloid plaque cores composed of fibrillar Aβ in a β‐sheet conformation are surrounded by a halo of diffuse Aβ. This neuropathological feature of AD is faithfully recapitulated in transgenic mouse models such as APPPS1 (Radde et al, 2006).
Neuroimmune changes are tightly linked to the pathology of AD, as well as other neurodegenerative disorders (Amor & Woodroofe, 2014; Gjoneska et al, 2015; Guillot‐Sestier et al, 2015b). This link has been strengthened by recent discoveries of genes implicated in microglial function such as triggering receptor expressed on myeloid cells 2 (TREM2), CD33, or complement receptor 1 (CR1) as susceptibility loci for late‐onset AD (Harold et al, 2009; Lambert et al, 2009; Naj et al, 2011; Guerreiro et al, 2013; Jonsson et al, 2013). Interestingly, these newly identified risk factors may be functionally linked to microglial phagocytosis and Aβ clearance (Griciuc et al, 2013; Thambisetty et al, 2013; Kleinberger et al, 2014; Wang et al, 2015). Although microglia are well known for their phagocytic capacity and are found to surround amyloid plaques in mouse models of amyloidosis as well as in AD patients (Dickson et al, 1988; Haga et al, 1989; Itagaki et al, 1989; Wegiel & Wisniewski, 1990; Frautschy et al, 1998; Stalder et al, 1999), their role in plaque clearance is still under debate. Several studies proposed that microglia/macrophages are involved in the clearance of Aβ (Rogers et al, 2002; Simard et al, 2006; El Khoury et al, 2007; Bolmont et al, 2008; Lee & Landreth, 2010; Liu et al, 2010; Krabbe et al, 2013). However, microglial depletion experiments in AD mouse models argued against the role of myeloid cells in restricting plaque growth (Grathwohl et al, 2009; Prokop et al, 2015). Nevertheless, microglia‐mediated phagocytosis and reduction in Aβ burden have been observed upon Aβ immunotherapy (Schenk et al, 1999; Bard et al, 2000; Bacskai et al, 2001; Nicoll et al, 2003, 2006; Wilcock et al, 2004; Boche & Nicoll, 2008; Sevigny et al, 2016). Moreover, reduction in TREM2 reduced immunotherapeutic clearance of amyloid plaques (Xiang et al, 2016). Thus, immune system activation may be beneficial in order to reduce amyloid load (Reardon, 2015). On the contrary, (pre‐)clinical trials including non‐steroidal anti‐inflammatory drugs (NSAIDs) suggested that inhibiting the immune system may also be of benefit in reducing the disease incidence or slowing down AD progression (Stewart et al, 1997; Anthony et al, 2000; in t' Veld et al, 2001; Zandi et al, 2002; Monsonego & Weiner, 2003; Heneka et al, 2005; Yip et al, 2005). Additionally, microglia can be activated by Αβ resulting in secretion of inflammatory cytokines that can harm neurons and provoke toxicity (Meda et al, 1995; Giulian et al, 1996; Combs et al, 1999; Manocha et al, 2016). It is therefore not clear whether microglial activation is beneficial or detrimental for disease progression (Morgan et al, 2005; Wyss‐Coray, 2006; Gandy & Heppner, 2013). Understanding microglial contribution to AD pathology and progression is of pivotal relevance since age‐dependent accumulation of Αβ in sporadic AD patients is apparently associated with reduced clearance (Saido, 1998; Mawuenyega et al, 2010; Wildsmith et al, 2013). Moreover, the major genetic risk factor for sporadic AD, apolipoprotein E, has been functionally linked to reduced clearance of Αβ (Deane et al, 2008; Castellano et al, 2011).
One of the major limitations to study microglial contribution to amyloid plaque phagocytosis is the lack of suitable model systems. Major attempts to study microglial phagocytosis of Αβ come from studies using cultured microglial cells to which Aβ has been exogenously added (Ard et al, 1996; Webster et al, 2000; D'Andrea et al, 2004; Mandrekar et al, 2009; Fleisher‐Berkovich et al, 2010). However, due to limited survival of microglial cells isolated from aged animals and cultured in the absence of growth factors (Moussaud & Draheim, 2010), mainly phagocytosis of young (neonatal) microglia could be examined. Furthermore, changes in microglial gene expression profile due to isolation and culturing conditions need to be considered (Butovsky et al, 2014). Alternatively, in vivo imaging studies in an amyloidosis mouse model provided some evidence for possible phagocytosis of Aβ by microglia surrounding amyloid plaques (Bolmont et al, 2008). However, age‐related decrease in phagocytic capacity of microglial cells, correlating with increased Aβ plaque burden, has been reported (Krabbe et al, 2013). Microglia recruited to amyloid plaques also show morphological changes, including increased soma size and thickening of processes (Itagaki et al, 1989; Baron et al, 2014; Derecki et al, 2014; Mosher & Wyss‐Coray, 2014). Transformation of microglia from ramified into cells with enlarged cell bodies and amoeboid morphology are morphological indicators of microglial activation (Kreutzberg, 1996). Thus, microglia in AD become chronically altered and this may compromise not only phagocytic, but also other physiological functions, including chemokine, cytokine, and growth factor secretion (Lue et al, 2001; Hanisch & Kettenmann, 2007; Hickman et al, 2008; Lai & McLaurin, 2012; Solito & Sastre, 2012; Derecki et al, 2014; Orre et al, 2014).
A key unresolved question is whether microglial dysfunction in AD is reversible and whether their phagocytic ability can be restored to limit amyloid accumulation. To this end, we developed a novel ex vivo model of amyloid plaque clearance by co‐culturing young WT brain slices together with brain slices from aged AD mice. We show that functional impairment of aged microglial cells in amyloid plaque‐bearing tissue can be reversed through factors secreted by young microglia, resulting in increased amyloid plaque clearance and thus reduced amyloid plaque load. Our results suggest a role of microglia in reducing the amyloid burden and support development of therapeutic approaches modulating microglial activity.
Results
Co‐culturing of old brain slices together with young slices triggers clearance of the plaque halo
To study the capability of microglial cells in amyloid plaque clearance, we developed a novel ex vivo model by co‐culturing organotypic brain slices from up to 20‐month‐old amyloid plaque‐bearing mice that co‐express mutant amyloid precursor protein (KM670/671NL) and mutant presenilin 1 (L166P) under the control of the neuron‐specific Thy1 promoter (APPPS1, line 21) (Radde et al, 2006) together with slices from young, neonatal WT mice (Fig 1A). To thoroughly characterize the newly introduced ex vivo model, we first investigated cell viability using propidium iodide, a dye permeable only to cells with compromised membrane integrity (Brana et al, 2002). In contrast to high cell viability in young brain slices at both 7 and 14 days in vitro (DIV), we observed poor cell viability in old brain slices already at 7 DIV (Fig 1B and C). Low survival of adult or aged brain slices ex vivo has also been reported by others (Staal et al, 2011; Mewes et al, 2012). Cell viability of young and old tissue in co‐cultures was not different compared to single cultures (Fig 1B and C). We detected a pronounced loss of neurons and astrocytes in the old tissue as judged by reduced NeuN and GFAP immunoreactivity (Figs 1D and E, and EV1A and B). In contrast to neurons and astrocytes, we detected an increased signal of the microglial marker CD68 in old brain slices which was even further elevated upon co‐culturing with young brain slices (Figs 1F and EV1C and D).
Figure 1. Characterization of the ex vivo model.
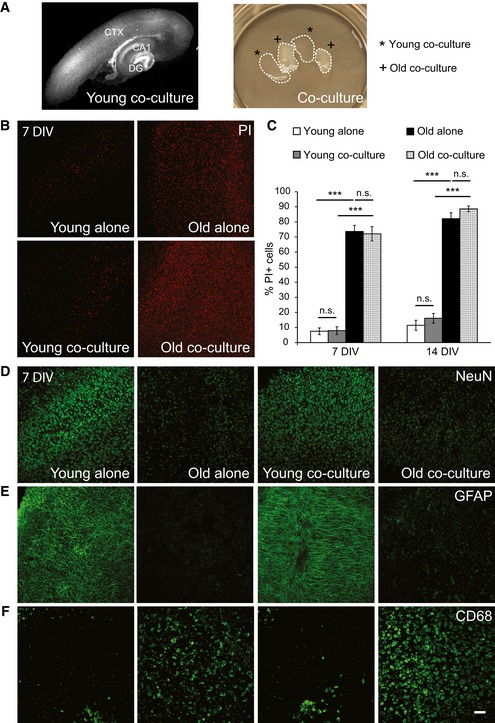
-
ARepresentative image of a typical young WT cortico‐hippocampal slice (350 μm) from co‐culture (14 DIV) stained with DAPI (gray) to visualize nuclei (left). Dentate gyrus (DG), cornus ammonis 1 (CA1), and cortical (CTX) regions are indicated. An example of the co‐culture dish is shown on the right. Co‐culture includes two young WT (*, young co‐culture) and two old APPPS1 (+, old co‐culture) brain slices.
-
BImmunofluorescence analysis of young WT and old APPPS1 slices cultured alone (young alone, old alone) or in co‐culture (young co‐culture, old co‐culture) at 7 days in vitro (DIV) stained with propidium iodide (PI, red) reveals reduced cell viability in old brain slices compared to young.
-
CQuantitative analysis of PI‐positive cells in young WT and old APPPS1 slices cultured alone (white and black bars, respectively) or in co‐culture (gray and white squared bars, respectively) at 7 and 14 DIV. The values are expressed as percentages of PI‐positive cells from the total number of DAPI‐positive cells. The values represent mean ± SEM from three independent experiments, each experiment including two independent slice culture dishes (n.s. = not significant; ***P < 0.001; unpaired two‐tailed Student's t‐test).
-
D–FImmunofluorescence analysis of young WT and old APPPS1 slices cultured alone or in co‐culture at 7 DIV and immunostained using neuronal (NeuN), astrocytic (GFAP), and microglial (CD68) markers (green) reveals reduced NeuN and GFAP and increased CD68 immunosignal. Scale bar: 75 μm (applies to panels B, D, E, F).
Figure EV1. Characterization of the ex vivo model at 14 DIV .
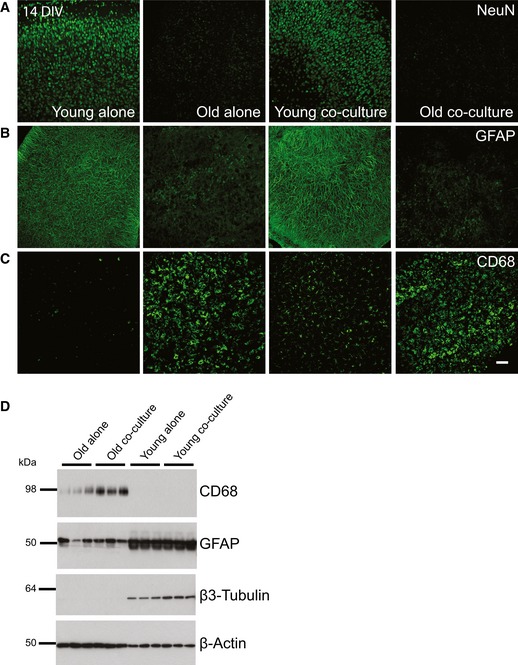
-
A–CImmunofluorescence analysis of young WT and old APPPS1 slices cultured alone or in co‐culture at 14 DIV and immunostained using neuronal (NeuN), astrocytic (GFAP), and microglial (CD68) cellular markers (green) reveals reduced NeuN and GFAP and increased CD68 immunosignal. Scale bar: 75 μm.
-
DWestern blot analysis of microglial (CD68), astrocytic (GFAP), and neuronal (β3‐Tubulin) markers in old APPPS1 and young WT slices cultured alone or in co‐culture at 14 DIV. β‐actin was used as a loading control.
Source data are available online for this figure.
Next, we analyzed amyloid plaques in the ex vivo co‐culture model and observed striking changes in their morphology over 14 DIV. Amyloid plaque cores visualized by co‐staining of the human anti‐Aβ antibody 6E10 and thiazine red (TR) and the plaque halo of diffuse Aβ visualized only by 6E10 are depicted in Fig 2A. Surprisingly, we observed clearance of the plaque halo in the co‐culture model, resulting almost exclusively in core‐only plaques at 14 DIV (Figs 2A and EV2A). We observed a significantly increased number of core‐only plaques upon co‐culturing of old slices together with young slices at 7 and 14 DIV compared to old slices alone (Figs 2B and EV2B and C). Moreover, Western blot analysis revealed an almost 60% reduced levels of Aβ upon co‐culturing of old and young brain slices (Fig 2C and D). A comparable beneficial effect on clearance of the amyloid plaque halo was observed independently of whether we co‐cultured young WT or young APPPS1 brain slices together with old brain slices (Fig EV2D). Furthermore, upon co‐culturing of old WT brain slices together with old APPPS1 brain slices, we observed no significant effect on clearance of amyloid plaque halos (Fig EV2D). Taken together, our data suggest that tissue age, rather than AD pathology, is instrumental for the observed differences in plaque phagocytosis. Thus, the presence of young tissue stimulates clearance of Aβ deposits in the old, amyloid‐bearing tissue.
Figure 2. Amyloid plaque clearance is enhanced in the ex vivo co‐culture model of old and young brain slices.
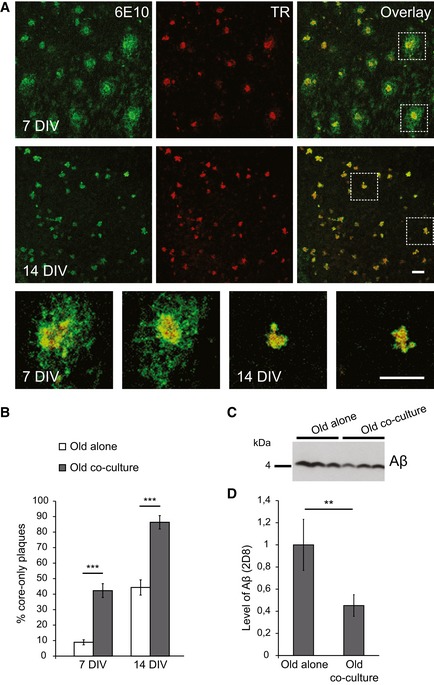
- Immunofluorescence analysis of the old APPPS1 slice in co‐culture using antibody 6E10 to detect amyloid plaques (green) and thiazine red (TR, red) to visualize fibrillar amyloid cores. At 7 DIV, the majority of amyloid plaques contain 6E10‐ and TR‐positive dense core surrounded by 6E10‐positive halo of diffuse Aβ. In contrast, at 14 DIV, the majority of amyloid plaques are core‐only plaques co‐stained with 6E10 and TR without detectable 6E10‐positive halo. Lower panels are higher magnification images of boxed regions. Scale bars: 50 μm.
- Quantitative analysis of core‐only plaques in old APPPS1 slices cultured alone (white bars) or in co‐culture (gray bars) at 7 and 14 DIV reveals an increased number of core‐only plaques upon co‐culturing of old APPPS1 brain slices together with young WT slices. The values are expressed as percentages of core‐only plaques from the total number of amyloid plaques. The values represent mean ± SEM from three independent experiments, and each experiment including three independent slice culture dishes (***P < 0.001; unpaired two‐tailed Student's t‐test).
- Western blot analysis of Aβ levels (triplicates) in the old APPPS1 tissue cultured alone or in co‐culture with the young WT (14 DIV) using 2D8 antibody.
- Quantification of Western blot signals reveals decreased levels of Aβ upon co‐culturing compared to old tissue cultured alone. The values are normalized to the levels of Aβ in the old tissue cultured alone and represent mean ± SEM from two independent experiments, and each experiment was performed in triplicates (**P < 0.01; unpaired two‐tailed Student's t‐test).
Source data are available online for this figure.
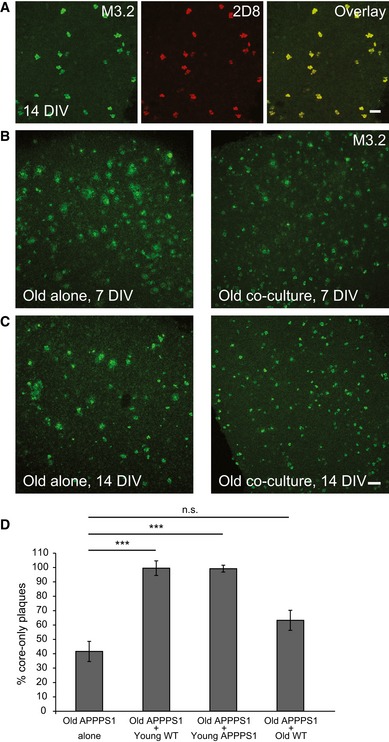
Figure EV2. Amyloid plaque clearance is enhanced in the co‐culture of old APPPS1 and young WT brain slices.
-
AImmunofluorescence analysis of core‐only plaques in the old APPPS1 slice in co‐culture (14 DIV) immunostained with mouse (M3.2, green) and human (2D8, red) Aβ antibodies reveals co‐localization due to co‐deposition of murine and human Aβ. Scale bar: 50 μm.
-
B, CImmunofluorescence analysis of old APPPS1 slices cultured alone or in co‐culture at 7 (B) and 14 (C) DIV and immunostained using M3.2. Amyloid plaque clearance is accelerated when old brain slices are co‐cultured together with young slices. Scale bar: 100 μm.
-
DQuantitative analysis of core‐only plaques in old APPPS1 brain slices cultured alone (old APPPS1 alone) or co‐cultured with young WT (old APPPS1 + young WT), young APPPS1 (old APPPS1 + young APPPS1), or old WT brain slices (old APPPS1 + old WT) at 14 DIV. Significantly enhanced amyloid plaque clearance is only observed upon co‐culturing of old APPPS1 brain slices with young slices (either WT or APPPS1). The values are expressed as percentages of core‐only plaques from the total number of amyloid plaques and represent mean ± SEM from three independent experiments, each experiment including three independent slice culture dishes (n.s. = not significant, ***P < 0.001; unpaired two‐tailed Student's t‐test).
Microglial recruitment correlates with the reduction in plaque size in the co‐culture model
The above‐described findings suggest that clearance of the plaque halo is enhanced in an ex vivo co‐culture model. We therefore examined microglial localization using the CD68 marker that identifies lysosomes of microglia/macrophages (Zotova et al, 2013; Walker & Lue, 2015). We observed a strong CD68 immunoreactivity in the old APPPS1 tissue (Fig 3A). CD68‐positive cells clustered around 6E10‐positive amyloid plaques (Fig 3B). We observed a trend toward increased number of CD68‐positive cells clustered around amyloid plaques from 7 to 14 DIV upon co‐culturing with the young tissue (Fig 3C), paralleled by a pronounced reduction in plaque size, mainly reflected by the clearance of the plaque halo (Fig 3D). Moreover, we also found a twofold increase in the proportion of amyloid plaques surrounded by at least four CD68‐positive cells (clustered plaques) from 7 to 14 DIV, in line with a reduction in plaque size (Fig 3E). These results suggest engulfment of amyloid plaques by CD68‐positive cells. Indeed, when phagocytosis was blocked by cytochalasin D (CytoD) (Schliwa, 1982), clearance of plaque halos was almost completely inhibited (Fig 3F and G). Thus, CD68‐positive microglial cells that cluster around amyloid plaques in the co‐culture model may reduce the amyloid burden by actively phagocytosing amyloid fibers.
Figure 3. Recruitment and clustering of CD68‐positive cells at amyloid plaques is paralleled by a decrease in plaque size.
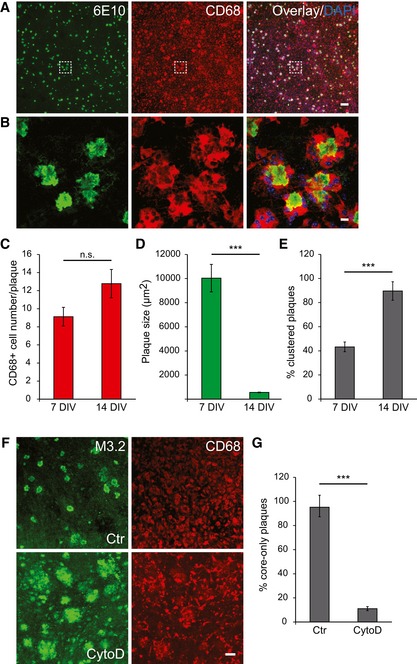
-
A, BImmunofluorescence analysis of the old APPPS1 slice in co‐culture (14 DIV) using antibodies 6E10 (green) and CD68 (red). Nuclei were counterstained using DAPI (blue). Image of boxed region in (A) is depicted at higher magnification in (B) and reveals clustering of several CD68‐positive cells around amyloid plaques. Scale bars: 100 μm (A) and 10 μm (B).
-
CQuantitative analysis of plaque‐associated CD68‐positive cells in co‐culture samples at 7 and 14 DIV reveals a trend toward increased number of CD68‐positive cells. The values represent mean ± SEM from at least three independent experiments, including total of at least six independent slice culture dishes (n.s. = not significant; unpaired two‐tailed Student's t‐test).
-
DQuantitative analysis of co‐culture samples at 7 and 14 DIV reveals a decrease in plaque size. The values represent mean ± SEM from at least three independent experiments, including total of at least six replicates (***P < 0.001; unpaired two‐tailed Student's t‐test).
-
EQuantitative analysis of plaques surrounded by CD68‐positive cells (clustered plaques) in co‐culture samples at 7 and 14 DIV reveals an increase in the number of clustered plaques. The values are expressed as percentages of clustered plaques from the total number of amyloid plaques. The values represent mean ± SEM from at least three independent experiments, including total of at least six independent slice culture dishes (***P < 0.001; unpaired two‐tailed Student's t‐test).
-
FImmunofluorescence analysis of the old APPPS1 slice in co‐culture (14 DIV) treated with cytochalasin D (CytoD) and vehicle control (Ctr) and immunostained with M3.2 (green) and CD68 (red). Inhibition of phagocytosis by CytoD blocks amyloid plaque clearance. Scale bar: 50 μm.
-
GQuantitative analysis of core‐only plaques in the old APPPS1 slice in co‐culture (14 DIV) treated with CytoD and Ctr reveals a decreased number of core‐only plaques upon CytoD treatment. The values are expressed as percentages of core‐only plaques from the total number of amyloid plaques. The values represent mean ± SEM from three independent experiments, including total of six independent slice culture dishes (***P < 0.001; unpaired two‐tailed Student's t‐test).
Amyloid plaque clearance depends on both young and old microglia
To analyze the contribution of microglial cells in amyloid plaque clearance, we pharmacologically depleted either young or old microglia with the macrophage toxin clodronate using established protocols (Kohl et al, 2003; Kreutz et al, 2009; Vinet et al, 2012; Ji et al, 2013; Hellwig et al, 2015). Clodronate treatment of young brain slices isolated from CX3CR1+/GFP microglia/macrophage reporter mice (Jung et al, 2000) revealed an almost complete removal of microglial cells without affecting astrocytic or neuronal cell counts (Fig EV3A–C). Next, we pre‐treated young slices with clodronate, stopped the treatment by exchanging the media, and then added old APPPS1 slices as indicated in Fig 4A. This resulted in a significant reduction in the amyloid plaque clearance efficacy, confirming involvement of young microglia in this process (Fig 4B). However, pre‐treatment of old slices with clodronate, monitored by a substantial reduction in CD68 coverage, and subsequent addition of young brain slices completely abolished amyloid plaque clearance (Fig 4C–E). This result suggests that microglia from the old APPPS1 tissue are the actual effector mediating amyloid plaque phagocytosis. Taken together, our results support a functional contribution of both young and old microglia in amyloid plaque clearance.
Figure EV3. Clodronate treatment of young brain slices efficiently depletes microglial cells.
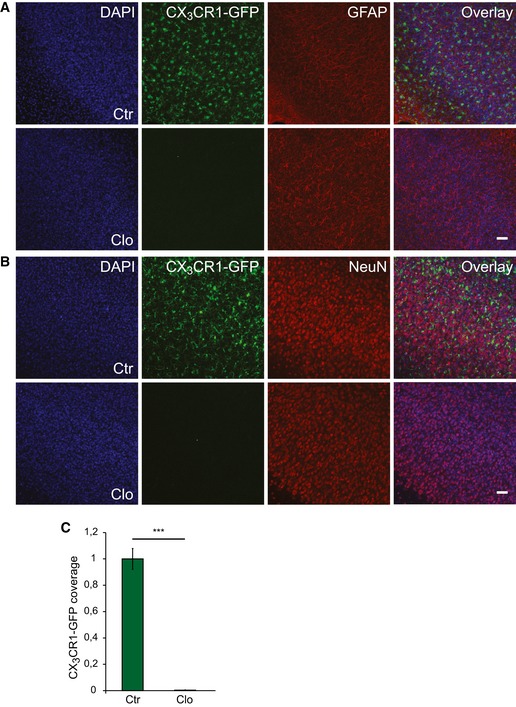
-
A, BImmunofluorescence analysis of the young CX3CR1+/GFP slice treated with Clo and Ctr from 1 until 7 DIV and immunostained with GFP (green) and the astrocytic marker GFAP (red) in (A), and with GFP (green) and the neuronal marker NeuN (red) in (B). DAPI (blue) was used to counterstain nuclei. A GFP antibody was used to amplify the signal of GFP‐expressing microglial cells. Scale bars: 50 μm.
-
CArea of CX3CR1‐GFP‐positive cells (CX3CR1‐GFP coverage) in the young CX3CR1+/GFP tissue treated with Clo and Ctr. CX3CR1‐GFP coverage demonstrates complete removal of young microglial cells upon Clo treatment. The values are normalized to CX3CR1‐GFP coverage of the Ctr and represent mean ± SEM from three independent experiments, including total of six independent slice culture dishes (***P < 0.001; unpaired two‐tailed Student's t‐test).
Figure 4. Amyloid plaque clearance depends on both young and old microglia.
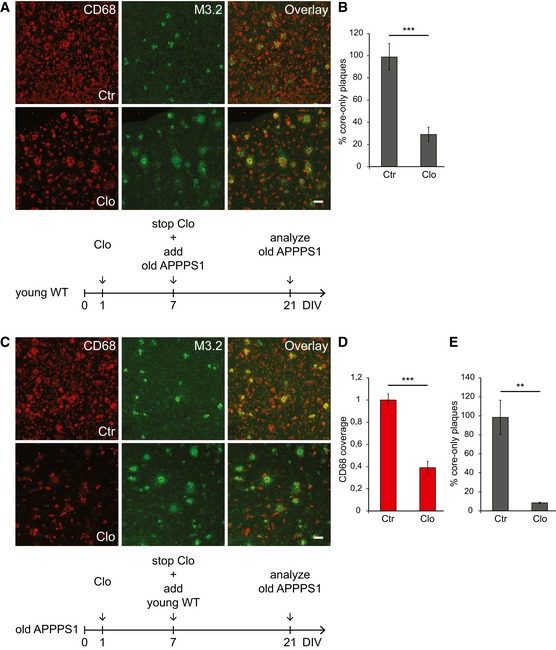
- Immunofluorescence analysis of the old APPPS1 slice co‐cultured with the young WT slice pre‐treated with Clo and Ctr and immunostained with CD68 (red) and M3.2 (green). Scale bar: 50 μm.
- Quantitative analysis of core‐only plaques in the old APPPS1 tissue co‐cultured with the young WT tissue pre‐treated with Clo and Ctr as indicated in (A) reveals a decreased number of core‐only plaques upon Clo treatment. The values are expressed as percentages of core‐only plaques from the total number of amyloid plaques and represent mean ± SEM from three independent experiments, including total of six independent slice culture dishes (***P < 0.001; unpaired two‐tailed Student's t‐test).
- Immunofluorescence analysis of the old APPPS1 slice treated with Clo and Ctr and subsequently co‐cultured with the young WT slice and immunostained with CD68 (red) and M3.2 (green). Scale bar: 50 μm.
- Area of CD68‐positive cells (CD68 coverage) in the old APPPS1 tissue treated with Clo and Ctr and subsequently co‐cultured with the young WT tissue as indicated in (C). CD68 coverage is reduced upon Clo treatment. The values are normalized to CD68 coverage of the Ctr and represent mean ± SEM from three independent experiments, including total of six independent slice culture dishes (***P < 0.001; unpaired two‐tailed Student's t‐test).
- Quantitative analysis of core‐only plaques in the old APPPS1 tissue treated with Clo and Ctr and subsequently co‐cultured with the young WT tissue as indicated in (C) reveals a decreased number of core‐only plaques upon Clo treatment. The values are expressed as percentages of core‐only plaques from the total number of amyloid plaques and represent mean ± SEM from three independent experiments, including total of six independent slice culture dishes (**P < 0.01; unpaired two‐tailed Student's t‐test).
Old microglia are Aβ engulfing cells
To further reveal whether CD68‐positive cells found to cluster around amyloid plaques originate from young or old tissue, we co‐cultured young brain slices from CX3CR1+/GFP animals with the old APPPS1 brain slices. At 14 DIV, we detected numerous GFP‐positive cells migrating toward the old tissue, but only very few reached the edge of the old tissue and almost none migrated deeper into the slice (Fig EV4A and B). Consequently, CD68‐positive cells surrounding M3.2‐labeled amyloid plaques were not positive for GFP, suggesting that the plaque‐surrounding microglia are derived from the old tissue (Fig 5A). To validate this assumption, we co‐cultured old APPPS1/CX3CR1+/GFP brain slices together with young WT slices. Indeed, we readily detected GFP and CD68 double‐positive microglial cells surrounding amyloid plaques (Fig 5B). Furthermore, we detected M3.2‐positive Aβ within intracellular compartments of old, GFP‐positive microglial cells (Fig 5C). We also observed morphological changes of old microglial cells ex vivo, illustrated by increased number of amoeboid cells. We compared microglial morphology in co‐cultures of young CX3CR1+/GFP and old APPPS1/CX3CR1+/GFP brain slices versus single cultures and freshly cut brain slices (Fig 5D and E). Both young and old microglia display ramified morphology in freshly cut brain slices (0 DIV). While young microglia mostly preserved their architecture after 10 DIV, old microglia presented mainly with amoeboid shape (Fig 5D and E). Amoeboid morphology of old microglial cells was further potentiated by the presence of young tissue, correlating well with increased phagocytosis upon co‐culturing of old and young brain slices. Thus, our data demonstrate that old APPPS1‐derived microglial cells acquire amoeboid morphology, cluster around amyloid plaques, and engulf Aβ.
Figure EV4. Young microglia do not infiltrate into the old APPPS1 brain slice at 14 DIV .
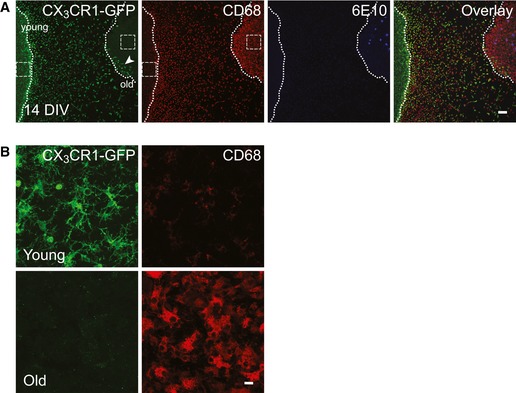
-
A, BImmunofluorescence analysis of co‐cultured young CX3CR1+/GFP and old APPPS1 brain slices at 14 DIV immunostained with GFP (green), CD68 (red), and 6E10 (blue). GFP‐positive young microglial cells are migrating toward the old tissue and sparse GFP‐positive cells are detectable within the old slice (arrowhead), although still restricted to the region in proximity to the young tissue. Dotted lines delineate borders of young and old brain slices. Images of boxed regions in (A) are depicted at higher magnification in (B) and reveal that GFP‐positive microglial cells are indeed present in the young (upper panels) but not in the old (lower panels) brain slices. A GFP antibody was used to amplify the signal of CX3CR1‐GFP‐positive young microglial cells. Scale bars: 100 μm (A) and 10 μm (B).
Figure 5. Old CD68‐positive cells acquire amoeboid morphology, cluster around amyloid plaques, and engulf Aβ.
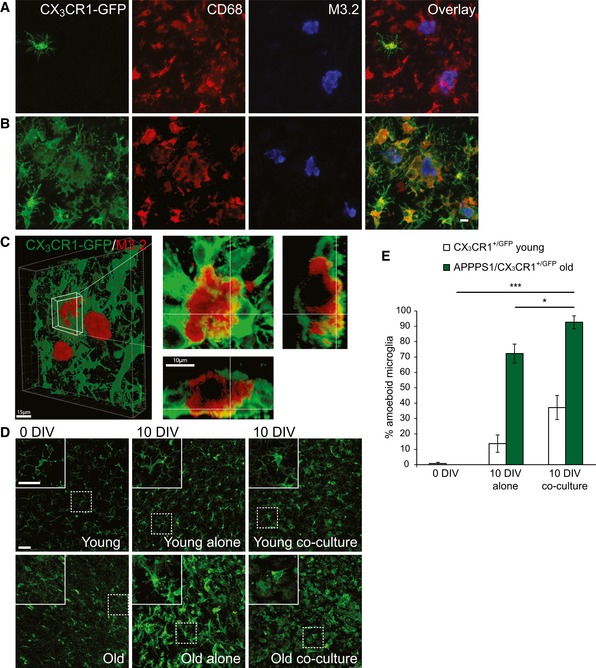
- Immunofluorescence analysis of the old APPPS1 slice in co‐culture with the young CX3CR1+/GFP slice (14 DIV) and immunostained using GFP (green), CD68 (red), and M3.2 (blue) reveals no co‐localization of young, GFP‐positive microglial cells with CD68‐positive cells surrounding amyloid plaques. A GFP antibody was used to amplify the signal of GFP‐expressing young microglial cells.
- Immunofluorescence analysis of the old APPPS1/CX3CR1+/GFP slice in co‐culture with the young WT slice (14 DIV) and immunostained using GFP (green), CD68 (red), and M3.2 (blue) reveals co‐localization of old, GFP‐positive microglial cells with CD68‐positive cells surrounding amyloid plaques. A GFP antibody was used to amplify the signal of GFP‐expressing old microglial cells. Scale bar: 10 μm (applies to panels A and B).
- 3D reconstruction of the old APPPS1/CX3CR1+/GFP slice in co‐culture with the young WT slice (10 DIV) and immunostained using GFP (green) and M3.2 (red). GFP‐positive old microglial cells surrounding amyloid plaques exhibit intracellular M3.2‐positive immunoreactivity indicative of Aβ uptake. Overview at low magnification is shown in the left panel. Scale bar: 15 μm. A detailed plaque is shown at higher magnification in the right panels. Scale bar: 10 μm.
- Immunofluorescence analysis of young CX3CR1+/GFP and old APPPS1/CX3CR1+/GFP slices freshly cut (0 DIV), cultured alone or in co‐culture (10 DIV), and immunostained using GFP (green) shows alterations in microglial morphology of old cells upon culturing. Upper left panels are enlargements of boxed regions. A GFP antibody was used to amplify the signal of GFP‐expressing microglial cells. Scale bars: 50 μm.
- Quantitative analysis of amoeboid microglial cells in young CX3CR1+/GFP (white bars) and old APPPS1/CX3CR1+/GFP (green bars) brain slices freshly cut (0 DIV), cultured alone or in co‐culture (10 DIV). The values are expressed as percentages of amoeboid microglia from the total number of microglial cells. The values represent mean ± SEM from three independent experiments, each experiment including at least 250 cells per condition (*P < 0.05, ***P < 0.001; unpaired two‐tailed Student's t‐test).
Soluble factors released by young microglia trigger Aβ uptake of old microglia
As suggested above, old microglial cells are responsible for clearance of Aβ halos while young microglia may secrete soluble factor(s) that stimulate this process. To investigate the contribution of soluble factors released by young slices in promoting Aβ uptake, we collected conditioned media of cultured WT brain slices and added them to old APPPS1 slices. This was sufficient to increase amyloid clearance and indistinguishable from the effect observed in co‐cultures of old and young brain slices at all investigated time points (Fig 6A). Addition of conditioned media collected from old APPPS1 slices did not result in increased numbers of core‐only plaques compared to old slices cultured alone (Fig 6A). Next, we tested conditioned media collected from microglia‐depleted young slices which failed to increase numbers of core‐only plaques in contrast to conditioned media from control‐treated young slices (Fig 6B). These results suggest that young microglial cells produce a soluble factor, which stimulates Aβ uptake by the old APPPS1 microglial cells. To further strengthen this hypothesis, we co‐cultured old APPPS1 slices in the presence or absence of conditioned media obtained from young primary microglia. Again, we observed a significantly increased number of core‐only plaques, consistent with the requirement of a soluble factor derived from young microglia that is able to stimulate Aβ uptake of the old microglia (Fig 6C).
Figure 6. Factors released by young microglia promote amyloid plaque clearance.
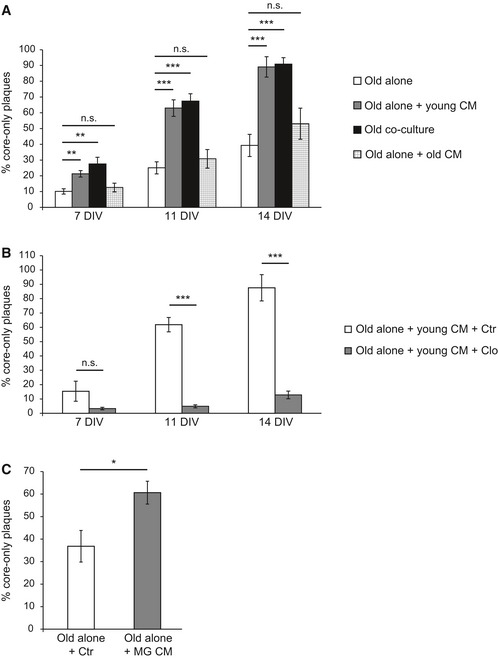
- Old APPPS1 tissue was cultured alone (old alone, white bars) or incubated with conditioned media (CM) collected from young WT slices (old alone + young CM, gray bars). Analysis included co‐culture of old APPPS1 and young WT brain slices (old co‐culture, black bars) as a positive control and old APPPS1 tissue incubated with conditioned media collected from old APPPS1 slices (old alone + old CM, white squared bars) as a negative control. Quantitative analysis at 7, 11, and 14 DIV reveals increased numbers of core‐only plaques upon incubation of the old APPPS1 tissue with conditioned media collected from young WT slices, fully recapitulating core‐only plaque numbers observed in the co‐culture. The values are expressed as percentages of core‐only plaques from the total number of amyloid plaques and represent mean ± SEM from three independent experiments, each experiment including at least two independent slice culture dishes (n.s. = not significant, **P < 0.01, ***P < 0.001; unpaired two‐tailed Student's t‐test).
- Old APPPS1 tissue was incubated with conditioned media collected from young WT slices previously treated with clodronate (old alone + young CM + Clo, gray bars) and vehicle control (old alone + young CM + Ctr, white bars). Quantitative analysis at 7, 11, and 14 DIV reveals decreased numbers of core‐only plaques upon incubation of the old APPPS1 tissue with conditioned media collected from young WT slices pre‐treated with Clo. The values are expressed as percentages of core‐only plaques from the total number of amyloid plaques and represent mean ± SEM from three independent experiments, each experiment including at least two independent slice culture dishes (n.s. = not significant, ***P < 0.001; unpaired two‐tailed Student's t‐test).
- Old APPPS1 tissue was incubated with conditioned media collected from cultured WT primary microglial cells (old alone + MG CM, gray bar) or with unconditioned slice culture media as a control (old alone + Ctr, white bar). Quantitative analysis of core‐only plaques at 14 DIV reveals enhanced amyloid plaque clearance upon incubation of the old APPPS1 tissue with conditioned media collected from cultured primary microglia. The values are expressed as percentages of core‐only plaques from the total number of amyloid plaques and represent mean ± SEM from three independent experiments, each experiment including two independent slice culture dishes (*P < 0.05; unpaired two‐tailed Student's t‐test).
GM‐CSF stimulates Aβ uptake
Previous data support the regulatory role of several pro‐ and anti‐inflammatory cytokines in phagocytosis of Aβ (Lai & McLaurin, 2012; Wilcock, 2012; Chakrabarty et al, 2015; Guillot‐Sestier et al, 2015a). To test whether direct addition of soluble factors to old APPPS1 tissue would be sufficient to enhance microglial Aβ uptake, we screened for the effects of several pro‐ and anti‐inflammatory cytokines. The key anti‐inflammatory cytokines interleukin‐10 (IL‐10) and transforming growth factor β (TGF‐β), as well as the prototypic pro‐inflammatory cytokines IL‐6 and IL‐12/p40, were not able to trigger clearance of the plaque halo or affect amyloid plaque morphology (Fig 7A–E). In contrast, GM‐CSF treatment stimulated Aβ uptake of old microglia and resulted in enhanced appearance of core‐only plaques (Figs 7F and 8B) as observed upon co‐culturing of young and old brain slices (Fig 2B).
Figure 7. The pro‐inflammatory factor GM‐CSF stimulates amyloid plaque clearance.
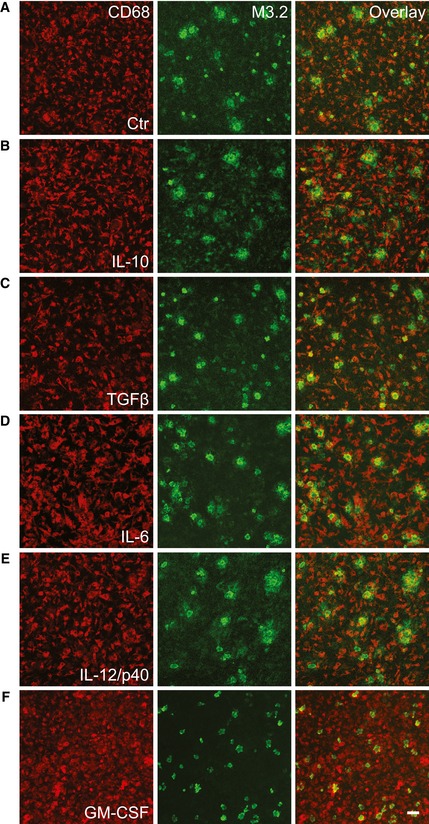
-
A–FImmunofluorescence analysis of the old APPPS1 slice (14 DIV) treated with vehicle Ctr in (A), anti‐inflammatory factors IL‐10 and TGF‐β in (B, C) and pro‐inflammatory factors IL‐6, IL‐12/p40, and GM‐CSF in (D, E, and F), respectively, and immunostained with CD68 (red) and M3.2 (green). In contrast to increased number of core‐only plaques upon GM‐CSF treatment, other factors tested did not induce amyloid plaque clearance, resulting in a better preservation of the M3.2‐positive halos of Aβ surrounding dense core of amyloid plaques. Scale bar: 50 μm.
Figure 8. Amyloid plaque clearance is enhanced in the co‐culture of old APPPS1 and young GM‐CSF −/− brain slices.
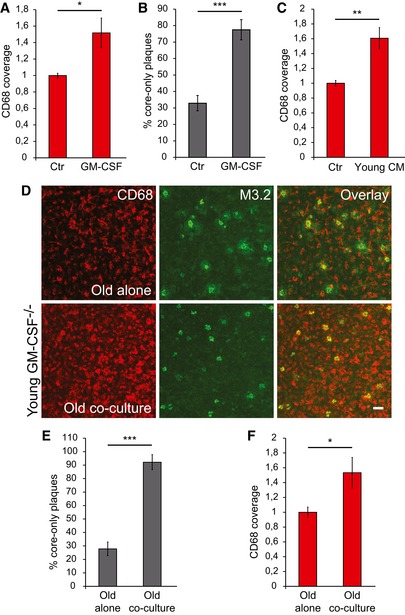
- CD68 coverage in the old APPPS1 tissue (14 DIV) treated with GM‐CSF and vehicle Ctr. CD68 coverage is increased upon treatment with GM‐CSF. The values are normalized to CD68 coverage of the Ctr and represent mean ± SEM from three independent experiments, each experiment including two independent slice culture dishes (*P < 0.05; unpaired two‐tailed Student's t‐test).
- Quantitative analysis of core‐only plaques in the old APPPS1 tissue (14 DIV) treated with GM‐CSF and Ctr reveals an increased number of core‐only plaques upon treatment with GM‐CSF. The values are expressed as percentages of core‐only plaques from the total number of amyloid plaques and represent mean ± SEM from three independent experiments, each experiment including three independent slice culture dishes (***P < 0.001; unpaired two‐tailed Student's t‐test).
- CD68 coverage in the old APPPS1 tissue (14 DIV) incubated with conditioned media collected from young WT slices (young CM) or with non‐conditioned slice culture media (Ctr). CD68 coverage is increased upon incubation of the APPPS1 tissue with the young CM. The values are normalized to CD68 coverage of the Ctr and represent mean ± SEM from three independent experiments, each experiment including two independent slice culture dishes (**P < 0.01; unpaired two‐tailed Student's t‐test).
- Immunofluorescence analysis of the old APPPS1 slice cultured alone (old alone) or in co‐culture with young GM‐CSF−/− brain slices (old co‐culture) at 14 DIV and immunostained with CD68 (red) and M3.2 (green) reveals an increased number of core‐only plaques upon co‐culturing with young GM‐CSF−/− brain slices. Scale bar: 50 μm.
- Quantitative analysis of core‐only plaques in the old APPPS1 slice cultured alone or in co‐culture with young GM‐CSF−/− brain slices (14 DIV) reveals an increased number of core‐only plaques upon co‐culturing of old APPPS1 brain slices together with young GM‐CSF−/− brain slices. The values are expressed as percentages of core‐only plaques from the total number of amyloid plaques. The values represent mean ± SEM from three independent experiments, each experiment including three independent slice culture dishes (***P < 0.001; unpaired two‐tailed Student's t‐test).
- CD68 coverage in the old APPPS1 tissue (14 DIV) cultured alone or in co‐culture with young GM‐CSF−/− brain slices. CD68 coverage is increased upon co‐culturing of old APPPS1 brain slices together with young GM‐CSF−/− brain slices. The values are normalized to CD68 coverage of the old slice cultured alone and represent mean ± SEM from three independent experiments, each experiment including two independent slice culture dishes (*P < 0.05; unpaired two‐tailed Student's t‐test).
As GM‐CSF is a well‐known mitogen capable of triggering proliferation of microglial cells (Giulian & Ingeman, 1988; Lee et al, 1994; Suh et al, 2005), we investigated the density of CD68‐positive cells. Indeed, this revealed a significantly increased CD68 coverage upon GM‐CSF treatment of old APPPS1 slices accompanied by increased engulfment of the plaque halo (Fig 8A and B). Moreover, increased CD68 coverage triggered by GM‐CSF treatment is also recapitulated by the addition of conditioned media from young WT slices to old APPPS1 slices (Fig 8C) indicating the possibility that GM‐CSF is the responsible factor secreted by microglia. To test whether GM‐CSF is necessary to drive microglial proliferation and Aβ uptake, we co‐cultured young brain slices isolated from GM‐CSF knockout mice (GM‐CSF−/−) with old APPPS1 slices (Fig 8D). However, even in the absence of GM‐CSF release from young brain slices we observed a significantly increased number of core‐only plaques (Fig 8D and E). In line with that, CD68 coverage was also increased upon co‐culturing of young GM‐CSF−/− and old APPPS1 brain slices (Fig 8F). Thus, GM‐CSF is clearly not the only factor stimulating microglial proliferation and Aβ uptake.
Proliferation of old microglial cells is required for amyloid clearance
As we observed an increased CD68 coverage upon GM‐CSF treatment (Fig 8A), as well as upon addition of young conditioned media to old brain slices (Fig 8C), we investigated microglial proliferation and its relevance for amyloid clearance in our newly established ex vivo model. We used a cell proliferation marker Ki67 (Scholzen & Gerdes, 2000) to assess the numbers of proliferating cells. Treatment of old APPPS1 slices with GM‐CSF strongly increased numbers of Ki67 and CD68 double‐positive microglial cells (Figs 9A and B, and EV5A) that is in line with the described mitogenic potential of GM‐CSF (Giulian & Ingeman, 1988; Lee et al, 1994; Suh et al, 2005). Next, we analyzed whether increased proliferation also occurs upon co‐culturing conditions. Similar to the effect of GM‐CSF, we also observed an increase in number of proliferating microglial cells upon co‐culturing of old together with young brain slices (Figs 9C and D, and EV5B). Finally, to test whether proliferation of old microglial cells is necessary for amyloid clearance, we incubated old APPPS1 slices with the young WT conditioned media, in the presence or absence of cytosine arabinoside (AraC), a widely used inhibitor of proliferation (Doetsch et al, 1999; Gomez‐Nicola et al, 2013). AraC treatment abolished amyloid plaque clearance, suggesting that proliferation of microglial cells is required for stimulation of Aβ uptake (Fig 9E and F). Taken together, our results revealed that factors secreted by the young microglia stimulate proliferation of old microglial cells, which is a pre‐requisite for enhanced amyloid clearance.
Figure 9. Microglial proliferation is a pre‐requisite for amyloid plaque clearance.
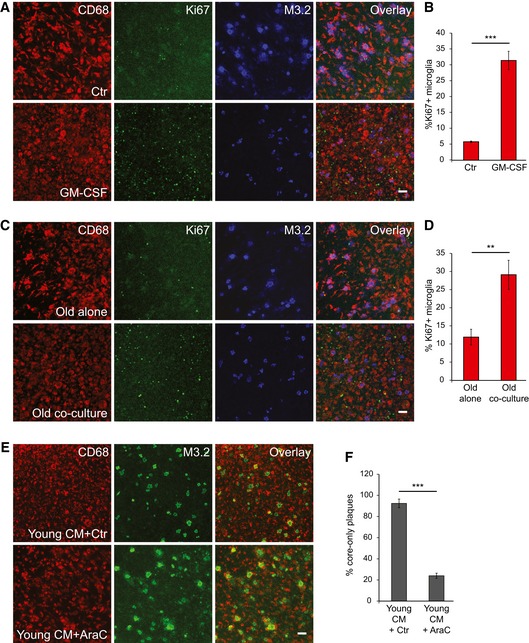
- Immunofluorescence analysis of the old APPPS1 slice (14 DIV) treated with GM‐CSF and vehicle Ctr and immunostained with CD68 (red), cell proliferation marker Ki67 (green) and M3.2 (blue). Scale bar: 50 μm.
- Quantitative analysis of Ki67 and CD68 double‐positive microglial cells in the old APPPS1 tissue (14 DIV) treated with GM‐CSF and Ctr reveals increased microglial proliferation upon treatment with GM‐CSF. The values are expressed as percentages of CD68 and Ki67 double‐positive microglial cells from the total number of CD68‐positive cells and represent mean ± SEM from two independent experiments, each experiment including four independent slice culture dishes (***P < 0.001; unpaired two‐tailed Student's t‐test).
- Immunofluorescence analysis of the old APPPS1 tissue cultured alone or in co‐culture with young WT brain slices (14 DIV) and immunostained with CD68 (red), Ki67 (green) and M3.2 (blue) Scale bar: 50 μm.
- Quantitative analysis of Ki67 and CD68 double‐positive microglial cells in the old APPPS1 tissue cultured alone or in co‐culture with young brain slices (14 DIV) reveals increased microglial proliferation upon co‐culturing of old and young brain slices. The values are expressed as percentages of CD68 and Ki67 double‐positive microglial cells from the total number of CD68‐positive cells and represent mean ± SEM from three independent experiments, each experiment including two independent slice culture dishes (**P < 0.01; unpaired two‐tailed Student's t‐test).
- Old APPPS1 tissue was incubated with conditioned media collected from young WT slices and in addition treated with proliferation inhibitor AraC (young CM + AraC) or vehicle Ctr (young CM + Ctr). Immunofluorescence analysis was performed at 14 DIV using CD68 (red) and M3.2 (green). Scale bar: 50 μm.
- Quantitative analysis of core‐only plaques in the old APPPS1 tissue (14 DIV) incubated with conditioned media from young WT slices and in addition treated with AraC or Ctr reveals a decreased number of core‐only plaques upon AraC treatment. The values are expressed as percentages of core‐only plaques from the total number of amyloid plaques and represent mean ± SEM from three independent experiments, each including two independent slice culture dishes (***P < 0.001; unpaired two‐tailed Student's t‐test).
Figure EV5. Proliferating microglial cells can easily be detected in the co‐culture model and upon GM‐CSF treatment.
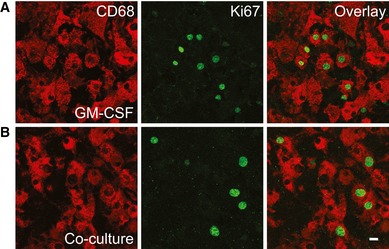
-
A, BImmunofluorescence analysis of proliferating microglial cells in old APPPS1 tissue treated with GM‐CSF (A) or in co‐culture (B) at 14 DIV and immunostained with the cell proliferation marker Ki67 (green) and microglial marker CD68 (red). Proliferating microglial cells can be detected in both conditions and are double‐positive for CD68 and Ki67. Scale bar: 10 μm.
Discussion
It has been a matter of debate whether microglial cells can modulate the Aβ burden during the course of AD. As failure to clear Aβ is assumed to be a culprit in sporadic AD patients, it is of great relevance to gain more insight into understanding clearance mechanisms (Saido, 1998; Mawuenyega et al, 2010; Wildsmith et al, 2013). Investigations of this important question have so far been hampered by the lack of suitable models. Therefore, we developed a novel ex vivo model of amyloid plaque clearance, which is allowing us to monitor microglial activity over several weeks. Our results show that the phagocytic function of aged microglial cells in Aβ clearance is not irreversibly impaired, but can be restored even at advanced stages of amyloid deposition.
Microglial role in Aβ clearance
In this study, we observed a reduction in amyloid plaque size when old APPPS1 brain slices were cultured together with young WT slices. Surprisingly, we found that only old APPPS1‐derived microglial cells are the cells responsible for Aβ uptake in our ex vivo co‐culture model. However, the Aβ uptake capacity of old microglia is stimulated by factors secreted from young microglia.
As shown in amyloidosis mouse models, plaques are growing over time (Hefendehl et al, 2011; Bittner et al, 2012) and microglial cells seem to be inefficient in phagocytosing and clearing Aβ (Hickman et al, 2008; Meyer‐Luehmann et al, 2008; Krabbe et al, 2013). Along these lines, it has been reported that myeloid cells of AD patients show reduced phagocytosis of Aβ compared to healthy controls (Fiala et al, 2005). Moreover, a reduced expression of genes involved in phagocytosis has been detected in aged AD microglia (Orre et al, 2014). Similarly, while young microglia from organotypic slice cultures were capable of ingesting exogenously added Aβ, this capacity was severely compromised in microglia isolated from adult AD mice (Hellwig et al, 2015). Our data are fully in line with above‐mentioned studies implicating phagocytosis defects in aged AD microglia.
Similar to the ex vivo co‐culture model described here, activation and clustering of microglial cells at amyloid plaques and reduction in amyloid plaque load have been observed in AD mouse models and humans after Aβ immunotherapy (Schenk et al, 1999; Bard et al, 2000; Bacskai et al, 2001; Nicoll et al, 2003, 2006; Wilcock et al, 2004; Boche & Nicoll, 2008; Sevigny et al, 2016; Xiang et al, 2016). Moreover, core‐only plaques were indeed observed in human AD cases after Aβ immunotherapy (Nicoll et al, 2006; Boche & Nicoll, 2008; Wisniewski & Goni, 2015; Sevigny et al, 2016). In agreement with our data, it has been reported that diffuse Aβ (found at the plaque halo) can be cleared more effectively compared to fibrillar (plaque core) Aβ deposits (Nicoll et al, 2006; Mandrekar et al, 2009; Serrano‐Pozo et al, 2010). As soluble Aβ species have been strongly implicated in synaptic dysfunction in AD, reducing their levels may indeed be beneficial for cognition (Haass & Selkoe, 2007; Koffie et al, 2009).
Collectively, immunotherapy studies, as well as our work, put forward the idea of immune system activation as a strategy to reduce amyloid load (Reardon, 2015). However, due to numerous pro‐ and anti‐inflammatory pathways that are directly controlled by microglia and often affected in AD (Lucin & Wyss‐Coray, 2009; Solito & Sastre, 2012; Heneka et al, 2015), titration of microglial activity appears challenging.
Effects of immune system modulation and microglial proliferation on amyloid clearance
Various studies have shown that modulation of the inflammatory state induced by cytokines, including TGF‐β, IL‐10, IL‐4, TNF‐α, and others can modify plaque load and improve AD outcome (Town et al, 2008; Chakrabarty et al, 2011, 2012, 2015; Wilcock, 2012; Guillot‐Sestier & Town, 2013; Guillot‐Sestier et al, 2015a). We achieved enhanced phagocytosis upon direct addition of GM‐CSF to old APPPS1 brain slices. Addition of other pro‐inflammatory (IL‐6 or IL‐12/p40) or anti‐inflammatory (TGF‐β and IL‐10) cytokines did not mimic the same phenotype. GM‐CSF‐mediated amyloid plaque clearance observed in our study was associated with induced microglial proliferation. Accordingly, in contrast to treatment with IL‐6, IL‐12/p40, TGF‐β or IL‐10, CD68 coverage was increased only upon treatment of the old tissue with GM‐CSF. Of note, TGF‐β treatment even appeared to reduce numbers of CD68‐positive cells which would be in agreement with its potential to decrease cell proliferation (Suzumura et al, 1993; Grainger et al, 1994).
Our finding that proliferation of old microglial cells is a pre‐requisite for increased amyloid clearance strengthens the link between microglial proliferation and Aβ clearance proposed by other studies (Liu et al, 2010; Smith et al, 2013; Neniskyte et al, 2014). In contrast, increased microglial proliferation in AD and beneficial effect of its inhibition on disease progression have recently been reported (Olmos‐Alonso et al, 2016). However, the amyloid burden was not modified in this study. Although proliferation seems to be necessary in our co‐culture model, we consider unlikely that increased phagocytosis is only due to increased microglial numbers. Often, microglial activation is linked to increased proliferation, but also enhanced phagocytic capacity of individual cells suggesting that regulation of proliferation and phagocytosis may be controlled by the same signaling pathway (Giulian & Ingeman, 1988). Similarly, macrophage colony‐stimulating factor (M‐CSF) was shown to increase both microglial proliferation and lysosomal activity (Majumdar et al, 2007; Boissonneault et al, 2009; Sanchez‐Ramos et al, 2009; Smith et al, 2013). In that regard, some experimental evidence suggested that microglia are not efficiently degrading engulfed Aβ (Frackowiak et al, 1992; Paresce et al, 1997; Chung et al, 1999), possibly due to poor hydrolytic activity of lysosomes (Majumdar et al, 2007), and it may therefore be required to enhance lysosomal degradation in order to successfully remove amyloid. However, further studies toward clarifying complex functions of particular cytokines are needed to strengthen their link with phagocytosis and amyloid clearance. GM‐CSF may be of relevance as its injection into an AD mouse model induced microglial proliferation and reduced amyloid and is currently also evaluated in phase II clinical trials for AD (Boissonneault et al, 2009; Boyd et al, 2010; Lai & McLaurin, 2012). However, also opposite reports exist that suggested neutralization of GM‐CSF to be beneficial and decrease amyloid load (Manczak et al, 2009). Although we could show that direct administration of GM‐CSF to old slices induces microglial proliferation and enhances phagocytosis, release of GM‐CSF by the young tissue is not a pre‐requisite for plaque phagocytosis in our ex vivo co‐culture model. Accordingly, we still detected increased CD68 coverage when old APPPS1 slices were co‐cultured with young GM‐CSF−/− brain slices suggesting that microglial numbers can be increased in the absence of GM‐CSF secretion from the young tissue and another mitogen may be involved in mediating this effect. Of note, upregulation of M‐CSF has been reported to occur as a compensatory mechanism in GM‐CSF−/− mice (Shibata et al, 2001; Bonfield et al, 2008). Although this effect has not yet been shown to occur in the brain parenchyma, we cannot rule out that M‐CSF upregulation is responsible for increased microglial numbers and consequently enhanced phagocytosis. Taken together, our work suggests that microglial modulation may be of benefit for AD. Their potential should be explored further to understand molecular changes occurring in diseased microglia and the potential to reverse those pathological changes by enhancing young and protective functions of healthy microglia.
Reduced amyloid clearance of aged microglia in AD is reversible
Previous reports described AD microglia as burned‐out, dystrophic (senescent) or degenerating (Streit et al, 2004, 2014), suggesting a low potential for functional repair. Intriguingly, the amyloid plaque clearance potential of aged AD microglia could be fully restored in our ex vivo co‐culture model despite their unfavorable cellular environment due to continuous loss of neurons and astrocytes. We want to note that is important to assess microglia not only in the context of amyloid plaques, but also in the context of neurodegeneration. Microglial response to amyloid pathology can be mimicked in transgenic mouse models, such as APPPS1 used here. However, microglial reaction to significant cell loss cannot be examined as no overt neurodegeneration has been observed in this and other commonly used mouse models of amyloidosis (Ashe & Zahs, 2010). Thus, our newly developed ex vivo model may more closely recapitulate microglial environment in AD brains. Attempts to rejuvenate old microglia toward promoting beneficial functions of young microglia may help understanding pathological changes in AD and open novel therapeutic avenues. Rejuvenation as the idea to restore brain functions that decline with aging has been proposed upon systemic manipulations such as caloric restriction, exercise or, as recently shown, heterochronic parabiosis (Villeda et al, 2011, 2014; Bouchard & Villeda, 2015; Smith et al, 2015). Our ex vivo model can be explored further to search for additional factors that enhance amyloid clearance and reduce accumulation of Aβ.
Materials and Methods
Animals
Hemizygous APPPS1 mice (line 21) were kindly provided by Mathias Jucker, Hertie‐Institute for Clinical Brain Research, University of Tübingen, and DZNE‐Tübingen (Radde et al, 2006). The APPPS1 mice are overexpressing human APPKM670/671NL and PS1L166P under the control of the Thy‐1 promoter and were bred in a C57BL/6J background. The CX3CR1GFP/GFP reporter line (Jung et al, 2000) was obtained from the Jackson Laboratory and bred with C57BL/6J WT and hemizygous APPPS1 line to obtain CX3CR1+/GFP and APPPS1/CX3CR1+/GFP mice, respectively. The GM‐CSF knockout mice (GM‐CSF−/−) were also obtained from the Jackson Laboratory (Dranoff et al, 1994). Both male and female mice were used in this study. Mice were group housed under specific pathogen‐free conditions. Mice had access to water and standard mouse chow (Ssniff® Ms‐H, Ssniff Spezialdiäten GmbH, Soest, Germany) ad libitum and were kept under a 12/12 h light–dark cycle in IVC System Type II L‐cages (528 cm2) equipped with solid floors and a layer of bedding. All animal experiments were performed in compliance with the German animal welfare law and have been approved by the government of Upper Bavaria.
Organotypic slice cultures
Organotypic slice cultures were prepared according to the membrane interface method described by Stoppini et al (1991) with some modifications. Brains were removed from either young neonatal (postnatal days 5–7) or old (10–20 months) mice. Young neonatal pups were decapitated, and old mice were sacrificed by CO2 inhalation, all according to animal handling laws. Hippocampi and neocortices of the old mice (hippocampal or cortical slice cultures) or hippocampi together with the adjacent neocortices of young mice (cortico‐hippocampal slice cultures, depicted in Fig 1A) were dissected. Sagittal sections, 350 μm thick, were cut using a Mcllwain tissue chopper (Model TC752, Mickle Laboratory Engineering Company), intact sections carefully selected under a dissection microscope (SZ61, Olympus) and incubated for 30 min at 4°C in pre‐cooled dissection media (50% HEPES‐buffered MEM, 1% penicillin–streptomycin, 10 mM Tris, pH 7.2). Subsequently, four slices were plated onto each 0.4‐μm porous polytetrafluoroethylene (PTFE) membrane insert (PICM ORG 050, Millipore) placed in a 3.5‐cm dish filled with 1 ml of slice culture media containing 50% HEPES‐buffered MEM (Gibco), 25% heat‐inactivated horse serum (Sigma‐Aldrich), 25% HBSS (Gibco), and 1 mM l‐glutamine (Gibco) at pH 7.4 and maintained in a cell‐culture incubator at 37°C, 5% CO2. In the co‐culture paradigm, two slices of young WT tissue (cortico‐hippocampal) were plated together with two slices of old APPPS1 tissue (cortical and hippocampal) in the same culture dish (Fig 1A). Media were exchanged 1 day after preparation and subsequently every 3–4 days.
Drug and cytokine treatments of slice cultures
Drugs were applied directly to the slice culture media. Slice cultures were treated as indicated and retreated at every media exchange. Treatments were as follows: cytochalasin D (CytoD, Sigma‐Aldrich, 1 mM stock in DMSO) at 1 μM; clodronate (Clo, Millipore, 50 mg/ml clodronate disodium salt stock in H2O) at 100 μg/ml; recombinant mouse IL‐10 (R&D systems, 100 μg/ml stock in PBS) at 100 ng/ml; recombinant mouse TGF‐β1 (R&D systems, 50 μg/ml stock in PBS) at 100 ng/ml; recombinant mouse IL‐6 (R&D systems, 100 μg/ml stock in PBS) at 100 ng/ml; recombinant mouse IL‐12/p40 (R&D systems, 100 μg/ml stock in PBS) at 100 ng/ml; recombinant mouse GM‐CSF (R&D systems, 100 ng/ml stock in PBS) at 1 ng/ml; and cytosine arabinoside (AraC, Merck Millipore, 1 mM stock in H2O) at 5 μM.
Biochemical characterization of slice cultures
For each sample, eight brain slices were pooled (from four independent slice culture dishes), and this was performed in triplicates and in three independent experiments. RIPA lysates were prepared, and the remaining pellet after 14,000 g centrifugation (60 min at 4°C) was homogenized in 70% formic acid. The formic acid fraction was neutralized with 20× 1 M Tris–HCl buffer at pH 9.5 and used for Aβ analysis. For Aβ detection, proteins were separated on Tris‐Tricine (10–20%, Thermo Fisher Scientific) gels, transferred to nitrocellulose membranes (0.1 μm, GE Healthcare) which were boiled for 5 min in PBS and subsequently incubated with the blocking solution containing 0.2% I‐Block (Thermo Fisher Scientific) and 0.1% Tween‐20 (Merck) in PBS for 1 h, followed by overnight incubation with 2 μg/ml 2D8 antibody in the blocking solution. The rat monoclonal 2D8 antibody against Aβ was described before (Shirotani et al, 2007). RIPA lysates (10–20 μg of protein) were separated on Tris‐Glycine (7–8%) or Tris‐Tricine (10–20%, Thermo Fisher Scientific) gels and blotted onto PVDF (Immobilon‐P; Millipore) membranes for the detection of CD68 (Serotec, MCA1957GA) 1:600, GFAP (Dako Cytomation, Z 0334) 1:1,500 and β3‐tubulin (Covance, MMS‐435P) 1:1,000. Antibody detection was performed using the corresponding anti‐HRP‐conjugated secondary antibodies (Santa Cruz) and chemiluminescence detection reagent ECL (Thermo Fisher Scientific) and quantified by ImageJ.
Slice culture conditioned media: collection and incubation
Conditioned media (CM) from young WT and old APPPS1 tissue (4 slices/dish) were collected in 1.5‐ml tubes starting from 4 to 5 DIV and collected every 3–4 days for a total of six times, centrifuged at 0.9 g for 5 min at 4°C, and the supernatants stored at −20°C. CM from different aliquots were thawed, pooled, and distributed into new 3.5‐cm dishes (1 ml/dish) and pre‐equilibrated at 37°C, 5% CO2 for at least 2 h before incubating the old APPPS1 tissue.
For collection of CM from microglia‐depleted young WT tissue, slices (4 slices/dish) were treated with 100 μg/ml of bisphosphonate clodronate (Clo) from 1 DIV until 7 DIV. After stopping the treatment, CM were collected (starting from 10 to 11 DIV) every 3–4 days for a total of five times. CM were filtered through a 0.2‐μm pore size sterile filter (VWR International) and stored at −20°C until usage. As a control, media were collected from vehicle (H2O)‐treated young WT tissue and subjected to the same experimental procedure. Slice cultures were incubated in CM, starting from 1 DIV, and re‐incubated every 3–4 days with freshly thawed and pre‐equilibrated CM. Samples were fixed at 7, 11 and 14 DIV and immunostained.
For analysis of cell proliferation, old APPPS1 slices were incubated in CM from young WT tissue in combination with 5 μM AraC or H2O as a vehicle control, starting from 1 DIV and re‐incubated every 3–4 days with freshly thawed and pre‐equilibrated CM and retreated. Samples were fixed at 14 DIV and immunostained.
Primary microglial cultures, conditioned media: collection and incubation
Primary microglia were isolated from postnatal day 5 WT mouse brains using MACS Technology (Miltenyi Biotec) and according to manufacturer′s instructions. Briefly, brain cortices were dissected, freed from meninges, and dissociated by enzymatic digestion using a Neural Tissue Dissociation Kit P (Miltenyi Biotec). Cd11b‐positive microglia were magnetically labeled using Cd11b MicroBeads, loaded onto a MACS Column (Miltenyi Biotec) and subjected to magnetic separation. Isolated microglia were plated onto 6‐cm tissue culture dish at density 1 × 106cells/dish and cultured for 24 h in DMEM/F12 media (Invitrogen) supplemented with 10% heat‐inactivated FCS (Sigma) and 1% penicillin–streptomycin (Invitrogen) and maintained in a humidified 5% CO2 incubator at 36.5°C. After 24 h, plating media were replaced with fresh slice culture media described above. Conditioned media (CM) were collected after additional 48 h, and filtered aliquots (0.45 μm, Millipore) have been stored at −20°C. Old APPPS1 slices were incubated in CM from young primary microglia or in slice culture media as a control, starting from 1 DIV and re‐incubated every 3–4 days with freshly thawed and pre‐equilibrated CM. Samples were fixed at 14 DIV and immunostained.
Immunohistochemistry
Slices were fixed in 4% paraformaldehyde (PFA)/sucrose, pH 7.4 for 15 min at room temperature (RT), washed in PBS (three times), and permeabilized for 30 min in PBS containing 0.5% Triton X‐100. Slices were cut from the membrane insert using a scalpel and transferred in a wet chamber, followed by 1‐h incubation in a blocking solution (PBS containing 0.5% Triton X‐100 and 5% goat serum). Slices were incubated overnight at RT in blocking solution containing primary antibodies: CD68 (Serotec, MCA1957GA) 1:1,000; Aβ antibodies M3.2 (Covance, SIG‐39155) 1:500, 6E10 (Covance, SIG‐39320) 1:500, and 2D8 (Shirotani et al, 2007) 0.011 μg/μl; GFAP (Dako Cytomation, Z 0334) 1:200; NeuN (Millipore, MAB377) 1:200; either GFP (NeuroMab, 75‐132) or GFP (Fitzgerald, 20R‐GR011) 1:200; and Ki67 (Cell Signaling Technology, 12202) 1:100. Slices were washed three times for 10 min with PBS containing 0.5% Triton X‐100 and incubated for 3–5 h at RT in blocking solution containing DAPI (Invitrogen) 1:20,000 and appropriate secondary antibodies conjugated to Alexa Fluor 488, 555, and 647 (Life Technologies) 1:200 and (Immunological Sciences) 1:350. Slices were subsequently washed three times for 10 min. Fibrillar dense core plaques were stained with thiazine red (Sigma‐Aldrich, 2 μM solution in PBS) for 20 min in the dark at RT followed by three PBS washing steps. Slices were mounted using Gel Mount media (Sigma‐Aldrich) and analyzed by confocal microscopy.
Image acquisition, analysis, and quantification
Confocal images were acquired using a Plan‐Apochromat 10×/0.45 M27 and 40×/1.4 oil differential interference contrast (DIC) objectives at 0.6‐ and 1.5‐fold magnifications on a LSM 710 confocal microscope (Zeiss) using ZEN 2011 software package. 3D image rendering was performed using Imaris x64 software (version 7.6.0, Bitplane). 3D images were analyzed using “surpass volume” mode. Representative images were acquired using the “snapshot” tool.
For quantification of amyloid plaques, samples were immunostained with M3.2 or 6E10 anti‐Aβ antibodies to visualize plaques. Images were acquired using a Plan‐Apochromat 10×/0.45 M27 objective on a fluorescence microscope (Zeiss AxioImager A2) equipped with AxioCam MRm using AxioVision software package. Total number of amyloid plaques and number of core‐only plaques were manually counted from acquired images and normalized to the tissue surface (in square micrometers). Imaged surface was calculated using the ZEN lite 2011 software package. The values are expressed as percentages of core‐only plaques from the total number of amyloid plaques. A minimum of 18 images per experiment, from at least three independent experiments, were used for quantification analysis (exceptions: Figs 6B and 9F where 12 images per experiment, from three independent experiments, were used for quantification analysis). Core‐only plaques were classified as plaques containing fibrillar, TR‐positive amyloid core, and no detectable 6E10‐positive halo (as depicted in Figs 2A, 14 DIV and EV2A). For quantification of the number of plaque‐associated CD68‐positive cells and plaque size, samples were immunostained with CD68 and 6E10 antibodies and confocal images acquired with 40×/1.4 oil DICIII objective at 1.5‐fold magnification. The number of plaque‐associated CD68‐positive cells was counted manually taking into account CD68 and DAPI signal, used to visualize nuclei. Results were expressed as numbers of CD68‐positive cells per plaque. The plaque area was determined from acquired images by drawing manually its outline and surface calculated using the ZEN lite 2011 software package and expressed in square micrometers. A minimum of 50 plaques, from at least three independent experiments, were used for quantification analysis. For quantification of the area of CD68‐positive cells (CD68 coverage), samples immunostained with CD68 Ab were used and confocal images acquired with Plan‐Apochromat 10×/0.45 M27 objective at 1.5‐fold magnification. CD68 coverage was analyzed by the thresholding technique using ImageJ software (NIH). The values were normalized to CD68 coverage of the Ctr. For each condition analyzed, a total of 12 regions of interest (ROI) were recorded from cortical areas. Twelve images per experiment, from three independent experiments, were used for the analysis. For quantification of the area of CX3CR1‐GFP‐positive cells (CX3CR1‐GFP coverage), samples were immunostained with GFP Ab and subjected to the same experimental procedure as described above. For quantification of the number of proliferating CD68‐positive microglial cells, samples were immunostained with CD68 and Ki67 antibodies and confocal images of cortical areas were acquired with Plan‐Apochromat 10×/0.45 M27 objective at 1.5‐fold magnification. The number of CD68‐positive and CD68 and Ki67 double‐positive microglial cells was determined by the thresholding technique and the “Analyze Particle” function using ImageJ (NIH) software (within size of 0.01‐Infinity for CD68‐positive cells and 0.0015‐Infinity for CD68 and Ki67 double‐positive cells). For the determination of the number of CD68‐positive cells, thresholded images were analyzed using the “watershed” module. A minimum of eight images per experiment, from three independent experiments, were used for the analysis in Fig 9B, and 12 images per experiment, from two independent experiments, were used for the analysis in Fig 9D. The values are expressed as percentages of Ki67 and CD68 double‐positive cells from the total number of CD68‐positive cells.
Cell viability assay
Cell viability was assessed by incubating brain slices (7 and 14 DIV) with 5 μg/ml propidium iodide (PI, Sigma‐Aldrich) for 15 min at 37°C, 5% CO2. Afterward, PFA‐/sucrose‐fixed samples have been stained with rabbit anti‐GFAP, rabbit anti‐NeuN antibodies, and DAPI as described before. Representative pictures have been acquired using a Leica SP5 confocal microscope with a 20× dry objective. PI‐positive cells have been quantified and normalized to the total cell number (DAPI‐positive) using an automated counting of single‐color images on ImageJ software (NIH). Quantification has been based on at least three pictures per sample from three independent experiments.
Microglial morphology
In order to investigate microglial morphology ex vivo, slice cultures from young CX3CR1+/GFP and old APPPS1/CX3CR1+/GFP mice have been prepared. Only cells not directly in contact with amyloid plaques have been analyzed in APPPS1/CX3CR1+/GFP mice. Brain slices at 0 DIV (young and old) and 10 DIV (young and old cultured alone and in co‐culture) have been PFA/sucrose fixed and stained using rabbit anti‐GFP, mouse anti‐Aβ antibody M3.2, and DAPI as described before. At least five representative pictures from each sample, from three independent experiments, have been acquired using a Leica SP5 confocal microscope with a 20× dry objective and 1.7‐fold magnification. Microglial cells have been classified by a rater blinded to treatment groups into two distinct categories (ramified versus amoeboid) according to cell body volume with a cut‐off of 523 μm3 (radius = 5 μm). Data are expressed as percentages of amoeboid cells versus total number of analyzed microglial cells.
Statistical analysis
The data are presented as mean ± standard error of the mean (± SEM) from at least three independent experiments. Statistical significance (P‐value) was calculated using the unpaired two‐tailed Student's t‐test (P‐value of < 0.05 was considered to be statistically significant; *P < 0.05, **P < 0.01 and ***P < 0.001, n.s. = not significant).
Author contributions
ST and CH designed and supervised the study. ST wrote the manuscript with the input from all authors. AD prepared all slice cultures and performed most of the experiments. AC prepared primary microglial cultures, performed imaging and quantifications of cellular markers, cell viability, and microglial morphology. AL and GL performed morphological (3D) analysis of microglial cells and measurements of microglial coverage. MW and HH established the aged APPPS1 mouse colony and performed biochemical analysis. Correspondence and requests for materials should be addressed to ST or CH.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 2
Acknowledgements
The authors thank Matthias Prestel and Dieter Edbauer for critically reading the manuscript. The APPPS1 colony was established from a breeding pair kindly provided by Mathias Jucker (Hertie‐Institute for Clinical Brain Research, University of Tübingen, and DZNE‐Tübingen). This work was supported by the European Research Council under the European Union's Seventh Framework Program (FP7/2007–2013)/ERC Grant Agreement No. 321366‐Amyloid to C.H., by the Excellence Cluster for Systems Neurology (SyNergy) to C.H. and A.L. and by the German Research Foundation (DFG, LI2534/1‐1) to A.L.
The EMBO Journal (2017) 36: 583–603
See also: K Biber (March 2017)
Contributor Information
Christian Haass, Email: christian.haass@mail03.med.uni-muenchen.de.
Sabina Tahirovic, Email: sabina.tahirovic@dzne.de.
References
- Amor S, Woodroofe MN (2014) Innate and adaptive immune responses in neurodegeneration and repair. Immunology 141: 287–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony JC, Breitner JC, Zandi PP, Meyer MR, Jurasova I, Norton MC, Stone SV (2000) Reduced prevalence of AD in users of NSAIDs and H2 receptor antagonists: the Cache County study. Neurology 54: 2066–2071 [DOI] [PubMed] [Google Scholar]
- Ard MD, Cole GM, Wei J, Mehrle AP, Fratkin JD (1996) Scavenging of Alzheimer's amyloid beta‐protein by microglia in culture. J Neurosci Res 43: 190–202 [DOI] [PubMed] [Google Scholar]
- Ashe KH, Zahs KR (2010) Probing the biology of Alzheimer's disease in mice. Neuron 66: 631–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacskai BJ, Kajdasz ST, Christie RH, Carter C, Games D, Seubert P, Schenk D, Hyman BT (2001) Imaging of amyloid‐beta deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat Med 7: 369–372 [DOI] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson‐Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B et al (2000) Peripherally administered antibodies against amyloid beta‐peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med 6: 916–919 [DOI] [PubMed] [Google Scholar]
- Baron R, Babcock AA, Nemirovsky A, Finsen B, Monsonego A (2014) Accelerated microglial pathology is associated with Abeta plaques in mouse models of Alzheimer's disease. Aging Cell 13: 584–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittner T, Burgold S, Dorostkar MM, Fuhrmann M, Wegenast‐Braun BM, Schmidt B, Kretzschmar H, Herms J (2012) Amyloid plaque formation precedes dendritic spine loss. Acta Neuropathol 124: 797–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boche D, Nicoll JA (2008) The role of the immune system in clearance of Abeta from the brain. Brain Pathol 18: 267–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissonneault V, Filali M, Lessard M, Relton J, Wong G, Rivest S (2009) Powerful beneficial effects of macrophage colony‐stimulating factor on beta‐amyloid deposition and cognitive impairment in Alzheimer's disease. Brain 132: 1078–1092. [DOI] [PubMed] [Google Scholar]
- Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, Kohsaka S, Jucker M, Calhoun ME (2008) Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci 28: 4283–4292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfield TL, Thomassen MJ, Farver CF, Abraham S, Koloze MT, Zhang X, Mosser DM, Culver DA (2008) Peroxisome proliferator‐activated receptor‐gamma regulates the expression of alveolar macrophage colony‐stimulating factor. J Immunol 181: 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard J, Villeda SA (2015) Aging and brain rejuvenation as systemic events. J Neurochem 132: 5–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd TD, Bennett SP, Mori T, Governatori N, Runfeldt M, Norden M, Padmanabhan J, Neame P, Wefes I, Sanchez‐Ramos J, Arendash GW, Potter H (2010) GM‐CSF upregulated in rheumatoid arthritis reverses cognitive impairment and amyloidosis in Alzheimer mice. J Alzheimers Dis 21: 507–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brana C, Benham C, Sundstrom L (2002) A method for characterising cell death in vitro by combining propidium iodide staining with immunohistochemistry. Brain Res Brain Res Protoc 10: 109–114 [DOI] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, Fanek Z, Liu L, Chen Z, Rothstein JD, Ransohoff RM, Gygi SP, Antel JP, Weiner HL (2014) Identification of a unique TGF‐beta‐dependent molecular and functional signature in microglia. Nat Neurosci 17: 131–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, Goate AM, Bales KR, Paul SM, Bateman RJ, Holtzman DM (2011) Human apoE isoforms differentially regulate brain amyloid‐beta peptide clearance. Sci Transl Med 3: 89ra57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarty P, Herring A, Ceballos‐Diaz C, Das P, Golde TE (2011) Hippocampal expression of murine TNFalpha results in attenuation of amyloid deposition in vivo . Mol Neurodegener 6: 16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarty P, Tianbai L, Herring A, Ceballos‐Diaz C, Das P, Golde TE (2012) Hippocampal expression of murine IL‐4 results in exacerbation of amyloid deposition. Mol Neurodegener 7: 36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarty P, Li A, Ceballos‐Diaz C, Eddy JA, Funk CC, Moore B, DiNunno N, Rosario AM, Cruz PE, Verbeeck C, Sacino A, Nix S, Janus C, Price ND, Das P, Golde TE (2015) IL‐10 alters immunoproteostasis in APP mice, increasing plaque burden and worsening cognitive behavior. Neuron 85: 519–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H, Brazil MI, Soe TT, Maxfield FR (1999) Uptake, degradation, and release of fibrillar and soluble forms of Alzheimer's amyloid beta‐peptide by microglial cells. J Biol Chem 274: 32301–32308 [DOI] [PubMed] [Google Scholar]
- Combs CK, Johnson DE, Cannady SB, Lehman TM, Landreth GE (1999) Identification of microglial signal transduction pathways mediating a neurotoxic response to amyloidogenic fragments of beta‐amyloid and prion proteins. J Neurosci 19: 928–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Andrea MR, Cole GM, Ard MD (2004) The microglial phagocytic role with specific plaque types in the Alzheimer disease brain. Neurobiol Aging 25: 675–683 [DOI] [PubMed] [Google Scholar]
- Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB, Holtzman DM, Zlokovic BV (2008) apoE isoform‐specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Investig 118: 4002–4013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derecki NC, Katzmarski N, Kipnis J, Meyer‐Luehmann M (2014) Microglia as a critical player in both developmental and late‐life CNS pathologies. Acta Neuropathol 128: 333–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW, Farlo J, Davies P, Crystal H, Fuld P, Yen SH (1988) Alzheimer's disease. A double‐labeling immunohistochemical study of senile plaques. Am J Pathol 132: 86–101 [PMC free article] [PubMed] [Google Scholar]
- Doetsch F, Garcia‐Verdugo JM, Alvarez‐Buylla A (1999) Regeneration of a germinal layer in the adult mammalian brain. Proc Natl Acad Sci USA 96: 11619–11624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dranoff G, Crawford AD, Sadelain M, Ream B, Rashid A, Bronson RT, Dickersin GR, Bachurski CJ, Mark EL, Whitsett JA, Mulligan RC (1994) Involvement of granulocyte‐macrophage colony‐stimulating factor in pulmonary homeostasis. Science 264: 713–716 [DOI] [PubMed] [Google Scholar]
- El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD (2007) Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer‐like disease. Nat Med 13: 432–438 [DOI] [PubMed] [Google Scholar]
- Fiala M, Lin J, Ringman J, Kermani‐Arab V, Tsao G, Patel A, Lossinsky AS, Graves MC, Gustavson A, Sayre J, Sofroni E, Suarez T, Chiappelli F, Bernard G (2005) Ineffective phagocytosis of amyloid‐beta by macrophages of Alzheimer's disease patients. J Alzheimers Dis 7: 221–232 [DOI] [PubMed] [Google Scholar]
- Fleisher‐Berkovich S, Filipovich‐Rimon T, Ben‐Shmuel S, Hulsmann C, Kummer MP, Heneka MT (2010) Distinct modulation of microglial amyloid beta phagocytosis and migration by neuropeptides (i). J Neuroinflammation 7: 61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frackowiak J, Wisniewski HM, Wegiel J, Merz GS, Iqbal K, Wang KC (1992) Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce beta‐amyloid fibrils. Acta Neuropathol 84: 225–233 [DOI] [PubMed] [Google Scholar]
- Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM (1998) Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol 152: 307–317 [PMC free article] [PubMed] [Google Scholar]
- Gandy S, Heppner FL (2013) Microglia as dynamic and essential components of the amyloid hypothesis. Neuron 78: 575–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulian D, Ingeman JE (1988) Colony‐stimulating factors as promoters of ameboid microglia. J Neurosci 8: 4707–4717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulian D, Haverkamp LJ, Yu JH, Karshin W, Tom D, Li J, Kirkpatrick J, Kuo LM, Roher AE (1996) Specific domains of beta‐amyloid from Alzheimer plaque elicit neuron killing in human microglia. J Neurosci 16: 6021–6037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjoneska E, Pfenning AR, Mathys H, Quon G, Kundaje A, Tsai LH, Kellis M (2015) Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer's disease. Nature 518: 365–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez‐Nicola D, Fransen NL, Suzzi S, Perry VH (2013) Regulation of microglial proliferation during chronic neurodegeneration. J Neurosci 33: 2481–2493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grainger DJ, Kemp PR, Witchell CM, Weissberg PL, Metcalfe JC (1994) Transforming growth factor beta decreases the rate of proliferation of rat vascular smooth muscle cells by extending the G2 phase of the cell cycle and delays the rise in cyclic AMP before entry into M phase. Biochem J 299(Pt 1): 227–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grathwohl SA, Kalin RE, Bolmont T, Prokop S, Winkelmann G, Kaeser SA, Odenthal J, Radde R, Eldh T, Gandy S, Aguzzi A, Staufenbiel M, Mathews PM, Wolburg H, Heppner FL, Jucker M (2009) Formation and maintenance of Alzheimer's disease beta‐amyloid plaques in the absence of microglia. Nat Neurosci 12: 1361–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griciuc A, Serrano‐Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, Hooli B, Choi SH, Hyman BT, Tanzi RE (2013) Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78: 631–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K et al (2013) TREM2 variants in Alzheimer's disease. N Engl J Med 368: 117–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillot‐Sestier MV, Town T (2013) Innate immunity in Alzheimer's disease: a complex affair. CNS Neurol Disord Drug Targets 12: 593–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillot‐Sestier MV, Doty KR, Gate D, Rodriguez J Jr, Leung BP, Rezai‐Zadeh K, Town T (2015a) Il10 deficiency rebalances innate immunity to mitigate Alzheimer‐like pathology. Neuron 85: 534–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillot‐Sestier MV, Doty KR, Town T (2015b) Innate immunity fights Alzheimer's disease. Trends Neurosci 38: 674–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta‐peptide. Nat Rev Mol Cell Biol 8: 101–112 [DOI] [PubMed] [Google Scholar]
- Haga S, Akai K, Ishii T (1989) Demonstration of microglial cells in and around senile (neuritic) plaques in the Alzheimer brain. An immunohistochemical study using a novel monoclonal antibody. Acta Neuropathol 77: 569–575 [DOI] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H (2007) Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci 10: 1387–1394 [DOI] [PubMed] [Google Scholar]
- Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC et al (2009) Genome‐wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet 41: 1088–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefendehl JK, Wegenast‐Braun BM, Liebig C, Eicke D, Milford D, Calhoun ME, Kohsaka S, Eichner M, Jucker M (2011) Long‐term in vivo imaging of beta‐amyloid plaque appearance and growth in a mouse model of cerebral beta‐amyloidosis. J Neurosci 31: 624–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellwig S, Masuch A, Nestel S, Katzmarski N, Meyer‐Luehmann M, Biber K (2015) Forebrain microglia from wild‐type but not adult 5xFAD mice prevent amyloid‐beta plaque formation in organotypic hippocampal slice cultures. Sci Rep 5: 14624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Sastre M, Dumitrescu‐Ozimek L, Hanke A, Dewachter I, Kuiperi C, O'Banion K, Klockgether T, Van Leuven F, Landreth GE (2005) Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1‐42 levels in APPV717I transgenic mice. Brain 128: 1442–1453. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss‐Coray T, Vitorica J, Ransohoff RM, Herrup K, Frautschy SA, Finsen B, Brown GC, Verkhratsky A, Yamanaka K, Koistinaho J, Latz E, Halle A, Petzold GC et al (2015) Neuroinflammation in Alzheimer's disease. Lancet Neurol 14: 388–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SE, Allison EK, El Khoury J (2008) Microglial dysfunction and defective beta‐amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci 28: 8354–8360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Morris JC, Goate AM (2011) Alzheimer's disease: the challenge of the second century. Sci Transl Med 3: 77sr71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itagaki S, McGeer PL, Akiyama H, Zhu S, Selkoe D (1989) Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol 24: 173–182 [DOI] [PubMed] [Google Scholar]
- Ji K, Akgul G, Wollmuth LP, Tsirka SE (2013) Microglia actively regulate the number of functional synapses. PLoS One 8: e56293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, Rujescu D, Hampel H, Giegling I, Andreassen OA, Engedal K, Ulstein I, Djurovic S, Ibrahim‐Verbaas C, Hofman A, Ikram MA et al (2013) Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med 368: 107–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR (2000) Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol 20: 4106–4114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinberger G, Yamanishi Y, Suarez‐Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger‐Weinzierl A, Mazaheri F, Tahirovic S, Lleo A, Alcolea D, Fortea J, Willem M, Lammich S, Molinuevo JL, Sanchez‐Valle R, Antonell A, Ramirez A et al (2014) TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med 6: 243ra286 [DOI] [PubMed] [Google Scholar]
- Koffie RM, Meyer‐Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia‐Alloza M, Micheva KD, Smith SJ, Kim ML, Lee VM, Hyman BT, Spires‐Jones TL (2009) Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci USA 106: 4012–4017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl A, Dehghani F, Korf HW, Hailer NP (2003) The bisphosphonate clodronate depletes microglial cells in excitotoxically injured organotypic hippocampal slice cultures. Exp Neurol 181: 1–11 [DOI] [PubMed] [Google Scholar]
- Krabbe G, Halle A, Matyash V, Rinnenthal JL, Eom GD, Bernhardt U, Miller KR, Prokop S, Kettenmann H, Heppner FL (2013) Functional impairment of microglia coincides with Beta‐amyloid deposition in mice with Alzheimer‐like pathology. PLoS One 8: e60921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutz S, Koch M, Bottger C, Ghadban C, Korf HW, Dehghani F (2009) 2‐Arachidonoylglycerol elicits neuroprotective effects on excitotoxically lesioned dentate gyrus granule cells via abnormal‐cannabidiol‐sensitive receptors on microglial cells. Glia 57: 286–294 [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW (1996) Microglia: a sensor for pathological events in the CNS. Trends Neurosci 19: 312–318 [DOI] [PubMed] [Google Scholar]
- Lai AY, McLaurin J (2012) Clearance of amyloid‐beta peptides by microglia and macrophages: the issue of what, when and where. Future Neurol 7: 165–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D, Bullido MJ, Tavernier B, Letenneur L, Bettens K, Berr C, Pasquier F, Fievet N, Barberger‐Gateau P, Engelborghs S, De Deyn P, Mateo I, Franck A et al (2009) Genome‐wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet 41: 1094–1099 [DOI] [PubMed] [Google Scholar]
- Lee SC, Liu W, Brosnan CF, Dickson DW (1994) GM‐CSF promotes proliferation of human fetal and adult microglia in primary cultures. Glia 12: 309–318 [DOI] [PubMed] [Google Scholar]
- Lee CY, Landreth GE (2010) The role of microglia in amyloid clearance from the AD brain. J Neural Transm 117: 949–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Condello C, Schain A, Harb R, Grutzendler J (2010) CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid‐beta phagocytosis. J Neurosci 30: 17091–17101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucin KM, Wyss‐Coray T (2009) Immune activation in brain aging and neurodegeneration: too much or too little? Neuron 64: 110–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue LF, Rydel R, Brigham EF, Yang LB, Hampel H, Murphy GM Jr, Brachova L, Yan SD, Walker DG, Shen Y, Rogers J (2001) Inflammatory repertoire of Alzheimer's disease and nondemented elderly microglia in vitro . Glia 35: 72–79 [DOI] [PubMed] [Google Scholar]
- Majumdar A, Cruz D, Asamoah N, Buxbaum A, Sohar I, Lobel P, Maxfield FR (2007) Activation of microglia acidifies lysosomes and leads to degradation of Alzheimer amyloid fibrils. Mol Biol Cell 18: 1490–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manczak M, Mao P, Nakamura K, Bebbington C, Park B, Reddy PH (2009) Neutralization of granulocyte macrophage colony‐stimulating factor decreases amyloid beta 1‐42 and suppresses microglial activity in a transgenic mouse model of Alzheimer's disease. Hum Mol Genet 18: 3876–3893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandrekar S, Jiang Q, Lee CY, Koenigsknecht‐Talboo J, Holtzman DM, Landreth GE (2009) Microglia mediate the clearance of soluble Abeta through fluid phase macropinocytosis. J Neurosci 29: 4252–4262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manocha GD, Floden AM, Rausch K, Kulas JA, McGregor BA, Rojanathammanee L, Puig KR, Puig KL, Karki S, Nichols MR, Darland DC, Porter JE, Combs CK (2016) APP regulates microglial phenotype in a mouse model of Alzheimer's disease. J Neurosci 36: 8471–8486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ (2010) Decreased clearance of CNS beta‐amyloid in Alzheimer's disease. Science 330: 1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meda L, Cassatella MA, Szendrei GI, Otvos L Jr, Baron P, Villalba M, Ferrari D, Rossi F (1995) Activation of microglial cells by beta‐amyloid protein and interferon‐gamma. Nature 374: 647–650 [DOI] [PubMed] [Google Scholar]
- Mewes A, Franke H, Singer D (2012) Organotypic brain slice cultures of adult transgenic P301S mice–a model for tauopathy studies. PLoS One 7: e45017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer‐Luehmann M, Spires‐Jones TL, Prada C, Garcia‐Alloza M, de Calignon A, Rozkalne A, Koenigsknecht‐Talboo J, Holtzman DM, Bacskai BJ, Hyman BT (2008) Rapid appearance and local toxicity of amyloid‐beta plaques in a mouse model of Alzheimer's disease. Nature 451: 720–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsonego A, Weiner HL (2003) Immunotherapeutic approaches to Alzheimer's disease. Science 302: 834–838 [DOI] [PubMed] [Google Scholar]
- Morgan D, Gordon MN, Tan J, Wilcock D, Rojiani AM (2005) Dynamic complexity of the microglial activation response in transgenic models of amyloid deposition: implications for Alzheimer therapeutics. J Neuropathol Exp Neurol 64: 743–753 [DOI] [PubMed] [Google Scholar]
- Mosher KI, Wyss‐Coray T (2014) Microglial dysfunction in brain aging and Alzheimer's disease. Biochem Pharmacol 88: 594–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussaud S, Draheim HJ (2010) A new method to isolate microglia from adult mice and culture them for an extended period of time. J Neurosci Methods 187: 243–253 [DOI] [PubMed] [Google Scholar]
- Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, Graff‐Radford NR, De Jager PL, Evans D, Schneider JA, Carrasquillo MM, Ertekin‐Taner N, Younkin SG et al (2011) Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late‐onset Alzheimer's disease. Nat Genet 43: 436–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neniskyte U, Vilalta A, Brown GC (2014) Tumour necrosis factor alpha‐induced neuronal loss is mediated by microglial phagocytosis. FEBS Lett 588: 2952–2956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO (2003) Neuropathology of human Alzheimer disease after immunization with amyloid‐beta peptide: a case report. Nat Med 9: 448–452 [DOI] [PubMed] [Google Scholar]
- Nicoll JA, Barton E, Boche D, Neal JW, Ferrer I, Thompson P, Vlachouli C, Wilkinson D, Bayer A, Games D, Seubert P, Schenk D, Holmes C (2006) Abeta species removal after abeta42 immunization. J Neuropathol Exp Neurol 65: 1040–1048 [DOI] [PubMed] [Google Scholar]
- Olmos‐Alonso A, Schetters ST, Sri S, Askew K, Mancuso R, Vargas‐Caballero M, Holscher C, Perry VH, Gomez‐Nicola D (2016) Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer's‐like pathology. Brain 139: 891–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orre M, Kamphuis W, Osborn LM, Jansen AH, Kooijman L, Bossers K, Hol EM (2014) Isolation of glia from Alzheimer's mice reveals inflammation and dysfunction. Neurobiol Aging 35: 2746–2760 [DOI] [PubMed] [Google Scholar]
- Paresce DM, Chung H, Maxfield FR (1997) Slow degradation of aggregates of the Alzheimer's disease amyloid beta‐protein by microglial cells. J Biol Chem 272: 29390–29397 [DOI] [PubMed] [Google Scholar]
- Prokop S, Miller KR, Drost N, Handrick S, Mathur V, Luo J, Wegner A, Wyss‐Coray T, Heppner FL (2015) Impact of peripheral myeloid cells on amyloid‐beta pathology in Alzheimer's disease‐like mice. J Exp Med 212: 1811–1818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, Calhoun ME, Jaggi F, Wolburg H, Gengler S, Haass C, Ghetti B, Czech C, Holscher C, Mathews PM, Jucker M (2006) Abeta42‐driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep 7: 940–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon S (2015) Antibody drugs for Alzheimer's show glimmers of promise. Nature 523: 509–510 [DOI] [PubMed] [Google Scholar]
- Rogers J, Strohmeyer R, Kovelowski CJ, Li R (2002) Microglia and inflammatory mechanisms in the clearance of amyloid beta peptide. Glia 40: 260–269 [DOI] [PubMed] [Google Scholar]
- Saido TC (1998) Alzheimer's disease as proteolytic disorders: anabolism and catabolism of beta‐amyloid. Neurobiol Aging 19: S69–S75 [DOI] [PubMed] [Google Scholar]
- Sanchez‐Ramos J, Song S, Sava V, Catlow B, Lin X, Mori T, Cao C, Arendash GW (2009) Granulocyte colony stimulating factor decreases brain amyloid burden and reverses cognitive impairment in Alzheimer's mice. Neuroscience 163: 55–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson‐Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C et al (1999) Immunization with amyloid‐beta attenuates Alzheimer‐disease‐like pathology in the PDAPP mouse. Nature 400: 173–177 [DOI] [PubMed] [Google Scholar]
- Schliwa M (1982) Action of cytochalasin D on cytoskeletal networks. J Cell Biol 92: 79–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholzen T, Gerdes J (2000) The Ki‐67 protein: from the known and the unknown. J Cell Physiol 182: 311–322 [DOI] [PubMed] [Google Scholar]
- Serrano‐Pozo A, William CM, Ferrer I, Uro‐Coste E, Delisle MB, Maurage CA, Hock C, Nitsch RM, Masliah E, Growdon JH, Frosch MP, Hyman BT (2010) Beneficial effect of human anti‐amyloid‐beta active immunization on neurite morphology and tau pathology. Brain 133: 1312–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevigny J, Chiao P, Bussiere T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, O'Gorman J, Qian F, Arastu M, Li M, Chollate S, Brennan MS, Quintero‐Monzon O, Scannevin RH, Arnold HM, Engber T et al (2016) The antibody aducanumab reduces Abeta plaques in Alzheimer's disease. Nature 537: 50–56 [DOI] [PubMed] [Google Scholar]
- Shibata Y, Berclaz PY, Chroneos ZC, Yoshida M, Whitsett JA, Trapnell BC (2001) GM‐CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity 15: 557–567 [DOI] [PubMed] [Google Scholar]
- Shirotani K, Tomioka M, Kremmer E, Haass C, Steiner H (2007) Pathological activity of familial Alzheimer's disease‐associated mutant presenilin can be executed by six different gamma‐secretase complexes. Neurobiol Dis 27: 102–107 [DOI] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien JP, Rivest S (2006) Bone marrow‐derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron 49: 489–502 [DOI] [PubMed] [Google Scholar]
- Smith AM, Gibbons HM, Oldfield RL, Bergin PM, Mee EW, Curtis MA, Faull RL, Dragunow M (2013) M‐CSF increases proliferation and phagocytosis while modulating receptor and transcription factor expression in adult human microglia. J Neuroinflammation 10: 85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LK, He Y, Park JS, Bieri G, Snethlage CE, Lin K, Gontier G, Wabl R, Plambeck KE, Udeochu J, Wheatley EG, Bouchard J, Eggel A, Narasimha R, Grant JL, Luo J, Wyss‐Coray T, Villeda SA (2015) beta2‐microglobulin is a systemic pro‐aging factor that impairs cognitive function and neurogenesis. Nat Med 21: 932–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solito E, Sastre M (2012) Microglia function in Alzheimer's disease. Front Pharmacol 3: 14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staal JA, Alexander SR, Liu Y, Dickson TD, Vickers JC (2011) Characterization of cortical neuronal and glial alterations during culture of organotypic whole brain slices from neonatal and mature mice. PLoS One 6: e22040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalder M, Phinney A, Probst A, Sommer B, Staufenbiel M, Jucker M (1999) Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am J Pathol 154: 1673–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart WF, Kawas C, Corrada M, Metter EJ (1997) Risk of Alzheimer's disease and duration of NSAID use. Neurology 48: 626–632 [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D (1991) A simple method for organotypic cultures of nervous tissue. J Neurosci Methods 37: 173–182 [DOI] [PubMed] [Google Scholar]
- Streit WJ, Sammons NW, Kuhns AJ, Sparks DL (2004) Dystrophic microglia in the aging human brain. Glia 45: 208–212 [DOI] [PubMed] [Google Scholar]
- Streit WJ, Xue QS, Tischer J, Bechmann I (2014) Microglial Pathology. Acta Neuropathol Commun 2: 142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh HS, Kim MO, Lee SC (2005) Inhibition of granulocyte‐macrophage colony‐stimulating factor signaling and microglial proliferation by anti‐CD45RO: role of Hck tyrosine kinase and phosphatidylinositol 3‐kinase/Akt. J Immunol 174: 2712–2719 [DOI] [PubMed] [Google Scholar]
- Suzumura A, Sawada M, Yamamoto H, Marunouchi T (1993) Transforming growth factor‐beta suppresses activation and proliferation of microglia in vitro . J Immunol 151: 2150–2158 [PubMed] [Google Scholar]
- Thambisetty M, An Y, Nalls M, Sojkova J, Swaminathan S, Zhou Y, Singleton AB, Wong DF, Ferrucci L, Saykin AJ, Resnick SM, Baltimore Longitudinal Study of A, the Alzheimer's Disease Neuroimaging I (2013) Effect of complement CR1 on brain amyloid burden during aging and its modification by APOE genotype. Biol Psychiatry 73: 422–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J, Duman RS, Flavell RA (2008) Blocking TGF‐beta‐Smad2/3 innate immune signaling mitigates Alzheimer‐like pathology. Nat Med 14: 681–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- in t' Veld BA, Ruitenberg A, Hofman A, Launer LJ, van Duijn CM, Stijnen T, Breteler MM, Stricker BH (2001) Nonsteroidal antiinflammatory drugs and the risk of Alzheimer's disease. N Engl J Med 345: 1515–1521 [DOI] [PubMed] [Google Scholar]
- Villeda SA, Luo J, Mosher KI, Zou B, Britschgi M, Bieri G, Stan TM, Fainberg N, Ding Z, Eggel A, Lucin KM, Czirr E, Park JS, Couillard‐Despres S, Aigner L, Li G, Peskind ER, Kaye JA, Quinn JF, Galasko DR et al (2011) The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 477: 90–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeda SA, Plambeck KE, Middeldorp J, Castellano JM, Mosher KI, Luo J, Smith LK, Bieri G, Lin K, Berdnik D, Wabl R, Udeochu J, Wheatley EG, Zou B, Simmons DA, Xie XS, Longo FM, Wyss‐Coray T (2014) Young blood reverses age‐related impairments in cognitive function and synaptic plasticity in mice. Nat Med 20: 659–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinet J, Weering HR, Heinrich A, Kalin RE, Wegner A, Brouwer N, Heppner FL, Rooijen N, Boddeke HW, Biber K (2012) Neuroprotective function for ramified microglia in hippocampal excitotoxicity. J Neuroinflammation 9: 27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DG, Lue LF (2015) Immune phenotypes of microglia in human neurodegenerative disease: challenges to detecting microglial polarization in human brains. Alzheimers Res Ther 7: 56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, Holtzman DM, Cirrito JR, Colonna M (2015) TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell 160: 1061–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster SD, Yang AJ, Margol L, Garzon‐Rodriguez W, Glabe CG, Tenner AJ (2000) Complement component C1q modulates the phagocytosis of Abeta by microglia. Exp Neurol 161: 127–138 [DOI] [PubMed] [Google Scholar]
- Wegiel J, Wisniewski HM (1990) The complex of microglial cells and amyloid star in three‐dimensional reconstruction. Acta Neuropathol 81: 116–124 [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Rojiani A, Rosenthal A, Levkowitz G, Subbarao S, Alamed J, Wilson D, Wilson N, Freeman MJ, Gordon MN, Morgan D (2004) Passive amyloid immunotherapy clears amyloid and transiently activates microglia in a transgenic mouse model of amyloid deposition. J Neurosci 24: 6144–6151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM (2012) A changing perspective on the role of neuroinflammation in Alzheimer's disease. Int J Alzheimers Dis 2012: 495243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildsmith KR, Holley M, Savage JC, Skerrett R, Landreth GE (2013) Evidence for impaired amyloid beta clearance in Alzheimer's disease. Alzheimers Res Ther 5: 33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski T, Goni F (2015) Immunotherapeutic approaches for Alzheimer's disease. Neuron 85: 1162–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss‐Coray T (2006) Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med 12: 1005–1015 [DOI] [PubMed] [Google Scholar]
- Xiang X, Werner G, Bohrmann B, Liesz A, Mazaheri F, Capell A, Feederle R, Knuesel I, Kleinberger G, Haass C (2016) TREM2 deficiency reduces the efficacy of immunotherapeutic amyloid clearance. EMBO Mol Med 8: 992–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip AG, Green RC, Huyck M, Cupples LA, Farrer LA, Group MS (2005) Nonsteroidal anti‐inflammatory drug use and Alzheimer's disease risk: the MIRAGE Study. BMC Geriatr 5: 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandi PP, Anthony JC, Hayden KM, Mehta K, Mayer L, Breitner JC, Cache County Study I (2002) Reduced incidence of AD with NSAID but not H2 receptor antagonists: the Cache County Study. Neurology 59: 880–886. [DOI] [PubMed] [Google Scholar]
- Zotova E, Bharambe V, Cheaveau M, Morgan W, Holmes C, Harris S, Neal JW, Love S, Nicoll JA, Boche D (2013) Inflammatory components in human Alzheimer's disease and after active amyloid‐beta42 immunization. Brain 136: 2677–2696 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 2