

ABSTRACT
Simian immunodeficiency viruses (SIVs) use their Nef proteins to counteract the restriction factor tetherin. However, a deletion in human tetherin prevents antagonism by the Nef proteins of SIVcpz and SIVgor, which represent the ape precursors of human immunodeficiency virus type 1 (HIV-1). To promote virus release from infected cells, pandemic HIV-1 group M strains evolved Vpu as a tetherin antagonist, while the Nef protein of less widespread HIV-1 group O strains acquired the ability to target a region adjacent to this deletion. In this study, we identified an unusual HIV-1 group O strain (RBF206) that evolved Vpu as an effective antagonist of human tetherin. While both RBF206 Vpu and Nef exert anti-tetherin activity in transient-transfection assays, mainly Vpu promotes RBF206 release in infected CD4+ T cells. Although mutations distinct from the adaptive changes observed in group M Vpus (M-Vpus) were critical for the acquisition of its anti-tetherin activity, RBF206 O-Vpu potently suppresses NF-κB activation and reduces CD4 cell surface expression. Interestingly, RBF206 Vpu counteracts tetherin in a largely species-independent manner, degrading both the long and short isoforms of human tetherin. Downmodulation of CD4, but not counteraction of tetherin, by RBF206 Vpu was dependent on the cellular ubiquitin ligase machinery. Our data present the first example of an HIV-1 group O Vpu that efficiently antagonizes human tetherin and suggest that counteraction by O-Nefs may be suboptimal.
IMPORTANCE Previous studies showed that HIV-1 groups M and O evolved two alternative strategies to counteract the human ortholog of the restriction factor tetherin. While HIV-1 group M switched from Nef to Vpu due to a deletion in the cytoplasmic domain of human tetherin, HIV-1 group O, which lacks Vpu-mediated anti-tetherin activity, acquired a Nef protein that is able to target a region adjacent to the deletion. Here we report an unusual exception, identifying a strain of HIV-1 group O (RBF206) whose Vpu protein evolved an effective antagonism of human tetherin. Interestingly, the adaptive changes in RBF206 Vpu are distinct from those found in M-Vpus and mediate efficient counteraction of both the long and short isoforms of this restriction factor. Our results further illustrate the enormous flexibility of HIV-1 in counteracting human defense mechanisms.
KEYWORDS: HIV-1 group O, Vpu, antagonism, tetherin
INTRODUCTION
Human immunodeficiency virus type 1 (HIV-1) is the result of at least four independent transmissions of simian immunodeficiency viruses (SIVs) infecting chimpanzees and gorillas in the wild (1, 2). In contrast to most SIVs infecting other primate species, SIVs of great apes seem preadapted to most human innate defenses. For example, SIVcpz is resistant to the antiviral effects of human, chimpanzee, and gorilla orthologs of the restriction factor TRIM5α (3) that mediate untimely uncoating (4) and play a relevant role in the species specificity of primate lentiviruses (5, 6). Furthermore, the Vif protein of SIVcpz is active against human APOBEC3G (7), which causes lethal hypermutation of the viral genome (8–11). Nonetheless, the four known cross-species transmission events generated HIV-1 lineages that differ significantly in their spread and prevalence in the human population. While the major M group is responsible for the global AIDS pandemic, HIV-1 group N has been found in fewer than 20 individuals (12). Similarly, HIV-1 group O has spread throughout West Central Africa, infecting several tens of thousands of individuals (13), while HIV-1 group P has been found only in two Cameroonians (14, 15). A number of studies suggested that differences in the ability to counteract the antiviral factor tetherin may play a role in the differential spread of the four groups of HIV-1 (16, 17).
Tetherin (BST-2) is an interferon (IFN)-inducible factor blocking the release of enveloped viral particles from infected cells (18, 19). Tetherin has an unusual topology, with an N-terminal transmembrane domain and a C-terminal glycosylphosphatidylinositol (GPI) anchor connected via long extracellular helical regions, and acts as direct physical tether with one transmembrane domain anchored in the virion and the other remaining in the cellular membrane (18–21). Studies with humanized mice showed that tetherin restriction contributes to the control of viral replication (22–24). In addition, tetherin antagonism appears to contribute to the relative resistance of transmitted-founder HIV-1 strains to type I IFN inhibition (25). Thus, tetherin has the potential to restrict replication and spread of HIV-1 in humans. Importantly, the human ortholog of tetherin contains a deletion in its cytoplasmic domain that was already present in archaic hominins (26) but is absent from tetherin proteins of nonhuman primates (27). This deletion usually confers resistance to the accessory Nef protein used by most SIVs, including the precursors of HIV-1 and HIV-2, to counteract tetherin in their respective primate hosts (27–29). Pandemic HIV-1 group M strains switched from Nef to Vpu to clear the tetherin barrier in the human host (27, 30). Vpu proteins of group N HIV-1 strains also acquired some anti-tetherin activity (27, 30, 31) but are usually less active than group M Vpus (M-Vpus) and lack other functions, including the ability to degrade CD4 (30).
Early studies suggested that HIV-1 groups O and P have not yet evolved an effective human tetherin antagonism (27, 32–35). However, we recently showed that HIV-1 O Nef proteins evolved the ability to target a region adjacent to the deletion to downmodulate human tetherin (36). Group O Nefs enhanced virus release from primary CD4+ T cells but exhibited little if any effect in transient-transfection assays, which explains why this activity was initially overlooked (27, 35, 36). In contrast, O-Vpus generally failed to show significant activity against human tetherin. In this study, we confirmed that most HIV-1 O strains use Nef to counteract tetherin but also identified an exception and show that the Vpu protein of RBF206, an HIV-1 O strain isolated from a 47-year-old woman in France (37), is a potent antagonist of human tetherin. RBF206 Vpu and Nef both display anti-tetherin activity in transient-transfection assays and in the context of the HIV-1 M NL4-3 molecular clone. However, an infectious molecular clone of HIV-1 O RBF206 uses primarily Vpu to antagonize human tetherin in virus-infected CD4+ T cells. Thus, although HIV-1 O viruses evolved Nef-mediated tetherin antagonism early after zoonotic transmission (36), continuing selection pressure by human tetherin resulted in the evolution of at least one O-Vpu that counteracts human tetherin as effectively as Vpus of pandemic HIV-1 M strains.
RESULTS
Identification of an HIV-1 group O Vpu that counteracts human tetherin.
Early studies suggested that HIV-1 group O strains lack Vpu-mediated anti-tetherin activity (27, 33–35) and instead evolved Nef as an antagonist of human tetherin (36). To determine whether these functional properties were general features of O-Vpus and O-Nefs, we analyzed vpu and nef alleles from 18 genetically diverse HIV-1 O strains. Transfection of HEK293T cells with vectors coexpressing Vpu or Nef and enhanced green fluorescent protein (eGFP) together with constructs expressing human tetherin confirmed that O-Nefs efficiently reduced cell surface expression of tetherin, while coexpression of most O-Vpus had significantly weaker effects (examples shown in Fig. 1A). One O-Vpu, however, downmodulated human tetherin as efficiently (>60%) as O-Nefs or M-Vpu (Fig. 1A). The associated vpu allele was derived from HIV-1 O RBF206, isolated from a 47-year-old Cameroonian woman living in France. Western blot analyses showed that the anti-tetherin activity of RBF206 Vpu was not due to particularly high expression levels (Fig. 1B). Some Vpu proteins migrated as smears because they tend to aggregate and form membrane-associated multimers. Notably, RBF206 Nef was as effective as RBF206 Vpu but weaker than many other O-Nefs in reducing cell surface expression of human tetherin in transfected 293T cells (Fig. 1A).
FIG 1.

Functional characterization of HIV-1 RBF206 Vpu and Nef proteins. (A) Effects of Nefs and Vpus on surface expression of human tetherin. Shown is flow cytometry analysis of HEK293T cells cotransfected with a tetherin expression vector and pCG plasmids expressing eGFP alone or together with the indicated vpu (U) or nef (N) alleles. The left side shows examples of primary flow cytometry data obtained 24 h posttransfection. The right side shows the levels of tetherin surface expression in the presence of Vpu or Nef relative to that in cells transfected with the control vector (100%, shown in black). The NL4-3 controls are shown in pink (Vpu) and orange (Nef), the SIVgor controls are shown in light blue (Vpu) and dark blue (Nef), and the O-Vpus are shown in turquoise and the O-Nefs in green. eGFP expression ranges used to calculate receptor downmodulation and the mean fluorescence intensities (MFIs) are indicated in the primary data. (B) Expression of Vpu and Nef proteins. HEK293T cells were transfected with plasmids encoding the indicated Vpu or Nef proteins, tagged with AU1, and analyzed by Western blotting. An empty vector and mock-transfected cells were used as controls. (C) Effects of various Vpus and the O-MRCA Nef on infectious virus yield. HEK293T cells were cotransfected with an HIV-1 NL4-3 Δvpu Δnef construct, pCG vectors coexpressing eGFP and Vpu or Nef, and increasing amounts of a construct expressing human tetherin. Viral supernatants were obtained 2 days later and used to measure infectious HIV-1 yield in the culture supernatants by infection of TZM-bl indicator cells. Shown are average infectious virus yields relative to those detected in the absence of tetherin (100%). (D) Effects of various Vpus on p24 release. HEK293T cells were transfected as described for panel C, and the levels of p24 in the culture supernatant and cell extracts were determined by ELISA. p24 release represents the p24 quantity in the supernatant relative to the total p24 amount in the supernatant and cellular extracts (100%). Values in all bar diagrams represent averages (± SEM) from three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To verify the anti-tetherin activity of RBF206 O-Vpu, we cotransfected HEK293T cells with an HIV-1 NL4-3 construct lacking intact vpu and nef genes and vectors expressing human tetherin and various Vpus or the inferred Nef protein of the most recent common ancestor (MRCA) of HIV-1 group O strains (36). Viral supernatants were obtained 2 days later and used to measure infectious HIV-1 yield in the culture supernatants by infection of TZM-bl indicator cells and to quantify virus release by determining the p24 levels in the supernatant, compared to the total p24 amount via enzyme-linked immunosorbent assay (ELISA). RBF206 O-Vpu significantly increased infectious virus yield (Fig. 1C) and virus release (Fig. 1D). In agreement with published data on HIV-1 group O Vpu proteins (33, 34, 35), the Vpu of the related strain HJ389 had no significant enhancing effect. Thus, in contrast to other O-Vpus, RBF206 Vpu efficiently counteracts human tetherin in transient-transfection assays.
Phylogeny of HIV-1 O RBF206.
Phylogenetic analyses showed that the central region of HIV-1 O RBF206 (3′ end of pol to 5′ end of env) falls well within the group O clade but is not particularly closely related to any other group O strain (Fig. 2A). This is also apparent in a distance plot, where RBF206 is more similar to other HIV-1 O strains than to SIVgor or HIV-1 M reference strains (Fig. 2B). Notably, the greatest distance between RBF206 and the reference sequences is found in the vpu gene, with the transmembrane domain (TMD)-encoding locus being the most divergent region. These data show that the anti-tetherin activity of HIV-1 O RBF206 Vpu is not the result of a recombination event with an HIV-1 group M strain, although several M/O recombinants have been described (38–40).
FIG 2.

Evolutionary relationship of RBF206 to other HIV-1 group O strains. (A) A maximum likelihood tree illustrating the phylogenetic relationship of RBF206 (highlighted in red) to other HIV-1 group O strains is shown for the central region spanning from the 3′ end of pol to the 5′ end of env (HXB2 coordinates 4730 to 6459; 1,638 bp). The tree is rooted using SIVgor sequences as outgroups. Asterisks indicate bootstrap values of ≥95% (the scale bar represents 5 substitutions per 100 sites). (B) The percent sequence identity of RBF206 to selected HIV-1 group O (orange and red), group M (blue), and SIVgor (green) sequences is plotted for a 1,619-bp region located between the 3′ end of pol and the 5′ end of env (HXB2 coordinates 4730 to 6459). Pairwise sequence distances were calculated for a 100-bp window, moved in increments of 10 bp along the alignment. The vpu coding region is shown (coordinates 6065 to 6327). The x axis indicates the genome position along the alignment (HXB2 coordinates), while the y axis shows sequence identity. Regions that could not be unambiguously aligned and sites with gaps were removed from the analysis. GenBank or DDBJ accession numbers for the sequences are for HIV-1 group O are as follows: LA33YBF37, KU168285; ANT70, L20587; MVP5180, L20571; and VAU, AF407418. Accession numbers for SIVgor are as follows: BQID2, KP004991; CP684, FJ424871; and BPID1, KP004989. Accession numbers for HIV-1 group M are as follows: subtype A U455A, M62320; subtype B HXB2, K03455; and subtype C ETH2220, U46016.
RBF206 O-Vpu inhibits NF-κB activation.
Recent studies have shown that Vpu inhibits activation of the transcription factor NF-κB to suppress expression of type I IFNs and other antiviral factors (41–44). This activity is conserved among primate lentiviral Vpu proteins and independent of its anti-tetherin activity (41, 45, 46). To determine the effect of RBF206 Vpu on NF-κB activation in comparison to other O-Vpus, we cotransfected HEK293T cells with vectors coexpressing Vpu and eGFP together with an NF-κB-dependent firefly luciferase reporter construct (2 μg). The canonical NF-κB signaling cascade was activated by cotransfection of a constitutively active mutant of IκB kinase β (IKKβ) (800 ng). All tested Vpu proteins suppressed IKKβ-mediated NF-κB activation at high levels of expression (2 μg) (Fig. 3, upper left). Only the SIVgor and RBF206 O-Vpus, however, potently inhibited NF-κB at a low dose (125 ng, upper right) of expression plasmid, while Vpus from the HIV-1 O strains HJ389, HJ162, HJ256, HJ001, HJ428, CMO2.5, and 13127 did not. These results were confirmed in the absence of fetal calf serum (FCS), thereby excluding any bias by growth factors or other stimuli in the cell culture medium (Fig. 3, lower graphs). Thus, RBF206 O-Vpu is not only active against human tetherin but also highly effective in inhibiting NF-κB activation.
FIG 3.
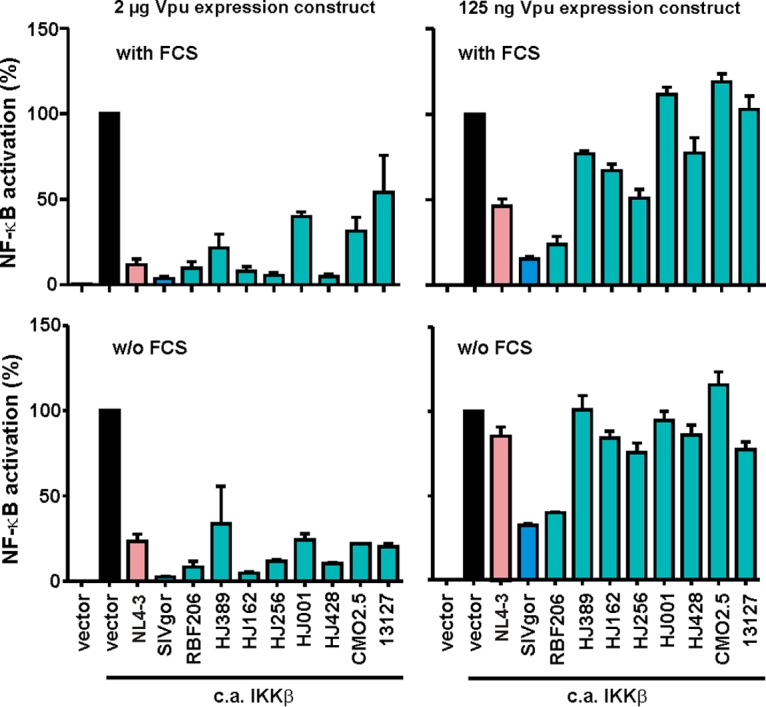
Effects of various Vpus on NF-κB activation. HEK293T cells were cotransfected with 2 μg or 125 ng of vectors expressing the indicated vpu alleles, a firefly luciferase reporter construct under the control of three NF-κB binding sites (2 μg), a Gaussia luciferase construct (500 ng) for normalization, and expression vectors for a constitutively active (c.a.) mutant of IKKβ (800 ng) as an inducer of NF-κB. Experiments were performed under standard cell culture conditions in the presence of 10% FCS (upper graphs) or in the absence of FCS (lower graphs), and luciferase activities were determined 48 h posttransfection. Values in all bar graphs represent averages (±SEM) from three independent experiments, each performed in triplicate.
RBF206 O-Vpu is active against both isoforms and different orthologs of tetherin.
Human tetherin contains a dual-tyrosine motif close to its N terminus (Fig. 4A) that acts as an endocytic recycling signal (47, 48) and affects its sensitivity to counteraction by M-Vpu proteins (49). Alanine substitutions of these tyrosine residues, however, did not significantly affect the susceptibility of human tetherin to downmodulation by RBF206 O-Vpu (Fig. 4B). To further examine the impact of alterations in the N-terminal cytoplasmic tail of human tetherin on antagonism by RBF206 O-Vpu, we next examined the short (S) isoform of this restriction factor. The S isoform is expressed by alternative translational initiation (42) and lacks the first 12 amino acids, including the tyrosine motif (Fig. 4A). It is efficiently expressed and inhibits virus release (42) but is resistant to O-Nef proteins (36) and less sensitive to downmodulation by M-Vpus than the long (L) isoform (49). RBF206 Vpu, however, was equally effective in downmodulating cell surface expression of both isoforms (Fig. 4C), which were expressed at similar levels (Fig. 4D). Functional studies confirmed that RBF206 Vpu enhanced infectious HIV-1 yield (Fig. 4E) and p24 antigen release (Fig. 4F) in the presence of S-tetherin more efficiently than NL4-3 M-Vpu. Thus, the N-terminal cytoplasmic domain of human tetherin is less critical for effective counteraction by RBF206 O-Vpu than by O-Nefs or M-Vpus. Interestingly, the Vpu proteins encoded by SIVgsn and SIVmon infecting related Cercopithecus species and the recently identified HIV-1 group N N1FR2011 strain also counteract both tetherin isoforms to similar extents (47). These data suggested that RBF206 Vpu might be a more broad-based tetherin antagonist than M-Vpus (27, 28, 50) and O-Nef proteins (36). Indeed, RBF206 O-Vpu protein efficiently downmodulated tetherin orthologs from different primate species (Fig. 4G) that were all efficiently expressed (Fig. 4D, right lanes).
FIG 4.

Effect of HIV-1 RBF206 Vpu on different tetherin isoforms and orthologs. (A) Amino acid alignment of the long and short isoforms of human tetherin. The region targeted by O-Nefs is indicated. A dual tyrosine endocytosis motif is shown in red. (B and C) Downmodulation of wild-type and Y6AY8A mutant (B) and the long and short isoforms of human tetherin (C). HEK293T cells were cotransfected with plasmids expressing the indicated tetherin and constructs expressing the indicated vpu alleles. Twenty-four hours later, cells were stained for tetherin and analyzed via flow cytometry. Shown are the levels of cell surface expression relative to those obtained in the presence of the vector expressing only eGFP (100%). (D) Expression of various tetherin isoforms and orthologs. HEK293T cells were cotransfected with plasmids expressing the indicated tetherin variants and examined by Western blotting 2 days later. All tetherin variants show variations in electrophoretic mobility due to differences in glycosylation. (E and F) Effects of various Vpus on infectious virus yield (E) and p24 release (F) in the presence of short tetherin. HEK293T cells were cotransfected with an HIV-1 NL4-3 Δvpu Δnef construct and vectors expressing the indicated Vpu proteins and the short isoform of human tetherin. Infectious virus yield was determined by infection of TZM-bl cells, and p24 release was determined via ELISA. The values obtained in the absence of tetherin represent 100%. (G) Downmodulation of the indicated tetherin orthologs by NL4-3 and RBF206 Vpus was determined as described for panels B and C. (H) Expression of the long and short forms of human tetherin in the presence of the indicated Vpu or Nef proteins. HEK293T cells were cotransfected with plasmids expressing the indicated tetherin variants and Vpu or Nef proteins and examined by Western blotting 2 days later. (I) Levels of L and S tetherin expression determined by Western blotting as shown in panel H. Values are mean percentages (±SEM, n = 3) obtained for the Vpu and Nef expression constructs relative to the vector control (100%). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To obtain our first insights into the underlying mechanisms, we examined whether RBF206 Vpu and Nef proteins affect total cellular protein levels of the short and long isoforms of tetherin. To determine this, we cotransfected HEK293T cells with plasmids expressing the L and S variants of the restriction factor and various Vpu or Nef expression constructs and analyzed them by Western blotting 2 days later (Fig. 4H). RBF206 Vpu efficiently degraded both isoforms, whereas O-Nefs and NL4-3 Vpu had only negligible effects (Fig. 4H and I). Thus, unlike M-Vpus and O-Nefs, RBF206 O-Vpu is equally active against the two isoforms of tetherin and counteracts this restriction factor in a largely species-independent manner.
Complex determinants of the anti-tetherin activity of RBF206 O-Vpu.
To identify determinants of the anti-tetherin activity of RBF206 O-Vpu, we first aligned its amino acid sequence with those of other O-Vpus (Fig. 5A). This viral accessory protein is highly variable, and the RBF206 Vpu sequence differs from those of other O-Vpus by at least 30 amino acids. One notable difference between RBF206 and other O-Vpus is a 4-amino-acid change, ESNG to GTTE, located between the two α-helical cytoplasmic domains (Fig. 5B). Notably, these changes generate a DYGxxE motif, which is similar to the DSphosGxxSphos motif mediating the recruitment of β-TrCP and an E3 ubiquitin ligase complex by HIV-1 M-Vpus. Mutation of these residues, however, did not impair expression of RBF206 or HJ389 O-Vpus (Fig. 5C) or their effect on tetherin cell surface expression (Fig. 5D). The GTTE-to-ESNG change also had no significant effect on CD4 downmodulation efficiency by RBF206 Vpu, while the reciprocal change impaired this activity of the HJ389 Vpu (Fig. 5E). Interestingly, mutation of GTTE to ESNG clearly reduced the activity of RBF206 Vpu in enhancing virus release, whereas the opposite alteration had no enhancing effect on HJ389 Vpu function (Fig. 5F). Thus, the effects of these amino acid residues on Vpu function are context dependent and other motifs in RBF206 O-Vpu contribute to its anti-tetherin activity.
FIG 5.

Mapping of residues critical for the anti-tetherin activity of RBF206 Vpu. (A) Alignment of HIV-1 O Vpu amino acid sequences. The NL4-3 M-Vpu sequence is shown at the bottom for comparison. The TMD, the cytoplasmic part, α-helix 1, and important motifs and residues are indicated. The AxxxAxxxW motif residues in the TMD important for anti-tetherin activity of M-Vpus, the consensus DSGxxS β-TrCP interaction site, and the (E/D)xxxL(L/I/V/M) motif involved in targeting of tetherin for endosomal degradation and recruitment of AP-1 and AP-2 are indicated in the well characterized NL4-3 Vpu. (B) Localization of specific sequence motifs in the RBF206 structure. A structural model of RBF206 Vpu is based on homology modeling using the NMR structure 2N28 (85). The LxxxSxxxW motif within the transmembrane domain of RBF206, which degenerates from the typical AxxxAxxxW consensus, the cytosolic DSDY and GTTE motifs, and the ExxxLL internalization motif are indicated. (C) Expression of analyzed mutant RBF206 and HJ389 variants. HEK293T cells were cotransfected with constructs expressing the indicated Vpus or empty vector and analyzed by Western blotting. β-Actin was included as a loading control. (D and E). Effect of wild-type and GTTE/ESSG mutant RBF206 and HJ389 Vpu proteins on tetherin (D) and CD4 (E) cell surface expression. PHA-activated PBMCs were transduced with VSV-G-pseudotyped nef-defective HIV-1 NL4-3 IRES-eGFP constructs containing the indicated vpu alleles and analyzed by flow cytometry 2 days later. The levels of tetherin or CD4 surface expression in the presence of Vpu relative to that of cells transduced with the vpu-deficient control (100%) are shown. (F) HEK293T cells were cotransfected with an HIV-1 NL4-3 Δvpu Δnef construct, vectors expressing the indicated Vpu proteins, and a construct expressing human tetherin. Viral supernatants were obtained 2 days later and used to quantify infectious HIV-1 in the culture supernatants by infection of TZM-bl indicator cells. Values in all bar graphs represent averages (±SEM) from three experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Next, we focused on the TMD of Vpu since it determines tetherin interaction in M- and N-Vpus (30, 51, 52) and because the TMD of M-Vpus is sufficient to confer anti-tetherin activity to SIVcpz and SIVgor Vpus (53). Notably, the TMD of RBF206 O-Vpu contains neither the AxxxAxxxW cluster found to be critical for tetherin antagonism by M-Vpus nor the AxxxAxxxxxLL motif of N-Vpus (30, 52). At the position corresponding to this motif, RBF206 Vpu contains LxxxSxxxW and differs only in single amino acid substitutions from other O-Vpus that do not counteract tetherin (Fig. 6A). Structural analysis of the tetherin TMD suggested N-terminal expansion of this motif to 10AxxxAxxxAxxxW in group M Vpu proteins (Fig. 6A, right). All residues are located on one side of the helical TMD, which aligns in an antiparallel manner to the TMD of tetherin, facing the hydrophobic residues V30, I34, L37, and L41. Whereas RBF206 Vpu contains the sequence motif V11, L15, S19, and W23, HJ389 Vpu exposes A11, L15, S19, and W23 to tetherin, while HJ001Vpu contains I11, L15, N19, and W23. While we do not fully understand from these sequence comparisons what drives the Vpu-mediated tetherin downmodulation, at least the spacious isoleucine at position 11 of HJ001 Vpu could counteract a tight interaction of these two anti-parallel-running helices (Fig. 6A). However, the TMD of RBF206 Vpu contains multiple additional alterations compared to other (inactive) O-Vpus that might contribute to its ability to counteract tetherin (Fig. 5A).
FIG 6.

Anti-tetherin activities of various wild-type and chimeric Vpu proteins. (A) Structural models illustrating possible interactions between tetherin and the TMDs of various Vpus. The assembly of the anti-parallel helices is based on the complex formation between human tetherin and the Vpu LW123 allele containing the classical AxxxAxxxAxxxW sequence motif reported by Skasko et al. (86) (right). Side chains of amino acid residues lining the interaction face of Vpu with tetherin are shown. Whereas RBF206 exposes a Vx3Lx3Sx3W motif, the poorly downmodulating HJ398 allele contains an Ax3Lx3Sx3W sequence and HJ001 an Ix3Lx3Nx3W motif. (B) Effect of mutant and chimeric Vpus on tetherin cell surface expression. Shown is flow cytometry analysis of HEK293T cells cotransfected with a tetherin expression vector and pCG plasmids expressing eGFP alone or together with the indicated wt or mutated vpu alleles. (C) HEK293T cells were cotransfected and virus yield was determined as described for Fig. 5F. Values in all bar graphs represent averages (±SEM) from three experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
An exchange of the TMD of RBF206 O-Vpu with that of HJ389 (HRR) significantly impaired tetherin downmodulation (Fig. 6B) and enhancement of virion release (Fig. 6C). Conversely, the RBF206 Vpu TMD conferred some tetherin downmodulation but no virion release activity on the HJ389 Vpu. Replacement of the central (RHR) and C-terminal (RRH) parts of RBF206 Vpu with those of HJ389 Vpu resulted in a phenotype intermediate between those of the two parental Vpu proteins. Thus, the TMD of RBF206 Vpu is critical for its ability to counteract tetherin, but residues in the cytoplasmic domain also play a role. Nonetheless, this activity was context dependent, since the RBF206 Vpu TMD did not confer anti-tetherin activity to SIVgor Vpu and disrupted the ability of NL4-3 Vpu to counteract human tetherin (Fig. 6B and C). As expected (53), the TMD of NL4-3 Vpu allowed SIVgor BQ644 Vpu to efficiently promote virus release (Fig. 6C) without markedly reducing tetherin cell surface expression levels (Fig. 6B). This is in agreement with previous data showing that tetherin downmodulation and virus release do not always correlate (54–57). Furthermore, the results show that the TMD and cytoplasmic domains of Vpu functionally cooperate and that unlike the TMD of NL4-3 Vpu, the TMD of RBF206 Vpu conferred effective tetherin antagonism only in its original context.
Tetherin downmodulation by RBF206 O-Vpu is cullin-RING E3 ligase independent.
It has been shown that HIV-1 M Vpu hijacks the cellular Skp1-Cul1-F-box protein (SCF) (β-TrCP) E3 ubiquitin ligase complex to mark CD4 for proteasomal degradation (58). In contrast, HIV-1 M Vpu uses cullin-RING ligase (CRL)-independent mechanisms to decrease the cell surface levels of tetherin. To obtain insights into the pathways underlying tetherin downmodulation by RBF206 O-Vpu, we examined its ability to modulate this restriction factor in the presence of the small-molecule inhibitor MLN4924, which blocks the NEDDylation and thus activation of the cullin-RING E3 ubiquitin ligase machinery (59, 60). CD4+ T cells were transduced with vesicular stomatitis virus glycoprotein (VSV-G)-pseudotyped NL4-3 internal ribosome entry site (IRES)-eGFP constructs expressing different Vpu and/or Nef proteins. Two days posttransduction, the infected peripheral blood mononuclear cell (PBMC) cultures were treated with 500 nM MLN4924 or dimethyl sulfoxide (DMSO) (as a control), and they were examined for tetherin and CD4 cell surface expression by flow cytometry 24 h later. Tetherin downmodulation by NL4-3 and RBF206 Vpus as well as by RBF206 Nef was not affected by MLN4924 treatment (Fig. 7A). In contrast, Vpu- but not Nef-mediated CD4 downmodulation was blocked by MLN4924 (Fig. 7B). Thus, CD4 but not tetherin downmodulation by M-Vpu and RBF206 O-Vpu requires the cullin-RING E3 ligase complex, suggesting that tetherin antagonism by RBF206 Vpu is not dependent on proteasomal degradation.
FIG 7.

CD4 but not tetherin downmodulation is impaired by an inhibitor of the cullin-RING E3 ubiquitin ligase machinery. CD4+ T cells activated with CD3/CD28 beads were transduced with VSV-G-pseudotyped HIV-1 NL4-3 UEOel IRES-eGFP constructs containing the indicated vpu and/or nef alleles. Two days postransduction, either DMSO or the small-molecule inhibitor MLN4924 (500 nM) was added. Cells were examined for eGFP and tetherin (A) or CD4 (B) surface expression via flow cytometry 24 h later. Values are averages (±SEM) from three independent experiments. ***, P < 0.001.
RBF206 Vpu and Nef counteract tetherin in a proviral HIV-1 M context.
To test whether RBF206 O-Vpu counteracts tetherin in the proviral context in primary HIV-1 target cells, we cloned this vpu allele into a nef-defective HIV-1 NL4-3 proviral construct in which the vpu/env overlap had been removed. Phytohemagglutinin (PHA)-activated PBMCs were transduced with VSV-G-pseudotyped HIV-1 NL4-3 IRES-eGFP constructs and examined for eGFP and tetherin or CD4 surface expression. The NL4-3 and RBF206 Vpus reduced the levels of tetherin surface expression by about 60%, whereas other O-Vpus (HJ001, HJ162, HJ256, HJ389, and HJ428) had only marginal effects (Fig. 8A). In comparison, all O-Vpus reduced the levels of CD4 surface expression, with RBF206 Vpu being slightly more active than the remaining five O-Vpus investigated (Fig. 8A).
FIG 8.

Modulation of surface molecules by RBF206 Vpu and Nef proteins in the context of NL4-3. (A) PHA-activated PBMCs were transduced with VSV-G-pseudotyped nef-defective HIV-1 NL4-3 UEOel IRES-eGFP constructs containing the indicated vpu alleles and examined for eGFP and tetherin or CD4 surface expression by flow cytometry 3 days later. The left side provides examples for primary data, and the right side shows average levels (±SEM) of surface expression in virally infected (eGFP+) cells relative to uninfected cells. The sample infected with a vpu-defective construct represents 100%. Values were derived from three independent infections of three donors. (B) PHA-stimulated PBMCs were transduced with VSV-G-pseudotyped NL4-3 IRES-eGFP constructs expressing the RBF206 Vpu and/or Nef proteins and analyzed by flow cytometry as described for panel A. Samples were additionally examined for MHC-I and CD74 surface expression. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
To further dissect the role of RBF206 Vpu and Nef in modulating tetherin and other cellular receptors, such as CD4 and class I MHC (MHC-I), we generated proviral NL4-3 constructs containing the two associated accessory genes individually or in combination. Analyses of these HIV-1 constructs showed that both RBF206 Vpu and Nef downmodulate tetherin as well as CD4 from the surface of virally infected PBMCs (Fig. 8B). In contrast, only Nef efficiently downmodulated MHC-I. We also examined modulation of the MHC-II-associated invariant chain (Ii, CD74), which affects peptide presentation and has been reported to be targeted by both Vpu and Nef (61–63). We found that RBF206 Vpu slightly reduced (by 21%) and Nef strongly increased (3-fold) CD74 cell surface expression (Fig. 8B). The presence of an intact RBF206 vpu gene reduced Nef-mediated CD74 induction from 3-fold to 1.5-fold (Fig. 8B). Thus, RBF206 Vpu and Nef proteins cooperate in the downmodulation of tetherin and CD4 but have opposing effects on CD74 surface expression.
To determine the contribution of intact RBF206 vpu and nef alleles to HIV-1 release, we performed Western blot analyses of cell-free and cell-associated viral antigens at different levels of tetherin expression. The results confirmed that the RBF206 Vpu and Nef proteins contribute individually to tetherin antagonism, although RBF206 Vpu was substantially more active than RBF206 Nef (Fig. 9). Moreover, both RBF206 Vpu and Nef acted specifically at the level of virion release into the culture supernatants but did not influence total levels of p24 antigen expression. Thus, in the context of the HIV-1 M NL4-3 proviral clone, RBF206 Vpu and Nef reduce tetherin cell surface expression with similar efficacies, but Vpu is more effective in promoting virus release.
FIG 9.

Effects of HIV-1 RBF206 Vpu and Nef proteins on virus release. Shown are average virus release (top) and one representative Western blot (bottom) from HEK293T cells transfected with NL4-3 UEOel constructs expressing the RBF206 Vpu and/or Nef proteins and different amounts of a tetherin expression plasmid (0, 6.25, 25, 100, and 250 ng). Viral and cellular protein expression in the cellular extracts and the culture supernatant was determined at 2 days postinfection. The upper portion shows the levels of Gag antigen (p24 plus p55) in the supernatant relative to the total amount of Gag in the supernatant and cellular extracts. The values obtained in the absence of tetherin represent 100%. Shown are mean values from three independent experiments (±SEM).
HIV-1 O RBF206 uses mainly Vpu to counteract tetherin in primary CD4+ T cells.
To examine the function of RBF206 Vpu and Nef proteins in their original genomic context, we generated an infectious molecular clone of this HIV-1 O strain (see Materials and Methods) and introduced inactivating mutations into its vpu and/or nef gene. Since the HIV-1 RBF206 construct did not express a fluorescent reporter gene, we measured receptor modulation by flow cytometric analysis of permeabilized cells stained for p24 antigen expression (Fig. 10). Surprisingly, only Vpu, and not Nef, significantly (∼70%) reduced tetherin cell surface expression in HIV-1 RBF206-infected CD4+ T cells (Fig. 10). Importantly, the lack of Nef-mediated anti-tetherin activity was not due to a lack of functional protein expression: Nef downmodulated CD4 by ∼70% and MHC-I by ∼50% and induced a 3-fold enhancement of CD74 expression in the absence of Vpu (Fig. 10B). In comparison, Vpu reduced tetherin expression by about 70% while also reducing CD4 and (less effectively) MHC-I expression but abolishing the enhancing effect of Nef on CD74 expression. The finding that Nef downmodulates tetherin in the context of HIV-1 NL4-3 but has little, if any, effect in the RBF206 backbone (48% versus 9% downmodulation) was confirmed in PBMC culture (Fig. 10C). Since no suitable antibodies are available and tagging is difficult in the proviral context, it remains to be determined whether differences in Vpu and/or Nef expression levels are responsible for these differences. Most importantly, the results clearly show that HIV-1 RBF206 uses predominantly Vpu to counteract human tetherin.
FIG 10.

Modulation of tetherin, CD4, MHC-I, and CD74 by RBF206 Vpu and Nef proteins in their original HIV-1 O context. CD4+ T cells activated with CD3/CD28 beads were transduced with the infectious molecular clone RBF206 or the indicated nef- and/or vpu-defective mutants thereof and examined for tetherin, CD4, MHC-I, and CD74 surface expression 3 days later. The left side shows examples of primary data. Bar graphs show average levels (±SEM) of surface expression in virally infected (eGFP+) cells relative to uninfected cells. The mean fluorescence intensities (MFIs) used to calculate receptor downmodulation are indicated in the primary data. The sample infected with a vpu- and nef-defective construct represents 100%. Values were derived from independent infections of three donors. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Role of Vpu and Nef in HIV-1 RBF206 replication and release in CD4+ T cells.
To analyze the importance of intact vpu and nef genes for HIV-1 RBF206 replication and virion release, we monitored virus production in primary CD4+ T cells infected with wild-type (wt) and mutant viral constructs in the absence and presence of type I interferon (IFN-α2). Predictably, all RBF206 constructs exhibited significantly higher levels of cell-free virus production in the absence of IFN-α (Fig. 11A and B). Disruption of vpu had only modest effects on the replicative capacity of HIV-1 RBF206, whereas the loss of an intact nef gene severely impaired viral replication (Fig. 11A and B). The lack of both intact vpu and nef genes abolished virus replication in CD4+ T cells almost entirely. To assess the effect of Vpu and Nef on the efficiency of HIV-1 RBF206 release, we determined the levels of cell-free and cell-associated p24 antigen in the infected CD4+ T cell cultures. IFN-α treatment usually reduced the efficiency of virus release by about 6% to 10% (Fig. 11C). A functional vpu gene significantly increased the efficiency of virus release in both the presence and absence of IFN-α, whereas mutation of nef had no significant effect (Fig. 11C and D). The differences in virion release efficiency were modest but were highly reproducible in CD4+ T cells from six independent blood donors. Thus, Nef is more important than Vpu for effective replication of HIV-1 RBF206 in primary CD4+ T cells, although only Vpu enhances virion release.
FIG 11.

Role of intact vpu and/or nef genes in HIV-1 O RBF206 replication and release. (A) Virus replication in CD4+ T cells in the presence and absence of IFN-α. Shown are the replication kinetics, measured as cell-free (cf) p24 in the supernatant, of the infectious molecular clone RBF206 and the corresponding nef- and/or vpu-defective mutants in the absence and presence of 500 U/ml of IFN-α. Data are mean values (±SEM) for p24 amounts in the culture supernatant over 10 days derived from infection of CD4+ T cells of six donors. (B) Cumulative (Cum) p24 amounts of the viral replication kinetics shown in panel A in the absence (left) and presence (right) of 500 U/ml of IFN-α. (C and D) Efficiencies of p24 release in the presence and absence of IFN-α by wt and nef- and/or vpu-defective mutants of RBF206. Values are percentages of cell-free p24 out of the total p24 detected via ELISA (±SEM) in the absence and presence of 500 U/ml of IFN-α and were derived from infection of T cells from six donors. Shown are release over time (C) and average release (D). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
DISCUSSION
For several years, HIV-1 group O strains were thought to lack anti-tetherin antagonism because their Vpu and Nef proteins failed to exhibit this activity in transient-transfection assays (27, 33–35). Recently, however, we showed that HIV-1 O Nef proteins efficiently reduce cell surface expression of human tetherin by targeting an alternative region in the N-terminal cytoplasmic domain and enhance virus release in infected CD4+ T cells (36). In this study, we identified the first HIV-1 group O strain encoding a Vpu protein that is an effective antagonist of human tetherin. Similar to Vpu proteins of primary HIV-1 M strains and unlike those of rare group N viruses (27, 30), RBF206 O-Vpu maintained full activity in other functions, i.e., downmodulation of CD4 (Fig. 8) and inhibition of NF-κB activation (Fig. 3). However, the changes that conferred anti-tetherin activity on this O-Vpu are clearly distinct from those of M-Vpus. In contrast to M-Vpus, for example, RBF206 O-Vpu is equally active against the long and short isoforms of human tetherin and seems to counteract this restriction factor in a largely species-independent manner (Fig. 4).
The HIV-1 O strain RBF206 is not the first example of a virus that uses both Vpu and Nef to counteract tetherin. Interestingly, the HIV-1 M strain JC16 acquired the ability to use Nef to counteract CPZ tetherin after inoculation into chimpanzees within a single passage in that host, while maintaining anti-tetherin activity of Vpu (64). In this case, just two amino acid changes in the C-loop of HIV-1 Nef were sufficient to confer full activity against CPZ tetherin. In contrast, the present example of Vpu-mediated anti-tetherin activity by the HIV-1 O strain RBF206 appears to have required a larger number of changes in different domains of this accessory protein (Fig. 5 and 6). This is somewhat surprising since changes in the TMDs of some SIVcpz and SIVgor Vpus are sufficient to confer potent activity against human tetherin (53). For example, the NL4-3 Vpu TMD rendered the SIVgor CP2139 and BQ664 Vpu proteins active against human tetherin. Similarly, N-Vpus also acquired alterations in their TMDs, allowing effective tetherin interaction, but they usually also contain mutations in their cytoplasmic parts that impair most functional activities (30). Thus, HIV-1 does not always seem to “find” the most direct and successful way to acquire anti-tetherin activity. It is of interest that M-Vpus, N-Vpus, and RBF206 O-Vpu differ considerably in the sequences of their TMD domains, with RBF206 Vpu lacking the canonical Ax3Ax3W and Ax3Ax5LL motifs found in both M- and N-Vpus (30, 52). Instead, the sequence motif Vx3Lx3Sx3W is found in RBF206 Vpu, whereas the HJ398 and HJ001 O-Vpus, which show little, if any, anti-tetherin activity, contain Ax3Lx3Sx3W or Ix3Lx3Nx3W sequence motifs, respectively (Fig. 5A). This does not fully explain why RBF206 Vpu is able to counteract tetherin but HJ389 and HJ001 Vpus are not. However, there are multiple additional sequence changes in the TMDs of these O-Vpus, suggesting that residues besides the Ax3Ax3W motif also contribute to tetherin interaction. While the RBF206 Vpu TMD is critical for anti-tetherin activity in its original context, it disrupted this function in the context of the NL4-3 Vpu (Fig. 6), indicating that transmembrane and cytoplasmic domains functionally cooperate.
Group M Vpus and O Nefs counteract tetherin in a species-specific manner (27, 36, 50, 65) and are more active against the long than against the short isoform of this protein (42). Interestingly, this was not the case for RBF206 O-Vpu, which was highly active against both isoforms, and all other primate orthologs investigated. Whether this function renders this O-Vpu superior to M-Vpus (or O-Nefs) remains to be determined. For example, it has been proposed that redistributing the short isoform of tetherin away from viral assembly sites may allow HIV-1 M Vpu to counteract the inhibitory effect on virus release while maintaining negative tetherin/ILT7 signaling to plasmacytoid dendritic cells (pDC) to attenuate the antiviral IFN response (66). Whether RBF206 O-Vpu exerts such a redistributing activity or rather resembles O-Nef proteins, which do not displace short tetherin from viral assembly sites (67), remains to be investigated. Furthermore, it is possible that tetherin counteraction by Nef, which is expressed early in the viral replication cycle, results in a faster removal of tetherin from the cell surface and thus a more efficient enhancement of viral release. Finally, it is unknown whether tetherin directly binds either of its viral antagonists Vpu and Nef, thereby preventing these proteins to exert other important functions, such as the downmodulation of CD4.
When expressed from its cognate provirus, the Vpu protein of RBF206 was more effective in counteracting human tetherin than its Nef counterpart (Fig. 10 and 11). In fact, Nef clearly contributed to tetherin counteraction only in the context of the NL4-3 backbone (Fig. 8 and 9). The underlying reasons for these context-dependent differences require further investigation. Although a direct comparison of Nef expression levels in the context of the two proviral constructs by Western blotting is not feasible due to the lack of suitable antibodies, efficient Nef expression by both NL4-3 and RBF206 was evident from the downmodulation of CD4 and MHC-I as well as upregulation of CD74 (Fig. 8 and 10). Whether acquisition of Vpu-mediated anti-tetherin activity was a cause or consequence of the lack of Nef to counteract this factor in the context of the HIV-1 O strain RBF206 remains a matter of speculation. It has recently been shown that some group M HIV-1 strains (particularly those belonging to subtype C) lack Vpu-mediated anti-tetherin activity and instead express Nef proteins that confer this function (68). Thus, some group M Nefs may be able to acquire anti-tetherin activity to compensate for the loss of this function by Vpu, and the reverse may have happened in the case of HIV-1 RBF206.
It has been shown that Vpu and Nef can cooperate in counteracting cellular factors that suppress efficient viral replication. For example, Nef downmodulates CD4 from the cell surface, whereas Vpu induces proteasomal degradation of newly synthesized CD4 (69–71). Furthermore, Nef inhibits transport of MHC-I to the cell surface (72, 73), while Vpu may suppress MHC-I expression by inhibiting NF-κB activity (41). While it may seem counterintuitive that HIV/SIV accessory proteins exert opposing effects on some receptors, such as CD74, this may be required to fine-tune virus replication in the face of a vigorous immune response. Vpu-mediated inhibition of NF-κB activation is clearly advantageous for HIV-1 because it suppresses the expression of many antiviral genes (41). However, this transcription factor is also involved in the expression of numerous cellular genes, including some (like the CD74 gene) that are advantageous for viral immune evasion. Thus, Nef may have evolved to compensate the Vpu-mediated inhibition of NF-κB by efficiently upmodulating CD74 cell surface expression to impair MHC-II antigen presentation to impair CD4+ helper T cell responses (61).
In summary, we provide compelling evidence that some HIV-1 O strains may acquire Vpu-mediated anti-tetherin activity, similar to pandemic HIV-1 group M strains. This observation, together with similar findings for the Nef proteins of some group M strains (68), further highlights the enormous plasticity of HIV-1 in counteracting tetherin. Whether O-Nefs are generally less potent tetherin antagonists than M-Vpus and may evolve improved counteraction mechanisms in the future remains to be determined. In agreement with the recent characterization of an HIV-1 N strain whose Vpu protein counteracts human tetherin as effectively as M-Vpus (30), the current findings support the notion that nonpandemic HIV-1 groups are still evolving toward increased replication and transmission fitness.
MATERIALS AND METHODS
Proviral HIV-1 NL4-3 constructs.
To generate nef-deficient HIV-1 NL4-3 IRES-eGFP molecular clones expressing heterologous vpu genes, the vpu/env overlap was eliminated (pBR_NL4-3 UEOel IRES eGFP) and vpu alleles were exchanged using SacII and NcoI restriction sites as described previously (27). An additional NcoI site in eGFP was silenced to facilitate cloning. For infection of primary cells, the NL4-3 UEOel constructs were pseudotyped with VSV-G as elimination of the vpu/env overlap abrogated env expression (27). The HIV-1 NL4-3 nef was replaced by the RBF206 nef allele using splice overlap extension PCR and the HpaI and MluI restriction sites as described previously (74).
Construction of an infectious molecular clone of HIV-1 RBF206.
HIV-1 O group strain RBF206 was isolated from a 47-year-old woman living in France by coculture of the subject's plasma with healthy donor PBMCs. At the time of sampling, the patient was treatment naive and classified at CDC stage C with 20 CD4+ T cells/mm3 and a plasma viral load of 4.8 log. Single genome amplification (SGA) of env C2V3 sequences revealed very limited within-isolate diversity, indicating that the RBF strain was isolated under limiting dilution conditions (data not shown). To generate a full-length proviral clone, SGA was used to amplify overlapping 5′ (R to vpr) and 3′ (pol to R) genome halves, and the resulting consensus sequence was then synthesized as four partially overlapping fragments (purchased from BlueHeron). An infectious molecular clone was constructed using unique BstEII (nucleotide position 2121), DraIII (nucleotide position 4152), and NheI (nucleotide position 8067) restriction enzyme sites and inserted between the MluI and NotI restriction sites of the pSMART-topo vector. Notably, the sequence of this RBF206 clone differs from a second independently derived consensus sequence of the same isolate, which was generated after an additional PBMC passage (37), by four nucleotides; however, none of these changes fell within the vpu gene, which is identical between the two consensus sequences. The pSMART-topo vector is a derivative of pSMART-LC-Amp containing the multiple-cloning site of pCR-XL TOPO from MluI to NotI. To generate vpu- and/or nef-deficient derivatives of HIV-1 RBF206, we used the QuikChange II XL site-directed mutagenesis kit according to the protocol provided by the manufacturer (Agilent Technologies) and introduced premature stop codons after the vpu and/or nef initiation codon.
Evolutionary analyses.
Nucleotide sequences from the accessory gene region of HIV-1 group O and three SIVgor strains were aligned using CLUSTALW v. 2 (75). This region, which spans HXB2 coordinates 4730 to 6459, was chosen because it includes the vpu gene within a genome segment that is not recombinant in the closest SIVgor relative (SIVgorBQID2) (2). Regions that could not be unambiguously aligned were removed from subsequent analyses. A maximum likelihood tree with bootstrap support (1,000 replicates) was constructed using PhyML v. 3.1 (76), with a GTR+G+I evolutionary model selected using jModelTest v. 2.1.4 (77). Sequence distance from the RBF206 nucleotide sequence was plotted for a subset of HIV-1 group O, HIV-1 group M, and SIVgor strains over the same region using SimPlot v. 3.5.1 (78). GenBank or DDBJ accession numbers for the sequences are as follows: SIVgorBPID1, KP004989; SIVgorBQID2, KP004991; SIVgorCP684, FJ424871; 10GAB1190, JX245014; 11GAB6352, JX245015; 18O04, AB485666; 95CAMP448, Y16028; 95SNMP331, Y16025; 96CAMP539, Y16027; 96CMA102, AY169803; 96CMABB009, AY169806; 96CMABB637, AY169810; 97CMABB447, AY169813; 97CMABB497, AY169809; 97US08692A, AY169805; 98CMA104, AY169802; 98CMA105, AY169816; 98CMABB141, AY169807; 98CMABB197, AY169811; 98CMABB212, AY169804; 98CMU2901, AY169812; 98CMU5337, AY169808; 99CMU4122, AY169815; 99USTWLA, AY169814; ANT70, L20587; BCF01, Y16030; BCF06, Y16026; BCF07, Y16029; BCF08, Y16024; BCF11, Y16018; BCF12, Y16031; DEOXXDE004, KF859742; DEOXXES001, KF859743; DEOXXUS001, KF859744; DSC1320.O, AY489739; GAVI686, Y16019; I-2478B, AB485668; LA29YBF26, KU168281; LA30RBF125, KU168282; LA31BCF108, KU168283; LA32RBF140, KU168284; LA33YBF37, KU168285; LA34RBF20, KU168286; LA45BCF02, KU168288; LA46BCF03, KU168289; LA47BCF09, KU168290; LA48BCF13, KU168291; LA49RBF189, KU168292; LA50YBF16, KU168293; LA51YBF35, KU168294; LA52YBF39, KU168295; LA53BCF109, KU168296; LA54BCF120, KU168297; LTNP, JN571034; MVP5180, L20571; pCMO2-3, AY618998; SEMP1299, AJ302646; SEMP1300, AJ302647; and VAU, AF407418.
Expression vectors.
Bicistronic cytomegalovirus (CMV) promoter-based pCG expression vectors coexpressing vpu, nef, or tetherin with either eGFP or DsRed have been described previously (27). Splice-overlap extension PCR, with primers introducing XbaI and MluI restriction sites flanking the reading frame, was used to generate chimeric or mutant vpu alleles. PCR fragments were purified from agarose gels and inserted into the pCG vector using standard cloning techniques. The human CD4 gene was cloned into pcDNA3.1(+) via HindIII/XbaI. The pHIT/G vector expressing vesicular stomatitis glycoprotein (VSV-G) has been described previously (79). An NF-êB firefly luciferase reporter plasmid containing three NF-êB binding sites was kindly provided by Bernd Baumann. A minimal promoter Gaussia luciferase construct was used for normalization. It contains the TATA-like promoter (pTAL) region from the herpes simplex virus thymidine kinase (HSV-TK) that is not responsive to NF-êB and was generated by exchanging the firefly luciferase gene of a reporter plasmid purchased from Clontech (631909). All PCR-derived inserts were sequenced to confirm their accuracy. Generation of the HIV-1 O MRCA Nef and Vpu expression vectors has been reported previously (36).
Cell culture and transfections.
HEK293T cells and TZM-bl cells were cultured in Dulbecco's modified Eagle medium (DMEM) containing 10% fetal calf serum (FCS) plus 2 mM glutamine, streptomycin (120 mg/ml), and penicillin (120 mg/ml) and were transfected using the calcium phosphate method (80). TZM-bl cells were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH from John C. Kappes, Xiaoyun Wu and Tranzyme, Inc (81). PBMCs and CD4+ T cells were isolated using lymphocyte separation solution (Biochrom) and cultured in RPMI 1640 medium with 10% FCS plus 2 mM glutamine, streptomycin (120 mg/ml), penicillin (120 mg/ml), and 10 ng/ml of interleukin 2 (IL-2). CD4+ T cells were negatively selected (RosetteSep) prior to density gradient centrifugation. PBMCs were activated using PHA (1 μg/ml), and CD4+ T cells were activated using CD3/CD28 beads (Dynabeads) at a ratio of 3:1.
Flow cytometric analysis.
HEK293T cells were transfected with 1 μg of tetherin or CD4 expression vector and 5 μg of a construct expressing eGFP alone or together with Vpu or Nef. One day posttransfection, CD4 and tetherin cell surface levels were analyzed via flow cytometry by staining of CD4 (anti-CD/allophycocyanin [APC], clone S3.5; Invitrogen) and tetherin (anti-tetherin/APC, clone RS38E; BioLegend) and gating for eGFP+ cells. The mean fluorescence intensity (MFI) of the APC signal in cells expressing Vpu or Nef was compared to that of the signal of cells transfected with the vector control expressing eGFP only.
PBMCs and CD4+ T cells were transduced with VSV-G-pseudotyped NL4-3 or RBF206 HIV-1 particles. Two days postransduction, cell surface levels of tetherin, CD4, CD74 (anti-CD74/R-phycoerythrin [PE], clone M-B741; Ancell) and MHC-I (anti-HLA-ABC/R-PE, clone W6/32; Dako) were determined via flow cytometry analysis. Cells transduced with RBF206 constructs were also permeabilized and stained intracellularly with an anti-p24 antibody (anti-p24/fluorescein isothiocyanate [FITC], clone KC57; Beckman Coulter). The ratios of the MFIs of infected cells (eGFP/FITC positive) and uninfected cells (eGFP/FITC negative) were used to calculate changes in cell surface expression.
Tetherin antagonism.
HEK293T cells seeded in 6-well plates were transfected with increasing amounts of a tetherin expression construct (0, 6.25, 25, 100, and 250 ng), together with 5 μg of a vpu- and nef-deficient proviral NL4-3 construct and 1 μg of a Vpu or Nef expression plasmid. Two days posttransfection, supernatants and cells were harvested. The infectious virus yield in the supernatant was determined via infection of TZM-bl reporter cells and measurement of the β-galactosidase activity 3 days later. The amount of p24 in the supernatant and the cells was determined by ELISA (82).
NF-κB activation.
Dual-luciferase assays with an NF-κB-dependent firefly luciferase and a Gaussia luciferase construct under the control of a minimal pTAL promoter for normalization were performed to determine the effect of Vpu on NF-κB activity as described previously (41), except that the experiments were performed in the presence and absence of FCS.
Western blotting.
The effect of RBF206 Vpu and Nef on viral particle release was determined by cotransfection of HEK293T cells with increasing amounts of a tetherin expression plasmid (0, 6.25, 25, 100, and 250 ng) and 5 μg of NL4-3 IRES-eGFP constructs expressing the RBF206 Vpu and/or Nef or neither. Two days posttransfection, viral particles in the supernatant were harvested by centrifugation at 14,000 rpm for 90 min. Cells and viral particles were lysed with Western blot lysis buffer (83) containing a protease inhibitor.
To verify expression of Vpu and Nef, HEK293T cells were transfected with 4 μg of a pCG construct expressing Vpu or Nef. To analyze degradation of tetherin, cells were additionally transfected with 1 μg of a pCG construct expressing this restriction factor, and cells were harvested and lysed with Western blot lysis buffer 24 h posttransfection. To verify expression of human and simian tetherin, HEK293T cells were cotransfected with 1 μg of a pCG construct expressing the respective tetherin and 4 μg of empty vector. Lysates were separated in 4 to 12% bis-Tris gels (Invitrogen) and transferred to polyvinylidene difluoride (PVDF) membranes. Blots were probed with antibodies against p24 and tetherin (to monitor release) or against AU1 (to monitor expression). The monoclonal anti-p24 antibody was purchased from Abcam (ab9071; dilution, 1:2,000), the anti-tetherin antibody from Abnova (B02P; dilution, 1:1,000) and the anti-AU1 antibody from Novus Biologicals (NB600-453; dilution, 1:10,000). For internal control, blots were incubated with antibodies specific for β-actin (Abcam; ab8227; dilution, 1:2,000) or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (G-9; sc-365062; dilution 1:500). Subsequently, membranes were incubated with anti-mouse or anti-rabbit IRDye Odyssey antibodies (dilution, 1:20,000) and proteins were detected using a Li-Cor Odyssey scanner.
Analysis of IFN-α sensitivity.
A total of 500,000 negatively selected CD4+ T cells, stimulated with CD3/CD28 beads at a ratio of 3:1, were either pretreated or not with 500 U/ml of IFN-α2 24 h prior to infection with viral stocks containing 500 ng of p24 (separate samples for each sample day). Virus production was monitored by p24 ELISA (82) every 2 days. Every second day, half of the medium was replaced with fresh medium containing 1,000 U/ml of IFN-α2, or not, to make up for loss in activity.
Sequence analyses.
Vpu amino acid sequences were aligned using multiple-sequence alignment with hierarchical clustering (http://multalin.toulouse.inra.fr/multalin/) followed by some manual editing to optimize the alignment.
Structure modeling.
Vpu structures of RBF206 were modeled using the SWISS-MODEL Workspace approach (84). As a template for model building, the structure of full-length Vpu determined by solid-state nuclear magnetic resonance (NMR) spectroscopy was used (85). Structure diagrams were rendered with PyMOL (http://www.pymol.org).
Statistical analysis.
Statistical calculations were performed with a two-tailed unpaired Student t test using Graph Pad Prism version 5.0. P values of <0.05 were considered significant. Correlations were calculated using the linear regression module.
Accession number(s).
The GenBank accession number of the HIV-1 RBF206 molecular clone is KY112585.
ACKNOWLEDGMENTS
We thank Daniela Krnavek, Martha Mayer, and Susanne Engelhart for excellent technical assistance. TZM-bl cells were obtained through the NIH AIDS Research and Reference Reagent Program.
This work was supported by grants from the National Institutes of Health (R01 AI 114266, R01 AI 120810, R01 AI 111789, R37 AI 050529, and P30 AI045008) and the Deutsche Forschungsgemeinschaft (DFG) and an ERC Advanced grant to F.K. K.M. was funded by the International Graduate School in Molecular Medicine (IGradU) of Ulm University.
REFERENCES
- 1.Keele BF, Van Heuverswyn F, Li Y, Bailes E, Takehisa J, Santiago ML, Bibollet-Ruche F, Chen Y, Wain LV, Liegeois F, Loul S, Ngole EM, Bienvenue Y, Delaporte E, Brookfield JFY, Sharp PM, Shaw GM, Peeters M, Hahn BH. 2006. Chimpanzee reservoirs of pandemic and nonpandemic HIV-1. Science 313:523–526. doi: 10.1126/science.1126531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.D'arc M, Ayouba A, Esteban A, Learn GH, Boué V, Liegeois F, Etienne L, Tagg N, Leendertz FH, Boesch C, Madinda NF, Robbins MM, Gray M, Cournil A, Ooms M, Letko M, Simon VA, Sharp PM, Hahn BH, Delaporte E, Mpoudi Ngole E, Peeters M. 2015. Origin of the HIV-1 group O epidemic in western lowland gorillas. Proc Natl Acad Sci U S A 112:E1343–E1352. doi: 10.1073/pnas.1502022112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kratovac Z, Virgen CA, Bibollet-Ruche F, Hahn BH, Bieniasz PD, Hatziioannou T. 2008. Primate lentivirus capsid sensitivity to TRIM5 proteins. J Virol 82:6772–6777. doi: 10.1128/JVI.00410-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stremlau M, Owens CM, Perron MJ. 2004. The cytoplasmic body component TRIM5α restricts HIV-1 infection in Old World monkeys. Nature 427. [DOI] [PubMed] [Google Scholar]
- 5.Sawyer SL, Wu LI, Emerman M, Malik HS. 2005. Positive selection of primate TRIM5 alpha identifies a critical species-specific retroviral restriction domain. Proc Natl Acad Sci U S A 102:2832–2837. doi: 10.1073/pnas.0409853102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stremlau M, Perron M, Welikala S, Sodroski J. 2005. Species-specific variation in the B30.2(SPRY) domain of TRIM5 alpha determines the potency of human immunodeficiency virus restriction. J Virol 79:3139–3145. doi: 10.1128/JVI.79.5.3139-3145.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gaddis NC, Sheehy AM, Ahmad KM, Swanson CM, Bishop KN, Beer BE, Marx PA, Gao F, Bibollet-Ruche F, Hahn BH, Malim MH. 2004. Further investigation of simian immunodeficiency virus Vif function in human cells. J Virol 78:12041–12046. doi: 10.1128/JVI.78.21.12041-12046.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sheehy AM, Gaddis NC, Choi JD, Malim MH. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- 9.Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, Malim MH. 2003. DNA deamination mediates innate immunity to retroviral infection. Cell 113:803–809. doi: 10.1016/S0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- 10.Mangeat B, Turelli P, Caron G, Friedli M. 2003. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 424:99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- 11.Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, Gao L. 2003. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 424:94–98. doi: 10.1038/nature01707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delaugerre C, De Oliveira F, Lascoux-Combe C, Plantier JC, Simon F. 2011. HIV-1 group N: travelling beyond Cameroon. Lancet 378:1894. doi: 10.1016/S0140-6736(11)61457-8. [DOI] [PubMed] [Google Scholar]
- 13.Mourez T, Simon F, Plantier J-C. 2013. Non-M variants of human immunodeficiency virus type 1. Clin Microbiol Rev 26:448–461. doi: 10.1128/CMR.00012-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Plantier J-CC, Leoz M, Dickerson JE, De Oliveira F, Cordonnier F, Lemée V, Damond F, Robertson DL, Simon F. 2009. A new human immunodeficiency virus derived from gorillas. Nat Med 15:871–872. doi: 10.1038/nm.2016. [DOI] [PubMed] [Google Scholar]
- 15.Vallari A, Holzmayer V, Harris B, Yamaguchi J, Ngansop C, Makamche F, Mbanya D, Kaptué L, Ndembi N, Gürtler L, Devare S, Brennan CA. 2011. Confirmation of putative HIV-1 group P in Cameroon. J Virol 85:1403–1407. doi: 10.1128/JVI.02005-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sauter D, Specht A, Kirchhoff F. 2010. Tetherin: holding on and letting go. Cell 141:392–398. doi: 10.1016/j.cell.2010.04.022. [DOI] [PubMed] [Google Scholar]
- 17.Sharp PM, Hahn BH. 2011. Origins of HIV and the AIDS epidemic. Cold Spring Harb Perspect Med 5:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neil SJD, Zang T, Bieniasz PD. 2008. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451:425–430. doi: 10.1038/nature06553. [DOI] [PubMed] [Google Scholar]
- 19.Van Damme N, Goff D, Katsura C, Jorgenson RL, Mitchell R, Johnson MC, Stephens EB, Guatelli J. 2008. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 3:245–252. doi: 10.1016/j.chom.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perez-Caballero D, Zang T, Ebrahimi A, Mcnatt MW, Gregory DA, Johnson MC, Bieniasz PD. 2009. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 139:499–511. doi: 10.1016/j.cell.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kupzig S, Korolchuk V, Rollason R, Sugden A, Wilde A, Banting G. 2003. Bst-2/HM1.24 is a raft-associated apical membrane protein with an unusual topology. Traffic 4:694–709. doi: 10.1034/j.1600-0854.2003.00129.x. [DOI] [PubMed] [Google Scholar]
- 22.Liberatore RA, Bieniasz PD. 2011. Tetherin is a key effector of the antiretroviral activity of type I interferon in vitro and in vivo. Proc Natl Acad Sci U S A 108:18097–18101. doi: 10.1073/pnas.1113694108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dave VP, Hajjar F, Dieng MM, Haddad É, Cohen ÉA. 2013. Efficient BST2 antagonism by Vpu is critical for early HIV-1 dissemination in humanized mice. Retrovirology 10:128. doi: 10.1186/1742-4690-10-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ikeda H, Nakaoka S, de Boer RJ, Morita S, Misawa N, Koyanagi Y. 2016. Quantifying the effect of Vpu on the promotion of HIV-1 replication in the humanized mouse model. Retrovirology 13:23. doi: 10.1186/s12977-016-0252-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kmiec D, Iyer SS, Stürzel CM, Sauter D, Hahn BH, Kirchhoff F. 2016. Vpu-mediated counteraction of tetherin is a major determinant of HIV-1 interferon resistance. mBio 7:e00934-16. doi: 10.1128/mBio.00934-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sauter D, Vogl M, Kirchhoff F. 2011. Ancient origin of a deletion in human BST2/tetherin that confers protection against viral zoonoses. Hum Mutat 32:1243–1245. doi: 10.1002/humu.21571. [DOI] [PubMed] [Google Scholar]
- 27.Sauter D, Schindler M, Specht A, Landford WN, Münch J, Kim KA, Votteler J, Schubert U, Bibollet-Ruche F, Keele BF, Takehisa J, Ogando Y, Ochsenbauer C, Kappes JC, Ayouba A, Peeters M, Learn GH, Shaw G, Sharp PM, Bieniasz P, Hahn BH, Hatziioannou T, Kirchhoff F. 2009. Tetherin-driven adaptation of Vpu and Nef function and the evolution of pandemic and nonpandemic HIV-1 strains. Cell Host Microbe 6:409–421. doi: 10.1016/j.chom.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jia B, Serra-Moreno R, Neidermyer W, Rahmberg A, Mackey J, Fofana Ben I, Johnson WE, Westmoreland S, Evans DT. 2009. Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by tetherin/BST2. PLoS Pathog 5:e1000429. doi: 10.1371/journal.ppat.1000429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang F, Wilson SJ, Landford WC, Virgen B, Gregory D, Johnson MC, Munch J, Kirchhoff F, Bieniasz PD, Hatziioannou T. 2009. Nef proteins from simian immunodeficiency viruses are tetherin antagonists. Cell Host Microbe 6:54–67. doi: 10.1016/j.chom.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sauter D, Unterweger D, Vogl M, Usmani SM, Heigele A, Kluge SF, Hermkes E, Moll M, Barker E, Peeters M, Learn GH, Bibollet-Ruche F, Fritz JV, Fackler OT, Hahn BH, Kirchhoff F. 2012. Human tetherin exerts strong selection pressure on the HIV-1 group N Vpu protein. PLoS Pathog 8:e1003093. doi: 10.1371/journal.ppat.1003093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang SJ, Lopez LA, Hauser H, Exline CM, Haworth KG, Cannon PM. 2010. Anti-tetherin activities in Vpu-expressing primate lentiviruses. Retrovirology 7:13. doi: 10.1186/1742-4690-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sauter D, Hué S, Petit SJ, Plantier J-C, Towers GJ, Kirchhoff F, Gupta RK. 2011. HIV-1 group P is unable to antagonize human tetherin by Vpu, Env or Nef. Retrovirology 8:103. doi: 10.1186/1742-4690-8-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petit SJ, Blondeau C, Towers GJ. 2011. Analysis of the human immunodeficiency virus type 1 M group Vpu domains involved in antagonizing tetherin. J Gen Virol 92:2937–2948. doi: 10.1099/vir.0.035931-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vigan R, Neil SJD. 2011. Separable determinants of subcellular localization and interaction account for the inability of group O HIV-1 Vpu to counteract tetherin. J Virol 85:9737–9748. doi: 10.1128/JVI.00479-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang SJ, Lopez LA, Exline CM, Haworth KG, Cannon PM. 2011. Lack of adaptation to human tetherin in HIV-1 group O and P. Retrovirology 8:78. doi: 10.1186/1742-4690-8-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kluge SF, Mack K, Iyer SS, Pujol M, Heigele A, Learn GH, Usmani SM, Sauter D, Joas S, Hotter D, Bibollet-Ruche F, Plenderleith LJ, Peeters M, Geyer M, Sharp PM, Fackler OT, Hahn BH, Kirchhoff F. 2014. Nef proteins of epidemic HIV-1 group O strains antagonize human tetherin. Cell Host Microbe 16:639–650. doi: 10.1016/j.chom.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berg MG, Yamaguchi J, Alessandri-Gradt E, Tell RW, Plantier J-C, Brennan CA. 2016. A pan-HIV strategy for complete genome sequencing. J Clin Microbiol 54:868–882. doi: 10.1128/JCM.02479-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peeters M, Liegeois F, Torimiro N, Bourgeois A, Mpoudi E, Vergne L, Saman E, Delaporte E, Saragosti S. 1999. Characterization of a highly replicative intergroup M/O human immunodeficiency virus type 1 recombinant isolated from a Cameroonian patient. J Virol 73:7368–7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ngoupo PA, Sadeuh-Mba SA, De Oliveira F, Ngono V, Ngono L, Tchendjou P, Penlap V, Mourez T, Njouom R, Kfutwah A, Plantier J-C. 2016. First evidence of transmission of an HIV-1 M/O intergroup recombinant virus. AIDS 30:1–8. [DOI] [PubMed] [Google Scholar]
- 40.Takehisa J, Zekeng L, Ido E, Yamaguchi-Kabata Y, Mboudjeka I, Harada Y, Miura T, Kaptu L, Hayami M. 1999. Human immunodeficiency virus type 1 intergroup (M/O) recombination in Cameroon. J Virol 73:6810–6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sauter D, Hotter D, Van Driessche B, Stürzel CM, Kluge SF, Wildum S, Yu H, Baumann B, Wirth T, Plantier JC, Leoz M, Hahn BH, Van Lint C, Kirchhoff F. 2015. Differential regulation of NF-kB-mediated proviral and antiviral host gene expression by primate lentiviral nef and vpu proteins. Cell Rep 10:586–600. doi: 10.1016/j.celrep.2014.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cocka LJ, Bates P. 2012. Identification of alternatively translated tetherin isoforms with differing antiviral and signaling activities. PLoS Pathog 8:e1002931. doi: 10.1371/journal.ppat.1002931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Galão RP, Le Tortorec A, Pickering S, Kueck T, Neil SJD. 2012. Innate sensing of HIV-1 assembly by tetherin induces N-kB-dependent proinflammatory responses. Cell Host Microbe 12:633–644. doi: 10.1016/j.chom.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tokarev A, Suarez M, Kwan W, Fitzpatrick K, Singh R, Guatelli J. 2013. Stimulation of NF-κB activity by the HIV restriction factor BST2. J Virol 87:2046–2057. doi: 10.1128/JVI.02272-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Akari H, Bour S, Kao S, Adachi A, Strebel K. 2001. The human immunodeficiency virus type 1 accessory protein Vpu induces apoptosis by suppressing the nuclear factor κB-dependent expression of antiapoptotic factors. J Exp Med 194:12991312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bour S, Perrin C, Akari H, Strebel K. 2001. The human immunodeficiency virus type 1 Vpu protein inhibits NF-κB activation by interfering with βTrCP-mediated degradation of IκB. J Biol Chem 276:15920–15928. doi: 10.1074/jbc.M010533200. [DOI] [PubMed] [Google Scholar]
- 47.Rollason R, Korolchuk V, Hamilton C, Schu P, Banting G. 2007. Clathrin-mediated endocytosis of a lipid-raft-associated protein is mediated through a dual tyrosine motif. J Cell Sci 120:3850–3858. doi: 10.1242/jcs.003343. [DOI] [PubMed] [Google Scholar]
- 48.Masuyama N, Kuronita T, Tanaka R, Muto T, Hirota Y, Takigawa A, Fujita H, Aso Y, Amano J, Tanaka Y. 2009. HM1.24 is internalized from lipid rafts by clathrin-mediated endocytosis through interaction with α-adaptin. J Biol Chem 284:15927–15941. doi: 10.1074/jbc.M109.005124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weinelt J, Neil SJD. 2014. Differential sensitivities of tetherin isoforms to counteraction by primate lentiviruses. J Virol 88:5845–5858. doi: 10.1128/JVI.03818-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McNatt MW, Zang T, Hatziioannou T, Bartlett M, Fofana Ben I, Johnson WE, Neil SJD, Bieniasz PD. 2009. Species-specific activity of HIV-1 Vpu and positive selection of tetherin transmembrane domain variants. PLoS Pathog 5:e1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kobayashi T, Ode H, Yoshida T, Sato K, Gee P, Yamamoto SP, Ebina H, Strebel K, Sato H, Koyanagi Y. 2011. Identification of amino acids in the human tetherin transmembrane domain responsible for HIV-1 Vpu interaction and susceptibility. J Virol 85:932–945. doi: 10.1128/JVI.01668-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McNatt MW, Zang T, Bieniasz PD. 2013. Vpu Binds Directly to Tetherin and Displaces It from Nascent Virions. PLoS Pathog 9:e1003299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kluge SF, Sauter D, Vogl M, Peeters M, Li Y, Bibollet-Ruche F, Hahn BH, Kirchhoff F. 2013. The transmembrane domain of HIV-1 Vpu is sufficient to confer anti-tetherin activity to SIVcpz and SIVgor Vpu proteins: cytoplasmic determinants of Vpu function. Retrovirology 10:32. doi: 10.1186/1742-4690-10-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miyagi E, Andrew AJ, Kao S, Strebel K. 2009. Vpu enhances HIV-1 virus release in the absence of Bst-2 cell surface down-modulation and intracellular depletion. Proc Natl Acad Sci U S A 106:2868–2873. doi: 10.1073/pnas.0813223106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dubé M, Bego MG, Paquay C, Cohen ÉA. 2010. Modulation of HIV-1-host interaction: role of the Vpu accessory protein. Retrovirology 7:114. doi: 10.1186/1742-4690-7-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goffinet C, Homann S, Ambiel I, Tibroni N, Rupp D, Keppler OT, Fackler OT. 2010. Antagonism of CD317 restriction of human immunodeficiency virus type 1 (HIV-1) particle release and depletion of CD317 are separable activities of HIV-1 Vpu. J Virol 84:4089–4094. doi: 10.1128/JVI.01549-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tervo H-M, Homann S, Ambiel I, Fritz JV, Fackler OT, Keppler OT. 2011. β-TrCP is dispensable for Vpu's ability to overcome the CD317/tetherin-imposed restriction to HIV-1 release. Retrovirology 8:9. doi: 10.1186/1742-4690-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Margottin F, Bour SP, Durand H, Selig L, Benichou S, Richard V, Thomas D, Strebel K, Benarous R. 1998. A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol Cell 1:565–574. doi: 10.1016/S1097-2765(00)80056-8. [DOI] [PubMed] [Google Scholar]
- 59.Soucy TA, Smith PG, Rolfe M. 2009. Targeting NEDD8-activated cullin-RING ligases for the treatment of cancer. Clin Cancer Res 15:3912–3916. doi: 10.1158/1078-0432.CCR-09-0343. [DOI] [PubMed] [Google Scholar]
- 60.Ramirez PW, Beatriz A, Silva D, Szaniawski M, Barker E, Bosque A. 2015. HIV-1 Vpu utilizes both cullin-RING ligase (CRL) dependent and independent mechanisms to downmodulate host proteins. Retrovirology 12:65. doi: 10.1186/s12977-015-0192-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stumptner-Cuvelette P, Morchoisne S, Dugast M, Le Gall S, Raposo G, Schwartz O, Benaroch P. 2001. HIV-1 Nef impairs MHC class II antigen presentation and surface expression. Proc Natl Acad Sci U S A 98:12144–12149. doi: 10.1073/pnas.221256498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schindler M, Würfl S, Benaroch P, Greenough TC, Daniels R, Easterbrook P, Brenner M, Münch J, Kirchhoff F. 2003. Down-modulation of mature major histocompatibility complex class II and up-regulation of invariant chain cell surface expression are well-conserved functions of human and simian immunodeficiency virus nef alleles. J Virol 77:10548–10556. doi: 10.1128/JVI.77.19.10548-10556.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hussain A, Wesley C, Khalid M, Chaudhry A, Jameel S. 2008. Human immunodeficiency virus type 1 Vpu protein interacts with CD74 and modulates major histocompatibility complex class II presentation. J Virol 82:893–902. doi: 10.1128/JVI.01373-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Götz N, Sauter D, Usmani SM, Fritz JV, Goffinet C, Heigele A, Geyer M, Bibollet-Ruche F, Learn GH, Fackler OT, Hahn BH, Kirchhoff F. 2012. Reacquisition of nef-mediated tetherin antagonism in a single in vivo passage of HIV-1 through its original chimpanzee host. Cell Host Microbe 12:373–380. doi: 10.1016/j.chom.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Goffinet C, Allespach I, Homann S, Tervo H, Habermann A, Rupp D, Oberbremer L, Kern C, Tibroni N, Welsch S, Krijnse-locker J, Fackler OT, Keppler OT, Banting G, Kra H. 2009. HIV-1 antagonism of CD317 is species specific and involves Vpu-mediated proteasomal degradation of the restriction factor. Cell Host Microbe 5:285–297. doi: 10.1016/j.chom.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 66.Bego MG, Côté É, Aschman N, Mercier J, Weissenhorn W, Cohen ÉA. 2015. Vpu exploits the cross-talk between BST2 and the ILT7 receptor to suppress anti-HIV-1 responses by plasmacytoid dendritic cells. PLoS Pathog 11:e1005024. doi: 10.1371/journal.ppat.1005024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bego MG, Cong L, Mack K, Kirchhoff F. 31 August 2016. Differential control of BST2 restriction and plasmacytoid dendritic cell antiviral response by antagonists encoded by HIV-1 group M and O strains. J Virol doi: 10.1128/JVI.01131-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Arias JF, Colomer-Lluch M, von Bredow B, Greene JM, MacDonald J, O'Connor DH, Serra-Moreno R, Evans DT. 21 September 2016. Tetherin antagonism by HIV-1 group M Nef proteins. J Virol doi: 10.1128/JVI.01465-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Aiken C, Konner J, Landau NR, Lenburg ME, Trono D. 1994. Nef induces CD4 endocytosis: requirement for a critical dileucine motif in the membrane-proximal CD4 cytoplasmic domain. Cell 76:853–864. doi: 10.1016/0092-8674(94)90360-3. [DOI] [PubMed] [Google Scholar]
- 70.Rhee SS, Marsh JW. 1994. Human immunodeficiency virus type 1 Nef-induced down-modulation of CD4 is due to rapid internalization and degradation of surface CD4. J Virol 68:5156–5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Willey RL, Maldarelli F, Martin MA, Strebel K. 1992. Human immunodeficiency virus type 1 Vpu protein induces rapid degradation of CD4. J Virol 66:7193–7200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kasper MR, Collins KL. 2003. Nef-mediated disruption of HLA-A2 transport to the cell surface in T cells. J Virol 77:3041–3049. doi: 10.1128/JVI.77.5.3041-3049.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kasper MR, Roeth JF, Williams M, Filzen TM, Fleis RI, Collins KL. 2005. HIV-1 Nef disrupts antigen presentation early in the secretory pathway. J Biol Chem 280:12840–12848. doi: 10.1074/jbc.M413538200. [DOI] [PubMed] [Google Scholar]
- 74.Schindler M, Münch J, Kutsch O, Li H, Santiago ML, Bibollet-Ruche F, Müller-Trutwin MC, Novembre FJ, Peeters M, Courgnaud V, Bailes E, Roques P, Sodora DL, Silvestri G, Sharp PM, Hahn BH, Kirchhoff F. 2006. Nef-mediated suppression of T cell activation was lost in a lentiviral lineage that gave rise to HIV-1. Cell 125:1055–1067. doi: 10.1016/j.cell.2006.04.033. [DOI] [PubMed] [Google Scholar]
- 75.Larkin MA, Blackshields G, Brown NP, Chenna R, Mcgettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 76.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 77.Darriba D, Taboada GL, Doallo R, Posada D. 2012. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC. 1999. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol 73:152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fouchier RAM, Meyer BE, Simon JHM, Fischer U, Malim MH. 1997. HIV-1 infection of non-dividing cells: evidence that the amino-terminal basic region of the viral matrix protein is important for Gag processing but not for post-entry nuclear import. EMBO J 16:4531–4539. doi: 10.1093/emboj/16.15.4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Münch J, Rajan D, Schindler M, Specht A, Ru E, Novembre FJ, Nerrienet E, Mu MC, Peeters M, Hahn BH, Kirchhoff F. 2007. Nef-mediated enhancement of virion infectivity and stimulation of viral replication are fundamental properties of primate lentiviruses. J Virol 81:13852–13864. doi: 10.1128/JVI.00904-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D. 1998. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J Virol 72:2855–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Langer SM, Hopfensperger K, Iyer SS, Kreider EF, Learn H, Lee L, Hahn BH, Sauter D. 2015. A naturally occurring rev1-vpu fusion gene does not confer a fitness advantage to HIV-1. PLoS One 10:e0142118. doi: 10.1371/journal.pone.0142118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Krapp C, Hotter D, Gawanbacht A, McLaren PJ, Kluge SF, Stürzel CM, Mack K, Reith E, Engelhart S, Ciuffi A, Hornung V, Sauter D, Telenti A, Kirchhoff F. 2016. Guanylate binding protein (GBP) 5 is an interferon-inducible inhibitor of HIV-1 infectivity. Cell Host Microbe 19:504–514. doi: 10.1016/j.chom.2016.02.019. [DOI] [PubMed] [Google Scholar]
- 84.Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. 2009. Protein structure homology modelling using SWISS-MODEL workspace. Nat Protoc 4:1–13. [DOI] [PubMed] [Google Scholar]
- 85.Zhang H, Lin EC, Das BB, Tian Y, Opella SJ. 2015. Structural determination of virus protein U from HIV-1 by NMR in membrane environments. Biochim Biophys Acta 1848:3007–3018. doi: 10.1016/j.bbamem.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Skasko M, Wang Y, Tian Y, Tokarev A, Munguia J, Ruiz A, Stephens EB, Opella SJ, Guatelli J. 2012. HIV-1 Vpu protein antagonizes innate restriction factor BST-2 via lipid-embedded helix-helix interactions. J Biol Chem 287:58–67. doi: 10.1074/jbc.M111.296772. [DOI] [PMC free article] [PubMed] [Google Scholar]