

ABSTRACT
Human adenoviruses (Ads) generally cause mild self-limiting infections but can lead to serious disease and even be fatal in high-risk individuals, underscoring the importance of understanding how the virus counteracts host defense mechanisms. This study had two goals. First, we wished to determine the molecular basis of cholesterol homeostatic responses induced by the early region 3 membrane protein RIDα via its direct interaction with the sterol-binding protein ORP1L, a member of the evolutionarily conserved family of oxysterol-binding protein (OSBP)-related proteins (ORPs). Second, we wished to determine how this interaction regulates innate immunity to adenovirus. ORP1L is known to form highly dynamic contacts with endoplasmic reticulum-resident VAP proteins that regulate late endosome function under regulation of Rab7-GTP. Our studies have demonstrated that ORP1L-VAP complexes also support transport of LDL-derived cholesterol from endosomes to the endoplasmic reticulum, where it was converted to cholesteryl esters stored in lipid droplets when ORP1L was bound to RIDα. The virally induced mechanism counteracted defects in the predominant cholesterol transport pathway regulated by the late endosomal membrane protein Niemann-Pick disease type C protein 1 (NPC1) arising during early stages of viral infection. However, unlike NPC1, RIDα did not reconstitute transport to endoplasmic reticulum pools that regulate SREBP transcription factors. RIDα-induced lipid trafficking also attenuated proinflammatory signaling by Toll-like receptor 4, which has a central role in Ad pathogenesis and is known to be tightly regulated by cholesterol-rich “lipid rafts.” Collectively, these data show that RIDα utilizes ORP1L in a way that is distinct from its normal function in uninfected cells to fine-tune lipid raft cholesterol that regulates innate immunity to adenovirus in endosomes.
IMPORTANCE Early region 3 proteins encoded by human adenoviruses that attenuate immune-mediated pathology have been a particularly rich source of information regarding intracellular protein trafficking. Our studies with the early region 3-encoded RIDα protein also provided fundamental new information regarding mechanisms of nonvesicular lipid transport and the flow of molecular information at membrane contacts between different organelles. We describe a new pathway that delivers cholesterol from endosomes to the endoplasmic reticulum, where it is esterified and stored in lipid droplets. Although lipid droplets are attracting renewed interest from the standpoint of normal physiology and human diseases, including those resulting from viral infections, experimental model systems for evaluating how and why they accumulate are still limited. Our studies also revealed an intriguing relationship between lipid droplets and innate immunity that may represent a new paradigm for viruses utilizing these organelles.
KEYWORDS: adenoviruses, cholesterol, endocytic pathway, endoplasmic reticulum, lipid droplet
INTRODUCTION
Adenovirus (Ad) is the one of the most common causes of respiratory tract infections in adults and children (1). Although Ad infections are usually self-limiting, these viral agents can also cause serious respiratory disease in both high-risk and healthy individuals, especially in young adults such as military recruits living under crowded conditions (2). Respiratory viruses, including Ad, are also associated with a significant risk of morbidity and mortality for hematopoietic cell transplant recipients, particularly in children (3, 4). Ads typically infect epithelial cells lining the lungs or other enteric organs, where virus uptake induces inflammatory cytokine and cytotoxic T-cell responses that protect the host and represent a major barrier to using Ads as therapeutic interventions in humans (5). Ad counteracts these antiviral effects by virtue of several viral proteins produced prior to the onset of viral replication (6, 7). Early region 3 (E3), which is dispensable for viral replication in cell culture, has a prominent role in immune evasion and also prevents excessive innate immunity, leading to tissue injury and Ad disease (8–10). Several E3 proteins shield infected cells from elimination in the host by coopting intracellular protein trafficking pathways that downregulate receptors for the innate immune system (11, 12). Other E3 proteins target downstream signal transduction pathways without directly affecting cell surface receptor expression by unknown mechanisms (13). Further mapping of pathway components intersecting with E3 proteins will provide important information on potential new targets for regulating innate immunity in populations at high risk for Ad disease and in individuals receiving engineered Ad vectors.
Our studies were the first to link an Ad-E3 protein to lipid trafficking (14–16). The E3 membrane protein RIDα regulates the transport of LDL-cholesterol from endosomes to the endoplasmic reticulum (ER), where it is converted to cholesteryl esters (CEs) by acyl-coenzyme A:cholesterol acyltransferase (ACAT) and stored in lipid droplets (LDs) that bud off the ER (16–18). This lipid transport pathway is typically regulated in the host by two proteins, called NPC1 (localized to limiting membranes) and NPC2 (present in the lumen), of late endosomes and lysosomes (19, 20). The predominant model proposes that NPC2 transfers LDL-cholesterol from intraluminal vesicles to NPC1 followed by export to multiple intracellular destinations, including the ER, by unknown mechanisms (21). NPC1/NPC2 mutations cause LDL-cholesterol to accumulate in abnormal lysosomal storage organelles (LSOs), resulting in false-compensatory activation of de novo cholesterol synthesis by SREBP transcription factors that are ordinarily downregulated by LDL-cholesterol trafficking to regulatory sterol pools in the ER (22, 23). The outcome of this self-perpetuating process is a massive intracellular deposition of cholesterol throughout the cytoplasm, which is a hallmark of Niemann-Pick disease type C (NPC) (23). Cells with NPC1 and NPC2 mutations exhibit a significant reduction in LD accumulation because excess cholesterol is not esterified by ACAT in the ER (24). Cholesterol transfer is also inhibited by acute gene silencing of ORP5, a member of the evolutionarily conserved family of oxysterol-binding protein (OSBP)-related proteins (ORPs) tethered on ER membranes, and the endocytic regulatory protein HRS (hepatocyte growth factor-regulated tyrosine kinase substrate), which initiates protein sorting in early endosomes (25–28). Although ORP5 forms a molecular complex with NPC1, its precise role in cholesterol trafficking remains uncertain since ORP5 regulates lipid exchange between the plasma membrane and the ER (25, 29). HRS regulates cholesterol transport upstream of NPC1 and NPC2, perhaps by organizing membrane subdomains required for cholesterol transport or initiating formation of stable membrane contacts where NPC1-ORP5 protein complexes eventually assemble in late endosomes (26, 30).
The original insight into the lipid trafficking properties of RIDα arose from studies performed with NPC1-deficient cell models, including patient fibroblasts, where expression of the viral protein was sufficient to alleviate LSO formation by diverting excess free cholesterol to LDs (14, 16). Acute gene silencing studies then led to identification of ORP1L as an essential host factor supporting RIDα-induced lipid trafficking in NPC1-deficient cells. Similarly to other members of the ORP protein family, ORP1L has a lipid-binding domain (ORD, for OSBP-related domain) that binds sterol and the phosphoinositide PI(4)P (see Fig. 2B) (28, 31). ORP1L also has a pleckstrin homology (PH) domain targeting it to late endosomes and an FFAT motif that interacts with ER vesicle-associated membrane protein-associated proteins (VAPs) (28, 32, 33). ORP1L has been analyzed in detail regarding its ability to regulate vesicle motility as part of a tripartite complex with the small GTPase Rab7 and a Rab7 effector called RILP that couples late endosomes to minus-end-directed dynein-dynactin microtubule motors (31, 34). The predominant model is that sterol sensing residues in the lid located at the entrance to the ORP1L-ORD control the affinity of the ORP1L FFAT motif for integral ER membrane VAP proteins (34). ORP1L and VAP form protein contacts that interfere with the interaction between RILP and dynein-dynactin motors when there is relatively little cholesterol in endosomal membranes. Rising levels of cholesterol sequester ORD-lids on endosomal membranes, initiating a different ORP1L conformation that disrupts ORP1L-VAP protein contacts, leading to persistent dynein motor activity. The role of ORP1L as a sterol sensor whose interaction with ER membranes is inhibited by increasing levels of endosomal cholesterol is seemingly at odds with its putative role in sterol transport downstream of RIDα. The ability of RIDα to reverse cholesterol storage defects in NPC1-deficient cells also raises questions regarding the status of the canonical cholesterol trafficking pathway, as well as of the innate immune signaling system pathways that are critically dependent on intracellular cholesterol flux, in acutely infected cells (35). The current study therefore had three goals. First, we wished to determine the effect of an acute infection on NPC1-dependent sterol transport. Second, we wished to understand how the adenovirus RIDα protein utilizes ORP1L in a way that is distinct from its normal function in uninfected cells. Third, we wished to determine how RIDα affects activation of a prototypical proinflammatory NK-κB signaling pathway by innate immune Toll-like receptor 4 (TLR4), which has a central role in Ad pathogenesis and is tightly regulated by cholesterol-rich “lipid rafts” (13, 35–37).
FIG 2.

Molecular requirements for RIDα-induced cholesterol trafficking. (A) Equal protein aliquots immunoblotted with antibodies to specific siRNA targets listed in the figure or actin following treatment with noncoding (NC) or target-specific siRNAs. (B) CE quantification (in nanograms of cholesterol per milligram of protein) in shNPC1-RIDα cells treated with target-specific siRNAs. Data are normalized to NC siRNA-treated cells (means ± s.e.m.; n = 3; **, P < 0.005). (C and D) Representative confocal images of all hepatocyte-derived cells following treatment with NC siRNA (C) and shNPC1-RIDα cells treated with the target-specific siRNAs listed in the figure (D). Cells were stained with Bodipy 493/503 (green) and DAPI (blue) and FLAG epitope antibody to detect FLAG-RIDα (red). (E and F) Representative confocal images of all hepatocyte-derived cells following treatment with NC siRNA (E) or of shNPC1-RIDα cells treated with target-specific siRNAs. Cells were stained with LAMP1 antibody (red), filipin (blue), and FLAG-RIDα antibody (green). Boxed areas indicate regions are magnified at ×2 to visualize individual and merged channels to the right of each image. (C to F) All size bars, 5 μm.
RESULTS
RIDα compensated for aberrant NPC1 function in acutely infected cells.
The effect of acute Ad infection on the morphology of NPC1-positive late endosomes and lysosomes in adenocarcinomic human alveolar basal epithelial A549 cells, which represent a standard tissue culture model for Ad studies (38–41), was evaluated. Cells were infected with the group C Ad2 lung pathogen using either wild-type (WT) virus or a mutant virus with an internal deletion in the E3 gene encoding RIDα (“RIDα-null”) in the presence of cytosine arabinoside (ara-C) to inhibit viral DNA replication (42, 43). Both viruses elicited robust expression of the early region 1A protein encoded by one of the first viral genes to be transcribed during an acute Ad infection (Fig. 1A, top panels) (44). Consistent with the genotype of the mutant virus, RIDα was produced only in cells infected with wild-type Ad2 (Fig. 1A, bottom panels). Mock-treated cells and cells infected with wild-type or RIDα-null viruses displayed similar levels of total NPC1 protein (Fig. 1B) (15, 45). However, confocal imaging revealed marked differences in the phenotype of NPC1-positive late endosomes and lysosomes that depended on the virus used to infect the cells. NPC1 was localized to uniformly sized LAMP1-positive late endosomes/lysosomes dispersed throughout the cytosol in cells infected with wild-type Ad2 similarly to the results seen with mock-treated cells (Fig. 1C). In contrast, NPC1-positive LAMP1 vesicles were enlarged and filled with free cholesterol based on the results of staining with the fluorescent antibiotic filipin in cells infected with RIDα-null virus (Fig. 1C). This phenotype is characteristic of the aberrant LSOs that are a hallmark of NPC1-deficient cells (22). We next assessed whether this aberrant morphology was associated with defects in normal NPC1 function by monitoring cholesterol esterification in infected cells stained with the lipophilic dye Bodipy 493/503, which is used extensively for fluorescent detection of LDs with stored CEs (46). Similarly to many cancer cells, mock-treated A549 cells displayed abundant LDs under normal tissue culture conditions (Fig. 1D). Although cells infected with wild-type Ad2 had a similar phenotype, LD density was significantly reduced in cells infected with the RIDα-null virus (Fig. 1D). Ad infection studies were also carried out in a stable human hepatocyte cell line with reduced NPC1 expression (shNPC1 cells) to determine whether Ad2 also compensated for loss of NPC1 function (Fig. 1E). Although Ads do not normally encounter the liver during a natural infection, the liver is a major site of infection in Ad-based gene therapy recipients and immunocompromised patients (47, 48). Wild-type Ad2 but not the RIDα-null virus was associated with LD accumulation in acutely infected shNPC1 cells (Fig. 1F). We next compared the effects of various sterol conditions on the ER-resident SREBP-1 transcription factor which is normally processed and transported to the nucleus under low-cholesterol conditions in parent hepatocytes versus shNPC1 cell lines without and with the RIDα protein (Fig. 1G). Cells were treated with the cysteine protease inhibitor ALLN for 4 h prior to cell harvest to block the rapid catabolism of nuclear SREBP-1 (49). The three cell lines exhibited similar levels of precursor SREBP-1 under sterol-depleted and LDL-loaded conditions in cytosolic cell fractions (Fig. 1G, top panel). Sterol deprivation also induced SREBP-1 trafficking and processing, giving rise to the nuclear SREBP-1 transcription factor in all three cell lines (Fig. 1G, middle panel). NPC1 protein levels are reduced by approximately 70% in the knockdown cells used in this study (50). While this amount of NPC1 expression was sufficient to substantially reduce SREBP-1 processing following LDL loading similarly to the results seen in parent cells with physiological NPC1 protein levels, SREBP-1 processing was not inhibited in LDL-loaded NPC1 knockdown cells expressing the viral protein (Fig. 1G, middle panel). Nuclear fractions were reprobed with an antibody to the nuclear envelope marker lamin B1 to verify equal protein loading (Fig. 1G, bottom panel). Collectively, these data suggested that Ad infection disrupted NPC1 function in late endosomes/lysosomes prior to the onset of viral replication. This aberrant phenotype was masked in cells infected with wild-type Ad2 because RIDα activated an alternative mechanism for selectively feeding into cholesterol pools and/or transport pathways that regulate ACAT but not SREBP within the ER independently of NPC1 (51).
FIG 1.

RIDα compensated for loss of NPC1 function following an acute infection. (A) A549 cells infected with Ad2 (wild type or RIDα-null) metabolically labeled from 1 to 3 h postinfection and harvested 8 h postinfection to recover E1A and RIDα immune complexes for detection by fluorography. IP, immunoprecipitation. (B) Equal protein aliquots from mock-treated or Ad-infected cells immunoblotted (IB) with antibodies to NPC1 (top) and actin (bottom) for protein loading control. (C) Representative confocal images from A549 cells mock treated or Ad infected for 18 h and stained with antibodies to LAMP1 (red) and NPC1 (green) and filipin (blue) to visualize free cholesterol. Boxed areas are magnified 2× to visualize individual channels beneath each image. Nu, nucleus. (D) A549 cells mock treated or Ad infected and stained with Bodipy 493/503 (green) and DAPI (blue) to detect LDs and nuclei, respectively. (E) Equal protein aliquots from NPC1-positive hepatocytes (shControl) and NPC1 knockdown hepatocytes without or with stable expression of FLAG-tagged RIDα (shNPC1 and shNPC1-RIDα, respectively) immunoblotted with antibodies to NPC1 (top) and FLAG (middle) and actin for protein loading control (bottom). (F) Mock-treated or Ad-infected shNPC1 cells stained with Bodipy 493/503 (green) and DAPI (blue). (C, D, and F) All size bars, 5 μm. (G) Equal protein aliquots of cytosol and nuclear fractions from sterol-depleted (−) and LDL-loaded (+) cells listed in the figure probed with SREBP-1 antibody to detect precursor (P) and nuclear (N) forms of the molecule. Nuclear fractions were reprobed for lamin B1 to verify equal protein loading. Representative immunoblots from 2 independent experiments are shown.
Molecular requirements for RIDα-regulated cholesterol homeostasis.
The role of ORP1L in the RIDα-induced LD pathway was originally discovered by expressing the viral protein in NPC1-deficent cells (16). This model mimics the LSO phenotype induced in cells infected with a RIDα-null virus and also allows pathway characterization by small interfering RNA (siRNA) gene silencing independently of other viral proteins. Using a similar approach, analysis was performed with several additional molecules that are known to regulate communication between endosomes and ER: (i) resident ER membrane VAP proteins that interact with multiple organelle-specific proteins, including ORP1L, which bind both isoforms (VAP-A and VAP-B) expressed in mammalian cells (34, 52); (ii) ORP5, which partners with NPC1 at endosome-ER membrane contact sites (25); and (iii) HRS, which is implicated in cholesterol transport upstream of NPC1-NPC2 (26) (Fig. 2A). CE storage in LDs was evaluated two ways. First, total CE levels were quantified following sterol depletion/loading. Similarly to ORP1L, both VAP-A gene silencing and HRS gene silencing led to a significant reduction in total CE levels in shNPC1-RIDα cells compared to treatment with a noncoding (NC) control siRNA (Fig. 2B). In contrast, ORP5 gene silencing did not interfere with the ability of RIDα to enhance CE levels in shNPC1 cells (Fig. 2B). Second, siRNA-treated cells were examined for LD accumulation by confocal imaging after sterol-loaded cells were stained with Bodipy 493/503. NC siRNA treatment did not have a significant impact on LD accumulation in stable human control (shControl) cells or shNPC1 cells expressing the viral RIDα protein (Fig. 2C). Furthermore, the targeted siRNA treatments did not significantly reduce LD accumulation in shControl cells with an intact NPC1 pathway (Fig. 3). Consistent with quantitative CE data, however, ORP1L, VAP-A, and HRS gene silencing led to a marked reduction in LD accumulation, in contrast to ORP5 gene silencing, which did not have a discernible effect in shNPC1-RIDα (Fig. 2D). The outcome of gene silencing on the ability of RIDα to attenuate LSO formation in shNPC1 cells was also evaluated by confocal microscopy. These phenotypes were unaffected by treatment with NC siRNA (Fig. 2E). ORP5 and HRS were both originally linked to the NPC1 cholesterol transport pathway because their targeted disruption led to LSO formation in HeLa cells with normal NPC1 expression (25, 26). Similar results were obtained in NPC1-expressing shControl hepatocytes, in contrast to targeted ORP1L and VAP-A gene silencing, which did not produce a noticeable phenotype (Fig. 3). In contrast, reduced expression of VAP-A as well as of HRS blocked the ability of RIDα to prevent cholesterol accumulation in LSOs in shNPC1 cells similarly to ORP1L depletion, while ORP5 depletion that did not have a discernible effect (Fig. 2F). Collectively, these results suggested that the RIDα/ORP1L pathway induced LDL-cholesterol transport to ACAT in the ER independently of ORP5, which facilitates the canonical NPC1 pathway (25). In contrast, HRS was required for the RIDα/ORP1L pathway, similarly to its published effect in the NPC1-mediated cholesterol transport from endosomes to the ER (26). While these data are also consistent with a role for ORP1L-VAP complexes in RIDα-mediated cholesterol transport, they do not exclude roles for other FFAT-containing proteins implicated in nonvesicular lipid trafficking that also bind ER VAP proteins (53).
FIG 3.
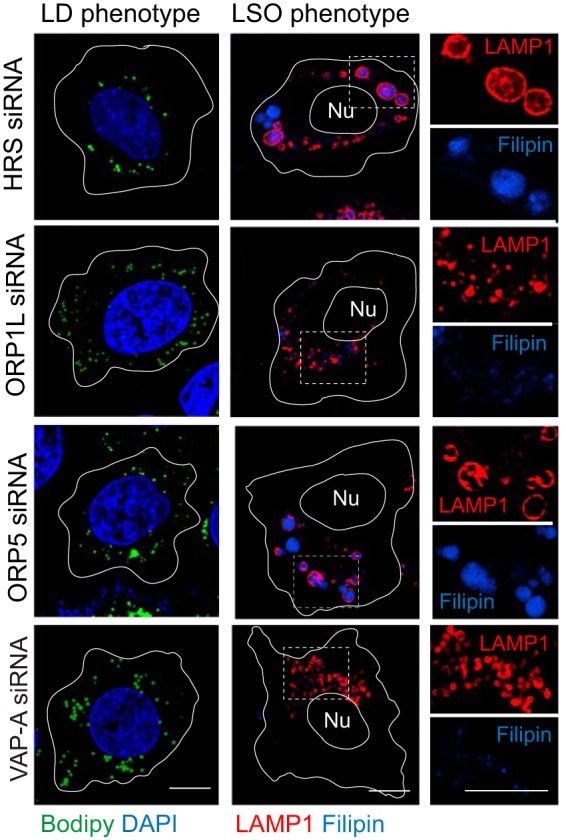
Gene silencing phenotypes in NPC1-containing shControl hepatocytes. Representative confocal images from cells treated with siRNAs listed in the figure were stained with Bodipy 493/503 (green) and DAPI (blue) to detect LDs and nuclei, respectively (left) or a LAMP1 antibody and filipin to image LSOs (right).
ORP1L molecular requirements for RIDα-induced cholesterol trafficking.
These experiments had two objectives. The first objective was to determine whether RIDα and ORP1L formed molecular complexes in cells. shNPC1-RIDα cells were transfected with a green fluorescent protein (GFP)-tagged ORP1L construct, and GFP immune complexes were probed with a FLAG-epitope antibody to detect coimmunoprecipitated RIDα protein, which is known to form a disulfide-linked dimer (Fig. 4A, left panel) (54). Interestingly, ORP1L was present in a complex with a population of RIDα dimers that was unusually resistant to reduction with dithiothreitol (Fig. 4A, right panel). These data suggested that the RIDα-ORP1L interaction could be regulated by the oxidizing potential of endosomes, which is known to limit the intracellular reduction of disulfide bonds (55). The second objective was to assess roles for two ORP1L structural features that are not directly involved in RIDα binding: the ORP1L FFAT motif enabling a direct interaction between ORP1L and ER VAP proteins and the sterol-sensing lid at the entrance to the ORP1L-ORD (Fig. 4B). These mutations did not have a significant impact on RIDα binding in an established pulldown assay with a glutathione S-transferase (GST) fusion protein encoding ORP1L binding sequences in the RIDα C terminus (RIDα-CT) (Fig. 4C) (56). The roles of FFAT and ORD-lid domains were evaluated in shNPC1-RIDα cells treated with a 3′-untranscribed-region (3′-UTR)-specific ORP1L siRNA and then reconstituted with siRNA-resistant GFP-ORP1L constructs encoding the wild-type protein or mutant variants with (i) a D478A substitution in the FFAT motif that eliminates VAP binding; (ii) a 4-amino-acid deletion (Δ560–563) that attenuates the sterol sensing capacity of the ORD-lid downstream of Rab7; and (iii) a dual D478A/Δ560–563 substitution (Fig. 4D) (34, 57). Transport of LDL-cholesterol to the ER for reesterification was monitored by measuring [3H]cholesteryl oleate synthesis following endocytosis of [3H]cholesteryl palmitate-labeled LDL (Fig. 4E). Similarly to the shNPC1 cell results, [3H]cholesteryl oleate synthesis was significantly reduced in shNPC1-RIDα cells treated with the 3′-UTR-specific ORP1L siRNA compared to untreated cells (Fig. 4F, left panel). ORP1L-depleted shNPC1-RIDα cells reconstituted with ORP1L (D478A) or the double ORP1L (D478A/Δ560–563) mutant were both associated with significantly reduced levels of [3H]cholesteryl oleate synthesis compared to cells reconstituted with wild-type ORP1L or ORP1L (Δ560-563) (Fig. 4F, right panel). Consistent with results obtained in the cholesterol trafficking assay, ORP1L (D478A) and ORP1L (D478A/Δ560-563) were not effective at reconstituting total CE levels in ORP1L-depleted cells compared to WT-ORP1L and ORP1L (Δ560-563) (Fig. 5A). Cells expressing GFP-ORP1L constructs were also stained with LipidTOX neutral lipid stain to image LDs with stored CEs. LD accumulation was clearly evident in shControl cells expressing WT-ORP1L or ORP1L (D478A) but not with ORP1L (Δ560-563), supporting publications in the literature reporting that sterol sensing by the ORP1L ORD-lid promotes cholesterol export by NPC1 (Fig. 5B, top panels) (58, 59). A majority of shNPC1 cells, on the other hand, lacked significant LD accumulation following treatment with any of the ORP1L constructs (Fig. 5B, middle panels). Consistent with the quantitative CE results, there was also marked LD accumulation in sterol-loaded shNPC1-RIDα cells reconstituted with WT-ORP1L or ORP1L (Δ560-563) but not in those reconstituted with ORP1L (D478A) (Fig. 5B, bottom panels).
FIG 4.

Molecular requirements for ORP1L-dependent cholesterol transport in RIDα pathway. (A) Left panel: equal protein aliquots from shNPC1-RIDα cells resolved under reducing (+DTT [dithiothreitol]) or nonreducing (−DTT) conditions for immunological detection of FLAG-RIDα. Right panel: shNPC-RIDα cells mock treated or transfected with GFP-ORP1L expression plasmid. GFP-tagged ORP1L proteins were affinity purified on anti-GFP-Sepharose beads, and immobilized proteins were resolved by SDS-PAGE for immunological detection of FLAG-tagged RIDα. Blots were reprobed for GFP to verify GFP-ORP1L affinity purification. (B) ORP1L protein schematic showing amino-terminal ankyrin repeats (blue), pleckstrin homology domain (PH; brown), FFAT motif (green), OSBP-related domain (ORD; orange) with bound sterol ligand (yellow), and cholesterol-binding lid encompassing amino acid residues 560-ELSK-563 (red). Inactivating mutations in FFAT and cholesterol-binding lid are italicized in the legend. RIDα binds ORD amino acid residues 755 to 923 in contrast to the amino terminal ankyrin repeats recognized by Rab7-GTP (15, 112). (C) Lysates from CHO cells transfected with GFP-tagged ORP1L constructs incubated with GST or GST fused to the C-terminal 30-amino-acid tail of RIDα (GST-RIDα-CT) immobilized on glutathione beads. Bound proteins were resolved by SDS-PAGE for immunological detection of GFP-ORP1L. (D) Left panel: shNPC1-RIDα cells were treated with NC or 3′-UTR ORP1L-specific siRNAs. Right panel: ORP1L-depleted shNPC1-RIDα cells were reconstituted with plasmids encoding siRNA-insensitive GFP-tagged WT-ORP1L or ORP1L mutant proteins with defective FFAT motifs (D478A) or ORD-lids (Δ560-563). Both panels: equal protein aliquots were immunoblotted for ORP1L or actin. (E) Schematic of cholesterol trafficking assay. Cells incubated with LDL particles radiolabeled with [3H]cholesteryl palmitate that were taken up by receptor-mediated endocytosis (step 1) and oleate-BSA that spontaneously distributes to intracellular membranes, including the ER (step 2). [3H]Cholesteryl palmitate was deesterified by lysosomal acid lipase in endosomes (LAL; step 3) and [3H]cholesterol transported to ER (step 4) for conversion to [3H]cholesteryl oleate by ACAT and storage in LDs (step 5). (F) Left panel: [3H]Cholesteryl oleate production in sterol-loaded shNPC1 cells, shNPC1-RIDα cells, and ORP1L-depleted shNPC1-RIDα cells. Right panel: [3H]Cholesteryl oleate production in ORP1L-depleted shNPC1-RIDα cells reconstituted with GFP-tagged ORP1L constructs. Both panels: data = mean disintegrations per minute per milligram of protein ± s.e.m. n = 3; **, P < 0.005; *, P < 0.05. (A, C, and D) Representative immunoblots from 2 to 3 independent experiments.
FIG 5.

RIDα stabilized ORP1L-VAP protein contacts. (A) Left panel: CE quantification in LDL-loaded shNPC1 cells or in shNPC1-RIDα cells following treatment with NC or 3′-UTR ORP1L-specific siRNAs using an Amplex Red assay. Right panel: ORP1L-depleted cells reconstituted with the siRNA-resistant GFP-tagged ORP1L constructs described in the Fig. 3 legend. Both panels: values were normalized to total cellular protein (means ± s.e.m.; n = 3; **, P < 0.005). (B) Representative confocal images from ORP1L-depleted cells reconstituted with siRNA-resistant GFP-ORP1L constructs (green) prior to sterol depletion/LDL loading. Cells were stained with LipidTOX reagent (red) to detect LDs. (C) GST or GST-RIDα-CT immobilized on glutathione beads incubated with recombinant GFP-ORP1L protein followed by recombinant Myc-tagged VAP-B protein. Reactions were also carried out in reverse order with Myc-tagged VAP-B added to GST fusion protein immobilized on glutathione beads first followed by GFP-ORP1L. Bound proteins were resolved by SDS-PAGE for immunological detection of recombinant proteins with native antibodies or GST to verify equal protein loading. (D) Left panels: cells listed in the figure expressing GFP-tagged WT-ORP1L. Right panels: shNPC1-RIDα cells expressing GFP-tagged ORP1L mutant constructs listed in figure. Both panels: GFP-tagged ORP1L proteins were affinity purified on anti-GFP-Sepharose beads, and immobilized proteins were resolved by SDS-PAGE for immunological detection of VAP-A isoforms I and II. Blots were reprobed for GFP to verify GFP-ORP1L affinity purification. (C and D) Immunoblots are representative of results of 2 independent experiments. (E) Representative confocal images from shNPC1-RIDα cells stained with antibodies to FLAG-RIDα (green) and ORP1L (left panel) or VAP (right panel) (both in red). (B and E) Boxed areas enlarged 2× to highlight LDs (B) or individual, merged, and masked (green and red pixels above the mean red or green intensity) images (E). Arrows indicate examples of colocalized signals. All size bars, 5 μm.
RIDα stabilized ORP1L-VAP protein complexes under high-cholesterol conditions.
Data in the previous section suggested that RIDα function was regulated by an interaction between ORP1L and ER VAP proteins under high-cholesterol conditions that normally suppress formation of stable ORP1L/VAP complexes (34). This hypothesis was tested by first establishing that ORP1L served as a bridge connecting RIDα-CT and VAP using recombinant proteins. RIDα/ORP1L/VAP tripartite complexes were formed when recombinant ORP1L was added first to RIDα-CT attached to glutathione beads followed by VAP but not when VAP was added to the beads prior to ORP1L (Fig. 5C). Next, previously described ORP1L constructs were expressed in shControl cells and NPC1 knockdown cells with and without stable RIDα expression to determine their ability to form molecular complexes with ER VAP proteins in coimmunoprecipitation assays. ORP1L-VAP complex formation was not detected in LDL-loaded shControl cells with functional NPC1 machinery or in NPC1 knockdown cells (Fig. 5D, left panel). However, RIDα stabilized ORP1L-VAP complex formation in LDL-loaded shNPC1 cells reconstituted with wild-type ORP1L or ORP1L (Δ560-563) (Fig. 5D, right panel). The inactivating D478A mutation in ORP1L-FFAT blocked ORP1L-VAP complex formation and also provided the dominant phenotype in cells expressing the double D478A/Δ560-563 mutant (Fig. 5D, right panel). Consistent with the results of the coimmunoprecipitation studies, confocal imaging revealed that ORP1L colocalized with RIDα-positive membrane compartments and that RIDα compartments were also associated with a subset of VAP-B-positive ER vesicles (Fig. 5E). Collectively, these data support a model proposing that RIDα induces cholesterol reesterification by a mechanism requiring a stable interaction between ORP1L-FFAT and VAP. Furthermore, RIDα alleviates sterol inhibition of this interaction since ORP1L-VAP complex formation and LDL-cholesterol transport to the ER were both reconstituted by the ORP1L (Δ560-563) mutant protein which attenuates sterol sensing capacity downstream of Rab7-GTP (34).
RIDα regulated LD accumulation in a novel endocytic compartment independently of Rab7 and RILP.
The objective of these experiments was to determine if the RIDα-ORP1L pathway was regulated in endocytic compartments that were the same as or fundamentally different from those of the late endosomes/lysosomes where ORP1L forms a multiprotein complex with Rab7 and RILP (34, 57). Given the requirement of HRS for RIDα function, we tested the hypothesis that HRS which is normally recruited to early endosomes also accumulated on RIDα vesicles (60). However, confocal imaging revealed only modest overlap between RIDα and HRS, suggesting that the viral protein transited through HRS-positive early endosomes en route to its final destination (Fig. 6A) (26). RIDα was also excluded from late endosomes/lysosomes marked with LAMP1 or LAMP2 (Fig. 6B). In addition, the viral protein did not cosediment with conventional markers for early or late endosomes following cell fractionation on Percoll gradients (Fig. 6C). Rab7 function was further evaluated by acute siRNA gene silencing (Fig. 6D). Consistent with its role in NPC1 function in late endosomes and lysosomes, reduced Rab7 expression led to a significant reduction in LD accumulation in A549 cells compared to cells treated with NC siRNA (Fig. 6E, top panels) (61). However, LD accumulation was not blocked by Rab7 depletion in A549 cells with stable RIDα expression (Fig. 6E, bottom panels). These results suggested that RIDα not only regulates sterol transport independently of Rab7 but also compensates for reduced Rab7 function. We have shown previously that the interaction between RIDα and RILP is regulated by two histidine residues (His75-His76) located in the RIDα cytosolic tail and that histidine-to-serine substitutions (75-HH to 75-SS) block the interaction (15). Cells with stable RIDα (75-SS) expression were therefore produced to evaluate a role for RILP in Ad-induced cholesterol transport (Fig. 5F). RIDα (75-SS) colocalized with β-COP, which is required for the formation of the endosome maturation intermediates responsible for transporting cargo from early to late endosomes, similarly to wild-type RIDα (Fig. 6G) (14, 62). Also similarly to the wild-type protein, the RILP-defective RIDα mutant protein promoted LD accumulation in NPC1 knockdown cells (Fig. 6H). Collectively, these findings suggested that RIDα coopts a previously unrecognized cholesterol trafficking pathway by diverting ORP1L to endosomal compartments that are fundamentally different from those of the late endosomes/lysosomes regulated by Rab7-RILP-ORP1L multiprotein complexes.
FIG 6.

RIDα regulated LD accumulation independently of Rab7 and RILP. (A and B) Representative confocal images from shNPC1-RIDα cells stained with antibodies to FLAG-RIDα (green) and HRS (red) (A) or late endosome/lysosome markers LAMP1 and LAMP2 (red) (B). (C) Postnuclear supernatants fractionated on 27% Percoll density gradients immunoblotted for RIDα and organelle-specific marker proteins (EGFR, plasma membrane; Rab5 and Rab7, early and late endosomes, respectively). (D) Equal protein aliquots immunoblotted with antibodies to Rab7 to monitor siRNA knockdown efficiency and to tubulin for protein loading control. (E) Representative confocal images from A549 cells without (top panels) or with (bottom panels) stable RIDα expression following treatment with NC or Rab7-specific siRNAs. Cells were stained with Bodipy 493/503 (green), DAPI (blue), and FLAG antibody (red). (F) RIDα immune complexes from stable cell lines expressing wild-type RIDα or RIDα (75-SS) resolved by SDS-PAGE for immunological detection with FLAG-epitope antibody. (G) Representative confocal images from stable cell lines expressing wild-type RIDα or RIDα (75-SS) stained with antibodies to β-COP (red) and FLAG-RIDα (green). (H) Representative confocal image of sterol-loaded shNPC1 cells expressing RILP-defective RIDα (75-SS) stained with FLAG-RIDα antibody (red), Bodipy 493-503 (green), and DAPI (blue). (A, G, and H) Boxed areas are magnified 2× to visualize individual and merged images to the right of each image. (A, B, E, G, and H) All size bars, 5 μm. (C and F) Representative immunoblots from two independent experiments.
The RIDα-induced LD pathway attenuated TLR4 signaling.
The goal of these experiments was to determine the effect of RIDα-induced cholesterol flux on TLR4 signaling. We first tested the hypothesis that a C67S mutation in the RIDα cytosolic tail which prevents palmitoylation was required for the ability of the viral protein to reconstitute lipid transport in NPC1-deficient cells. Although we had shown previously that a palmitoylation-blocking C67S substitution did not affect the subcellular localization of the viral protein, this posttranslational modification could stabilize a protein conformation that is critical for ORP1L binding by anchoring the cytosolic tail of RIDα to the lipid bilayer (14, 63). RIDα (C67S) formed disulfide-linked dimers comparable to those seen with the wild-type protein (Fig. 7A), and the two RIDα-expressing cell lines exhibited equivalent levels of total ORP1L protein relative to the parent hepatocytes (Fig. 7B). However, in contrast to the wild-type RIDα protein results (see Fig. 2C and E), stable RIDα (C67S) expression failed to reconstitute LD accumulation or attenuate LSO formation in NPC1-deficient cells (Fig. 7C), supporting our hypothesis that RIDα (C67S) was a dominant-negative mutant that blocked sterol transport in the virally induced pathway. Next, we investigated if RIDα expression was sufficient to modulate the TLR4/NF-κB signaling axis independently of other viral proteins by treating cells with the TLR4 ligand lipopolysaccharide (LPS). LPS similarly induced NF-κB/p65 phosphorylation at Ser536, which is required for optimal induction of NF-κB target genes, in hepatocytes with physiological levels (shControl) or reduced levels (shNPC1) of NPC1 (Fig. 7D). In contrast, while NF-κB activity was constitutively active independently of LPS stimulation, stable RIDα expression blocked inducible TLR/NF-κB activation (Fig. 7D). The effect of RIDα-induced cholesterol flux on TLR4 signaling following an acute infection was evaluated in parent hepatocytes infected with wild-type Ad2 versus Ad mutants affecting RIDα expression (Fig. 7E) (14, 45). Control studies were carried out by evaluating the effect of various Ads on epidermal growth factor receptor (EGFR), which is known to be selectively targeted for degradation by wild-type Ad2 (45). EGFR protein was downregulated in cells infected with RIDα (C67S) similarly to wild-type Ad2 compared to cells infected with the RIDα-null mutant virus (Fig. 7F). However, TLR4 expression was not significantly altered by acute infection with any of the Ads tested, similarly to the transferrin receptor included as a membrane protein loading control (Fig. 7F). Cells were mock treated or Ad infected for 24 h and then stimulated with LPS to monitor NF-κB activation by immunoblotting with the phosphospecific NF-κB/p65 antibody. LPS-induced NF-κB activation was significantly attenuated by infection of cells with wild-type Ad2 compared to mock-treated cells or cells infected with RIDα (C67S) Ad and actually enhanced in cells infected with RIDα-null Ad (Fig. 7G). Collectively, these results suggested that the RIDα-induced LD pathway attenuates inducible TLR4/NF-κB signaling but not constitutive NF-κB signaling, which are known to be under the control of different regulatory mechanisms (64).
FIG 7.

Ad2-induced cholesterol homeostasis regulated TLR4/NF-κB signaling. (A) Equal protein aliquots from shNPC1 cells with wild-type RIDα or palmitoylation-defective RIDα mutant (C67S) resolved under reducing (+DTT) or nonreducing (−DTT) conditions to detect RIDα monomers and dimers. (B) Stable cell lines listed in the figure analyzed for endogenous NPC1 and ORP1L expression by immunoblotting. (C) Representative confocal images of RIDα (C67S)-expressing shNPC1 cells stained with FLAG-RIDα antibody (red), Bodipy 493/503 (green), and DAPI (blue) (left) or LAMP1 antibody (red) and filipin (blue) (right), following standard LDL-loading protocol. The boxed area in the right panel is enlarged 2× to visualize individual channels. All size bars, 5 μm. (D) Equal protein aliquots from untreated (−) hepatocyte-derived cells or following treatment with LPS immunoblotted with activation-specific phospho-NF-κB/p65 (Ser536) antibody and reprobed for total NF-κB/p65 to verify equal protein loading. (E) Equal protein aliquots from parent hepatocytes infected with WT-Ad2 or an Ad2 mutant encoding RIDα (C67S) immunoblotted for RIDα at 6 or 12 h postinfection (p.i.). (F) Equal protein aliquots from parent hepatocytes that were mock treated or infected with wild-type or Ad2 mutants listed in the figure for 20 h immunoblotted with receptor-specific antibodies (EGFR, TLR4, and transferrin receptor [TfR]). (G) Equal protein aliquots from mock-treated cells or cells infected with Ads listed in the figure for 24 h and then left untreated (−) or stimulated with LPS for 30 or 60 min, immunoblotted with activation-specific phospho-NF-κB/p65 (Ser536) antibody, and reprobed for total NF-κB/p65 to verify equal protein loading. (D to G) Representative immunoblots from 2 independent experiments.
DISCUSSION
Ad E3 proteins have provided rich insights into novel membrane protein trafficking mechanisms that help to shape the host response to the innate immune system (12, 65). The studies reported here extend the scope of E3 biology to intracellular cholesterol trafficking by revealing a novel host-virus interaction that remodels lipid homeostasis in infected cells. Our findings also provide new insights into the intracellular pathogens that coopt the endosomal system to invade and replicate in the host as well as the role of lipids in innate antiviral defense.
Ad2 entry into epithelial cells is initiated by attachment of fiber knobs projecting from the viral capsid to CAR (Coxsackie and adenovirus receptor) followed by interaction of capsid hexon proteins with integrin receptors and clathrin-mediated endocytosis (66, 67). Internalized viral particles disrupt endosomal membranes, escape into the cytosol, and are transported along microtubules to the nuclear membrane, where viral DNA is imported into the cell nucleus followed by expression of its genetic program (66). Dynein-dynactin motors are switched away from Rab7-containing late endosomes and lysosomes to viral capsids in order to initiate transport to the nucleus by a mechanism involving a direct interaction between a hexon subunit of the capsid and dynein, leading to a partial inhibition of lysosome function (68). Consistent with these findings, we showed that Ad2 infection led to a significant reduction in cholesterol transport from late endosomes and lysosomes to other organelles by a mechanism that is normally masked by RIDα expression in cells infected with wild-type Ad2. However, rather than repairing the NPC1 pathway, the viral protein induces a novel trafficking route that fine-tunes cholesterol transport to ER sterol pools in infected cells. Although the exact nature of these pools remains relatively undefined, differences in cholesterol delivery kinetics and inhibitor sensitivity suggest that cholesterol esterification by ACAT is dissociable from cholesterol transport to the homeostatic SREBP transcription machinery (51, 69, 70). There is also increasing evidence that lipids, including ACAT-derived CEs, are mobilized to LDs under different metabolic conditions by distinct mechanisms and perhaps even in distinct regions in the cells (71). Our data show that RIDα diverts cholesterol to storage in CEs at the expense of the ER regulatory pools that fine-tune SREBP gene transcription and lipid homeostasis, in contrast to NPC1, which feeds into both ER regulatory pools.
Although the ability of ORP1L to form protein contacts with VAP is well established, these studies provide the first evidence that ORP1L-VAP complexes facilitate sterol transport at the interface between endosomes and ER (34). The RIDα binding site has been previously mapped to ∼150 amino acids in the ORP1L-ORD (see Fig. 4B) that is represented as a 3-dimensional molecular model with bound cholesterol generated using the structure of the yeast ORP homologue Osh4 as a template in Fig. 8 (15). The RIDα binding site(s) is located on the hydrophilic exterior surface of the ORD tunnel encompassing helix α7, which is conserved across the ORP family, and several amino acid insertions that are unique to ORP1L (Fig. 8) (15, 72). As described in the introduction, ORP1L is known to interconvert between two conformational states that facilitate sterol sensing on endosomal membranes via the ORD-lid versus interaction between ORP1L-FFAT and VAP on ER membranes downstream of Rab7 (Fig. 9) (34). We hypothesize that RIDα stabilizes ORP1L/VAP complexes by interacting with unique ORP1L insertions that control the switch between ORP1L conformations. DNA viruses such as Ad produce a range of “mimics” that resemble host components in function without significant sequence identity (73). Thus, RIDα probably recognizes a previously unknown ORP1L-ORD binding site for cellular effectors that modulate ORP1L sterol binding properties to fine-tune ER sterol pools in response to developmental and physiological cues.
FIG 8.
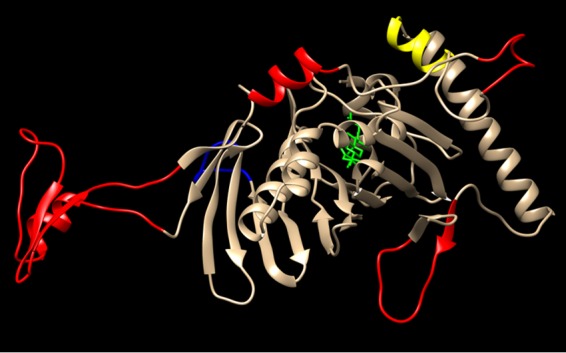
Potential RIDα binding site includes several unique ORP1L insertions. A three-dimensional model of human ORP1L-ORD (NCBI accession no. NP_542164.2) was generated using the structure of the yeast ORP homologue Osh4 as a template (PDB code 1ZHY) (72). Overlay was performed using UCSF Chimera Molecular Modeling software (http://www.cgl.ucsf.edu/chimera/). The ORD backbone is indicated in gold, the α7 helix in yellow, and the bound cholesterol ligand in green. The RIDα binding site has been mapped to ORP1L-ORD residues 755 to 923 encompassing the highly conserved α7 helix and four of the unique ORP1L insertions highlighted in red (15). A fifth unique insertion outside the RIDα binding site is highlighted in blue.
FIG 9.
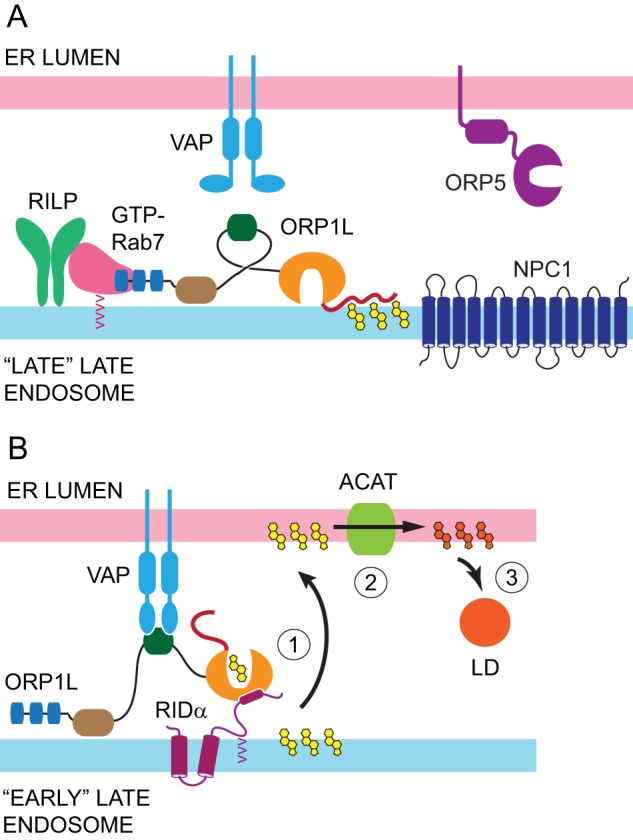
Ad reprograms ORP1L-VAP protein complexes to facilitate cholesterol transport in acutely infected cells. (A) ORP1L has been shown to form a tripartite complex with GTP-Rab7 and RILP that regulates late endosome motility of NPC1-containing “late-late endosomes.” The model depicts ORP1L conformation under high-cholesterol (yellow structures) conditions that tether the ORD-lid on endosomal membranes (34). This ORP1L conformation disrupts the interaction between ORP1L-FFAT and VAP and favors dynein motor activity downstream of RILP (34). (B) Our data support a model in which RIDα promotes cholesterol transport to ER (1) where ACAT catalyzes formation of CEs (orange structures) (2) stored in cytoplasmic LDs (3) via a direct interaction between RIDα-CT and the ORP1L-ORD in RIDα-induced endosome maturation intermediates (“early-late endosomes”). See Fig. 4B for a key to the ORP1L domain structure.
Similarly to other PH domains, ORP1L-PH binds phosphoinositides, but with only low specificity and affinity, and while ORP1L-PH may contribute to membrane binding, ORP1L ankyrin repeats that interact with Rab7-GTP are the main ORP1L targeting determinant for late endosomes (74). Our results suggest that RIDα is necessary and sufficient to recruit ORP1L to endocytic vesicles that are not traditionally classified as early or late endosomes. ORP1L was recently shown to interact with Oas1b, an interferon-inducible ER membrane protein that confers resistance to infection with flavivirus West Nile virus as well as several other types of RNA viruses in mice (75). Although the exact mechanism is incompletely understood, the interaction between ORP1L and Oas1b apparently blocks late endosome transport/fusion processes required for viral replication by interfering with normal function of RILP/Rab7/ORP1L complexes (76). In contrast, RIDα may promote formation of novel endocytic compartments capable of sterol transport to distinct ER metabolic pools by diverting ORP1L from its normal localization on Rab7-positive late endosomes. Further studies are needed to determine if RIDα-containing vesicles represent transition intermediates in the canonical Rab5-Rab7 endosome maturation pathway or unknown compartments in an alternative endocytic route that does not merge with NPC1-containing late endosomes.
Exactly how ORP1L-VAP complexes contribute to cholesterol trafficking remains to be determined. The ORP1L-ORD binds both cholesterol and PI(4)P (31, 74). Thus, the RIDα-induced ORP1L pathway could function similarly to those of other OSBP family members that facilitate a nonvesicular transfer of lipids whereby sterols are exchanged for PI(4)P at closely apposed membrane contact sites (77). For instance, LDL-cholesterol could be exchanged for PI(4)P, which is synthesized at ER exit sites under regulation of an ACAT-driven metabolic gradient (78). Alternatively, stable ORP1L-VAP membrane contacts could facilitate cholesterol transfer by another sterol binding protein located at the endosome-ER interface such as STARD3/MLN64. STARD3/MLN64 and ORP1L are thought to regulate sterol handling in two stages, with cholesterol first entering STARD3/MLN64 “early” late endosomes, from which it can be recycled to the plasma membrane before reaching the ORP1L/NPC1-positive “late” late endosomes that mediate cholesterol export to ER (79). However, a minor fraction of STARD3/MLN64 and ORP1L colocalizes in the same vesicles, suggesting that both molecules could be present in RIDα-induced endosome maturation intermediates (79).
TLR4 is a key element in the innate immune response of pulmonary epithelial cells to molecules derived from Gram-negative bacteria as well as endogenous TLR4 ligands released by tissue damage and cell lysis during viral infections (80, 81). Experimental interventions such as changes in cholesterol loading or cross-linking of raft lipids modulate TLR4 signaling (35). Cholesterol-rich lipid rafts are found throughout the endocytic system, including late endosomes, where they are recycled to plasma membrane by a mechanism involving NPC1 (82). Biochemical and cell biological studies have shown that the equilibrium between free cholesterol and raft cholesterol plays a critical role in lipid raft function and cell signaling (83, 84). So far, the mechanisms that regulate lipid raft cholesterol levels have been largely unknown. Our data suggest that a constitutively active RIDα-ORP1L-LD transport pathway modulates the lipid raft cholesterol required for innate immune signaling in endosomes. Similarly to the results seen with the nonimmune cells examined in these studies, expression of E3B encompassing the region encoding three E3 proteins, including RIDα, inhibits chemokine production in Ad-infected monocytes (85–87). Interestingly, knockdown studies have revealed an unexpected role for ORP1L in endosomal cholesterol transport in macrophages (88). Further studies will be required to determine whether RIDα activates a specialized cholesterol transport pathway that regulates lipid homeostasis and innate immunity in macrophages.
Although LDs are generally viewed as storage depots, these organelles have recently been shown to act as central nodes for the regulation of cellular lipids involved in signal transduction (36). Our studies have revealed a functional relationship between the RIDα-induced LD pathway and TLR/NF-κB signaling, which is known to be tightly regulated by dynamic membrane lipid domains in endosomes (35). While the wild-type RIDα protein was inhibitory, the palmitoylation-defective RIDα mutant that did not support LD accumulation also failed to inhibit TLR signaling responses. We speculate that RIDα impairs TLR/NF-κB signaling by altering the homeostatic flow of cholesterol and other lipids through rafts that regulate this signaling axis and possibly other innate immune responses. Although stable RIDα expression attenuated inducible activation of latent NF-κB/p65 complexes similarly to acute infection with wild-type Ad, our studies uncovered evidence that RIDα actually upregulated constitutive NF-κB/p65 activity. Both pathways are regulated by degradation of the NF-κB inhibitor IκBα (37). However, the inducible pathway requires degradation of IκBα by proteasomes, while basal NK-κB activity levels are maintained by a variety of proteasome-independent, calcium-dependent IκBα degradation pathways (64). The existence of distinct IκBα degradation mechanisms facilitates the switch from inducible to constitutive NF-κB transcription required for B-cell differentiation (64). Ads establish long-term, latent infections of mucosal lymphocytes associated with intermittent virus shedding in stool (89). It has been suggested that E3 proteins, including RIDα, prevent the elimination of persistently infected lymphocytes by downregulating expression of receptors involved in innate immune responses (90). Our results suggest that RIDα may also induce limited changes in gene expression that mimic a developmental program regulating B cell differentiation, by upregulating constitutive NF-κB activity in persistently infected lymphocytes. Further investigation is needed to understand how chronic RIDα expression upregulates IκBα stability in order to maintain constitutive NF-κB responses.
Numerous studies have identified viral mechanisms that remodel cellular membranes in order to generate replication organelles, control membrane fusion, or facilitate budding and release of enveloped viruses (91–93). Thus, RIDα-induced lipid homeostasis probably has other effects yet to be discovered. While RIDα is not required for viral replication in tissue culture, the ability of this E3 protein to remodel cholesterol trafficking pathways could have a broader role in modifying major lipid components of mammalian cell membranes required for proper membrane permeability, fluidity, organelle identity, and protein function that influence virus spread in the host. RIDα could also shift the metabolic state of the infected cell toward the generation of energy stored in LD-derived fatty acids to overcome host metabolic limitations on viral replication (94, 95).
The E3B-encoded RIDα protein was originally discovered because of its ability to selectively downregulate EGFR protein expression independently of other virally encoded proteins (42, 45, 96). Our previous studies have shown that RIDα redirects inactive EGFRs from recycling endosomes to lysosomes by usurping the ability of RILP to promote endosome-lysosome fusion (15, 97, 98). Several groups of investigators have reported that RIDα also forms molecular complexes with other E3 proteins that have been linked to downregulation of the tumor necrosis alpha (TNF-α) family of death receptors (90, 99, 100). In addition to its protein-centered roles, these studies demonstrated that RIDα regulates lipid homeostasis by a mechanism that is dissociable from its ability to downregulate EGFR. Although intracellular pathogens frequently inhibit endosome maturation in order to benefit their own replication, relatively little is known regarding how diverse infectious agents overcome subsequent deficits in late endosome/lysosome function (101–104). Understanding the coevolution of host-pathogen interactions that modulate cholesterol homeostasis provides a new avenue for understanding fundamental principles of this essential process and will also lead to the discovery of new therapies for limiting pathogen infections in mammalian cells.
MATERIALS AND METHODS
Antibodies and reagents.
Antibodies and reagents used in the study are described in Tables 1 and 2, respectively.
TABLE 1.
Description of antibodies used in the studya
| Target | Source | Vendor | Catalog no. |
|---|---|---|---|
| Actin | Rabbit | Sigma (St. Louis, MO) | A2066 |
| EGFR | Rabbit | Fitzgerald (Concord, MA) | 20R-ER004 |
| FLAG | Mouse | Sigma | F3165 |
| GFP | Mouse | Stressgen (San Diego, CA) | SAB-500 |
| HRS | Rabbit | Bethyl (Montgomery, TX) | A300-989A |
| Lamin B1 | Rabbit | Abcam (Cambridge, MA) | ab16048 |
| LAMP1 | Mouse | DSHBb | H4A3 |
| LAMP2 | Mouse | DSHB | H4B |
| NF-κB (p65) | Rabbit | Cell Signaling (Beverly, MA) | 8242 |
| NF-κB (pSer536) | Rabbit | Cell Signaling | 3033 |
| ORP1L | Mouse | Santa Cruz (San Diego, CA) | sc-376602 |
| ORP1L | Rabbit | Abcam | ab203352 |
| ORP5 | Rabbit | Novus (Littleton, CO) | NBP2-19683 |
| Rab5 | Mouse | BD Biosciences (San Jose, CA) | 610281 |
| Rab7 | Rabbit | Santa Cruz | sc6563 |
| SREBP1 | Mouse | Novus | NB600-582 |
| TLR4 | Mouse | R&D Systems (Minneapolis, MN) | MAB14782 |
| TfR | Mouse | Zymed (South San Francisco, CA) | 13-6800 |
| Tubulin | Rabbit | Cell Signaling | 2125 |
| VAP-A | Rabbit | Sigma | HPA009174 |
| VAP-B | Mouse | Proteintech (Rosemont, IL) | 14477-1-AP |
Antibodies were routinely used at a dilution of 1:1,000 to 1:2,000 for immunoblotting and at a dilution of 1:100 to 1:500 for immunostaining.
DSHB, Developmental Studies Hybridoma Bank (developed under the auspices of the Eunice Kennedy Shriver National Institute of Child Health and Human Development [NICHD] and maintained by The University of Iowa Department of Biological Sciences [Iowa City, IA]).
TABLE 2.
Reagents and kits used in this study
| Reagent | Company | Catalog no. |
|---|---|---|
| ALLN | Calbiochem (Billerica, MA) | 110044-82-1 |
| Bio-Rad protein assay kit | Bio-Rad (Hercules, CA) | 500-0001 |
| Cell fractionation kit | Cell Biolabs | AKR-171 |
| HRS siRNA SMARTpool | Dharmacon (Pittsburgh, PA) | L-016835 |
| ORP1L HRS siRNA SMARTpool | Dharmacon | M-008350 |
| ORP5 siRNA SMARTpool | Dharmacon | L-009274 |
| Rab7 siRNA SMARTpool | Dharmacon | L-010388 |
| ECL detection reagent | GE Life Sciences (Pittsburgh, PA) | RPN2236 |
| Percoll | GE Life Sciences | 17-0891-01 |
| Amplex Red cholesterol assay kit | Invitrogen (Grand Island, NY) | A12216 |
| Lipofectamine | Invitrogen | 11668-019 |
| Oligofectamine | Invitrogen | 12252-011 |
| Bodipy 493/503 | Invitrogen | D3922 |
| DAPI | Invitrogen | D1306 |
| LipidTOX | Invitrogen | H34157 |
| SlowFade antifade kit | Invitrogen | S2828 |
| Anti-GFP-agarose | MBL (Woburn, MA) | D153-8 |
| Glutathione-agarose | Pierce (Rockford, IL) | 16100 |
| AllStars negative-control siRNA | Qiagen (Valencia, CA) | 1027280 |
| Cab-O-Sil | Sigma (St. Louis, MO) | 13760 |
| Filipin | Sigma | F4767 |
| Lipopolysaccharide (LPS) | Sigma | L5293 |
| Poly-l-lysine | Sigma | P4707 |
| Protein A Sepharose | Sigma | P3391 |
Cells and tissue culture.
Human cell lines purchased from American Type Culture Collection (ATCC; Manassas, VA) are authenticated utilizing short tandem repeat (STR) profiling. Cells are routinely thawed every 10 passages to avoid unintentional cross-contamination. Human lung epithelial A549 cells (ATCC catalog number CCL-185) and Chinese hamster ovary (CHO) cells (ATCC catalog PTA9816) were grown in Ham's F12 and minimal essential medium (MEM)-alpha media, respectively, supplemented with 10% fetal bovine serum (FBS). Immortalized human hepatocyte cells (ATCC catalog number CRL-11233) stably expressing noncoding shRNA or NPC1 shRNA (denoted “shControl” or “shNPC1,” respectively) were grown in Dulbecco's minimal essential media (DME) supplemented with puromycin (10 μg/ml) and 10% FBS (50). A549 and shNPC1 cells with stable expression of a FLAG-tagged RIDα transgene are described in references 15 and 16. A stable human hepatocyte cell line expressing RIDα protein with substitutions of 75-HH to 75-SS was produced by infecting cells with a pantropic retrovirus encoding the mutant RIDα protein, followed by selection for drug resistance in DME supplemented with G418 (200 μg/ml), exactly as described in references 15 and 16. Cholesterol loading was carried out following a 24-h preincubation in DME supplemented with 5% delipidated FBS prepared using Cab-O-Sil according to methods described in references 105 and 106. Cholesterol-depleted cells were then incubated in the same media supplemented with freshly prepared LDL (50 μg/ml) for an additional 24 h. LDL was isolated from fresh human plasma by sequential ultracentrifugation followed by extensive dialysis against 0.9% NaCl–0.02% EDTA (pH 7.4) (gift of Richard Morton, Lerner Research Institute).
Adenovirus infections.
Adenovirus (Ad) stocks were grown in HEK293 cells, and titers were determined by plaque assay using standard techniques. Cells were infected with wild-type Ad2 or with previously described Ad2 mutant viruses encoding RIDα with an internal deletion (RIDα-null) or a C67S point mutation [RIDα (C67S)], using approximately 200 PFU per cell, as previously described (14, 42, 45). Cytosine arabinoside (ara-C) was added (20 μg/ml) at 1 h postinfection and replenished every 8 h to inhibit viral DNA replication (43). Some cells were metabolically labeled with 35S Express protein labeling mix (Perkin-Elmer, Wilmington, DE) exactly as described in reference 107 prior to infection. These cells were solubilized with a lysis buffer consisting of 50 mM Tris (pH 8.0) supplemented with 150 mM NaCl, 2 mM EGTA, 5 mM EDTA, 1% NP-40, 0.5% Na deoxycholate, and 0.1% SDS (buffer A) and a protease inhibitor cocktail with 0.2 mM phenylmethylsulfonyl fluoride and 1 μM leupeptin. Cell lysates were immunoprecipitated with antibodies absorbed to protein A-Sepharose CL-4B beads, and immune complexes were solubilized with 2× Laemmli buffer for SDS-PAGE and fluorographic detection of virally encoded proteins.
Confocal microscopy.
Cells were seeded on glass coverslips coated with poly-l-lysine, perforated using 0.5% β-escin, fixed with 3% paraformaldehyde, and incubated with primary and fluorescent-tagged secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA) for either 1 h at room temperature or 16 h at 4°C. Fixed cells were incubated with filipin (50 μg/ml) to detect free cholesterol, Bodipy 493/503, or LipidTOX (each at 10 μg/ml) to detect LDs or with DAPI (4′,6-diamidino-2-phenylindole) (10 μg/ml) to detect nuclei. Stained coverslips were mounted on glass slides using SlowFade anti-fade reagent for examination with a confocal laser scanning microscope (LSM 510 Meta; Carl Zeiss, Jena, Germany) using diode (excitation, 405 nm), argon (excitation, 488 nm), and HeNe (excitation, 543 and 633) lasers and 63× or 100× Plan-Apochromat numerical aperture 1.4 objectives. LD phenotypes were evaluated visually in approximately 20 cells in at least three independent experiments. Filipin was excited with the diode laser, and emissions were collected at between 411 and 486 nm. Bright-field images were also collected to visualize outlines of nuclear and cell boundaries that were overlaid onto respective confocal images.
siRNA and plasmid transfections.
Cells were transfected with siRNA duplexes using Oligofectamine transfection reagent. Two days later, cells were solubilized with buffer A and equal aliquots of total cellular protein (determined by Bradford assay) were resolved by SDS-PAGE and transferred to nitrocellulose for immunological detection by enhanced chemiluminescence (ECL) using horseradish peroxidase (HRP)-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories). Signals were quantified with ImageJ processing software (National Institutes of Health, Bethesda, MD) to estimate knockdown efficiency in cells treated with specific siRNAs versus a noncoding control siRNA. Knockdown efficiencies were approximately 70% to 90% in all instances. For ORP1L depletion/add-back experiments, cells were cotransfected with siRNA targeting the ORP1L 3′ open reading frame (ORF) and plasmids encoding GFP-tagged ORP1L proteins [wild-type ORP1L; ORP1L (D478A); ORP1L (Δ560-563); ORP1L (D478A/Δ560-563)] kindly provided by V. M. Olkkonen (Minerva Foundation Institute for Medical Research, Helsinki, Finland). All siRNAs were purchased from Dharmacon (Thermo Scientific, Pittsburgh, PA), except for the 3′-UTR ORP1L oligonucleotide, which was synthesized by Qiagen (Germantown, MD) using the sequence published in reference 108.
Coimmunoprecipitation assays.
Cells were transfected with GFP-tagged ORP1L constructs using Lipofectamine reagent according to the manufacturer's instructions, and the cholesterol depletion-loading protocol was initiated 5 h later. Cells were lysed with 0.2% NP-40–20 mM HEPES supplemented with 50 mM NaCl and 10% glycerol (buffer B) to monitor formation of RIDα-ORP1L complex formation and with 0.5% Triton X-100 in a 5 mM KCl solution supplemented with 2 mM EDTA and 2 mM EGTA to detect ORP1L-VAP complexes (buffer C). Both lysis buffers were also supplemented with previously described protease inhibitors. Clarified supernatants were incubated with anti-GFP-agarose beads overnight at 4°C with rotation, and beads were washed with phosphate-buffered saline (PBS) supplemented with 0.1% Triton. Immune complexes were resolved by SDS-PAGE and transferred to nitrocellulose filters for immunological detection by ECL.
Cholesterol quantification.
Cholesterol-loaded cells were washed twice, collected by scraping using PBS, and homogenized by 10 passages through a 22-gauge needle. Aliquots were analyzed for total (plus cholesterol esterase) and free (minus cholesterol esterase) cholesterol using an Amplex Red cholesterol assay kit according to the manufacturer's instructions, and fluorescence was monitored at 590 nm using a Tecan GENios Pro plate reader and xFluor4 GENios Pro software. CEs were quantified by subtracting free cholesterol from total cholesterol and then normalized to total protein (determined by Bradford assay). Data are represented as mean nanograms of CE per milligram of protein ± standard errors of the means (s.e.m.) from three independent experiments.
Analysis of LDL-cholesterol trafficking to ER.
Freshly prepared LDL was labeled with [1,2-3H]cholesteryl palmitate (American Radiolabeled Chemicals, St. Louis, MO) (50 Ci/mmol) by a lipid dispersion technique (109) to yield ∼15,000 dpm of 3H/μg protein. Cholesterol-depleted cells were incubated with [3H]cholesteryl palmitate-labeled LDL (50 μg/ml)–DME supplemented with 5% delipidated serum and 250 μM oleate-bovine serum albumin (BSA) complexes for 24 h. Cells collected by low-speed centrifugation were resuspended in PBS, and lipids were extracted using standard techniques (110). Lipids were fractionated on 5% silver nitrate-impregnated thin-layer chromatography plates in a developing system of hexane:diethyl ether (97:3 [vol/vol]), and bands were identified by comigration with lipid standards (cholesterol, cholesteryl oleate, and cholesteryl palmitate). Bands were scraped, and 3H content was determined by scintillation counting. Protein concentration was determined by the method described in reference 111. Data are presented as the mean numbers of [3H]cholesteryl oleate disintegrations per minute per milligram of protein ± s.e.m. from three independent experiments.
GST pulldown assays.
A bacterial expression plasmid encoding glutathione S-transferase (GST) fused to the 30-amino-acid C terminus of RIDα (RIDα-CT) has been described previously (56). BL21 cells were transformed with GST plasmids and cultured at 37°C to an optical density at 600 nm (OD600) of approximately 0.6, induced with 0.1 mM isopropyl-β-d-thiogalactopyranoside for 16 h at room temperature, and collected by low-speed centrifugation. Cell pellets were subjected to one freeze-thaw cycle and resuspended in STE buffer (10 mM Tris [pH 8.0], 150 mM NaCl, 1 mM EDTA) supplemented with lysozyme (20 μl/ml of 5 mg/ml stock), dithiothreitol (5 μl/ml of 1 mM stock) and the protease inhibitor cocktail, followed by a 30-min incubation at 4°C. Lysates were sonicated, and pellets collected by centrifugation were reextracted with STE buffer supplemented with 1% sarcosyl for 30 min at 4°C. Fusion proteins were retrieved by incubating the 1% sarcosyl supernatants with glutathione-Sepharose beads overnight at 4°C on a rotating platform. Beads were washed 4 times with PBS, and aliquots were resolved by SDS-PAGE to estimate fusion protein yield by Coomassie staining. CHO cells transfected with various GFP-tagged ORP1L constructs were lysed 2 days later with buffer B, and clarified lysates were incubated with immobilized fusion proteins overnight at 4°C. Beads were washed 4 times with buffer B, and proteins attached to beads were solubilized with 2× Laemmli buffer, resolved by SDS-PAGE, and transferred to nitrocellulose filters for immunological detection. The interaction between RIDα, ORP1L, and VAP-B was reconstituted by preequilibrating glutathione beads with attached GST fusion proteins with buffer B followed by sequential addition of recombinant GFP-tagged ORP1L protein and myc-tagged VAP-B protein. The order of incubation with fusion protein-loaded glutathione beads was also reversed by adding myc-tagged VAP-B first followed by GFP-ORP1L in some experiments. All incubations were carried overnight at 4°C with gentle rotation. Recombinant GFP-ORP1L protein was purified from mammalian cells transfected with the previously described ORP1L expression vector by anti-GFP affinity chromatography. Recombinant VAP-B protein was purified from mammalian cells transfected with a human VAP-B cDNA cloned in pCMV6-Entry (Origene Technologies, Rockville, MD) by anti-myc agarose affinity chromatography.
Cell fractionation.
Cell homogenates were fractionated on Percoll gradients essentially as described in reference 97. Briefly, cells were rinsed twice with PBS supplemented with 2 mM EDTA and 5 mM EGTA and then scraped into ice-cold homogenization buffer (HB), consisting of 10 mM HEPES (pH 7.5), 0.25 M sucrose, 1 mM EDTA, 0.2 mM phenylmethylsulfonyl fluoride (PMSF), and 1 μM leupeptin. Cells were collected by centrifugation, resuspended in HB buffer, and homogenized with 22 strokes of a Dounce homogenizer. The homogenate was diluted with an equal volume of fresh HB and centrifuged at 400 × g for 10 min at 4°C to precipitate unbroken cells and nuclei. Postnuclear supernatants were adjusted to a final concentration of 27% Percoll–0.25 M sucrose using a 90% Percoll stock solution and were then layered over a 1-ml sucrose cushion consisting of 10× HB. Gradients were centrifuged for 90 min at 25,000 × g in an SS34 fixed-angle rotor (Sorvall Instruments, Newtown, CT) without braking. A total of nine fractions were collected manually, starting from the top of the gradient, for analysis by immunoblotting.
SREBP processing.
Sterol-deprived and LDL-loaded cells were incubated with 25 mM N-acetyl-leu-leu-norleucinal (ALLN), a cysteine protease inhibitor that blocks degradation of nuclear SREBP (49), for the last 4 h of treatment. Nuclear and cytoplasmic fractions isolated using a kit from Cell Biolabs (see Table 2) were analyzed for the presence of uncleaved SREBP-1 precursor (∼120 kDa) or the mature cleaved form of nuclear SREBP-1 (∼ 65-kDa) by immunoblotting.
Statistical analysis.
Statistical analyses were performed using data from at least three independent experiments. The error bars in all figures are s.e.m. All P values were determined from a two-sided unpaired Student's t test using SigmaStat software. P values of <0.05 were considered statistically significant (**, P < 0.005; *, P < 0.05).
Image processing.
All images were processed with Photoshop CS5 and Illustrator CS5 software (Adobe).
ACKNOWLEDGMENTS
We thank Maryanne Pendergast and the Neurosciences Imaging Center for assistance with the confocal microscopy and Diane Greene and Richard Morton of the Department of Cellular and Molecular Medicine, Lerner Research Institute, Cleveland Clinic Foundation, for providing LDL and help with the cholesterol trafficking assays.
REFERENCES
- 1.Hayashi S, Hogg JC. 2007. Adenovirus infections and lung disease. Curr Opin Pharmacol 7:237–243. doi: 10.1016/j.coph.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ginsberg H. 1999. The life and times of adenoviruses. Adv Virus Res 54:1–13. doi: 10.1016/S0065-3527(08)60363-2. [DOI] [PubMed] [Google Scholar]
- 3.Fischer S. 2008. Emerging viruses in transplantation: there is more to infection after transplant than CMV and EBV. Transplantation 86:1327–1339. doi: 10.1097/TP.0b013e31818b6548. [DOI] [PubMed] [Google Scholar]
- 4.Shah D, Ghantoji S, Mulanovich V, Ariza-Heredia E, Chemaly R. 2012. Management of respiratory viral infections in hematopoietic cell transplant recipients. Am J Blood Res 2:203–218. [PMC free article] [PubMed] [Google Scholar]
- 5.Ginsberg HS, Prince GA. 1994. The molecular basis of adenovirus pathogenesis. Infect Agents Dis 3:1–8. [PubMed] [Google Scholar]
- 6.Schaack J. 2005. Induction and inhibition of innate inflammatory responses by adenovirus early region proteins. Viral Immunol 18:79–88. doi: 10.1089/vim.2005.18.79. [DOI] [PubMed] [Google Scholar]
- 7.Hendrickx R, Stichling N, Koelen J, Kuryk L, Lipiec A, Greber UF. 2014. Innate immunity to adenovirus. Hum Gene Ther 25:265–284. doi: 10.1089/hum.2014.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ginsberg H, Moldawer L, Sehgal P, Redington M, Kilian P, Chanock R, Prince G. 1991. A mouse model for investigating the molecular pathogenesis of adenovirus pneumonia. Proc Natl Acad Sci U S A 88:1651–1655. doi: 10.1073/pnas.88.5.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ginsberg HS, Lundholm-Beauchamp U, Horswoord RL, Pernis B, Wold WSM, Chanock RM, Prince GA. 1989. Role of early region 3 (E3) in pathogenesis of adenovirus disease. Proc Natl Acad Sci U S A 86:3823–3827. doi: 10.1073/pnas.86.10.3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, Hallden G, Hill R, Anand A, Liu T-C, Francis J, Brooks G, Lemoine N, Kirn D. 2003. E3 gene manipulations affect oncolytic adenovirus activity in immunocompetent tumor models. Nat Biotech 21:1328–1335. doi: 10.1038/nbt887. [DOI] [PubMed] [Google Scholar]
- 11.Fessler S, Delgado-Lopez F, Horwitz M. 2004. Mechanisms of E3 modulation of immune and inflammatory responses. Curr Top Microbiol Immunol 273:113–135. [DOI] [PubMed] [Google Scholar]
- 12.Windheim M, Hilgendorf A, Burgert H. 2004. Immune evasion by adenovirus E3 proteins: exploitation of intracellular trafficking pathways. Curr Top Microbiol Immunol 273:29–85. [DOI] [PubMed] [Google Scholar]
- 13.Delgado-Lopez F, Horwitz MS. 2006. Adenovirus RID complex inhibits lipopolysaccharide signaling without altering TLR4 cell surface expression. J Virol 80:6378–6386. doi: 10.1128/JVI.02350-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cianciola NL, Carlin CR. 2009. Adenovirus RID-α activates an autonomous cholesterol regulatory mechanism that rescues defects linked to Niemann-Pick disease type C. J Cell Biol 187:537–552. doi: 10.1083/jcb.200903039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shah AH, Cianciola NL, Mills JL, Sonnichsen FD, Carlin C. 2007. Adenovirus RIDα regulates endosome maturation by mimicking GTP-Rab7. J Cell Biol 179:965–980. doi: 10.1083/jcb.200702187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cianciola NL, Greene DJ, Morton RE, Carlin CR. 2013. Adenovirus RIDα uncovers a novel pathway requiring ORP1L for lipid droplet formation independent of NPC1. Mol Biol Cell 24:3309–3325. doi: 10.1091/mbc.E12-10-0760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown M, Goldstein J. 1986. A receptor-mediated pathway for cholesterol homeostasis. Science 232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- 18.Chang T-Y, Chang CCY, Ohgami N, Yamauchi Y. 2006. Cholesterol sensing, trafficking, and esterification. Annu Rev Cell Dev Biol 22:129–157. doi: 10.1146/annurev.cellbio.22.010305.104656. [DOI] [PubMed] [Google Scholar]
- 19.Neufeld EB, Wastney M, Patel S, Suresh S, Cooney AM, Dwyer NK, Roff CF, Ohno K, Morris JA, Carstea ED, Incardona JP, Strauss JF III, Vanier MT, Patterson MC, Brady RO, Pentchev PG, Blanchette-Mackie EJ. 1999. The Niemann-Pick C1 protein resides in a vesicular compartment linked to retrograde transport of multiple lysosomal cargo. J Biol Chem 274:9627–9635. doi: 10.1074/jbc.274.14.9627. [DOI] [PubMed] [Google Scholar]
- 20.Naureckiene S, Sleat DE, Lackland H, Fensom A, Vanier MT, Wattiaux R, Jadot M, Lobel P. 2000. Identification of HE1 as the second gene of Niemann-Pick C disease. Science 290:2298–2301. doi: 10.1126/science.290.5500.2298. [DOI] [PubMed] [Google Scholar]
- 21.Vance JE. 2010. Transfer of cholesterol by the NPC team. Cell Metab 12:105–106. doi: 10.1016/j.cmet.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 22.Patterson MC, Vanier MT, Suzuki K, Morris JA, Carstea E, Neufeld EB, Blanchette-Mackie JE, Pentchev PG. 2001. Niemann pick disease type C: a lipid trafficking disorder, p 3611–3633. In Scriver CR, Beaudet AL, Sly WS, Valle D (ed), The metabolic and molecular bases of inherited disease, 8th ed, vol 3 McGraw-Hill, New York, NY. [Google Scholar]
- 23.Chang T-Y, Reid PC, Sugii S, Ohgami N, Cruz JC, Chang CCY. 2005. Niemann-Pick type C disease and intracellular cholesterol trafficking. J Biol Chem 280:20917–20920. doi: 10.1074/jbc.R400040200. [DOI] [PubMed] [Google Scholar]
- 24.Wiegand V, Chang T-Y, Strauss JF, Fahrenholz F, Gimpl G. 2003. Transport of plasma membrane-derived cholesterol and the function of Niemann-Pick C1 protein. FASEB J 17:782–784. [DOI] [PubMed] [Google Scholar]
- 25.Du X, Kumar J, Ferguson C, Schulz TA, Ong YS, Hong W, Prinz WA, Parton RG, Brown AJ, Yang H. 2011. A role for oxysterol-binding protein-related protein 5 in endosomal cholesterol trafficking. J Cell Biol 192:121–135. doi: 10.1083/jcb.201004142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Du X, Kazim A, Brown AJ, Yang H. 2012. An essential role of Hrs/Vps27 in endosomal cholesterol trafficking. Cell Rep 1:29–35. doi: 10.1016/j.celrep.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 27.Raiborg C, Stenmark H. 2002. Hrs and endocytic sorting of ubiquitinated membrane proteins. Cell Struct Funct 27:403–408. doi: 10.1247/csf.27.403. [DOI] [PubMed] [Google Scholar]
- 28.Olkkonen VM, Beaslas O, Nissila E. 2012. Oxysterols and their cellular effectors. Biomolecules 2:76–103. doi: 10.3390/biom2010076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chung J, Torta F, Masai K, Lucast L, Czapla H, Tanner LB, Narayanaswamy P, Wenk MR, Nakatsu F, De Camilli P. 2015. PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ER-plasma membrane contacts. Science 349:428–432. doi: 10.1126/science.aab1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eden ER, White IJ, Tsapara A, Futter CE. 2010. Membrane contacts between endosomes and ER provide sites for PTP1B-epidermal growth factor receptor interaction. Nat Cell Biol 12:267–272. [DOI] [PubMed] [Google Scholar]
- 31.Vihervaara T, Uronen R-L, Wohlfahrt G, Björkhem I, Ikonen E, Olkkonen V. 2011. Sterol binding by OSBP-related protein 1L regulates late endosome motility and function. Cell Mol Life Sci 68:537–551. doi: 10.1007/s00018-010-0470-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Helle SCJ, Kanfer G, Kolar K, Lang A, Michel AH, Kornmann B. 2013. Organization and function of membrane contact sites. Biochim Biophys Acta 1833:2526–2541. doi: 10.1016/j.bbamcr.2013.01.028. [DOI] [PubMed] [Google Scholar]
- 33.Moser von Filseck J, Mesmin B, Bigay J, Antonny B, Drin G. 2014. Building lipid PIPelines throughout the cell by ORP/Osh proteins. Biochem Soc Trans 42:1465–1470. doi: 10.1042/BST20140143. [DOI] [PubMed] [Google Scholar]
- 34.Rocha N, Kuijl C, van der Kant R, Janssen L, Houben D, Janssen H, Zwart W, Neefjes J. 2009. Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7-RILP-p150Glued and late endosome positioning. J Cell Biol 185:1209–1225. doi: 10.1083/jcb.200811005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fessler MB, Parks JS. 2011. Intracellular lipid flux and membrane microdomains as organizing principles in inflammatory cell signaling. J Immunol 187:1529–1535. doi: 10.4049/jimmunol.1100253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lingwood D, Simons K. 2010. Lipid rafts as a membrane-organizing principle. Science 327:46–50. doi: 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- 37.Lawrence T. 2009. The nuclear factor NF-κB pathway in inflammation. Cold Spring Harb Perspect Biol 1:a001651. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bruder JT, Kovesdi I. 1997. Adenovirus infection stimulates the Raf/MAPK signaling pathway and induces interleukin-8 expression. J Virol 71:398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li E, Stupack DG, Brown SL, Klemke R, Schlaepfer DD, Nemerow GR. 2000. Association of p130CAS with phosphatidylinositol-3-OH kinase mediates adenovirus cell entry. J Biol Chem 275:14729–14735. doi: 10.1074/jbc.275.19.14729. [DOI] [PubMed] [Google Scholar]
- 40.Gastaldelli M, Imelli N, Boucke K, Amstutz B, Meier O, Greber UF. 2008. Infectious adenovirus type 2 transport through early but not late endosomes. Traffic 9:2265–2278. doi: 10.1111/j.1600-0854.2008.00835.x. [DOI] [PubMed] [Google Scholar]
- 41.Trinh HV, Lesage G, Chennamparampil V, Vollenweider B, Burckhardt CJ, Schauer S, Havenga M, Greber UF, Hemmi S. 2012. Avidity binding of human adenovirus serotypes 3 and 7 to the membrane cofactor CD46 triggers infection. J Virol 86:1623–1637. doi: 10.1128/JVI.06181-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carlin CR, Tollefson AE, Brady HA, Hoffman BL, Wold W. 1989. Epidermal growth factor receptor is down-regulated by a 10,400 MW protein encoded by the E3 region of adenovirus. Cell 57:135–144. doi: 10.1016/0092-8674(89)90179-7. [DOI] [PubMed] [Google Scholar]
- 43.Feldman L, Rapp F. 1966. Inhibition of adenovirus replication by 1-beta-d-arabinofuranosylcytosine. Proc Soc Exp Biol Med 122:243–247. doi: 10.3181/00379727-122-31100. [DOI] [PubMed] [Google Scholar]
- 44.Nevins JR, Ginsberg HS, Blanchard J-M, Wilson MC, Darnell JE. 1979. Regulation of the primary expression of the early adenovirus transcription units. J Virol 32:727–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoffman P, Carlin C. 1994. Adenovirus E3 protein causes constitutively internalized EGF receptors to accumulate in a prelysosomal compartment, resulting in enhanced degradation. Mol Cell Biol 14:3695–3706. doi: 10.1128/MCB.14.6.3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gocze P, Freeman D. 1994. Factors underlying the variability of lipid droplet fluorescence in MA-10 Leydig tumor cells. Cytometry 17:151–158. doi: 10.1002/cyto.990170207. [DOI] [PubMed] [Google Scholar]
- 47.Shayakhmetov DM, Li Z-Y, Ni S, Lieber A. 2004. Analysis of adenovirus sequestration in the liver, transduction of hepatic cells, and innate toxicity after injection of fiber-modified vectors. J Virol 78:5368–5381. doi: 10.1128/JVI.78.10.5368-5381.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ronan BA, Agrwal N, Carey EJ, De Petris G, Kusne S, Seville MT, Blair JE, Vikram HR. 2014. Fulminant hepatitis due to human adenovirus. Infection 42:105–111. doi: 10.1007/s15010-013-0527-7. [DOI] [PubMed] [Google Scholar]
- 49.Wang X, Sato R, Brown MS, Hua X, Goldstein JL. 1994. SREBP-1, a membrane-bound transcription factor released by sterol-regulated proteolysis. Cell 77:53–62. doi: 10.1016/0092-8674(94)90234-8. [DOI] [PubMed] [Google Scholar]
- 50.Ulatowski L, Parker R, Davidson C, Yanjanin N, Kelley TJ, Corey D, Atkinson J, Porter F, Arai H, Walkley SU, Manor D. 2011. Altered vitamin E status in Niemann-Pick type C disease. J Lipid Res 52:1400–1410. doi: 10.1194/jlr.M015560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kristiana I, Yang H, Brown AJ. 2008. Different kinetics of cholesterol delivery to components of the cholesterol homeostatic machinery: implications for cholesterol trafficking to the endoplasmic reticulum. Biochim Biophys Acta 1781:724–730. doi: 10.1016/j.bbalip.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 52.Lev S, Halevy DB, Peretti D, Dahan N. 2008. The VAP protein family: from cellular functions to motor neuron disease. Trends Cell Biol 18:282–290. doi: 10.1016/j.tcb.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 53.Raiborg C, Wenzel EM, Stenmark H. 2015. ER-endosome contact sites: molecular compositions and functions. EMBO J 34:1848–1858. doi: 10.15252/embj.201591481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoffman P, Yaffe MB, Hoffman BL, Yei S, Wold WSM, Carlin C. 1992. Characterization of the adenovirus E3 protein that down-regulates the epidermal growth factor receptor. J Biol Chem 267:13480–13487. [PubMed] [Google Scholar]
- 55.Austin CD, Wen X, Gazzard L, Nelson C, Scheller RH, Scales SJ. 2005. Oxidizing potential of endosomes and lysosomes limits intracellular cleavage of disulfide-based antibody-drug conjugates. Proc Natl Acad Sci U S A 102:17987–17992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cianciola NL, Crooks D, Shah AH, Carlin C. 2007. A tyrosine-based signal plays a critical role in the targeting and function of adenovirus RID{alpha} protein. J Virol 81:10437–10450. doi: 10.1128/JVI.00399-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van der Kant R, Fish A, Janssen L, Janssen H, Krom S, Ho N, Brummelkamp T, Carette J, Rocha N, Neefjes J. 2013. Late endosomal transport and tethering are coupled processes controlled by RILP and the cholesterol sensor ORP1L. J Cell Sci 126:3462–3474. doi: 10.1242/jcs.129270. [DOI] [PubMed] [Google Scholar]
- 58.Alpy F, Tomasetto C. 2006. MLN64 and MENTHO, two mediators of endosomal cholesterol transport. Biochem Soc Trans 34:343–345. doi: 10.1042/BST0340343. [DOI] [PubMed] [Google Scholar]
- 59.van der Kant R, Neefjes J. 2014. Small regulators, major consequences for Ca2+ and cholesterol at the endosome-ER interface. J Cell Sci 127:929–938. doi: 10.1242/jcs.137539. [DOI] [PubMed] [Google Scholar]
- 60.Bache KG, Brech A, Mehlum A, Stenmark H. 2003. Hrs regulates multivesicular body formation via ESCRT recruitment to endosomes. J Cell Biol 162:435–442. doi: 10.1083/jcb.200302131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bucci C, Thomsen P, Nicoziani P, McCarthy J, van Deurs B. 2000. Rab7: a key to lysosome biogenesis. Mol Biol Cell 11:467–480. doi: 10.1091/mbc.11.2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aniento F, Gu F, Parton R, Gruenberg J. 1996. An endosomal βCOP is involved in the pH-dependent formation of transport vesicles destined for late endosomes. J Cell Biol 133:29–41. doi: 10.1083/jcb.133.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Linder M, Deschenes R. 2007. Palmitoylation: policing protein stability and traffic. Nat Rev Mol Cell Biol 8:74–84. doi: 10.1038/nrm2084. [DOI] [PubMed] [Google Scholar]
- 64.Fields ER, Seufzer BJ, Oltz EM, Miyamoto S. 2000. A switch in distinct IκBα degradation mechanisms mediates constitutive NF-κB activation in mature B cells. J Immunol 164:4762–4767. doi: 10.4049/jimmunol.164.9.4762. [DOI] [PubMed] [Google Scholar]
- 65.Horwitz MS. 2004. Function of adenovirus E3 proteins and their interactions with immunoregulatory cell proteins. J Gene Med 6(Suppl 1):S172–S183. doi: 10.1002/jgm.495. [DOI] [PubMed] [Google Scholar]
- 66.Meier O, Greber UF. 2004. Adenovirus endocytosis. J Gene Med 6:S152–S163. doi: 10.1002/jgm.553. [DOI] [PubMed] [Google Scholar]
- 67.Nemerow G. 2000. Cell receptors involved in adenovirus entry. Virology 274:1–4. doi: 10.1006/viro.2000.0468. [DOI] [PubMed] [Google Scholar]
- 68.Scherer J, Yi J, Vallee RB. 2014. PKA-dependent dynein switching from lysosomes to adenovirus: a novel form of host-virus competition. J Cell Biol 205:163–177. doi: 10.1083/jcb.201307116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Edwards PA, Ericsson J. 1999. Sterols and isoprenoids: signaling molecules derived from the cholesterol biosynthetic pathway. Annu Rev Biochem 68:157–185. doi: 10.1146/annurev.biochem.68.1.157. [DOI] [PubMed] [Google Scholar]
- 70.Du X, Pham YH, Brown AJ. 2004. Effects of 25-hydroxycholesterol on cholesterol esterification and sterol regulatory element-binding protein processing are dissociable: implications for cholesterol movement to the regulatory pool in the endoplasmic reticulum. J Biol Chem 279:47010–47016. doi: 10.1074/jbc.M408690200. [DOI] [PubMed] [Google Scholar]
- 71.Meyers A, del Rio ZP, Beaver RA, Morris RM, Weiskittel TM, Alshibli AK, Mannik J, Morrell-Falvey J, Dalhaimer P. 2016. Lipid droplets form from distinct regions of the cell in the fission yeast Schizosaccharomyces pombe. Traffic 17:657–669. doi: 10.1111/tra.12394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Im YJ, Raychaudhuri S, Prinz WA, Hurley JH. 2005. Structural mechanism for sterol sensing and transport by OSBP-related proteins. Nature 437:154–158. doi: 10.1038/nature03923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Elde NC, Malik HS. 2009. The evolutionary conundrum of pathogen mimicry. Nat Rev Microbiol 7:787–797. doi: 10.1038/nrmicro2222. [DOI] [PubMed] [Google Scholar]
- 74.Johansson M, Lehto M, Tanhuanpaa K, Cover TL, Olkkonen VM. 2005. The oxysterol-binding protein homologue ORP1L interacts with Rab7 and alters functional properties of late endocytic compartments. Mol Biol Cell 16:5480–5492. doi: 10.1091/mbc.E05-03-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Courtney SC, Di H, Stockman BM, Liu H, Scherbik SV, Brinton MA. 2012. Identification of novel host cell binding partners of Oas1b, the protein conferring resistance to Flavivirus-induced disease in mice. J Virol 86:7953–7963. doi: 10.1128/JVI.00333-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stiasny K, Kiermayr S, Heinz F. 2006. Entry functions and antigenic structure of flavivirus envelope proteins. Novartis Found Symp 277:57–65. doi: 10.1002/0470058005.ch5. [DOI] [PubMed] [Google Scholar]
- 77.Mesmin B, Bigay J, Moser von Filseck J, Lacas-Gervais S, Drin G, Antonny B. 2013. A four-step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER-Golgi tether OSBP. Cell 155:830–843. doi: 10.1016/j.cell.2013.09.056. [DOI] [PubMed] [Google Scholar]
- 78.Blumental-Perry A, Haney CJ, Weixel KM, Watkins SC, Weisz OA, Aridor M. 2006. Phosphatidylinositol 4-phosphate formation at ER exit sites regulates ER export. Dev Cell 11:671–682. doi: 10.1016/j.devcel.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 79.van der Kant R, Zondervan I, Janssen L, Neefjes J. 2013. Cholesterol-binding molecules MLN64 and ORP1L mark distinct late endosomes with transporters ABCA3 and NPC1. J Lipid Res 54:2153–2165. doi: 10.1194/jlr.M037325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guillot Lc., Medjane S, Le-Barillec K, Balloy V, Danel C, Chignard M, Si-Tahar M. 2004. Response of human pulmonary epithelial cells to lipopolysaccharide involves Toll-like receptor 4 (TLR4)-dependent signaling pathways: evidence for intracellular compartmentalization of TLR4. J Biol Chem 279:2712–2718. doi: 10.1074/jbc.M305790200. [DOI] [PubMed] [Google Scholar]
- 81.Yu L, Wang L, Chen S. 2010. Endogenous toll-like receptor ligands and their biological significance. J Cell Mol Med 14:2592–2603. doi: 10.1111/j.1582-4934.2010.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lusa S, Blom TS, Eskelinen EL, Kuismanen E, Mansson JE, Simons K, Ikonen E. 2001. Depletion of rafts in late endocytic membranes is controlled by NPC1-dependent recycling of cholesterol to the plasma membrane. J Cell Sci 114:1893–1900. [DOI] [PubMed] [Google Scholar]
- 83.Golub T, Wacha S, Caroni P. 2004. Spatial and temporal control of signaling through lipid rafts. Curr Opin Neurobiol 14:542–550. doi: 10.1016/j.conb.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 84.Mansfield PJ, Hinkovska-Galcheva V, Borofsky MS, Shayman JA, Boxer LA. 2005. Phagocytic signaling molecules in lipid rafts of COS-1 cells transfected with FcgammaRIIA. Biochem Biophys Res Commun 331:132–138. doi: 10.1016/j.bbrc.2005.02.191. [DOI] [PubMed] [Google Scholar]
- 85.Zsengellér Z, Otake K, Hossain S-A, Berclaz P-Y, Trapnell BC. 2000. Internalization of adenovirus by alveolar macrophages initiates early proinflammatory signaling during acute respiratory tract infection. J Virol 74:9655–9667. doi: 10.1128/JVI.74.20.9655-9667.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Doronin K, Flatt JW, Di Paolo NC, Khare R, Kalyuzhniy O, Acchione M, Sumida JP, Ohto U, Shimizu T, Akashi-Takamura S, Miyake K, MacDonald JW, Bammler TK, Beyer RP, Farin FM, Stewart PL, Shayakhmetov DM. 2012. Coagulation factor X activates innate immunity to human species C adenovirus. Science 338:795–798. doi: 10.1126/science.1226625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Spurrell E, Gangeswaran R, Wang P, Cao F, Gao D, Feng B, Wold W, Tollefson A, Lemoine NR, Wang Y. 2014. STAT1 interaction with E3–14.7K in monocytes affects the efficacy of oncolytic adenovirus. J Virol 88:2291–2300. doi: 10.1128/JVI.02829-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vihervaara T, Kakela R, Liebisch G, Tarasov K, Schmitz G, Olkkonen VM. 2013. Modification of the lipidome in RAW264.7 macrophage subjected to stable silencing of oxysterol-binding proteins. Biochimie 95:538–547. doi: 10.1016/j.biochi.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 89.Fox JP, Hall CE, Cooney MK. 1977. The Seattle Virus Watch. VII. Observations of adenovirus infections. Am J Epidemiol 105:362–386. [DOI] [PubMed] [Google Scholar]
- 90.McNees AL, Garnett CT, Gooding LR. 2002. The adenovirus E3 RID complex protects some cultured human T and B lymphocytes from Fas-induced apoptosis. J Virol 76:9716–9723. doi: 10.1128/JVI.76.19.9716-9723.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schoggins J, Randall G. 2013. Lipids in innate antiviral defense. Cell Host Microbe 14:379–385. doi: 10.1016/j.chom.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Goodwin CM, Xu S, Munger J. 2015. Stealing the keys to the kitchen: viral manipulation of the host cell metabolic network. Trends Microbiol 23:789–798. doi: 10.1016/j.tim.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chukkapalli V, Heaton NS, Randall G. 2012. Lipids at the interface of virus-host interactions. Curr Opin Microbiol 15:512–518. doi: 10.1016/j.mib.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Birch E, Ruggero N, Covert M. 2012. Determining host metabolic limitations on viral replication via integrated modeling and experimental perturbation. PLoS Comput Biol 8:e1002746. doi: 10.1371/journal.pcbi.1002746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Welte M. 2015. Expanding roles for lipid droplets. Curr Biol 25:R470–R481. doi: 10.1016/j.cub.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hoffman BL, Ullrich A, Wold W, Carlin C. 1990. Retrovirus-mediated transfer of an adenovirus gene encoding an integral membrane protein is sufficient to down regulate the receptor for epidermal growth factor. Mol Cell Biol 10:5521–5524. doi: 10.1128/MCB.10.10.5521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Crooks D, Kil SJ, McCaffery JM, Carlin C. 2000. E3–13.7 integral membrane proteins encoded by human adenoviruses alter epidermal growth factor receptor trafficking by interacting directly with receptors in early endosomes. Mol Biol Cell 11:3559–3572. doi: 10.1091/mbc.11.10.3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hoffman PH, Rajakumar P, Hoffman B, Heuertz R, Wold WSM, Carlin CR. 1992. Evidence for intracellular down-regulation of the epidermal growth factor receptor during adenovirus infection by an EGF-independent mechanism. J Virol 66:197–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chin YR, Horwitz MS. 2005. Mechanism for removal of tumor necrosis factor receptor 1 from the cell surface by the adenovirus RID{alpha}/{beta} complex. J Virol 79:13606–13617. doi: 10.1128/JVI.79.21.13606-13617.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hilgendorf A, Lindberg J, Ruzsics Z, Honing S, Elsing A, Lofqvist M, Engelmann H, Burgert H-G. 2003. Two distinct transport motifs in the adenovirus E3 proteins act in concert to down-modulate apoptosis receptors and the epidermal growth factor receptor. J Biol Chem 278:51872–51884. doi: 10.1074/jbc.M310038200. [DOI] [PubMed] [Google Scholar]
- 101.Gruenberg J, van der Goot FG. 2006. Mechanisms of pathogen entry through the endosomal compartments. Nat Rev Mol Cell Biol 7:495–504. doi: 10.1038/nrm1959. [DOI] [PubMed] [Google Scholar]
- 102.Cossart P, Roy CR. 2010. Manipulation of host membrane machinery by bacterial pathogens. Curr Opin Cell Biol 22:547–554. doi: 10.1016/j.ceb.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liss V, Hensel M. 2015. Take the tube: remodelling of the endosomal system by intracellular Salmonella enterica. Cell Microbiol 17:639–647. doi: 10.1111/cmi.12441. [DOI] [PubMed] [Google Scholar]
- 104.Lozach P-Y, Huotari J, Helenius A. 2011. Late-penetrating viruses. Curr Opin Virol 1:35–43. doi: 10.1016/j.coviro.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 105.Lala DS, Syka PM, Lazarchik SB, Mangelsdorf DJ, Parker KL, Heyman RA. 1997. Activation of the orphan nuclear receptor steroidogenic factor 1 by oxysterols. Proc Natl Acad Sci U S A 94:4895–4900. doi: 10.1073/pnas.94.10.4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Haas D, Morgenthaler J, Lacbawan F, Long B, Runz H, Garbade SF, Zschocke J, Kelley RI, Okun JG, Hoffmann GF, Muenke M. 2007. Abnormal sterol metabolism in holoprosencephaly: studies in cultured lymphoblasts. J Med Gen 44:298–305. doi: 10.1136/jmg.2006.047258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.He C, Hobert M, Friend L, Carlin C. 2002. The epidermal growth factor receptor juxtamembrane domain has multiple basolateral plasma membrane localization determinants, including a dominant signal with a polyproline core. J Biol Chem 277:38284–38293. doi: 10.1074/jbc.M104646200. [DOI] [PubMed] [Google Scholar]
- 108.Johansson M, Rocha N, Zwart W, Jordens I, Janssen L, Kuijl C, Olkkonen V, Neefjes J. 2007. Activation of endosomal dynein motors by stepwise assembly of Rab7-RILP-p150Glued, ORP1L, and the receptor betalll spectrin. J Cell Biol 176:459–471. doi: 10.1083/jcb.200606077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Morton RE, Zilversmit DB. 1981. A plasma inhibitor of triglyceride and cholesteryl ester transfer activities. J Biol Chem 256:11992–11995. [PubMed] [Google Scholar]
- 110.Thompson J, Erdody P, Brien R, Murray T. 1971. Fluorometric determination of vitamin A in human blood and liver. Biochem Med 5:67–89. doi: 10.1016/0006-2944(71)90076-7. [DOI] [PubMed] [Google Scholar]
- 111.Peterson G. 1977. A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal Biochem 83:346–356. doi: 10.1016/0003-2697(77)90043-4. [DOI] [PubMed] [Google Scholar]
- 112.Johansson M, Olkkonen VM. 2005. Assays for interaction between Rab7 and oxysterol binding protein related protein 1L (ORP1L). Methods Enzymol 403:743–758. doi: 10.1016/S0076-6879(05)03065-X. [DOI] [PubMed] [Google Scholar]