

ABSTRACT
Functional analysis of T-cell responses in HIV-infected individuals has indicated that virus-specific CD8+ T cells with superior antiviral efficacy are well represented in HIV-1 controllers but are rare or absent in HIV-1 progressors. To define the role of individual T-cell receptor (TCR) clonotypes in differential antiviral CD8+ T-cell function, we performed detailed functional and mass cytometric cluster analysis of multiple CD8+ T-cell clones recognizing the identical HLA-B*2705-restricted HIV-1 epitope KK10 (KRWIILGLNK). Effective and ineffective CD8+ T-cell clones segregated based on responses to HIV-1-infected and peptide-loaded target cells. Following cognate peptide stimulation, effective HIV-specific clones displayed significantly more rapid TCR signal propagation, more efficient initial lytic granule release, and more sustained nonlytic cytokine and chemokine secretion than ineffective clones. To evaluate the TCR clonotype contribution to CD8+ T-cell function, we cloned the TCR α and β chain genes from one effective and two ineffective CD8+ T-cell clones from an elite controller into TCR-expressing lentivectors. We show that Jurkat/MA cells and primary CD8+ T cells transduced with lentivirus expressing TCR from one of the ineffective clones exhibited a level of activation by cognate peptide and inhibition of in vitro HIV-1 infection, respectively, that were comparable to those of the effective clonotype. Taken together, these data suggest that the potent antiviral capacity of some HIV-specific CD8+ T cells is a consequence of factors in addition to TCR sequence that modulate functionality and contribute to the increased antiviral capacity of HIV-specific CD8+ T cells in elite controllers to inhibit HIV infection.
IMPORTANCE The greater ex vivo antiviral inhibitory activity of CD8+ T cells from elite controllers than from HIV-1 progressors supports the crucial role of effective HIV-specific CD8+ T cells in controlling HIV-1 replication. The contribution of TCR clonotype to inhibitory potency was investigated by delineating the responsiveness of effective and ineffective CD8+ T-cell clones recognizing the identical HLA-B*2705-restricted HIV-1 Gag-derived peptide, KK10 (KRWIILGLNK). KK10-stimulated “effective” CD8+ T-cell clones displayed significantly more rapid TCR signal propagation, more efficient initial lytic granule release, and more sustained cytokine and chemokine secretion than “ineffective” CD8+ T-cell clones. However, TCRs cloned from an effective and one of two ineffective clones conferred upon primary CD8+ T cells the equivalent potent capacity to inhibit HIV-1 infection. Taken together, these data suggest that other factors aside from intrinsic TCR-peptide-major histocompatibility complex (TCR-peptide-MHC) reactivity can contribute to the potent antiviral capacity of some HIV-specific CD8+ T-cell clones.
KEYWORDS: T cells, TCR, clonotype, HIV
INTRODUCTION
We have previously demonstrated that differential immune control of HIV-1 infections is associated with particular T-cell receptor (TCR) clonotypes engaging peptide-major histocompatibility complex class I (pMHC-I) complexes on infected cells (1). The differences in potency and cross-reactive recognition of their cognate HIV-1 peptide and its variants among CD8+ T lymphocyte clones expressing different TCR clonotypes (1, 2) are compatible with the TCR-based modulation of effector cell subsets (3) and suggest that the fine specificity of the TCR may modulate their antiviral function (4–7). Recent crystallographic and computational studies revealed that specific structural and binding patterns among TCR-pMHC interactions are associated with enhanced antiviral efficacy and cross-reactivity of the CD8+ T-cell clones (6, 8).
Following TCR encounter of antigenic peptide bound to MHC-I in association with accessory molecules, such as CD8, CD28, and lymphocyte function-associated antigen-1 (LFA-1), the T cell is activated (9–11), and its functional antiviral activity may be commensurate to the ability of the TCR to initiate and sustain intracellular signal transduction (12, 13). T-cell stimulation involves TCR-induced activation of cellular kinases that phosphorylate multiple downstream protein targets (14–16) that induce the translocation of transcription factors into the nucleus to activate a network of genes whose level of expression is determined by the initial strength and duration of the TCR triggering (17). It is not known whether the antiviral differences observed among epitope-specific CD8+ T cells are due to differences in TCR signaling, which alters signal transduction and affects the expression of antiviral cytokines and chemokines, or to other intrinsic functions of the CD8+ T cells.
In this study, we used flow cytometry and mass cytometry (CyTOF) to investigate the fine-tuning of HIV-1 peptide-specific TCR signaling on the T-cell responsiveness, using a panel of well-characterized CD8+ T-cell clones. We took advantage of unique reagents and technologies: HIV-1-specific CD8+ T-cell clones with distinct TCR clonotypes generated in vivo during HIV-1 infection, all of which recognize the same HLA B*2705-restricted epitope, KK10 (KRWIILGLNK, Gag amino acids [aa] 263 to 272), but differ in antiviral function; CyTOF analysis for sensitive multiparameter phenotypic cellular analyses; and a CD4+ T-cell-derived CEM cell line expressing HLA B*2705 infected with HIV-1 viruses or loaded with the epitopic peptides for antigenic stimulation of the CD8+ T-cell clones and for use as target cells. This allowed the comparative assessment of CD8+ T-cell function and antiviral efficacy of different clones from the same donors in a setting in which the primary variable was the TCR clonotype. We further focused analysis on the functional activity conferred by specific TCR clonotypes by cloning the TCR α and β chain genes of the KK10-specific CD8+ T-cell clones with divergent functional activity into lentiviral vectors to express the TCRs in Jurkat/MA cells, a T-cell line engineered to measure TCR signaling using a nuclear factor of activated T cells (NFAT)-regulated luciferase reporter, to quantify TCR signal transduction, and in HIV-1-naive primary CD8+ lymphocytes to evaluate TCR-dependent anti-HIV-1 activity.
In the current study, we demonstrated that CD8+ T-cell clones with superior antiviral efficacy segregate phenotypically by mass-cytometric cluster analysis of TCR-specific antigen responses and are characterized by rapid TCR signal propagation and efficient initial cytotoxicity followed by sustained nonlytic cytokine and chemokine secretion. Focused evaluation of the functional activity of TCRs by cloning TCR from clones with divergent activities and using lentivirus to express them in Jurkat/MA cells and naive CD8+ T cells demonstrated that at least some effective and ineffective CTL clonotypes mediate equivalent TCR clonotype function in signal transduction and in anti-HIV activity. These findings, using defined TCR interacting with identical cognate pMHC complexes, indicated that factors in addition to the intrinsic structure of the TCR clonotype contribute to the divergent capacity of some patient-derived HIV-specific CD8+ T cells to control infection with antigenically variable pathogens such as HIV-1.
RESULTS
Antiviral function of KK10-specific clonotypes.
Eight previously established HLA-B*2705 KK10-specific CD8+ T-cell clones derived from HIV-1-infected persons were used in these studies and were characterized as effective CD8+ T-cell clones (EC) or ineffective CD8+ T-cell clones (IC) based on their KK10 peptide-specific cytotoxic activity (Table 1). The CD8+ T-cell clones had considerable sequence diversity in their TCR complementarity-determining regions (CDR) and differed in Vβ and Vα gene usage (1). First, we retested serially passaged, cryopreserved samples of these clones in the standard killing assay with virally infected target cells, confirming that despite recognizing the same peptide, KK10, in the context of the same MHC molecules, HLA-B*2705, these CD8+ T-cell clones maintained stable differences in their ability to kill CD4+ T cells infected with HIV-1; four were characterized as effective CD8+ T-cell clones (EC1 to EC4) and four as ineffective CD8+ T-cell clones (IC1 to IC4) (1). Based on these HIV-1-killing assays, two of the four effective CD8+ T-cell clones against the wild-type (WT) virus (EC2 and EC4) also recognized 3 different viral KK10 variants, whereas the ineffective CD8+ T-cell clones demonstrated only weak recognition of the WT and L6M variant (Fig. 1). These data indicate that the antiviral properties of these clones differ and are maintained with serial passage.
TABLE 1.
Clonotypes of HLA-B*2705-restricted KK10-specific CD8+ T-cell clones
| Clone (original designation)a | TCRBVb | CDR3 | TCRBJc | % Specific lysisd |
|---|---|---|---|---|
| EC1 (S-C003) | 25.1 | CASSEADFEAF | 1.1 | 74 |
| EC2 (S-T001) | 18 | CASSPGQFSHEQY | 2.7 | 77 |
| EC3 (B3) | 4.3 | CASRPGLASNEQF | 2.1 | 75 |
| EC4 (B5) | 6.5 | CASRPGQGATEAF | 1.1 | 78 |
| IC1 (S-C007) | 20.1 | CSARDGGEQY | 2.7 | 30 |
| IC2 (B6) | 20.1 | CSARDRGTREVADNYGYT | 1.2 | 22 |
| IC3 (002) | 5.6 | CASGGGTVYEQY | 2.7 | 26 |
| IC4 (013) | 2 | CASSAGPGQYGNTIY | 1.3 | 16 |
| IC5 (S-T002) | 7.9 | CASSLDRLEQF | 1.1 | ND |
The original designations are from reference 1.
TCRBV, TCR β-chain variable region.
TCRBJ, TCR β-chain joining (J) region.
The ability of KK10-specific effective CD8+ T-cell clones (EC) and ineffective CD8+ T-cell clones (IC) to kill GXR cells infected with NL4-3 wild-type virus was tested in the standard 4-h chromium release assay, and percent specific lysis was calculated as described in Materials and Methods. CD8+ T-cell clones EC1, EC2, IC1, and IC5 were obtained from elite controller CTR203, CD8+ T-cell clones EC3, EC4, and IC2 were obtained from elite controller FW56, and CD8+ T-cell clones IC3 and IC4 were obtained from chronic progressor CR540 (1). The ineffective functional phenotype of IC5 (as S-T002) has previously been established and reported (1). ND, not done.
FIG 1.
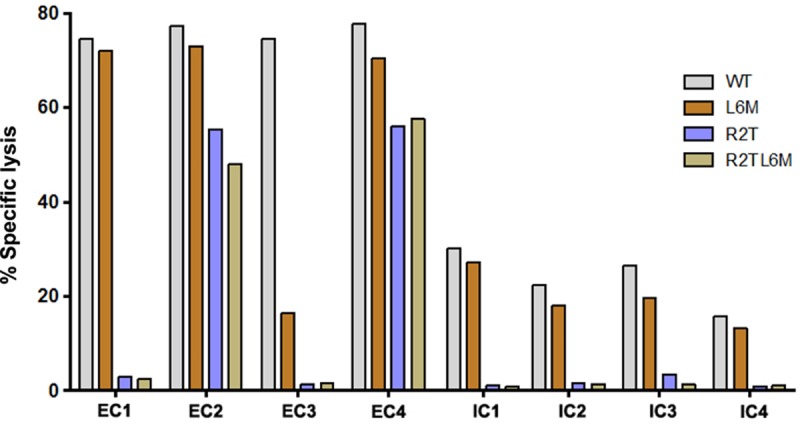
Antiviral function of KK10-specific clonotypes. The ability of KK10-specific CD8+ T-cell clones to recognize NL4-3 WT and variant viruses was tested in the standard 4-h chromium release assay with virally infected (wild-type and variant strains as indicated) HLA-B*2705-encoding green fluorescent protein (GFP) reporter GXR cells at an effector/target cell ratio of 1:1. Viable infected (GFP-positive) GXR cells were sorted by a FACSAria cell-sorting instrument after infection for 5 days and used as target cells.
Phenotypic profiles of KK10-specific effective and ineffective clonotypes.
We next used CyTOF to assess potential associations between CD8+ T-cell clone function and TCR-dependent and TCR-independent activation-induced expression of 20 phenotypic and 13 functional markers (18). Data were analyzed in a manner similar to that used for standard flow cytometry data by FlowJo software as shown in representative scatter plots (Fig. 2). The expression of phenotypic and functional markers was first analyzed by CyTOF after stimulation for 3 h with phorbol myristate acetate (PMA) and ionomycin, a TCR-independent activation signal that directly triggers downstream signal pathways. Effective CD8+ T-cell clones segregated together by cluster analysis, as did ineffective CD8+ T-cell clones, although the differences between effective and ineffective clones in the TCR-independent induction of expression of these markers were modest (Fig. 3A). In contrast, following antigen-specific TCR stimulation for 3 h with cognate KK10 peptide-loaded HLA-B*2705-expressing GXR cells, differences in phenotypic and functional markers between effective and ineffective clonotypes were readily apparent after subtracting the background from stimulations with GXR cells not loaded with peptide. The effective and ineffective clonotypes did not differ significantly in their expression of regulatory markers such as PD-1, CTLA-4, and KLRG-1, activation markers such as CD38, CD57, CD69, and HLA-DR, and differentiation markers such as CD45RA, CD45RO, CD27, CD28, CD62L, and CCR7. In contrast, we observed significantly increased production of the functional cytokines gamma interferon (IFN-γ), macrophage inflammatory protein 1β (MIP-1β), MIP-1α, tumor necrosis factor alpha (TNF-α), and granulocyte-macrophage colony-stimulating factor (GM-CSF) and the degranulation marker CD107a in the effective CD8+ T-cell clones compared to the responses seen in the ineffective CD8+ T-cell clones (P < 0.001) (Fig. 3B). These results indicate that although effective and ineffective clonotypes segregate based on both TCR-independent and TCR-dependent stimulation, the latter is associated with much greater differences in a subset of phenotypic and functional markers associated with antiviral functions.
FIG 2.

Representative CyTOF scatter plots. The effective CD8+ T-cell clone EC1 and ineffective CD8+ T-cell clone IC2 were incubated with either unloaded HLA-B*2705-expressing GXR cells, KK10 peptide-loaded HLA-B*2705-expressing GXR cells or PMA and ionomycin (Iono) for 3 h. The cells were then stained with the indicated metal-tagged antibody specific for IFN-γ and CD107 (upper panels), CD40L and GM-CSF (middle panels), and IL-2 and TNF-α (lower panels) and analyzed by mass cytometry. The data are presented as dot plots.
FIG 3.

CyTOF characterization of functional and phenotypic markers. Stimulation-induced changes in 20 phenotypic and 13 functional markers on clonotypic CD8+ T cells were measured by CyTOF after incubation for 3 h with PMA and ionomycin (A) or KK10 peptide-loaded HLA-B*2705-expressing GXR cells (B). Stimulation-induced changes in intensity of each marker were averaged, and background was subtracted. Similarity between clonotypes was compared by hierarchical cluster analysis. Similarities between the profiles of each marker tested were also clustered.
TCR signal transduction of KK10-specific clonotypes.
We next assessed immediate effector function following cognate epitope recognition by each CD8+ T-cell clone expressing a different TCR clonotype, using phosphoflow cytometry to assess the kinetics and amplitude of the phosphorylation of proteins crucial for TCR signal transduction, such as the p44/42 mitogen-activated protein kinase (MAPK) extracellular signal-regulated kinase 1/2 (ERK1/2) (19). Ten minutes after stimulation with KK10 peptide, the phosphorylated protein forms of p44/42 MAPK in the effective CD8+ T-cell clones were more rapidly mobilized than in the ineffective CD8+ T-cell clones (Fig. 4A and B) (P = 0.028), whereas the effective and ineffective CD8+ T-cell clones displayed a similar mobilization rate of phosphorylated MAPK following TCR-independent stimulation with PMA and ionomycin (P = 0.28). These data indicated marked differences in the kinetics of antigen-specific TCR-initiated signaling events between effective and ineffective CD8+ T-cell clones.
FIG 4.

TCR signal transduction and cytokine kinetics. (A) A representative flow cytometry histogram of phosphorylated protein forms of p44/42 MAPK examined by phosphoflow cytometry at the 10-min time point following stimulation with KK10 peptides or PMA and ionomycin of EC1 and IC1 CD8+ T-cell clones. (B) Comparison of phosphorylation of p44/42 MAPK between effective and ineffective CD8+ T-cell clones was made with the Mann-Whitney test. (C and D) Functional cytokine and chemokine expression was examined by flow cytometry after incubation with KK10 peptide-loaded HLA-B*2705-expressing GXR cells for 0.5 h, 1 h, 2 h, 3 h, and 5 h for effective CD8+ T-cell clone EC1 (C) and ineffective CD8+ T-cell clone IC1 (D).
Subsequent flow-cytometric analysis of dynamic intracellular cytokine profiles demonstrated that a representative effective CD8+ T-cell clone, EC1, rapidly upregulated antiviral cytokines and chemokines such as IFN-γ, MIP-1β, and TNF-α, an upregulation that was paralleled by declining levels of perforin expression (Fig. 4C), indicating the rapid transition from efficient initial lytic to sustained nonlytic function. In contrast to the effective CD8+ T-cell clone, only a fraction of the representative ineffective IC1 CD8+ T-cell clone cells displayed loading of lytic granules and upregulation of these antiviral cytokines and chemokines, and this response was delayed (Fig. 4D). Early perforin expression in the effective clonotypes declined and was followed by significant expression of degranulation marker CD107a (Fig. 4C), consistent with previous findings that effective clonotypes rapidly load and deliver perforin (1), mediating target cell lysis, which is associated with the acquisition of cell surface CD107a (20).
Direct expression of KK10-specific TCRs from representative effective and ineffective clonotypes using TCR expression lentiviral vectors.
To more precisely determine the contribution of the TCR clonotype to the differences observed in CD8+ T-cell clones specific for the same viral peptide but segregated into the effective or ineffective CD8+ T-cell clones, we directly evaluated the functional activity of TCR clonotypes by cloning the full-length TCR α and β chains from representative effective (EC1) and ineffective (IC1 and IC5) CD8+ T-cell clones (Table 1). These CD8+ T-cell clones were isolated from the same elite controller (CTR203) and were specific for KK10 and its L6M variant but not the R2T and R2T/L6M variants (1). The TCR α and β chains cloned from the EC1, IC1, and IC5 CD8+ T-cell clones were expressed in a lentiviral vector (Fig. 5A) encoding a single transcript linked by a P2A self-cleaving peptide that produces equimolar quantities of both TCR chains in transduced T cells and an internal ribosome entry site (IRES)-driven green fluorescent protein (GFP) marker gene to enable identification and quantification of transduced T cells, as described previously (21). A modified CD8+ Jurkat cell line, Jurkat/MA, that does not express endogenous surface TCR, was transduced with the TCR-expressing lentivirus and evaluated for the expression and functional properties of the lentivirus-encoded TCR (22). Greater than 90% of the Jurkat/MA cells were transduced with the individual TCR lentiviruses as indicated by expression of GFP and of surface TCR, which specifically bound to the HLA-B*2705-KK10 dextramer (Fig. 5B). Based on the geometric mean fluorescent intensity (MFI) of dextramer binding, EC1-TCR (MFI = 40,616 relative fluorescence units [RFU]) and IC1-TCR (MFI = 30,072 RFU) were expressed at moderately higher densities than IC5-TCR (MFI = 13,759 RFU), which may be due to the differential stability of different TCR α and β proteins (23).
FIG 5.

Phenotypic and functional analysis of the EC1, IC1, and IC5 TCRs expressed by the transduced Jurkat/MA cells. (A) The TCR α and β genes of effective (EC1) and ineffective (IC1 and IC5) CD8+ T-cell clones were cloned into a lentiviral transfer vector driven by a spleen focus-forming virus (SFFV) viral promoter. TCR α and β genes were linked by a self-cleaving P2A peptide followed by an IRES-driven GFP marker gene to allow visualization of the transduced cells by flow cytometry. (B) Efficiency of Jurkat/MA cell transduction by the GFP-expressing EC1-TCR, ICI-TCR, and IC5-TCR lentiviral vectors and their expression of a KK10-specific TCR were determined by flow-cytometric analysis 72 h after transduction by staining with HLA-B*2705-KK10 dextramer. (C) TCR responsiveness was measured by incubating mock-transduced and EC1-TCR-, IC1-TCR-, or IC5-TCR-transduced Jurkat/MA cells with CTR0075 cells pulsed with the indicated peptide (10 μM) for 16 h and quantifying luciferase activity in cellular lysates. (D) Functional activity of the EC1-TCR, IC1 TCR, and IC5-TCR expressed by the Jurkat/MA cells was determined by stimulating the transduced cells with CTR0075 cells pulsed with the indicated concentration of KK10 peptide or a control influenza virus peptide (10 μM) for 16 h and measuring luciferase activity. The data shown in panels C and D are normalized relative luciferase unit (RLU) values from 3 individual experiments performed in duplicate. We normalized the data from 3 experiments to aid in accurate comparison between individual experiments using the formula Xi, 0 to 1 = (Xi − Xmin)/(Xmax − Xmin), where Xi represents each data point, Xmin is the minimum among all data points in an experiment, Xmax is the maximum among all data points in an experiment, and Xi, 0 to 1 represents the data point i normalized between 0 and 1. The data shown are the mean normalized RLU ± standard errors of the means (SEM) from 3 experiments performed in duplicate. Statistical significance was evaluated using the 2-way analysis of variance (ANOVA) test. *, P < 0.05; ****, P < 0.0001.
Functional activity of the expressed TCR from effective versus ineffective clonotypes.
The stable integration of the TCR-induced NFAT luciferase reporter construct in the Jurkat/MA cell line permitted this cell line to be used to quantify the amplitude of the antigen-specific signal transduced by defined TCRs (21). Jurkat/MA cells were transduced with the indicated lentivirus and incubated with CTR0075 cells, allogeneic HLA-B*2705-expressing B-LCL cells, loaded with either the KK10 peptide, the KK10-L6M variant peptide, or an HLA-B27 control influenza virus peptide (influenza A virus nucleopeptide, SRYWAIRTR) for ∼16 h (24). The responsiveness of the lentivirus-encoded TCR to its cognate pMHC complex was quantified by measuring the amplitude of increased luciferase activity compared to that in mock-transduced cells. In contrast to the minimal reactivity of the Jurkat/MA cells expressing EC1-TCR, IC1-TCR, or IC5-TCR to CTR0075 cells pulsed with the control influenza virus peptide, CTR0075 cells pulsed with the KK10 peptide or the KK10-L6M variant peptide markedly stimulated luciferase activity in the Jurkat/Ma cells expressing EC1-TCR, IC1-TCR, or IC5-TCR (Fig. 5C). The level of luciferase activity induced after EC1-TCR KK10-specific activation was not significantly greater than after IC1-TCR activation (P = 0.30) but was significantly higher than after the IC5-TCR activation (P < 0.05).
We further evaluated the functional activities of the expressed EC1-TCR, IC1-TCR, and IC5-TCR by incubating Jurkat/MA cells transduced with the EC1-TCR, IC1-TCR, or IC5-TCR lentivirus with CTR0075 cells loaded with no peptide, a control influenza virus peptide (10 μM), or KK10 peptide at concentrations that ranged from 0.001 μM to 100 μM. The Jurkat/MA cells transduced with EC1-TCR, IC1-TCR, or IC5-TCR displayed a similar dose-response activation of the TCR-responsive luciferase reporter construct in the Jurkat/MA cells, and there were no differences in magnitude between EC1-TCR and IC1-TCR (Fig. 5D). However, at the higher peptide doses, EC1-TCR and IC1-TCR expressing Jurkat/MA cells displayed reproducibly higher maximal activation of the TCR-responsive luciferase reporter construct than IC5-TCR expressing Jurkat/MA cells, albeit the magnitude of differences was small. Overall, these data indicate a lack of significant differences in TCR-mediated activation between an effective clonotype (EC1) and one of the ineffective clonotypes (IC1), demonstrating that TCR alone does not account for the functional differences observed in the parental clones IC1 and IC5. The difference observed in the comparison of EC1 to IC5 is difficult to interpret, given that the clone with the lower activation (IC5) also had the lowest TCR expression levels.
Primary human CD8+ T cells transduced with lentivectors encoding the EC1, IC1, or IC5 TCR clonotypes display equivalent anti-HIV-1 activity.
We have previously reported that primary naive CD8+ T cells can be reprogrammed into HIV-specific CD8+ cells after transduction with lentivirus encoding the TCR α and TCR β genes from an HLA-A*02-restricted SL9-specific clone (21). We used this approach to evaluate the potency of anti-HIV activity conferred by the TCR clonotypes derived from the representative effective and ineffective HLA-B*27-restricted CD8+ T-cell clones by transducing primary CD8+ T cells isolated from two different HIV-1-naive donors with the EC1, IC1, or IC5-TCR-encoding lentivirus.
After transduction of primary CD8+ T cells with an EC1 or IC1 lentivirus that utilized an IRES to express the GFP marker gene, we observed consistently low levels of transduction and binding of the HLA-B*2705-KK10 dextramer by GFP+ transduced cells (Fig. 6). To increase transduction efficiency and TCR expression, we replaced the IRES sequence expressing the GFP marker gene with a 2A ribosomal “skip” peptide (T2A [Thosea asigna virus 2A]) (Fig. 6A) reported to increase the expression of multiple proteins by a single vector (25). Based on flow cytometric analysis, greater than 95% of Jurkat/MA cells transduced with EC1-TCR-, IC1-TCR-, or the IC5-TCR-encoding lentiviruses linked with a T2A peptide to the GFP marker gene expressed GFP and expressed TCR that bound the HLA-B*2705-KK10 dextramer (Fig. 6B). Primary CD8+ T cells were efficiently transduced with lentivirus encoding EC1, IC1, and EC5 TCR and expressed TCR that bound the HLA-B*2705-KK10 dextramer (Fig. 6C). The transduction efficiency and HLA-B*2705-KK10 dextramer binding in primary CD8+ T cells transduced with the EC1-TCR- and IC1-TCR-encoding lentivirus, which used a T2A-linked GFP marker gene (Fig. 6C), was significantly higher than with the EC1-TCR- and IC1-TCR-encoding lentivirus, which used an IRES (P < 0.05) (Fig. 6D and E). Multiple factors contribute to the discrepancy in transduced CD8+ primary T cells between GFP+ reporter gene expression and the expression of sufficient surface TCR for MHC-KK10 dextramer staining, including TCR expression cassette design, TCR α and TCR β chain gene codon optimization and orientation, the more-rapid degradation of the TCR α chain, the varying stability of individual TCR α and TCR β chains, and mispairing with the endogenous TCR α and TCR β chains (23).
FIG 6.

HIV-naive CD8+ T cells can be transduced and engineered to express HIV-specific TCRs that bind to KK10-specific dextramer. (A) The TCR α and β genes of effective (EC1) and ineffective (IC1 and IC5) CD8+ T-cell clones were cloned into a lentiviral transfer vector driven by a spleen focus-forming virus (SFFV) viral promoter. TCR α and β genes were linked by a self-cleaving P2A peptide followed by a T2A self-cleaving peptide-linked GFP reporter cassette to allow visualization of the transduced cells by flow cytometry. (B) Jurkat/MA cells were transduced with lentiviral vectors expressing EC1-TCR, ICI-TCR, IC5-TCR, or a control glutamic acid decarboxylase peptide (residues 555 to 567)-specific TCR linked with a T2A sequence to a GFP reporter gene or a control lentiviral vector expressing only the GFP reporter gene. Expression of a KK10-specific TCR was determined by flow-cytometric analysis 48 h after transduction after staining with HLA-B*2705-KK10 dextramer or anti-human TCR αβ antibody. (C) Primary human CD8+ T cells, isolated from PBMC of HIV-naive donors, were activated and transduced with lentiviral vectors expressing EC1-TCR, IC1-TCR, or IC5-TCR linked with a T2A sequence to a GFP reporter gene. After 8 days of culture, the CD8+ T cells were evaluated by flow cytometry for transduction by quantifying the fraction of GFP+ cells. After gating on the GFP+ cells, their expression of the KK10-specific TCR was determined by measuring their binding to HLA-B*2705-KK10 dextramer. Results shown are representative of 3 experiments with 2 different donors. SSC, side scatter. (D) Primary human CD8+ T cells were activated and transduced with the lentiviral vectors expressing EC1-TCR or IC1-TCR linked to a GFP reporter gene by a T2A peptide or expressed by an IRES-driven GFP reporter gene. After 4 days of culture, the CD8+ T cells were evaluated by flow cytometry for transduction by quantifying the fraction of GFP+ cells. The values presented are the means ± SEM of the percentage of GFP-positive CD8+ T cells from 9 different experiments using primary CD8+ T cells from 3 different HIV-naive donors. (E) After gating, the GFP+ cells were evaluated for their expression of the KK10-specific TCR by measuring their binding to HLA-B*2705-KK10 dextramer. The values presented are the means ± SEM of the percentage of HLA-B*2705-KK10 dextramer binding-CD8+ T cells from 3 different experiments using primary CD8+ T cells from 2 different HIV-naive donors. *, P < 0.05; **, P < 0.01.
The anti-HIV-1 activity of the primary CD8+ T cells transduced with the EC1, IC1, and IC5-TCR lentivectors was evaluated by using HIV-infected, HLA-B*2705-expressing GXR cells infected with HIV-LucR, an infectious HIV-1 molecular clone expressing a Renilla luciferase reporter gene, as target cells as described previously (26). CD8+ T cells transduced with EC1-TCR, IC1-TCR, and IC5-TCR lentivirus displayed equivalently potent HIV-1-specific inhibitory activity (∼80 to 90%) compared to mock-transduced CD8+ T cells and CD8+ T cells transduced with a control lentivirus expressing the GFP gene alone (Fig. 7A and B). To further evaluate the functional inhibitory capacity of EC1-TCR and IC1-TCR, we used syngeneic HLA-B*2705 CD8+ T-cell-depleted peripheral blood mononuclear cells (PBMC) infected with HIV-LucR as the target HIV-1-infected cells. Primary CD8+ T cells transduced with lentivirus expressing the EC1-TCR and IC1-TCR equivalently inhibited HIV-1 infection (∼60%) in the primary syngeneic B*2705 CD8+ T-cell-depleted PBMC (Fig. 7C). These results indicate that some ineffective CD8+ T-cell clones express TCR with the intrinsic capacity to mediate potent suppression of HIV-1 infection when expressed on primary CD8+ T cells.
FIG 7.

Antiviral capacity of TCR-engineered CD8+ T cells. CD8+ T cells were isolated from PBMC, activated, and mock transduced or transduced with lentivirus expressing EC1-TCR, IC1-TCR, or IC5-TCR and a GFP reporter linked by a T2A peptide or a control lentivirus expressing only the GFP reporter gene. (A) Five days after transduction, the cells were added to GXR cells previously infected with HIV-LucR for 24 h at a CD8+ T cell/GXR cell ratio of 1:1. After 3 days, the cells were harvested and the luciferase activities in the cellular lysates were determined and reported as RLU ± SEM. Results from a representative experiment performed in quadruplicate are shown. (B) The individual means of 4 experiments performed as described for panel A with the data for the experiments normalized as percent suppression of HIV-1 infection and the total means from the 4 independent experiments ± SEM are shown. (C) CD8+ T cells mock transduced or transduced with lentiviral vectors expressing EC1-TCR or IC1-TCR were added to syngeneic HLA-B27-expressing activated CD8+ T cell-depleted PMBC 2 days after they were infected with HIV-LucR. Six days later, the luciferase activity in the cellular lysates was determined and reported as RLU ± SEM. Statistical significance was evaluated using the ordinary one-way ANOVA test. ns, P > 0.05; **, P < 0.01; ****, P < 0.0001.
DISCUSSION
Multiple studies have sought to define quantitative and qualitative differences in CD8+ T-cell responses that may correlate with different outcomes in terms of disease course and immune control of HIV-1 (2). Simple quantitative measures of HIV-specific CD8+ T-cell populations have shown little correlation with viral control (27, 28), suggesting that qualitative features of HIV-specific CD8+ T cells may correlate with in vivo control. Qualitative factors potentially modulating CD8+ T-cell responses include, among others, polyfunctionality (29), antigen sensitivity or functional avidity (30, 31), proliferative capacity (32), loading of lytic granules (33, 34), specific targeting of conserved regions (35, 36), immunoregulatory mechanisms, including CD8+ T-cell exhaustion (37–39), concurrent responses to multiple epitopes restricted by different HLA alleles (40), and CD8+ T-cell-associated mutations that impair viral fitness (41, 42) and immune escape (43). Some studies also suggest that properties of the TCR-pMHC interaction may play a role in CD8+ T-cell functional activity (44, 45).
We previously reported that HLA-B*27-restricted and HLA-B*57-restricted CD8+ T-cell clones targeting the same epitope in elite controllers and progressors can be clearly differentiated based on potency and cross-reactivity of TCR recognition of HIV-1 and viral variants, which is in turn related to specific TCR clonotypes that are selected during natural HIV-1 infection (1). One factor that may contribute to the elite controller phenotype is the “fortunate” selection and expansion of anti-HIV CD8+ T cells expressing broadly reactive TCRs during the evolution of the KK10-specific CD8+ T-cell response that does not occur in progressors. Supporting this scenario was our isolation of effective clonotypes such as EC2 and EC4, which were also characterized by broad cross-reactivity against viral variants; these broadly reactive CD8+ T cells are well represented in controllers but rare or absent in progressors, whereas ineffective clonotypes such as IC1, IC2, IC3, IC4, and IC5 are present in both progressors and controllers. However, other factors likely contribute to the effective CD8+ T-cell phenotype as indicated by EC1, which reacted with only the KK10 and L6M variant, and EC3, which was predominately reactive with the unmutated KK10 peptide (Fig. 1). We and others have further demonstrated that the antiviral efficacy and cross-reactivity of the clonotypes were determined by different recognition and binding patterns among TCR-pMHC interactions by crystallographic and computational studies (6, 8). All-atom molecular dynamics simulations of TCR-KK10-MHC complexes revealed a structural association with the clonotypic differences in CD8+ T-cell phenotypes and functions. Although both effective and ineffective clonotypes bind to the N- and C-terminal portions of the KK10-MHC through similar salt bridges, specific hydrophobic side chain interactions with the TCR are the major force associated with the observed superior antiviral efficacy of certain TCR clonotypes (6, 8).
The nature of the TCR-pMHC interactions directs the physical recruitment of signaling pathways differentially inside the lymphocyte, which impacts on the kinetics of signal propagation in various segments of the TCR signaling network that ultimately influence the responsiveness of T cells (46–48), but the mechanistic basis for this difference in signal transduction due to TCR variants is not established. We observed that effective and ineffective clones were more greatly differentiated by KK10-specific activation than by TCR-independent activation and that effective clones phosphorylated p44/42 MAPK more rapidly than ineffective clones when stimulated with their cognate pMHC (Fig. 4A). This correlates with our previous findings that the effective CD8+ T-cell clones are able to rapidly upregulate perforin and granzyme B, polarize them at the immunologic synapse, and deliver them to the infected target cells after incubation with HIV-1-infected HLA-B*2705-encoding GXR cells for 30 min (1). This also explains why we did not observe much perforin and granzyme B in effective clonotypes by CyTOF after culture with KK10 peptide-loaded HLA-B*2705-encoding GXR cells for 3 h; this time point likely followed the rapid delivery of perforin and granzyme B, which is indicated by the rapid upregulated expression of CD107a that we observed on the effective CD8+ T-cell clones following stimulation for 3 h (Fig. 4C). CD107a expression is associated with loss of intracellular perforin, can be observed as early as 30 min following stimulation of primary CD8+ T cells, and reaches maximum expression by 4 h after activation (20). During this phase, effective CD8+ T-cell clones upregulate secretion of antiviral cytokines and chemokines, such as IFN-γ, MIP-1β, MIP-1α, and TNF-α, and readily transit from efficient initial lytic to sustained nonlytic function, whereas the ineffective CD8+ T-cell clones lack this ability.
In order to more precisely address the specific roles of TCR clonotypes in the observed functional differences, we directly cloned and expressed the TCRs from one effective CD8+ T-cell clone and two ineffective CD8+ T-cell clones from a single donor, all with an identical specificity for WT KK10 and L6M KK10 but not R2T KK10. We demonstrated using Jurkat/MA cells transduced with TCR-expressing lentiviral vectors that the EC1 and IC1 TCR clonotypes displayed comparable TCR responsiveness to HLA-B*2705-expressing target cells loaded with the KK10 peptide or the KK10 L6M variant peptide, while the maximal response of the IC5 TCR clonotype was less than that of EC1 and IC1 (Fig. 5C and D). Although this may reflect true TCR-specific modulation of effector function between the effective and ineffective clonotypes, it may also be due to the lower levels of expression of IC5 TCR than of EC1 and IC1 TCR (Fig. 6B), which may be a consequence of differential intrinsic stability of different TCR α and β chains (23). Our data do indicate that the phenotype of an ineffective clonotype is not transferred by the TCR alone in all cases, since there were no functional differences observed between cells transfected with the EC1 and IC1 TCRs, using either TCR-transduced Jurkat/MA cells (Fig. 6) or primary CD8+ T cells (Fig. 7).
While we clearly show that a TCR cloned from an ineffective clone has equivalent function to that of a TCR cloned from an effective clone, a limitation of this study is that it evaluates only cloned TCRs from one effective and two ineffective clones from the same individual. Another limitation is that we studied the function of HIV-specific TCRs expressed by lentiviral vectors in primary CD8+ T cells, which may not precisely recapitulate the behavior of HIV-specific CD8+ T cells because of inherent restrictions of lentivirus-encoded TCR expression, including vector design, orientation of the expression cassette, and the impact of mispairing with the endogenous TCR α and β chains of the transduced CD8+ T cell (23). The impact of TCR chain mispairing likely contributed to the disparity in TCR expression between Jurkat/MA cells, which do not express endogenous TCR and among which almost all GFP+ cells bound to the HLA-B*2705-KK10 dextramer (Fig. 6B), and the GFP+ primary CD8+ T cells, of which only about 20 to 25% bound dextramer (Fig. 6C). Nevertheless, an adequate number of primary CD8+ T cells expressed sufficient lentivirus-encoded TCR to enable us to demonstrate that the EC1 TCR and IC1 TCR, and perhaps to a lesser extent the IC5 TCR, conferred upon primary CD8+ T cells the capacity to potently inhibit HIV-1 infection.
Alternate TCR-independent mechanisms may modulate CD8+ T-cell function, including the introduction of epigenetic changes into gene-regulatory elements of CD8+ T-cell differentiation and function that can be transmitted to daughter cells and triggered by acute viral infection (49–53). For example, commitment of virus-specific CD8+ T cells to an exhausted phenotype is reinforced by persistent demethylation of the PD-1 locus as described for lymphocytic choriomeningitis virus (LCMV) and HIV infections (54–56). Thus, the effectiveness of the antiviral response of CD8+ T cells experiencing sustained high levels of TCR signaling during chronic virus infection may be compromised by epigenetic modifications that permanently repress transcription, which may be overcome by the lentivector-mediated expression of the ineffective CD8+ T-cell clone TCR in CD8+ T cells from an HIV-1-naive donor that are not carrying these epigenetic modifications. It is possible that the stimulation of epigenetic modifications by chronic in vivo stimulation that confer an ineffective phenotype upon CD8+ T cells may preferentially occur in CD8+ T cells expressing some TCR clonotypes but not others, enabling them to continue to function as effective CD8+ T cells.
The results reported here also support the capacity of lentiviral vectors to transform naive CD8+ T cells into potent HIV-specific CD8+ T cells as a possible adjunctive therapy to control and eliminate HIV-infected cells, potentially including reactivated latently infected cells, thereby contributing to achieving a functional cure. For optimal therapeutic efficacy, an approach needs to be developed to maximize expression of the lentivirus-encoded TCR in the transduced primary CD8+ T cells. Treatment of HIV-1-infected individuals with infusions of autologous CD8+ T cells transduced ex vivo with lentivectors encoding broadly reactive HIV-1-specific TCRs isolated from a broadly directed CD8+ T-cell clone such as EC2 and EC4 would be expected to delay the emergence of immune escape variants and thereby to confer increased immune control as observed in elite controllers. It is also possible that TCRs derived from these effective CD8+ T-cell clones may display prolonged in vivo anti-HIV-1 activity due to resistance to inhibitory and regulatory mechanisms, which would compromise their function as reported for the association between increased functional avidity of HIV-specific CD8+ T cells and T-cell exhaustion (57).
Taken together, these findings suggest that factors other than TCR structure likely contribute to the ineffective phenotype of some CD8+ T-cell clones. Determining these factors may enable the development of in vivo treatments to convert ineffective HIV-specific CD8+ T cells into effective CD8+ T cells and to convert infected individuals who are chronic progressors into elite controllers.
MATERIALS AND METHODS
Generation of CD8+ T-cell clones.
PBMC were isolated from HIV-infected individuals and stained with fluorophore-labeled HLA-B*2705 KK10 tetramer (ProImmune, Oxford, UK) and fluorophore-labeled anti-CD8 and anti-CD3 antibodies. Tetramer-positive, CD8+ T cells were sorted on a FACSAria cell-sorting instrument (BD Biosciences, San Jose, CA) at 70 lb/in2, and single cells were placed into each well of 96-well plates, using irradiated allogeneic PBMC and CD3-specific monoclonal antibody (MAb) 12F6 to stimulate T-cell proliferation (58). Developing HLA-B*2705-restricted, KK10-specific clones were identified by IFN-γ enzyme-linked immunospot (ELISPOT) assays after stimulation with optimal epitopes and by tetramer staining. Cloned HLA-B*2705-restricted, KK10-specific CD8+ T cells were maintained by restimulation every 14 to 21 days with an anti-CD3 MAb and irradiated allogeneic PBMC in RPMI 1640 medium with added heat-inactivated fetal bovine serum (10%, vol/vol), 100 U/ml penicillin, 10 μg/ml streptomycin, 2 mM glutamine, and 10 mM HEPES buffer (R-10) supplemented with 50 units/ml recombinant interleukin-2 (IL-2) (R-10-50) (58). TCR clonotypes were determined by TCR sequencing, as described previously (59).
Cell lines and isolation of CD8+ T cells from human peripheral blood mononuclear cells.
The Jurkat/MA cells are molecularly engineered Jurkat T cells that do not express endogenous surface TCR and express CD8 and an NFAT-regulated luciferase reporter gene (22) and were cultured as described previously (24). CTR0075 is an Epstein-Barr virus (EBV)-transformed B cell line isolated from an HIV-infected patient with the HLA type of A*0201/0301, B*1501/2705, Cw*0102/0304. GXR is an HLA-B*2705-expressing CEM-derived cell line stably transduced with a plasmid encoding green fluorescent protein (GFP) driven by the long terminal repeat of HIV-1 and was constructed as described previously (60). Purified CD8+ T cells were isolated from peripheral blood mononuclear cells by immunomagnetic sorting using CD8 microbeads (Miltenyi Biotec, San Diego, CA) according to the manufacturer's instructions.
Chromium release assay.
GXR cells were infected at the specified multiplicity of infection (MOI) with HIV-1NL4-3 wild-type or designated HIV-1NL4-3 KK10-variant viruses generated by site-directed mutagenesis to introduce one or more mutations in the region of the gene encoding the Gag p24 KK10 peptide (KRWIILGLNK, Gag amino acids [aa] 263 to 272) (61). On day 5 after infection, viable GFP-expressing HIV-1-infected cells were isolated on a FACSAria cell sorter and loaded with radiolabeled 51chromium for 1 h at 37°C. CD8+ T-cell clones were then mixed with the target GXR cells at the indicated effector-target ratios, and cytotoxic activity of the CD8+ T-cell clones was determined using a standard 4-hour chromium release assay as described previously (62). Percent specific lysis was calculated as [(mean experimental cpm − mean spontaneous cpm)/(mean maximum cpm − mean spontaneous cpm)] × 100, where cpm is counts per minute. Spontaneous and maximum releases were determined by incubating the labeled target cells with medium alone or 2% Triton X-100, respectively.
Phosphoflow cytometry.
KK10-specific CD8+ T-cell clones were stimulated with HLA-B*2705-expressing GXR cells loaded with KK10 peptides (1 μM) for 10 min and fixed in cold Cytofix buffer (BD Biosciences). Cells were permeabilized using Perm/Wash buffer III (BD Biosciences), stained with the appropriate phospho-specific antibodies (Cell Signaling Technology, Danvers, MA), washed thoroughly, and then analyzed on an LSRFortessa flow cytometer (BD Biosciences).
Flow cytometry.
Cells were stained with fluorochrome-labeled antibodies to the indicated phenotypic markers for 30 min at room temperature, washed, and fixed using the Cytofix/Cytoperm kit (BD Biosciences) according to the manufacturer's instructions. Following fixation, the cells were washed twice with the Cytofix/Cytoperm wash buffer and stained with antibodies against intracellular proteins. Following staining, the cells were fixed by resuspension in phosphate-buffered saline (PBS) containing 2% paraformaldehyde. Cellular fluorescence was evaluated by an LSRFortessa cytometer (BD Biosciences), and the data were analyzed with the FlowJo software package (FlowJo, Ashland, OR).
Mass cytometry (CyTOF).
Stimulation and staining of the KK10-specific CD8+ T-cell clones and analysis of data were performed as described previously (18). Briefly, CD8+ T-cell clones were cultured for 3 h in R-10 medium containing brefeldin (5 μg/ml), monensin (5 μg/ml), and anti-CD107a (2.5 μg/ml). For KK10 peptide stimulation, CD8+ T-cell clones were cultured with HLA-B*2705-expressing GXR cells preloaded with KK10 peptide (1 μM) for 45 min at an effector cell/stimulator cell ratio of 1:1 at 37°C or GXR cells not loaded with peptide as a negative control. At the end of the 3-h stimulation, cells were washed, resuspended in flow cytometry buffer (PBS–0.05% sodium azide–2 mM EDTA–2% fetal calf serum), and stained for 30 min on ice with a prepared cocktail of metal-conjugated surface marker antibodies. After surface staining, cells were washed, resuspended in DM-115 (20 μM; Fluidigm, San Francisco, CA) for 30 min on ice, washed three times in flow cytometry buffer, and then resuspended in PBS containing 2% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA). After fixation at 4°C overnight, the cells were washed twice in intracellular staining permeabilization buffer (eBioscience, San Diego, CA), stained with a cocktail of antibodies to phenotypic and functional markers (Table 2) on ice for 45 min, washed twice in flow cytometry buffer, and labeled for 20 min at room temperature with 250 nM iridium interchelator (Fluidigm) suspended in PBS containing 2% paraformaldehyde. The cells were washed twice in flow cytometry buffer, twice in PBS, and twice in distilled water before dilution to the appropriate concentration required to achieve an acquisition rate of <500 events/second on the CyTOF instrument. Data were analyzed in a manner similar to that used for standard flow cytometry data by FlowJo software (FlowJo). Bioexponential transformed intensity values of each marker were averaged, and similarity between KK10-specific CD8+ T-cell clonotypes was analyzed by hierarchical cluster analysis (using Matlab and Euclidean distance). After the average intensity values were plotted as a heat plot (maximal intensity in red, minimal intensity in blue), the differences in average between stimulated and unstimulated cells were normalized to the largest observed differences for each plot and represented as heat plots (large positive differences in red, small/negative differences in blue).
TABLE 2.
Specific antibodies used for CyTOF staining and analysis
| Antibody | Label atomic mass | Ab clone; source |
|---|---|---|
| CD3 | QDot (112, 114, etc.) | S1.4; Invitrogen Qdot655 |
| IL-8 | 139 | E8N1; Biolegend |
| CD45RA | 141 | HI100; eBioscience |
| CD69 | 142 | MCA1442; AbD Serotec |
| CD5 | 143 | UCHT2; Biolegend |
| CD45RO | 144 | UCHL1; Biolegend |
| CD57 | 145 | HCD57; Biolegend |
| CD8 | 146 | HIT8a; Biolegend |
| GM-CSF | 147 | BVD2-21C11; Biolegend |
| CD11a | 148 | HI111; Biolegend |
| CD4 | 149 | SK3; Biolegend |
| MIP-1β | 150 | D21-1351; BD custom order |
| Granzyme B | 151 | 2CF/F5; BD |
| TNF-α | 152 | MAb11; eBioscience |
| CD107a/b | 153 | H4A3, H4B4; BD |
| CD27 | 154 | LG.7F9; eBioscience |
| PD1 | 155 | EH12.2H7; Biolegend |
| CD13 | 156 | WM15; Biolegend |
| CD19 | 157 | HiB19; Biolegend |
| KLRG1 | 158 | 13F12F2; kind gift from Hanspeter Pircher |
| CD56 | 159 | HiB19; Biolegend |
| CD28 | 160 | CD28.2; BD |
| CD38 | 161 | HIT2; eBioscience |
| IL-4 | 162 | MP4-25D2 |
| cCAS3 | 163 | C92-605; BD |
| IL-17 | 164 | BL168; Biolegend |
| CD40L | 165 | 24-31; Biolegend |
| IL-2 | 166 | MQ1-17h12; eBioscience |
| Integrin B7 | 167 | FIB504; Biolegend |
| CCR7 | 168 | 150503; R&D |
| MIP-1α | 176 | 11A3; BD custom order |
| IFN-γ | 170 | 4S.B4; eBioscience |
| HLA-DR | 171 | L243; BD |
| CD49d | 172 | 9F10; Biolegend |
| CTLA-4 | 173 | BN13; Biolegend |
| CD62L | 174 | DREG-56; BD |
| Perforin | 175 | B-D48; AbCam |
| Granzyme A | 176 | CB9; Biolegend |
Cloning of TCR α and β chains and construction of lentiviral vectors for clonotype TCR expression.
The HLA-B*2705-restricted, KK10-specific TCR gene families and variable regions were sequenced from RNA isolated from a representative effective (EC1, previously reported as clone S-C003) and two ineffective CD8+ T-cell clones (IC1 and IC5, previously reported as clones S-C007 and S-T002) cloned from KK10-specific CD8+ T cells obtained from the same elite controller (CTR203) (1). The ineffective functional phenotype of IC5 (as S-T002) has previously been established and reported (1). EC1 (TRAV8-4*03/TRBV-25*01), IC1 (TRAV8-4*03/TRBV20*01), and IC5 (TRAV8-4*03/TRBV7-9*03) TCR α and TCR β sequences were cloned as a single transcript linked by a “self-cleaving” picornavirus 2A (P2A) peptide, which permits equimolar translation of the TCRα and TCRβ chains regulated by the spleen focus-forming virus (SFFV) promoter, followed by an IRES-driven or T2A peptide-linked GFP marker gene as previously described (21, 63, 64). As a control, we used a lentiviral vector expressing a human TCR specific for a peptide derived from the type 1 diabetes autoantigen glutamic acid decarboxylase (GAD555–567) linked to a GFP marker gene by a T2A peptide (65).
Third-generation vesicular stomatitis virus pseudotyped glycoprotein (VSV-g) lentiviral vectors expressing the TCR in transduced cells were generated by calcium-mediated cotransfection of 293T cells with four plasmids: a TCR expression cassette, a construct expressing Rev, a packaging construct expressing the gag and pol genes, and a construct expressing a cytomegalovirus (CMV) promoter-driven VSV-g envelope as described previously (66).
Transduction of Jurkat/MA cells and evaluation of TCR expression by flow cytometry.
Jurkat/MA cells were plated in 24-well plates (5 × 105/well) and the indicated lentivirus at an MOI of 10 in R-10. The plate was spinoculated at 24,000 rpm for 30 min at 24°C and incubated at 37°C overnight. Fresh R-10 was added, and the transduced cells were cultured for an additional 48 h. TCR expression by transduced Jurkat/MA cells was evaluated by flow cytometric analysis on an LSRII flow cytometer (BD Biosciences) using Flowjo software after staining with phycoerythrin (PE)-labeled-HLA*B2705-KK10 dextramer (Immudex, Copenhagen, Denmark) for 10 min at room temperature or PE-labeled anti-TCRαβ antibody (Biolegend, San Diego, CA) for 30 min at 4°C and washing with PBS.
Evaluation of TCR function using Jurkat/MA cells expressing an NFAT-regulated luciferase reporter gene.
The functional activity of the KK10-specific TCR clonotypes was evaluated by quantification of the activation of the NFAT-luciferase reporter gene in response to the KK10 (KRWIILGLNK) or KK10-L6M variant peptide (KRWIIMGLNK) (variation underlined) as described previously (21). Jurkat/MA cells were plated (2.5 × 105 cells/well) in a 48-well tissue culture plate or a 96-well plate (1 × 105 cells/well) in R-10 and mock transduced or transduced with EC1-TCR-, IC1-TCR-, or IC5 TCR-expressing lentivirus and cocultured with an equivalent number of CTR0075 HLA-B*2705 antigen-presenting cells pulsed with 10 μM the indicated peptide (24). For the peptide titration, the transduced Jurkat/MA cells were plated in a 96-well plate (1 × 105 cells/well) in R-10 and mock transduced or transduced with either the EC1-TCR-, IC1-TCR-, or IC5-TCR-expressing lentivirus and cocultured with an equivalent number of CTR0075 HLA-B*2705 antigen-presenting cells pulsed with 10-fold dilutions of the indicated KK10 peptide (24). Cells from duplicate cultures were harvested ∼16 h later, and the luciferase activity in the cellular lysates was determined using the firefly luciferase assay system (Promega, Madison, WI) and quantified using a Luminat Plus luminometer (Berthold Technologies, Oak Ridge, TN).
Evaluation of the capacity of the TCR clonotypes to inhibit in vitro HIV-1 infection.
CD8+ T cells isolated by immunomagnetic sorting (Miltenyi Biotec) from PBMC from an HIV-naive donor were plated in a 24-well plate (5 × 105 cells/well) and activated with anti-CD28 antibody (1 μg/ml), anti-CD3 antibody (100 ng/ml; Orthoclone OKT3), and IL-2 (100 units/ml). Two days later, the indicated lentivirus (MOI, 10) and Polybrene (4 μg/ml) were added to each well, and the plate was spinoculated at 24,000 rpm for 60 min at 24°C and then cultured at 37°C overnight. R-10-100 was added the following day, and the cells were cultured for an additional 3 days with a transduction efficiency ranging from ∼35% to 45% based on GFP expression. KK10-specific TCR expression by the transduced CD8+ T cells was determined by sequential staining with PE-labeled HLAB*2705/KK10 dextramer for 10 min at room temperature and with Pacific Blue-labeled anti-CD8a antibody (BD Biosciences) for 20 min at 4°C followed by washing with PBS and analysis by flow cytometry. In parallel, HLA-B*2705-expressing GXR cells (1 × 105 cells/well) were infected in 96-well plates with HIV-LucR (MOI, ∼0.5), an infectious HIV-1 molecular clone that expresses the HIV-1JR-CSF Env and a Renilla reniformis luciferase (LucR) reporter gene (67, 68). One day later, the HIV-1-infected GXR cells and either mock-transduced or lentivirus-transduced CD8+ T cells were added at an effector-to-target ratio of 1:1 in quadruplicate and cultured for an additional 3 days. HIV-1 infection was quantified by harvesting the cells, lysing them, and measuring luciferase activity in the cellular lysate using the Promega Renilla Luciferase Assay system (26).
Alternatively, PBMC were obtained from an HIV-naive HLA-B*2705 donor, CD8+ T cells were isolated by immunomagnetic sorting and transduced with the indicated lentivirus as described above, and the CD8+ T cell-depleted fraction was activated with phytohemagglutinin (PHA; 4 μg/ml), cultured (1 × 105 cells/well) in R-10-100 in a 96-well plate, and infected with HIV-LucR (MOI, ∼0.5). Two days after infection, the mock-transduced and lentivirus-transduced syngeneic CD8+ T cells (1 × 105 cells) were added to triplicate wells containing the infected CD8+ T-cell-depleted PBMC. Six days later, HIV-1 infection was quantified by measuring luciferase activity in the cellular lysates.
Study approval.
All the studies were performed under protocols approved by the Institutional Review Boards at the Albert Einstein College of Medicine and the Massachusetts General Hospital in compliance with the human experimentation guidelines of the U.S. Department of Health and Human Services.
ACKNOWLEDGMENTS
This work was supported by the National Institute of Drug Abuse at the National Institutes of Health (DA033788, R01DA036171 to H.G.), the National Institute of Allergy and Infectious Diseases at the National Institutes of Health (UM1AI26617 to H.G., T32-AI007501 to T.D.G.), the Charles Michael Chair in Autoimmune Diseases (to H.G.), the National Institute of Allergy and Infectious Diseases at the National Institutes of Health (AI030914 to B.D.W.), and the Howard Hughes Medical Institute (B.D.W.). Support was also provided by the Einstein-Rockefeller-CUNY Center for AIDS Research (P30-AI124414) and the Harvard University Center for AIDS Research (P30-AI060354), which are supported by the following NIH Co-Funding and Participating Institutes and Centers: NIAID, NCI, NICHD, NHBL, NIDA, NIMH, NIA, FIC, and OAR.
Christina Ochsenbauer (University of Alabama at Birmingham) kindly provided the plasmid for the LucR reporter gene-expressing HIV-1 infectious molecular clone.
REFERENCES
- 1.Chen H, Ndhlovu ZM, Liu D, Porter LC, Fang JW, Darko S, Brockman MA, Miura T, Brumme ZL, Schneidewind A, Piechocka-Trocha A, Cesa KT, Sela J, Cung TD, Toth I, Pereyra F, Yu XG, Douek DC, Kaufmann DE, Allen TM, Walker BD. 2012. TCR clonotypes modulate the protective effect of HLA class I molecules in HIV-1 infection. Nat Immunol 13:691–700. doi: 10.1038/ni.2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feinberg MB, Ahmed R. 2012. Born this way? Understanding the immunological basis of effective HIV control. Nat Immunol 13:632–634. doi: 10.1038/ni.2351. [DOI] [PubMed] [Google Scholar]
- 3.Tubo NJ, Pagan AJ, Taylor JJ, Nelson RW, Linehan JL, Ertelt JM, Huseby ES, Way SS, Jenkins MK. 2013. Single naive CD4+ T cells from a diverse repertoire produce different effector cell types during infection. Cell 153:785–796. doi: 10.1016/j.cell.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iglesias MC, Almeida JR, Fastenackels S, van Bockel DJ, Hashimoto M, Venturi V, Gostick E, Urrutia A, Wooldridge L, Clement M, Gras S, Wilmann PG, Autran B, Moris A, Rossjohn J, Davenport MP, Takiguchi M, Brander C, Douek DC, Kelleher AD, Price DA, Appay V. 2011. Escape from highly effective public CD8+ T-cell clonotypes by HIV. Blood 118:2138–2149. doi: 10.1182/blood-2011-01-328781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Janbazian L, Price DA, Canderan G, Filali-Mouhim A, Asher TE, Ambrozak DR, Scheinberg P, Boulassel MR, Routy JP, Koup RA, Douek DC, Sekaly RP, Trautmann L. 2012. Clonotype and repertoire changes drive the functional improvement of HIV-specific CD8 T-cell populations under conditions of limited antigenic stimulation. J Immunol 188:1156–1167. doi: 10.4049/jimmunol.1102610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ladell K, Hashimoto M, Iglesias MC, Wilmann PG, McLaren JE, Gras S, Chikata T, Kuse N, Fastenackels S, Gostick E, Bridgeman JS, Venturi V, Arkoub ZA, Agut H, van Bockel DJ, Almeida JR, Douek DC, Meyer L, Venet A, Takiguchi M, Rossjohn J, Price DA, Appay V. 2013. A molecular basis for the control of preimmune escape variants by HIV-specific CD8(+) T cells. Immunity 38:425–436. doi: 10.1016/j.immuni.2012.11.021. [DOI] [PubMed] [Google Scholar]
- 7.Mendoza D, Royce C, Ruff LE, Ambrozak DR, Quigley MF, Dang T, Venturi V, Price DA, Douek DC, Migueles SA, Connors M. 2012. HLA B*5701-positive long-term nonprogressors/elite controllers are not distinguished from progressors by the clonal composition of HIV-specific CD8+ T cells. J Virol 86:4014–4018. doi: 10.1128/JVI.06982-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xia Z, Chen H, Kang SG, Huynh T, Fang JW, Lamothe PA, Walker BD, Zhou R. 2014. The complex and specific pMHC interactions with diverse HIV-1 TCR clonotypes reveal a structural basis for alterations in CTL function. Sci Rep 4:4087. doi: 10.1038/srep04087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stinchcombe JC, Griffiths GM. 2003. The role of the secretory immunological synapse in killing by CD8+ CTL. Semin Immunol 15:301–305. doi: 10.1016/j.smim.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 10.Pipkin ME, Lieberman J. 2007. Delivering the kiss of death: progress on understanding how perforin works. Curr Opin Immunol 19:301–308. doi: 10.1016/j.coi.2007.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prakken B, Wauben M, Genini D, Samodal R, Barnett J, Mendivil A, Leoni L, Albani S. 2000. Artificial antigen-presenting cells as a tool to exploit the immune ‘synapse’. Nat Med 6:1406–1410. doi: 10.1038/82231. [DOI] [PubMed] [Google Scholar]
- 12.Lin J, Weiss A. 2001. T cell receptor signalling. J Cell Sci 114:243–244. [DOI] [PubMed] [Google Scholar]
- 13.van Leeuwen JE, Samelson LE. 1999. T cell antigen-receptor signal transduction. Curr Opin Immunol 11:242–248. doi: 10.1016/S0952-7915(99)80040-5. [DOI] [PubMed] [Google Scholar]
- 14.Myung PS, Boerthe NJ, Koretzky GA. 2000. Adapter proteins in lymphocyte antigen-receptor signaling. Curr Opin Immunol 12:256–266. doi: 10.1016/S0952-7915(00)00085-6. [DOI] [PubMed] [Google Scholar]
- 15.Cannons JL, Schwartzberg PL. 2004. Fine-tuning lymphocyte regulation: what's new with tyrosine kinases and phosphatases? Curr Opin Immunol 16:296–303. doi: 10.1016/j.coi.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 16.Marie-Cardine A, Schraven B. 1999. Coupling the TCR to downstream signalling pathways: the role of cytoplasmic and transmembrane adaptor proteins. Cell Signal 11:705–712. doi: 10.1016/S0898-6568(99)00047-9. [DOI] [PubMed] [Google Scholar]
- 17.Gett AV, Sallusto F, Lanzavecchia A, Geginat J. 2003. T cell fitness determined by signal strength. Nat Immunol 4:355–360. doi: 10.1038/ni908. [DOI] [PubMed] [Google Scholar]
- 18.Newell EW, Sigal N, Bendall SC, Nolan GP, Davis MM. 2012. Cytometry by time-of-flight shows combinatorial cytokine expression and virus-specific cell niches within a continuum of CD8+ T cell phenotypes. Immunity 36:142–152. doi: 10.1016/j.immuni.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stefanova I, Hemmer B, Vergelli M, Martin R, Biddison WE, Germain RN. 2003. TCR ligand discrimination is enforced by competing ERK positive and SHP-1 negative feedback pathways. Nat Immunol 4:248–254. doi: 10.1038/ni895. [DOI] [PubMed] [Google Scholar]
- 20.Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M, Koup RA. 2003. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods 281:65–78. doi: 10.1016/S0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 21.Joseph A, Zheng JH, Follenzi A, DiLorenzo T, Sango K, Hyman J, Chen K, Piechocka-Trocha A, Brander C, Hooijberg E, Vignali DA, Walker BD, Goldstein H. 2008. Lentiviral vectors encoding human immunodeficiency virus type I (HIV-1)-specific T-cell receptor genes efficiently convert peripheral blood CD8 T lymphocytes into cytotoxic T lymphocytes with potent in vitro and in vivo HIV-1-specific inhibitory activity. J Virol 82:3078–3089. doi: 10.1128/JVI.01812-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Calogero A, Hospers GA, Kruse KM, Schrier PI, Mulder NH, Hooijberg E, de Leij LF. 2000. Retargeting of a T cell line by anti MAGE-3/HLA-A2 alpha beta TCR gene transfer. Anticancer Res 20:1793–1799. [PubMed] [Google Scholar]
- 23.Banu N, Chia A, Ho ZZ, Garcia AT, Paravasivam K, Grotenbreg GM, Bertoletti A, Gehring AJ. 2014. Building and optimizing a virus-specific T cell receptor library for targeted immunotherapy in viral infections. Sci Rep 4:4166. doi: 10.1038/srep04166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scholten KBJ, Schreurs MWJ, Ruizendaal JJ, Kueter EWM, Kramer D, Veenbergen S, Meijer C, Hooijberg E. 2005. Preservation and redirection of HPV16E7-specific T cell receptors for immunotherapy of cervical cancer. Clin Immunol 114:119–129. doi: 10.1016/j.clim.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 25.de Felipe P, Luke GA, Hughes LE, Gani D, Halpin C, Ryan MD. 2006. E unum pluribus: multiple proteins from a self-processing polyprotein. Trends Biotechnol 24:68–75. doi: 10.1016/j.tibtech.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 26.Naarding MA, Fernandez N, Kappes JC, Hayes P, Ahmed T, Icyuz M, Edmonds TG, Bergin P, Anzala O, Hanke T, Clark L, Cox JH, Cormier E, Ochsenbauer C, Gilmour J. 2014. Development of a luciferase based viral inhibition assay to evaluate vaccine induced CD8 T-cell responses. J Immunol Methods 409:161–173. doi: 10.1016/j.jim.2013.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Betts MR, Ambrozak DR, Douek DC, Bonhoeffer S, Brenchley JM, Casazza JP, Koup RA, Picker LJ. 2001. Analysis of total human immunodeficiency virus (HIV)-specific CD4(+) and CD8(+) T-cell responses: relationship to viral load in untreated HIV infection. J Virol 75:11983–11991. doi: 10.1128/JVI.75.24.11983-11991.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Addo MM, Yu XG, Rathod A, Cohen D, Eldridge RL, Strick D, Johnston MN, Corcoran C, Wurcel AG, Fitzpatrick CA, Feeney ME, Rodriguez WR, Basgoz N, Draenert R, Stone DR, Brander C, Goulder PJ, Rosenberg ES, Altfeld M, Walker BD. 2003. Comprehensive epitope analysis of human immunodeficiency virus type 1 (HIV-1)-specific T-cell responses directed against the entire expressed HIV-1 genome demonstrate broadly directed responses, but no correlation to viral load. J Virol 77:2081–2092. doi: 10.1128/JVI.77.3.2081-2092.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, Lederman MM, Benito JM, Goepfert PA, Connors M, Roederer M, Koup RA. 2006. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood 107:4781–4789. doi: 10.1182/blood-2005-12-4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Almeida JR, Sauce D, Price DA, Papagno L, Shin SY, Moris A, Larsen M, Pancino G, Douek DC, Autran B, Saez-Cirion A, Appay V. 2009. Antigen sensitivity is a major determinant of CD8+ T-cell polyfunctionality and HIV-suppressive activity. Blood 113:6351–6360. doi: 10.1182/blood-2009-02-206557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bennett MS, Ng HL, Dagarag M, Ali A, Yang OO. 2007. Epitope-dependent avidity thresholds for cytotoxic T-lymphocyte clearance of virus-infected cells. J Virol 81:4973–4980. doi: 10.1128/JVI.02362-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Migueles SA, Laborico AC, Shupert WL, Sabbaghian MS, Rabin R, Hallahan CW, Van Baarle D, Kostense S, Miedema F, McLaughlin M, Ehler L, Metcalf J, Liu S, Connors M. 2002. HIV-specific CD8+ T cell proliferation is coupled to perforin expression and is maintained in nonprogressors. Nat Immunol 3:1061–1068. doi: 10.1038/ni845. [DOI] [PubMed] [Google Scholar]
- 33.Migueles SA, Osborne CM, Royce C, Compton AA, Joshi RP, Weeks KA, Rood JE, Berkley AM, Sacha JB, Cogliano-Shutta NA, Lloyd M, Roby G, Kwan R, McLaughlin M, Stallings S, Rehm C, O'Shea MA, Mican J, Packard BZ, Komoriya A, Palmer S, Wiegand AP, Maldarelli F, Coffin JM, Mellors JW, Hallahan CW, Follman DA, Connors M. 2008. Lytic granule loading of CD8+ T cells is required for HIV-infected cell elimination associated with immune control. Immunity 29:1009–1021. doi: 10.1016/j.immuni.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hersperger AR, Pereyra F, Nason M, Demers K, Sheth P, Shin LY, Kovacs CM, Rodriguez B, Sieg SF, Teixeira-Johnson L, Gudonis D, Goepfert PA, Lederman MM, Frank I, Makedonas G, Kaul R, Walker BD, Betts MR. 2010. Perforin expression directly ex vivo by HIV-specific CD8 T-cells is a correlate of HIV elite control. PLoS Pathog 6:e1000917. doi: 10.1371/journal.ppat.1000917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dahirel V, Shekhar K, Pereyra F, Miura T, Artyomov M, Talsania S, Allen TM, Altfeld M, Carrington M, Irvine DJ, Walker BD, Chakraborty AK. 2011. Coordinate linkage of HIV evolution reveals regions of immunological vulnerability. Proc Natl Acad Sci U S A 108:11530–11535. doi: 10.1073/pnas.1105315108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kiepiela P, Ngumbela K, Thobakgale C, Ramduth D, Honeyborne I, Moodley E, Reddy S, de Pierres C, Mncube Z, Mkhwanazi N, Bishop K, van der Stok M, Nair K, Khan N, Crawford H, Payne R, Leslie A, Prado J, Prendergast A, Frater J, McCarthy N, Brander C, Learn GH, Nickle D, Rousseau C, Coovadia H, Mullins JI, Heckerman D, Walker BD, Goulder P. 2007. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat Med 13:46–53. doi: 10.1038/nm1520. [DOI] [PubMed] [Google Scholar]
- 37.Elahi S, Dinges WL, Lejarcegui N, Laing KJ, Collier AC, Koelle DM, McElrath MJ, Horton H. 2011. Protective HIV-specific CD8+ T cells evade Treg cell suppression. Nat Med 17:989–995. doi: 10.1038/nm.2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C, Mncube Z, Duraiswamy J, Zhu B, Eichbaum Q, Altfeld M, Wherry EJ, Coovadia HM, Goulder PJ, Klenerman P, Ahmed R, Freeman GJ, Walker BD. 2006. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 39.Kaufmann DE, Kavanagh DG, Pereyra F, Zaunders JJ, Mackey EW, Miura T, Palmer S, Brockman M, Rathod A, Piechocka-Trocha A, Baker B, Zhu B, Le Gall S, Waring MT, Ahern R, Moss K, Kelleher AD, Coffin JM, Freeman GJ, Rosenberg ES, Walker BD. 2007. Upregulation of CTLA-4 by HIV-specific CD4+ T cells correlates with disease progression and defines a reversible immune dysfunction. Nat Immunol 8:1246–1254. doi: 10.1038/ni1515. [DOI] [PubMed] [Google Scholar]
- 40.Perez CL, Larsen MV, Gustafsson R, Norstrom MM, Atlas A, Nixon DF, Nielsen M, Lund O, Karlsson AC. 2008. Broadly immunogenic HLA class I supertype-restricted elite CTL epitopes recognized in a diverse population infected with different HIV-1 subtypes. J Immunol 180:5092–5100. doi: 10.4049/jimmunol.180.7.5092. [DOI] [PubMed] [Google Scholar]
- 41.Miura T, Brockman MA, Schneidewind A, Lobritz M, Pereyra F, Rathod A, Block BL, Brumme ZL, Brumme CJ, Baker B, Rothchild AC, Li B, Trocha A, Cutrell E, Frahm N, Brander C, Toth I, Arts EJ, Allen TM, Walker BD. 2009. HLA-B57/B*5801 human immunodeficiency virus type 1 elite controllers select for rare Gag variants associated with reduced viral replication capacity and strong cytotoxic T-lymphocyte recognition. J Virol 83:2743–2755. doi: 10.1128/JVI.02265-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lassen KG, Lobritz MA, Bailey JR, Johnston S, Nguyen S, Lee B, Chou T, Siliciano RF, Markowitz M, Arts EJ. 2009. Elite suppressor-derived HIV-1 envelope glycoproteins exhibit reduced entry efficiency and kinetics. PLoS Pathog 5:e1000377. doi: 10.1371/journal.ppat.1000377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goulder PJ, Phillips RE, Colbert RA, McAdam S, Ogg G, Nowak MA, Giangrande P, Luzzi G, Morgan B, Edwards A, McMichael AJ, Rowland-Jones S. 1997. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat Med 3:212–217. doi: 10.1038/nm0297-212. [DOI] [PubMed] [Google Scholar]
- 44.Lee K-H, Dinner AR, Tu C, Campi G, Raychaudhuri S, Varma R, Sims TN, Burack WR, Wu H, Wang J, Kanagawa O, Markiewicz M, Allen PM, Dustin ML, Chakraborty AK, Shaw AS. 2003. The immunological synapse balances T cell receptor signaling and degradation. Science 302:1218–1222. doi: 10.1126/science.1086507. [DOI] [PubMed] [Google Scholar]
- 45.Kosmrlj A, Read EL, Qi Y, Allen TM, Altfeld M, Deeks SG, Pereyra F, Carrington M, Walker BD, Chakraborty AK. 2010. Effects of thymic selection of the T-cell repertoire on HLA class I-associated control of HIV infection. Nature 465:350–354. doi: 10.1038/nature08997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alberola-Ila J, Takaki S, Kerner JD, Perlmutter RM. 1997. Differential signaling by lymphocyte antigen receptors. Annu Rev Immunol 15:125–154. doi: 10.1146/annurev.immunol.15.1.125. [DOI] [PubMed] [Google Scholar]
- 47.Sykulev Y. 2010. T cell receptor signaling kinetics takes the stage. Sci Signal 3:pe50. doi: 10.1126/scisignal.3153pe50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garcia KC, Degano M, Pease LR, Huang M, Peterson PA, Teyton L, Wilson IA. 1998. Structural basis of plasticity in T cell receptor recognition of a self peptide-MHC antigen. Science 279:1166–1172. doi: 10.1126/science.279.5354.1166. [DOI] [PubMed] [Google Scholar]
- 49.Araki Y, Fann M, Wersto R, Weng NP. 2008. Histone acetylation facilitates rapid and robust memory CD8 T cell response through differential expression of effector molecules (eomesodermin and its targets: perforin and granzyme B). J Immunol 180:8102–8108. doi: 10.4049/jimmunol.180.12.8102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Juelich T, Sutcliffe EL, Denton A, He Y, Doherty PC, Parish CR, Turner SJ, Tremethick DJ, Rao S. 2009. Interplay between chromatin remodeling and epigenetic changes during lineage-specific commitment to granzyme B expression. J Immunol 183:7063–7072. doi: 10.4049/jimmunol.0901522. [DOI] [PubMed] [Google Scholar]
- 51.Northrop JK, Thomas RM, Wells AD, Shen H. 2006. Epigenetic remodeling of the IL-2 and IFN-gamma loci in memory CD8 T cells is influenced by CD4 T cells. J Immunol 177:1062–1069. doi: 10.4049/jimmunol.177.2.1062. [DOI] [PubMed] [Google Scholar]
- 52.Russ BE, Olshanksy M, Smallwood HS, Li J, Denton AE, Prier JE, Stock AT, Croom HA, Cullen JG, Nguyen ML, Rowe S, Olson MR, Finkelstein DB, Kelso A, Thomas PG, Speed TP, Rao S, Turner SJ. 2014. Distinct epigenetic signatures delineate transcriptional programs during virus-specific CD8(+) T cell differentiation. Immunity 41:853–865. doi: 10.1016/j.immuni.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wherry EJ, Kurachi M. 2015. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 15:486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Youngblood B, Noto A, Porichis F, Akondy RS, Ndhlovu ZM, Austin JW, Bordi R, Procopio FA, Miura T, Allen TM, Sidney J, Sette A, Walker BD, Ahmed R, Boss JM, Sekaly RP, Kaufmann DE. 2013. Cutting edge: prolonged exposure to HIV reinforces a poised epigenetic program for PD-1 expression in virus-specific CD8 T cells. J Immunol 191:540–544. doi: 10.4049/jimmunol.1203161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Youngblood B, Oestreich KJ, Ha SJ, Duraiswamy J, Akondy RS, West EE, Wei Z, Lu P, Austin JW, Riley JL, Boss JM, Ahmed R. 2011. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8(+) T cells. Immunity 35:400–412. doi: 10.1016/j.immuni.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Youngblood B, Wherry EJ, Ahmed R. 2012. Acquired transcriptional programming in functional and exhausted virus-specific CD8 T cells. Curr Opin HIV AIDS 7:50–57. doi: 10.1097/COH.0b013e32834ddcf2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vigano S, Bellutti Enders F, Miconnet I, Cellerai C, Savoye AL, Rozot V, Perreau M, Faouzi M, Ohmiti K, Cavassini M, Bart PA, Pantaleo G, Harari A. 2013. Rapid perturbation in viremia levels drives increases in functional avidity of HIV-specific CD8 T cells. PLoS Pathog 9:e1003423. doi: 10.1371/journal.ppat.1003423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Walker BD, Flexner C, Birch-Limberger K, Fisher L, Paradis TJ, Aldovini A, Young R, Moss B, Schooley RT. 1989. Long-term culture and fine specificity of human cytotoxic T-lymphocyte clones reactive with human immunodeficiency virus type 1. Proc Natl Acad Sci U S A 86:9514–9518. doi: 10.1073/pnas.86.23.9514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Varadarajan N, Julg B, Yamanaka YJ, Chen H, Ogunniyi AO, McAndrew E, Porter LC, Piechocka-Trocha A, Hill BJ, Douek DC, Pereyra F, Walker BD, Love JC. 2011. A high-throughput single-cell analysis of human CD8(+) T cell functions reveals discordance for cytokine secretion and cytolysis. J Clin Invest 121:4322–4331. doi: 10.1172/JCI58653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brockman MA, Tanzi GO, Walker BD, Allen TM. 2006. Use of a novel GFP reporter cell line to examine replication capacity of CXCR4- and CCR5-tropic HIV-1 by flow cytometry. J Virol Methods 131:134–142. doi: 10.1016/j.jviromet.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 61.Schneidewind A, Brockman MA, Sidney J, Wang YE, Chen H, Suscovich TJ, Li B, Adam RI, Allgaier RL, Mothe BR, Kuntzen T, Oniangue-Ndza C, Trocha A, Yu XG, Brander C, Sette A, Walker BD, Allen TM. 2008. Structural and functional constraints limit options for cytotoxic T-lymphocyte escape in the immunodominant HLA-B27-restricted epitope in human immunodeficiency virus type 1 capsid. J Virol 82:5594–5605. doi: 10.1128/JVI.02356-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang OO, Sarkis PT, Trocha A, Kalams SA, Johnson RP, Walker BD. 2003. Impacts of avidity and specificity on the antiviral efficiency of HIV-1-specific CTL. J Immunol 171:3718–3724. doi: 10.4049/jimmunol.171.7.3718. [DOI] [PubMed] [Google Scholar]
- 63.Szymczak AL, Workman CJ, Wang Y, Vignali KM, Dilioglou S, Vanin EF, Vignali DA. 2004. Correction of multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide-based retroviral vector. Nat Biotechnol 22:589–594. doi: 10.1038/nbt957. [DOI] [PubMed] [Google Scholar]
- 64.Arnold PY, Burton AR, Vignali DA. 2004. Diabetes incidence is unaltered in glutamate decarboxylase 65-specific TCR retrogenic nonobese diabetic mice: generation by retroviral-mediated stem cell gene transfer. J Immunol 173:3103–3111. doi: 10.4049/jimmunol.173.5.3103. [DOI] [PubMed] [Google Scholar]
- 65.Ali R, Babad J, Follenzi A, Gebe JA, Brehm MA, Nepom GT, Shultz LD, Greiner DL, DiLorenzo TP. 2016. Genetically modified human CD4(+) T cells can be evaluated in vivo without lethal graft-versus-host disease. Immunology 148:339–351. doi: 10.1111/imm.12613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Follenzi A, Naldini L. 2002. Generation of HIV-1 derived lentiviral vectors. Methods Enzymol 346:454–465. doi: 10.1016/S0076-6879(02)46071-5. [DOI] [PubMed] [Google Scholar]
- 67.Edmonds TG, Ding H, Yuan X, Wei Q, Smith KS, Conway JA, Wieczorek L, Brown B, Polonis V, West JT, Montefiori DC, Kappes JC, Ochsenbauer C. 2010. Replication competent molecular clones of HIV-1 expressing Renilla luciferase facilitate the analysis of antibody inhibition in PBMC. Virology 408:1–13. doi: 10.1016/j.virol.2010.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ochsenbauer C, Kappes JC. 2009. New virologic reagents for neutralizing antibody assays. Curr Opin HIV AIDS 4:418–425. doi: 10.1097/COH.0b013e32832f011e. [DOI] [PubMed] [Google Scholar]