

ABSTRACT
Mouse adenovirus type 1 (MAV-1) infection causes encephalitis in susceptible strains of mice and alters the permeability of infected brains to small molecules, which indicates disruption of the blood-brain barrier (BBB). Under pathological conditions, matrix metalloproteinases (MMPs) can disrupt the BBB through their proteolytic activity on basement membrane and tight junction proteins. We examined whether MAV-1 infection alters MMP activity in vivo and in vitro. Infected MAV-1-susceptible SJL mice had higher MMP2 and MMP9 activity in brains, measured by gelatin zymography, than mock-infected mice. Infected MAV-1-resistant BALB/c mice had MMP activity levels equivalent to those in mock infection. Primary SJL mouse brain endothelial cells (a target of MAV-1 in vivo) infected ex vivo with MAV-1 had no difference in activities of secreted MMP2 and MMP9 from mock cells. We show for the first time that astrocytes and microglia are also infected in vivo by MAV-1. Infected mixed primary cultures of astrocytes and microglia had higher levels of MMP2 and MMP9 activity than mock-infected cells. These results indicate that increased MMP activity in the brains of MAV-1-infected susceptible mice may be due to MMP activity produced by endothelial cells, astrocytes, and microglia, which in turn may contribute to BBB disruption and encephalitis in susceptible mice.
IMPORTANCE RNA and DNA viruses can cause encephalitis; in some cases, this is accompanied by MMP-mediated disruption of the BBB. Activated MMPs degrade extracellular matrix and cleave tight-junction proteins and cytokines, modulating their functions. MAV-1 infection of susceptible mice is a tractable small-animal model for encephalitis, and the virus causes disruption of the BBB. We showed that MAV-1 infection increases enzymatic activity of two key MMPs known to be secreted and activated in neuroinflammation, MMP2 and MMP9, in brains of susceptible mice. MAV-1 infects endothelial cells, astrocytes, and microglia, cell types in the neurovascular unit that can secrete MMPs. Ex vivo MAV-1 infection of these cell types caused higher MMP activity than mock infection, suggesting that they may contribute to the higher MMP activity seen in vivo. To our knowledge, this provides the first evidence of an encephalitic DNA virus in its natural host causing increased MMP activity in brains.
KEYWORDS: encephalitis, matrix metalloproteinase, endothelial, astrocytes, endothelial cells, microglia
INTRODUCTION
Mouse adenovirus type 1 (MAV-1; also known as MAdV-1) causes acute and persistent infections, and it infects monocytes, macrophages, and endothelial cells throughout the animal (1–4). The highest viral loads are found in the brain, spleen, and spinal cord in susceptible mice after intraperitoneal (i.p.) infection (3–5). MAV-1 causes dose-dependent encephalitis by infecting endothelial cells in the brains of susceptible mice, which leads to an alteration of the permeability of the blood-brain barrier (BBB) (2, 5–7).
The BBB regulates access of molecules and cells to the brain parenchyma from the circulation, maintaining homeostasis and protecting the brain from pathogens (8, 9). Microvascular endothelial cells are the main functional component of the BBB. Endothelial cells (ECs) are tightly linked to one another through tight-junction proteins, and the BBB endothelium is distinct from other endothelium in the body due to interactions with other cells, particularly astrocytes, and noncellular elements (8). Astrocytes are glial cells that comprise more than 50% of the total cells in the brain (9). They are critically involved in neuron integrity and function, and they have a pivotal role in maintaining BBB integrity through their end feet, which surround the microvessels in the central nervous system (CNS). Together, the endothelium, astrocytes, pericytes, neurons, extracellular matrix, and microglia have been termed the neurovascular unit (10).
Under pathological conditions, cells of the neurovascular unit (including endothelial cells, astrocytes, and microglia) and neutrophils and macrophages recruited from the circulation produce increased matrix metalloproteinase (MMP) mRNAs, protein levels, and enzyme activity (11, 12). MMPs are a multigene-encoded family of zinc-dependent endopeptidases involved in the remodeling of the extracellular matrix. MMP activities are regulated in at least four ways: by alteration of gene expression, by activation of proenzyme through proteolytic cleavage, by compartmentalization, and by association with physiological inhibitors (13, 14). MMPs can cleave tight-junction proteins, directly affecting BBB integrity (15–17); they can also cleave cytokines, modulating inflammation (18). Of particular importance in CNS pathology are MMP2, a constitutively expressed MMP in the brain, and MMP3 and MMP9, which are the major inducible MMPs arising in neuroinflammatory responses (12). MMP protein and/or activity levels are altered by infections by viruses, including HIV-1, West Nile virus, Semliki Forest virus, and herpes simplex virus (reviewed in reference 14). We recently showed that MAV-1 infection of susceptible SJL mice results in increased levels of MMP2 and MMP9 activity in brains (19).
Outbred and inbred strains of mice differ in their susceptibilities to MAV-1 infection (4–6). We mapped a major quantitative trait locus (QTL) for susceptibility, Msq1, to the Ly6 locus on mouse chromosome (Chr) 15 in a backcross between susceptible SJL and resistant BALB/c mice (20, 21). Some phenotypes that are related to encephalitis and BBB disruption are linked to Msq1, including high brain viral loads and disruption of the BBB (measured by permeability to small-molecule dyes) (22). Here, we investigated whether the secretion of MMPs in vivo in response to MAV-1 infection correlated with mouse strain susceptibility. Brains of MAV-1-resistant BALB/c mice did not have increased MMP levels after MAV-1 infection, in contrast to susceptible SJL brains and brains of an interval-specific congenic susceptible mouse strain, C.SJL-Msq1SJL (BALB/c background with an SJL-derived Chr 15 region encompassing the Msq1 quantitative trait locus for susceptibility), indicating that viral induction of increased MMP activity is another phenotype linked to Msq1. To determine which cells capable of producing MMPs in the neurovascular unit might contribute to the increased MMP activity in susceptible mice, we infected endothelial cells, astrocytes, and microglia ex vivo. We show for the first time that astrocytes and microglia are also infected by MAV-1 in vivo and that mixed astrocyte-microglial cultures (MAMCs) produced increased MMP2 and MMP9 activity upon infection. Surprisingly, we found no increased MMP activity produced from infected endothelial cells unless we also included conditioned media from MAMCs. Increased MMP activity produced by MAMCs and their effects on endothelial cells likely contribute to the mechanism by which MAV-1 disrupts the BBB in mice.
RESULTS
MAV-1 infection results in increased MMP activity in brains.
To determine whether MAV-1 infection causes an increase in MMP activity that could contribute to disruption of the BBB, we infected MAV-1-susceptible SJL mice with 105 PFU and collected their brains 3 days postinfection (dpi), when viral infection peaks and BBB disruption occurs for this dose. We prepared whole-brain homogenates for analysis by gelatin zymography to assay the enzymatic (gelatinase) activities of MMP2 and MMP9. Infected brains showed an increase in MMP9 activity compared to mock-infected brains that was statistically significant (Fig. 1A). There was no difference in MMP2 activity between mock-infected and infected brains. All five infected brains had high viral loads by capture enzyme-linked immunosorbent assay (ELISA) (data not shown). SJL mice infected at a lower dose (102 PFU) and harvested 8 dpi had an apparent increase in MMP2 and MMP9 activity upon infection that did not reach statistical significance (Fig. 1B). We also assayed MAV-1-resistant BALB/c mice; there was no increase in MMP2 or MMP9 activity (Fig. 1B). The mouse brains shown in Fig. 1B were also assayed for viral load and sodium fluorescein uptake (a measure of BBB permeability). Viral loads were not elevated above those in mock infection for the BALB/c mice, and sodium fluorescein uptake was seen only in the infected SJL mice (data not shown), consistent with our previous report (22). These data suggest that MAV-1 infection increased MMP activity in brains of susceptible mice.
FIG 1.
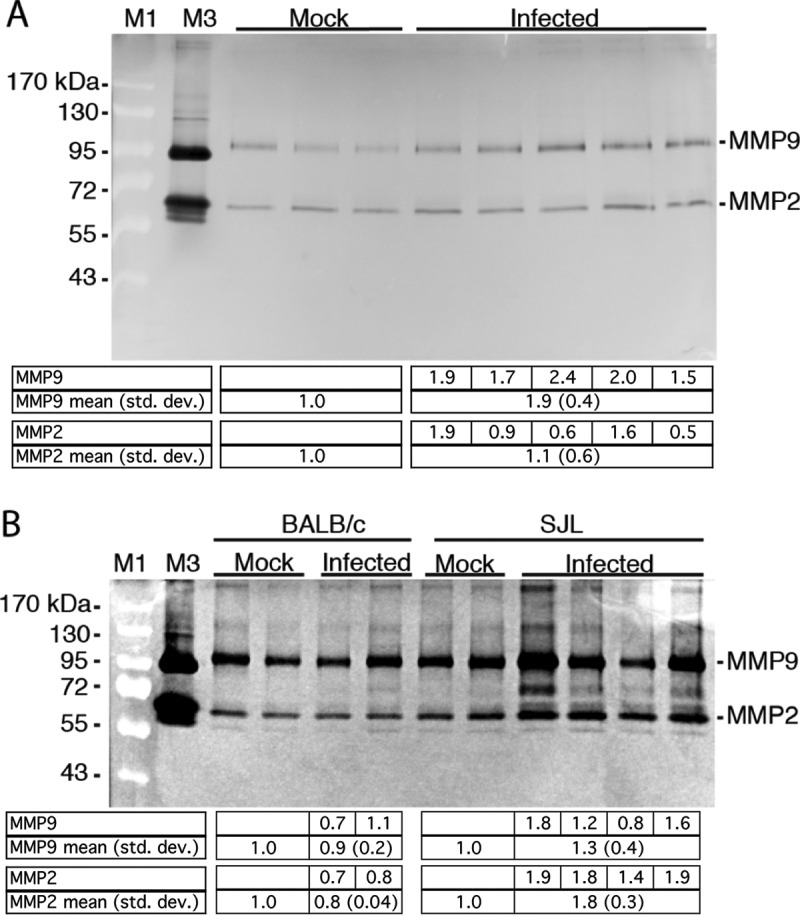
MAV-1 infection increases MMP activity in brains. (A) Five-week-old SJL mice were mock infected (n = 3) or infected i.p. with 105 PFU of MAV-1 (n = 5) and euthanized 3 dpi. Brain homogenates (200 mg) were analyzed by gelatin zymography. The higher level of MMP9 in infected brains was statistically significant (P = 0.04; Mann-Whitney test). (B) Six-week old BALB/c or SJL mice were mock infected (n = 2) or infected i.p. with 102 PFU MAV-1 (BALB/c, n = 2; SJL, n = 4) and euthanized 8 dpi. Brain homogenates (600 mg) were analyzed by zymography. The differences between mock-infected and infected or between infected BALB/c and SJL mice were not statistically significant (Mann-Whitney test). M1, molecular mass standards (indicated on the left); M3, human HT1080 cell supernatant (proMMP9, 92 kDa; MMP2, 72 and 62 kDa). The levels of MMP activity were quantitated by densitometry and normalized to the mean for the mock-infected samples. Individual values are listed for the infected mice, and the means and standard deviations (std. dev.) are listed for the corresponding lanes below the gels.
We previously mapped a major QTL, called Msq1, for susceptibility to MAV-1 to mouse Chr 15 (21). Using a congenic strain of mice carrying Msq1 from SJL (susceptible) mice in a BALB/c (resistant) mouse background, we demonstrated that some phenotypes of encephalitis map to Msq1 (high virus load and sodium fluorescein uptake, indicative of BBB disruption), while others do not (edema and survival) (22). To determine whether the MAV-1-induced increase in MMP activity seen in SJL brains mapped to Msq1, we infected the congenic mice (C.SJL-Msq1SJL) or BALB/c controls with 102 PFU and measured the virus load, sodium fluorescein uptake, and MMP activity levels from brains at 8 dpi. As expected and as shown previously (22), compared to mock-infected mice, MAV-1-infected BALB/c mice did not have high virus loads or sodium fluorescein uptake (Fig. 2A and B). Compared to mock-infected mice, infected BALB/c mice had no statistically significant differences in MMP2 or MMP9 activity (Fig. 2C). The infected congenic mice had high virus loads and high sodium fluorescein uptake compared to BALB/c mice (Fig. 2A and B), as shown previously (22). The infected congenic mice also had a statistically significantly higher level of MMP9 activity than mock-infected mice, and both MMP2 and MMP9 activities in the infected congenic mice were significantly higher than in control BALB/c mice (Fig. 2C). We also assayed MMP activity at 6 dpi in congenic and BALB/c control mice infected with 104 PFU (Fig. 2D) to determine whether increased MMP activity was seen in the congenic mice at a higher viral dose. The infected BALB/c mice had no difference in MMP2 or MMP9 activity compared to mock-infected mice, whereas congenic mice had a statistically significant increase in MMP2 and MMP9 activity. These data indicate that the virus-induced increase in MMP activity is linked to the susceptibility QTL and confirm our findings that MAV-1 infection increases MMP activity in brains of susceptible mice.
FIG 2.

MAV-1-induced increase in MMP activity is linked to the Msq1 susceptibility locus. Three- to 5-week-old BALB/c or C.SJL-Msq1SJL mice (n = 3 per group) were mock infected or infected with 102 PFU of MAV-1 and harvested 8 dpi (A to C) or with 104 PFU and harvested 6 dpi (D). Ten minutes prior to euthanasia, the mice were injected i.p. with sodium fluorescein. The mice were perfused, and each brain was divided into parts for various assays. Viral loads were measured by capture ELISA (A), sodium fluorescein uptake was quantitated (B), and MMP activity was visualized by gelatin zymography (C and D) and quantitated by densitometry. (A to C) Individual mouse brains are displayed in the same order for each assay. The levels of MMP activity were normalized to the mean for the mock-infected samples. Individual values are listed for the infected mice, and the means and standard deviations are listed for the corresponding lanes below the gels. The data from the gel in panel C were combined with mouse data from an independent experiment (n = 4 total) for statistical analysis (Mann-Whitney test). The levels of MMP9 activity in the infected congenic mice were significantly higher than those in uninfected congenic mice (P < 0.03), and levels of both MMP2 and MMP9 activity were significantly higher than those in both uninfected and infected BALB/c mice (P < 0.03; Mann-Whitney test). The data from the gel in panel D were combined with mouse data from an independent experiment (n = 6 total) for statistical analysis. The levels of MMP2 and MMP9 in infected mice were significantly higher than those in uninfected congenic mice (P < 0.009) and higher than those in both uninfected and infected BALB/c mice (P = 0.002; Mann-Whitney test). (C and D) The data are representative of the results of two independent experiments. Lanes M1 and M3 are as in Fig. 1. The last lane of panel C was from the same gel and image exposure as the rest of the image.
MMP activity in MAV-1-infected pmBECs.
We investigated which cell types in the neurovascular unit produce MMP activity and how the MMP activity changes after MAV-1 infection. MAV-1 infects endothelial cells in mice (2, 3) and primary mouse brain endothelial cells (pmBECs) in culture (7). We infected pmBECs from SJL mice with MAV-1 at a multiplicity of infection (MOI) of 2 or 5 and analyzed the culture supernatants for MMP2 and MMP9 activity by zymography 48 h postinfection (hpi). Contrary to our expectation, there was no significant change in MMP2 or MMP9 activity after infection of five independent pmBEC preparations, two of which are shown in Fig. 3. We also found no changes in MMP2 or MMP9 activity in pmBECs at 24 or 72 hpi (data not shown). This was surprising, since endothelial cells are a major target of MAV-1 in susceptible mice (2, 3) and can be infected ex vivo (7, 22), and they thus seemed the most likely cell type to produce the observed increased MMP activity upon infection.
FIG 3.
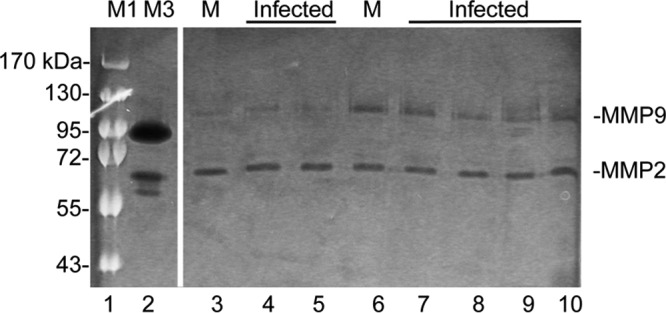
MMP expression in pmBECs. PmBECs were infected with MAV-1 at an MOI of 5 (lanes 4, 5, 9, and 10) or 2 (lanes 7 and 8) or mock infected (M) (lanes 3 and 6). Two independent preparations of pmBECs are shown; lanes 3 to 5 are from one and lanes 6 to 10 from the other. After 24 h, the medium was changed to serum-free medium, and the cell supernatants were harvested after an additional 24 h and processed for zymography. Similar results were obtained with three additional independent pmBEC preparations. There were no statistically significant differences between mock-infected and infected cells for MMP2 or MMP9 for either preparation. Lanes M1 and M3 are as in Fig. 1. All lanes are from the same gel and image exposure.
MAV-1 infects astrocytes and microglia ex vivo.
We next investigated whether MAV-1 replicates in other cell types in the brain that might be responsible for increased MMP activity. Although endothelial cells are the primary target of MAV-1, signs of microglial activation and astrocytes with swollen end feet were seen by electron microscopy of spinal cords of infected susceptible mice (5). Primary astrocytes and microglia can be enriched from a mixed culture from neonatal brains by a shake-off procedure, followed by culturing in cell-type-specific medium. We performed ex vivo MAV-1 infections of primary astrocyte- and microglia-enriched cultures to determine whether the virus replicates in these cells. We isolated mRNA and assayed its cDNA for early region 3 (E3) gene expression, using primers that span an E3 intron and thus specifically identify mRNA. Both infected astrocyte- and microglia-enriched cells showed E3 gene expression, indicating that MAV-1 entered the cells and was transcriptionally active (Fig. 4A). We assayed infected astrocyte- and microglia-enriched cultures for infectious virus by plaque assay, and they both showed increases in infectious-virus yield relative to input (0 hpi) at 48 and 96 hpi, demonstrating that the cells were productively infected (Fig. 4B). We also infected astrocyte- and microglia-enriched cultures with MAV-1 that expressed green fluorescent protein (GFP) as a fusion protein with the minor capsid protein pIX (MAV-1.IXeGFP) at an MOI of 5; production of this protein occurs at late times after infection and is visible by fluorescence microscopy in infected cells (19). By flow cytometry analysis, 61% of an astrocyte-enriched culture and 67% of a microglia-enriched culture were GFP positive (GFP+) at 48 hpi, evidence of infection of both by MAV-1.IXeGFP (data not shown).
FIG 4.
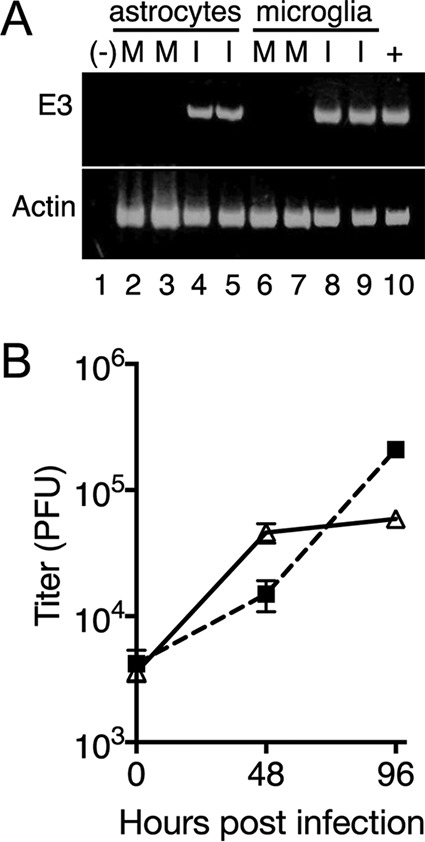
MAV-1 infects cultures enriched for astrocytes and microglia. Primary astrocytes and microglia were infected with MAV-1 at an MOI of 5 or mock infected and harvested at 48 hpi (A) or at the times shown (B). (A) cDNA was prepared and assayed by PCR for E3 and actin, as indicated. −, negative control (water); +, positive control (cDNA from mRNA from MAV-1-infected 3T6 cells 48 hpi). Lanes 2 to 5, astrocytes; lanes 6 to 9, microglia. M, mock infected; I, infected. Each condition was assayed on duplicate wells of cells from the same primary cell preparation. The experiment shown is representative of six experiments from independent primary cell preparations. (B) Cells and supernatants were harvested and titrated on mouse 3T6 cells. Each point indicates the mean and standard deviation of three plaque assay plates. The experiment shown is representative of four experiments from two independent primary cell preparations. Triangles, astrocytes; squares, microglia.
To confirm the cell-specific infection of astrocytes and microglia in vivo, we used two approaches. In the first, we infected mice i.p. with MAV-1.IXeGFP and harvested brain cells from whole-brain homogenates at 7 dpi. The cells were surface stained for the microglial marker CD11b or the astrocyte marker GLAST and then stained intracellularly for GFP and analyzed by flow cytometry. A 5.6-fold increase in GFP+ CD11b+ staining was seen after MAV-1 infection (Fig. 5A), indicating that MAV-1 infects microglia in vivo. A 9.1-fold increase in GFP+ GLAST+ staining was seen after infection (Fig. 5B), indicating that MAV-1 infects astrocytes in vivo.
FIG 5.
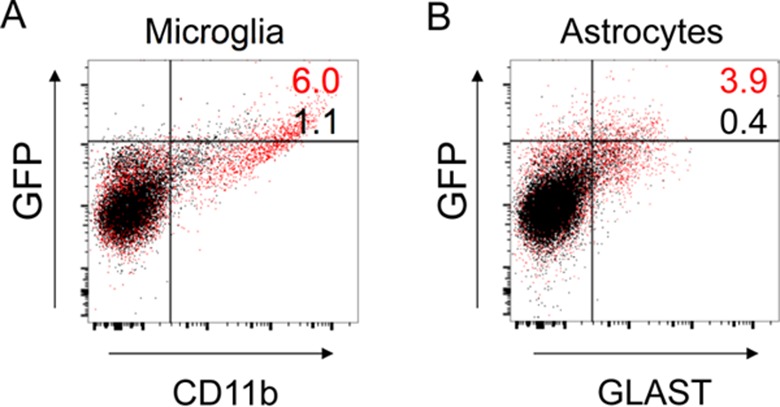
In vivo infection of microglia and astrocytes in SJL mouse brains infected intraperitoneally. Shown is flow cytometry analysis of whole-brain homogenates after i.p. inoculation of SJL mice with 104 PFU of MAV-1.IXeGFP or mock infection. A total of 30,000 events from five pooled brain homogenates were captured for each condition, and the events were gated to exclude debris and cell doublets. Infected-mouse data are represented by red dots and numerals, and mock-infected-mouse data are represented by black dots and numerals. MAV-1.IXeGFP-infected cells are GFP+, and the percentage of cells double positive for virus and cell type marker are indicated in the upper right quadrant. (A) Staining for GFP+ (infected) cells and CD11b+ cells (microglia). (B) Staining for GFP+ (infected) cells and GLAST+ cells (astrocytes). Of the gated populations, 6.0% of infected microglia were positive for both markers compared to 1.1% of mock-infected microglia, a 5.6-fold increase (A), and 3.9% of infected microglia were positive for both markers compared to 0.4% of mock-infected microglia, a 9.1-fold increase (B). The data shown are representative of the results of two independent experiments.
In the second approach to demonstrate infection of astrocytes and microglia in vivo, we performed immunofluorescence staining of brains of mice infected intracerebrally with wild-type (wt) MAV-1 or dlE102, a CR2 deletion of E1A. We used the dlE102 virus because we had a highly purified stock of it and could introduce a high dose with potentially fewer side effects than with our wt virus stock (a clarified infected cell lysate). We did not expect qualitative pathogenesis differences between wt and dlE102 (23). We found evidence of MAV-1 infection of astrocytes (identified by positive staining for glial fibrillary acidic protein [GFAP] and the MAV-1 E3 protein) and microglia (positive staining for IBA1 and MAV-1 E3) by both wt and E1A mutant virus (Fig. 6). We saw similar positive staining of astrocytes and microglia in C57BL/6 mice infected intracerebrally (data not shown). As expected, we did not see differences between infections with the wt and E1A mutant viruses. Thus, in addition to the previously known infection of endothelial cells in vivo and ex vivo, we have identified two other cell types in the neurovascular unit that MAV-1 infects in vivo, astrocytes and microglia. Summarizing Fig. 5 and 6, MAV-1-infected astrocytes and microglia were found in two mouse strains, with three MAV-1 strains inoculated by two different routes.
FIG 6.
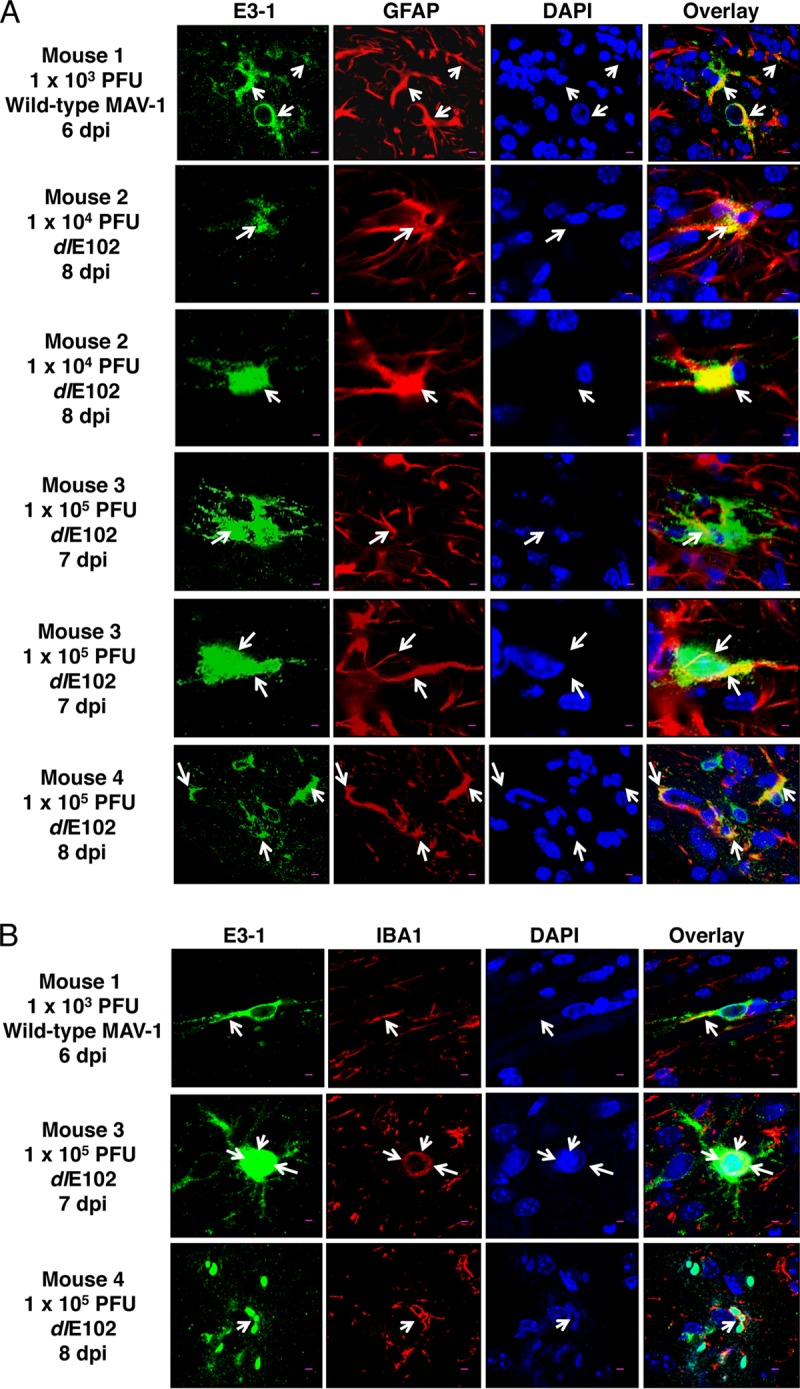
In vivo infection of microglia and astrocytes in SJL mouse brains infected intracerebrally. SJL mice were administered wt MAV-1 or dlE102 by intracerebral inoculation of the indicated dose and euthanized when moribund at the indicated times. MAV-1 E3 gp19K expression was detected by immunofluorescent staining with anti-E3-1 antibody. (A) GFAP-positive cells (astrocytes) were infected with MAV-1. Confocal fluorescence scanning images show six examples of cytoplasmic E3 gp19K expression (green) within GFAP+ astrocytes (red). DAPI nuclear staining is blue. Scale bars, 10 μm. The arrows point to the cell bodies of GFAP+-immunoreactive astrocytes. (B) IBA1-positive cells (microglia) are infected with MAV-1. E3 gp19K expression is green, Iba1 cells are red, and DAPI nuclear staining is blue. The arrows indicate IBA1+ cells. Scale bars, 10 μm.
MMP activity in astrocytes and microglia.
Astrocytes and microglia secrete MMPs (12), and since astrocytes and microglia were infected by MAV-1, we examined whether they produce increased MMP activity when infected ex vivo, using MAMCs. MMP9 activity was seen in multiple bands in MAMCs (Fig. 7). The upper MMP9 band is likely glycosylated MMP9 proenzyme present in MAMC cells (see Materials and Methods). Exposure to SDS during sample preparation for gelatin zymography exposes the catalytic domain of MMP proenzymes, and thus, its activity can be detected upon development of the zymogram. We performed infections with two different wt virus stocks (Fig. 7, lanes 2, 3, 8, and 9) and with a virus with a mutation in E3, pmE314 (lanes 4 and 10). We previously showed that the E3 mutant does not differ from the wt with respect to disruption of brain permeability to sodium fluorescein or alteration of tight-junction protein expression and transendothelial electrical resistance (TEER) (7). We expected that pmE314 and wt viruses would be similar in their effects on MMP activity. Indeed, at both 24 and 48 hpi, wt and mutant viruses appeared to have higher levels of MMP2 and MMP9 activity than mock-infected MAMCs (Fig. 7). We determined by quantitative PCR (qPCR) analysis for MMP2, MMP3, and MMP9 in astrocyte and microglial preparations whether higher steady-state mRNA levels paralleled the higher MMP activity in infected MAMCs than in mock infection. Like MMP2 and MMP9, MMP3 activity is induced in brains upon infection or neurological pathogenesis (14), and MMP3 can proteolytically activate MMP9 (12, 24). We infected four independent preparations of MAMCs in duplicate with wt MAV-1. Analysis of mRNAs of MMP2, MMP3, and MMP9 at 12, 24, and 48 hpi showed no difference in expression from mock infection (data not shown). We conclude that the apparent difference in MMP2 and MMP9 activity in MAMCs is most likely due to posttranscriptional events.
FIG 7.
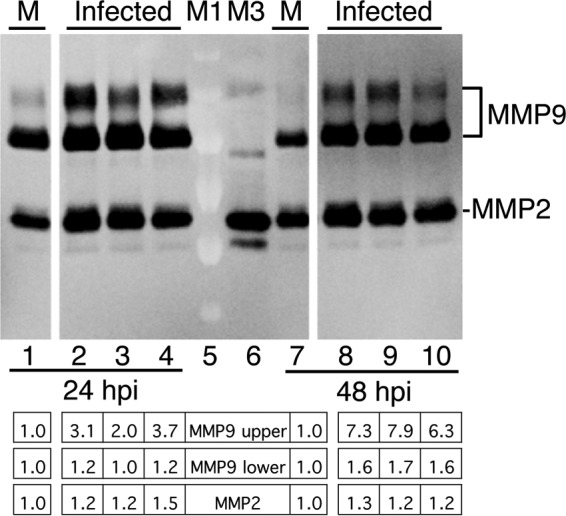
MMP activity in mixed astrocyte-microglia cultures. MAMC cells on a 12-well plate were infected with MAV-1 (three wells at each time point) at an MOI of 5 or mock infected (M) (one well). The supernatants were harvested from the cultures at 24 hpi (lanes 1 to 4) or 48 hpi (lanes 7 to 10) and analyzed by gelatin zymography. Lanes 2 and 8 were infected with one wt virus stock, lanes 3 and 9 were infected with an independent wt virus stock, and lanes 4 and 10 were infected with an E3 mutant virus, pmE314. The levels of MMP activity were quantitated by densitometry and normalized to the mock-infected samples. Individual values are listed for the infected wells below the gel. The statistical significance of the observed differences could not be determined because there was only one sample per condition. All the lanes are from the same gel and image exposure. Lanes M1 and M3 are as in Fig. 1.
Infected pmBECS produce increased MMP activity when cultured with MAMC-conditioned medium.
It is well established that in vitro brain endothelial cultures of a number of mammalian species have greater barrier integrity (e.g., higher TEER and lower permeability to low-molecular-weight tracers) when astrocytes or astrocyte-conditioned medium is present (25–30). Moreover, the presence of astrocytes or astrocyte-conditioned medium can help endothelial cells maintain their endothelial phenotypes in cell culture (31). We confirmed that inclusion of MAMC-conditioned medium resulted in higher levels of TEER in our pmBEC cultures (Fig. 8). We then tested whether MAV-1-infected pmBECs incubated in the upper chamber of a transwell plate, with MAMC-conditioned medium in the lower chamber, produced higher levels of MMP activity than mock-infected cells. At 12 hpi, the medium from the infected pmBECs appeared to have higher MMP2 and MMP9 activities than medium from mock-infected cells; this was true for media from both the upper and lower chambers (Fig. 9A), but the differences were not statistically significant. At 24 hpi, no MMP9 activity was detected in supernatant from either chamber; however, MMP2 activity appeared to be higher in the upper and lower chambers for infected wells than for mock-infected wells (Fig. 9B). A greater apparent increase in MMP activity was seen in the upper chamber than in the lower chamber at both time points, but the differences did not reach statistical significance. pmBECs cultured similarly in transwells but without MAMC-conditioned medium showed no differences in MMP activity between mock-infected and infected cultures (data not shown), consistent with the results shown in Fig. 3.
FIG 8.
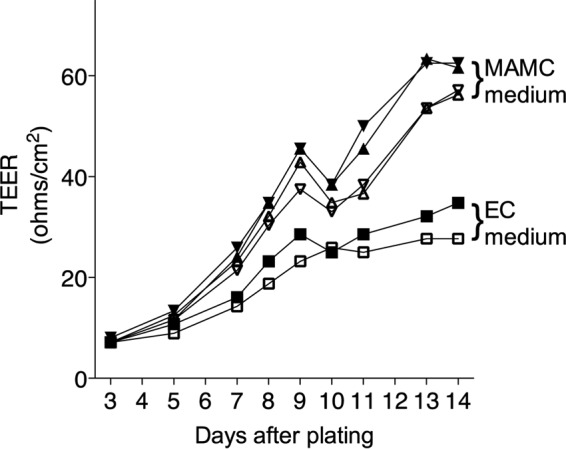
Conditioned medium from MAMCs increases TEER of pmBECs. TEER was measured in replicate transwells of pmBECs cultured with endothelial cell medium containing hydrocortisone in both the upper and lower chambers (squares) (2 wells) or with endothelial cell medium with hydrocortisone in the upper chamber and 20% endothelial cell medium-80% MAMC-conditioned medium in the lower chamber (triangles) (4 wells).
FIG 9.

MMP activity in pmBECs cultured with MAMC-conditioned medium. (A) pmBECs on transwells were infected with MAV-1 at an MOI of 5 or mock infected in triplicate. The medium in the upper chamber was endothelial medium with hydrocortisone. The medium in the lower chamber was 100% MAMC-conditioned medium. The supernatants were harvested from the cultures at 12 (A) or 24 (B) hpi. Medium from the upper or lower chamber of each culture, as indicated, was analyzed by gelatin zymography. The levels of MMP activity were quantitated by densitometry and normalized to the mean for the mock-infected samples. Individual values are listed for the infected wells, and the means and standard deviations are listed below the gels. There were no statistically significant differences between means for infected and mock-infected cells at either time point. Lanes M1 and M3 are as in Fig. 1. The results are representative of two experiments.
DISCUSSION
Infection of susceptible mice by MAV-1 results in dose-dependent encephalitis via infection of endothelial cells (2, 5, 6). In addition, we established that MAV-1 infection causes BBB disruption in brains of susceptible mice, demonstrated by permeability to small-molecule tracer dyes (7, 19, 22). To address the mechanism by which this disruption occurs, we assayed whether brains of infected mice had higher levels of MMP activity than those of mock-infected mice. MMP2 and MMP9 are gelatinases whose enzymatic activities are assayable by gelatin zymography. They frequently have altered activity in infections by both DNA and RNA viruses (reviewed in reference 14). Susceptible SJL mice and susceptible congenic C.SJL-Msq1SJL mice had increased levels of MMP2 and MMP9 activity upon MAV-1 infection. We note that even modest differences assayed in whole brains may be biologically relevant, especially if the increased MMP activity is localized to specific regions of the brain. In contrast to susceptible mice, infected BALB/c mice, resistant to MAV-1, did not show increased MMP2 and MMP9 activity levels compared to mock-infected mice. As expected, the increase in MMP activity correlated with increased brain viral loads and sodium fluorescein uptake in the susceptible mice. We noted larger MMP activity increases in the congenic mice than in SJL mice (compare Fig. 2C and D with Fig. 1). One possible explanation for this may be in the mouse strain backgrounds. The congenic mice have a BALB/c background, and only the introgressed genetic region encompassing the Msq1 locus is from SJL mice. It is possible that other genes in SJL mice not found in the congenic mice can negatively modulate MMP activity in the SJL mice. It is also possible that in congenic mice (which have a primarily BALB/c background), supporting cells that contribute to MMP activity, such as astrocytes and microglia, may provide better support for MMP activity induction than do supporting cells of the pure SJL background.
A number of cell types in the neurovascular unit can produce increased MMP mRNA, protein, and/or enzymatic activity in response to pathological conditions, including endothelial cells, astrocytes, microglia, and macrophages and neutrophils that are recruited from the circulation (11, 12). We investigated which cell types might contribute to increased MMP activity in MAV-1-infected brains by infecting primary cell cultures of endothelial cells, astrocytes, and microglia. MAV-1 infects endothelial cells throughout the animal, and the highest levels of virus are found in the brain, spleen, and spinal cord (reviewed in reference 32). Thus, while we expected that infected pmBECs would have increased MMP activity, in multiple experiments with independent pmBEC preparations, infection did not increase MMP activity. However, when we incubated pmBECs with conditioned medium from MAMCs, infection by MAV-1 resulted in increased levels of MMP9 at very early times after infection (12 hpi) and increased MMP2 levels at both 12 and 24 hpi. This is consistent with many reports that conditioned medium from MAMCs or coculture with them can help endothelial cells maintain their endothelial phenotypes in cell culture (31). These phenotypes include greater barrier integrity, such as higher TEER and lower permeability to low-molecular-weight tracers (25–30). Furthermore, Behzadian et al. (33) reported that production of MMP9 by bovine retinal endothelial cells is increased by contact with medium from glial cells expressing transforming growth factor beta (TGF-β) or direct cell-cell contact with glial cells. While we have not examined whether MAV-1 infection alters TGF-β expression in MAMCs, the findings of Behzadian et al. provide support for a possible cytokine mechanism for the increased MMP9 activity in MAV-1-infected pmBECs that we saw only when the pmBECs were cultured with MAMC-conditioned medium. We conclude that pmBECs likely contribute to increased MMP activity seen in infected mouse brains via close cell-cell contact with astrocytes.
Although MAV-1 has previously been thought to primarily infect endothelial cells in the brain, microglia and astrocytes may also be infected, since spinal cords of infected susceptible mice show histopathological signs in astroglial end feet and microglia (5). These signs could also be bystander effects of the host response to infection. Thus, we examined whether astrocytes and microglia could be infected by MAV-1. We showed production of an E3 mRNA (indicative of early gene expression) and production of infectious virus from both infected astrocyte- and microglia-enriched cultures. Infection by MAV-1 was confirmed in vivo using flow cytometry of brain cells from mice infected i.p. Infected brain cells costained for markers of astrocytes and microglia. We also confirmed infection of these cells in vivo by coimmunofluorescence staining of E3 protein and of GFAP and IBA1 in astrocytes and microglia, respectively, in brains of mice infected with wt MAV-1 and an E1A mutant, dlE102. Wt virus and the E1A mutant virus gave similar results, as we expected, because the function of the E1A protein is primarily nuclear and unrelated to virus entry. That is, we consider dlE102 to be like a reporter virus for virus entry and early gene expression in these experiments. The coimmunofluorescence staining provides strong evidence for MAV-1 infection of astrocytes and microglia by both viruses. We have thus expanded the types of cells known to be infected by MAV-1 in the brain to include astrocytes and microglia in mice infected i.p. and intracerebrally. We saw similar immunofluorescence staining in intracerebrally infected mice of a different strain, C57BL/6 (data not shown). MAV-1 infection of astrocytes and microglia may modulate production of proinflammatory mediators that are known to result in host innate and adaptive immune responses (34). This may lead to damaging inflammation in the CNS parenchyma, i.e., encephalitis.
Because astrocytes and microglia are infected by MAV-1 and they are known producers of MMPs, we tested whether they could produce increased MMP activity upon infection. MAMCs infected with two different wt virus stocks and an E3 mutant virus stock did produce higher levels of MMP2 and MMP9 activity than mock-infected cultures (Fig. 7). The E3 mutant virus induces BBB disruption similarly to wt virus (7), and thus, it is not surprising that it also induced higher MMP activity in MAMCs than in uninfected MAMCs. We examined whether factors produced in MAMCs could induce increased MMP activity when added to pmBEC cultures. Although the differences did not reach statistical significance for either time point alone (Fig. 9A and B), the results suggest that culturing pmBECs with MAMC-conditioned medium resulted in increased MMP activity, consistent with soluble factors produced by MAMCs contributing to increased MMP activation induced by MAV-1 infection.
In summary, MAV-1 infection of susceptible mice led to increased MMP2 and MMP9 activity in whole brains. A mouse strain difference was noted; MAV-1-resistant BALB/c mice did not show increased MMP activity after infection, whereas increased activity was seen in susceptible SJL mice or C.SJL-Msq1SJL congenic mice. This indicates that Msq1 is linked to the MMP activity induction. Cultures containing astrocytes and microglia produced increased MMP activity after infection, and cultured medium from MAMCs stimulated MMP production from primary brain endothelial cells. Together, the data support the hypothesis that disruption of the BBB by MAV-1 infection of susceptible mice is due in part to infection-induced increases in MMP activity.
MATERIALS AND METHODS
Cells and virus.
MAV-1 was the standard stock originally obtained from S. Larsen as described in reference 35. It was propagated and titrated by plaque assay on mouse NIH 3T6 cells as described previously (36). Mutant virus dlE102 has a deletion in early region 1A (E1A) conserved region 2 (CR2), and it was propagated as described previously (37). A dlE102 stock of high titer (2 × 109 PFU/ml) was used and was highly purified using cell factories (38). Mutant virus pmE314 has point mutations that eliminate expression of E3, and it was propagated as described previously (39). MAV-1 inp903 has the wt pIX minor structural protein gene replaced with a pIX-GFP fusion construct and is referred to here as MAV-1.IXeGFP (19). PmBECs were prepared and cultured in endothelial cell medium as described previously (19).
Primary astrocytes and microglia were derived from postnatal day 1 or 2 mouse brains (SJL/J) by standard methods (40). Briefly, cortexes were isolated and trypsinized. The brains were triturated and plated onto poly-l-lysine-coated T-75 flasks in DMEM with l-glutamine (Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum with 10 ng/ml mouse macrophage colony-stimulating factor and 5 ng/ml mouse granulocyte-macrophage colony-stimulating factor (Sigma, St. Louis, MO). The cells were fed by addition of 3 ml of medium on days 3, 6, and 9. At 14 days, the resulting mixed glial culture (MAMC) was shaken at 200 rpm for 2 h at 4°C to release presumptive microglia. The shake-off medium was centrifuged for 5 min at 200 × g, and the pelleted cells (enriched for microglia; referred to here as “microglia enriched”) were plated at 1 × 106 cells per well. Fresh medium was added to the original flask containing adherent cells (enriched for astrocytes, referred to here as “astrocyte enriched”) for 24 h; then, the cells were passaged in DMEM containing 5% heat-inactivated fetal bovine serum before use. MAMCs and astroctye- and microglia-enriched cells used in these experiments were passaged ≤2 times and were infected 21 to 30 days after isolation from mouse brains. MAMC-conditioned medium was obtained from uninfected cultures at 14 days of culturing and centrifuged to remove cells and cell debris.
Mice and infections.
All animal work complied with relevant federal and University of Michigan policies. Animals were housed in microisolator cages and provided with food and water ad libitum. Male SJL/J mice (3 to 5 weeks old) were obtained from Jackson Laboratory (Bar Harbor, ME); we used males because our pathogenesis studies in SJL mice have used males. Congenic mice (C.SJL-Msq1SJL/SJL) have the mouse Chr 15 Msq1 locus in a BALB/c background (22); we infected males and females from our breeding colony and observed no differences based on sex. Mice were infected i.p. with virus diluted in endotoxin-free phosphate-buffered saline (PBS) in a total volume of 100 μl, except as noted below for intracerebral infections. Mock-infected mice were injected with conditioned medium diluted in PBS. Organs were snap-frozen on dry ice and stored at −80°C until analysis.
Determination of MAV-1 brain viral loads and BBB permeability assays.
Brain viral loads were measured by capture ELISA as described previously (19). BBB permeability was quantitated by sodium fluorescein uptake assay as described previously (19).
Sample preparation for MMP2 and MMP9 assays.
After CO2 euthanasia, mice were perfused with cold PBS, and brains were removed and stored at −20°C or −80°C. MMP2 and MMP9 were purified from whole-brain homogenates as described previously (19). Briefly, protein concentrations were determined using a Pierce bicinchoninic assay (BCA) kit. Equal protein quantities of each sample (600 to 2,000 μg) were then concentrated using gelatin-Sepharose 4B beads (GE Healthcare). Samples were incubated with the beads for 1 to 2 h at 4°C with nutation and then centrifuged at 16,000 to 21,000 × g for 5 min at 4°C. Gelatinases were eluted in equal amounts (30 to 50 μl) of 10% dimethyl sulfoxide (DMSO) in PBS or in buffer containing 50 mM Tris-HCl, pH 7.6, 150 mM NaCl, 5 mM CaCl2, 0.05% Brij 35, 0.02% NaN3, 10% DMSO. Equal volumes of eluted samples (10 to 15 μl) were mixed with an equal volume of 2× Laemmli sample buffer (41) or 4× NuPAGE LDS sample buffer (Life Technologies) and stored at −20°C. For sample preparation of secreted MMPs from cultured cells, at 24 h prior to harvest, the cells were washed once with PBS and then serum-free medium was added to remove MMPs contributed by serum. Secreted MMP2 and MMP9 were isolated from cell supernatants, assayed by BCA assay prior to gelatin concentration, and concentrated as described above.
Gelatin zymography.
MMP2 and MMP9 activities were assayed by gelatin zymography. Samples were electrophoresed on an 8% or 10% SDS-polyacrylamide gel copolymerized with 1 mg/ml gelatin (0.1%). The gels were then rinsed briefly with water and then twice for 20 min in 2.5% Triton X-100, interspersed with 1- to 5-min washes in water. The gels were incubated for 4 to 5 days in developing buffer (50 mM Tris-HCl, pH 7.6, 200 mM NaCl, 10 mM CaCl2, 0.02% Brij 35, 0.02% NaN3) to enable gelatin digestion by the gelatinases. The gels were then stained in 0.125% Coomassie blue for 1 h and destained in 10% acetic acid overnight. Gels processed in parallel in developing buffer containing 5 mM EDTA had no MMP activity, as expected (MMP activity is calcium and zinc dependent [12]) (data not shown). Images were acquired using an AlphaImagerHP system (Alpha Innotech) and inverted (black/white) in Adobe Photoshop or acquired from a ChemiDoc XRS (Bio-Rad). Quantification was performed with spot density analysis using AlphaEaseFC software (Alpha Innotech) or ImageJ (42), respectively, for the two image acquisition methods. The gelatin zymography standards were MMP9 (Chemicon and R&D Systems) and supernatant from human HT1080 cells (ATCC). There are multiple isoforms of MMP9 detectable by zymography (43), in which the development step activates both prepro- and pro-MMP9 forms (44). We saw heterogeneous high-molecular-mass bands (e.g., ∼120 to 130 kDa, higher than the 105-kDa murine proMMP) when large amounts of commercial recombinant murine MMP9 (R&D Systems; 909-MM) were loaded and/or when gel development times were long (data not shown). These bands likely represent a multiply glycosylated form of murine MMP9 (43; E. Candelario, personal communication). Similar bands can be seen in the human HT1080 marker lanes (Fig. 1, 2, 7, and 9, M3) when large amounts or long developing times were used.
TEER measurement.
TEER was measured on pmBECs cultured on collagen IV-coated transwells (12-well; 0.4-μm pore size; Corning). Endothelial cell medium (19) was supplemented with 500 ng/ml hydrocortisone (Sigma H6909). As indicated in the figure legends, in some experiments, MAMC-conditioned medium was included in the lower chamber to facilitate tight-junction formation. TEER was measured on confluent cells using an Endohm-12 apparatus (World Precision Instruments, Inc.), and values were corrected by subtracting a blank well measured at the same time, as described previously (19).
RNA isolation, RT-PCR, and RT-qPCR.
Total RNA was isolated from primary astrocytes and microglia using Tri Reagent (Molecular Research Center Inc., Cincinnati, OH) according to the manufacturer's instructions. cDNA was synthesized using random hexamers and Moloney murine leukemia virus reverse transcriptase (Invitrogen, Carlsbad, CA). PCR for E3 and actin was performed as described previously (1). qPCR was performed for MMP mRNAs as described previously (19) using an Applied Biosystems (Foster City, CA) 7300 real-time (RT) PCR machine.
Brain cell isolation for flow cytometry.
We modified a published procedure to isolate brain cells for flow cytometry (45). After CO2 euthanasia, brains were removed from five MAV-1.IXeGFP-infected or mock-infected, 4- to 5-week-old SJL mice at 7 dpi and placed into ice-cold 30% sucrose in PBS (S/PBS). On dry petri dishes, the meninges were removed, and the brains were cut into small pieces and put back into S/PBS. The cut pieces were centrifuged for 8 min at 450 × g, and the supernatants were discarded. The cells were incubated at 37°C for 15 min in 2 to 3 ml of 0.25% trypsin (Invitrogen); 8 to 15 mg of collagenase-dispase (Roche) was added to 2 ml S/PBS and incubated at 37°C for 10 min prior to adding 5 μl of 2.5 μg/μl DNase I (Sigma). The collagenase-dispase-DNase I suspension was added to the cells and incubated at 37°C for 20 min, with pipetting through a Pasteur pipet 10 times every 10 min. Homogenates were passed through a 40-μm cell strainer, resuspended in 20 to 40 ml of 30% Percoll (GE Healthcare) in S/PBS, and centrifuged for 10 min at 650 × g. The supernatants were aspirated to remove myelin and resuspended in ACK cell lysis buffer (0.15 M NH4Cl, 1.0 mM KHCO3, 0.1 mM Na2EDTA, pH 7.2 to 7.4) for 2 min on ice; 10 ml of IMAG buffer (PBS, 0.5% bovine serum albumin [BSA], 2 mM EDTA) was added, and the cells were centrifuged for 5 min at 450 × g. The pellets were resuspended in 1 ml IMAG buffer and counted.
Flow cytometry.
Cells isolated from brains were washed once with IMAG buffer and preincubated for 20 min at room temperature with 3 μl anti-FcγR monoclonal antibody (MAb) 2.4G2 (BD Biosciences) to block nonspecific binding. Cells (106) were stained with 1 μl of allophycocyanin (APC)-conjugated antibodies (CD11b or GLAST [Miltenyi Biotec; catalog numbers 130-098-088 and 130-095-814, respectively]) for 30 min at 4°C, washed with IMAG buffer, and permeabilized and fixed (Foxp3/Transcription Factor Fix/Perm; Tonbo Biosciences) overnight at 4°C. After washing with 1× Perm Buffer (Tonbo Biosciences), the cells were stained intracellularly with 0.5 μl of anti-GFP antibody (Biolegend; catalog number 338002) for 10 min at room temperature and washed with 1× Perm Buffer, and 1 μl of secondary rabbit anti-rat fluorochrome-conjugated antibodies (Sigma) was added for 10 min at room temperature. Finally, the cells were washed with IMAG buffer, resuspended in IMAG buffer, and analyzed by flow cytometry on a FACSCanto (BD) flow cytometer. The data were analyzed with FlowJo v.10.2 software (Tree Star).
Infection and immunofluorescent-antibody analysis of brains of mice infected intracerebrally.
Anesthetized SJL mice were injected with wt MAV-1 or dlE102 in 5 μl into the right and left striatum using a 5-μl Hamilton syringe fitted with a 33-gauge needle. When moribund, at 6 to 8 dpi, the mice were euthanized and perfused with Tyrode's solution (137 mM NaCl, 1.8 mM CaCl2 · 2H2O, 0.32 mM NaH2PO4 · 2H2O, 5.6 mM d-glucose, 11.9 mM NaHCO3, 3.4 mM KCl, 100 U heparin [Stemcell Technologies; catalog number 7980]), followed by 4% paraformaldehyde (PFA). Brains were harvested and kept in PFA at 4°C for 2 days. Coronal 50-μm brain sections were taken using a Leica VT1000S vibratome.
Rabbit αE3-1 is a rabbit polyclonal antiserum to E3gp19K, an early viral protein (46). It was used at 1:10,000 dilution after preincubation overnight with brain sections for preabsorption of background signal. Purified mouse anti-GFAP (Millipore; catalog number AB5541) was used at 1:1,000. Anti-IBA1 (Abcam; catalog number ab5076) was used at 1:500.
Free-floating PFA-fixed brain sections were permeabilized in Tris-buffered saline (TBS)–0.05% Triton X-100 (TBS-Tx) for 30 min. Antigen retrieval was carried out by incubation in 0.1 M sodium citrate for 20 min at 90°C, followed by two 5-min TBS-Tx washes. The sections were blocked in 10% horse serum (ThermoFisher; catalog number 16050122)/TBS-Tx for >3 h at 4°C and then incubated with αE3-1 serum at 1:10,000, mouse anti-GFAP at 1:1,000, or IBA1 at 1:500 in 1% horse serum–TBS-Tx for >48 h at 4°C. The sections were then washed six times in TBS-Tx for 5 min each time.
Secondary staining for E3gp19K was done with highly cross-absorbed Alexa Fluor 488 goat anti-rabbit IgG(H+L) (Life Technologies; catalog number A11034) or Alexa Fluor 488 donkey anti-rabbit IgG(H+L) (ThermoFisher; catalog number A21206) at 1:1,000.
Secondary staining for GFAP was done with Alexa Fluor 594 goat anti-chicken (ThermoFisher; catalog number A11042) at 1:1,000 dilution. Secondary staining for IBA1 was done with Alexa Fluor 594 donkey anti-goat (ThermoFisher; catalog number 11058) at 1:1,000 dilution. Sections were washed six times in TBS-Tx for 5 min each time and incubated with 1:1,000 DAPI (4′,6-diamidino-2-phenylindole) in PBS, followed by three 5-min PBS washes. The sections were mounted onto electrostatic slides in PBS and coverslipped with ProLong Gold antifade reagent (Life Technologies; catalog number 36930). High-magnification images were obtained using confocal microscopy (Carl Zeiss; MIC system).
Statistical analysis.
Statistical analysis was carried out using Microsoft (Everett, WA) Excel v. 11.5.3 and Graph Pad (La Jolla, CA) Prism 6 software. Due to the small sample sizes, nonparametric analyses were used for comparisons between means. A P value of 0.05 was used as the cutoff for determination of significance.
ACKNOWLEDGMENTS
We thank Jason Weinberg, Mike Imperiale, and Danielle Goodman for comments on the manuscript. We thank Eduardo Candelario-Jalil and Gary Rosenberg for helpful advice and reagents for MMP zymography. We are grateful to Anuska Andjelkovic for assistance with primary cell preparations and helpful discussions. We thank Aaron Robida of the University of Michigan Flow Cytometry Core for advice and discussions on data analysis.
This work was supported by Brazil Science without Borders (Ciências sem Fronteiras) (2438/13-5) (Luiza Castro Jorge). It was also supported by HHS/NIH/National Institute of Allergy and Infectious Diseases (NIAID) 1 R01 AI068645, 1 R01 AI023762, and 1 R01 AI091721 (Katherine R. Spindler). Work in the laboratories of Pedro R. Lowenstein and Maria G. Castro is supported by HHS/NIH/National Institute of Neurological Disorders and Stroke (NINDS) grants R01 NS082311, R21 NS084275, R01 NS096756, R01 NS074387, R21 NS091555, R01 NS057711, and R37 NS094804. The University of Michigan Flow Cytometry Core, and thus this research, was supported by the National Cancer Institute of the National Institutes of Health under award number P30 CA046592.
REFERENCES
- 1.Ashley SL, Welton AR, Harwood KM, Van Rooijen N, Spindler KR. 2009. Mouse adenovirus type 1 infection of macrophages. Virology 390:307–314. doi: 10.1016/j.virol.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Charles PC, Guida JD, Brosnan CF, Horwitz MS. 1998. Mouse adenovirus type-1 replication is restricted to vascular endothelium in the CNS of susceptible strains of mice. Virology 245:216–228. doi: 10.1006/viro.1998.9180. [DOI] [PubMed] [Google Scholar]
- 3.Kajon AE, Brown CC, Spindler KR. 1998. Distribution of mouse adenovirus type 1 in intraperitoneally and intranasally infected adult outbred mice. J Virol 72:1219–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kring SC, King CS, Spindler KR. 1995. Susceptibility and signs associated with mouse adenovirus type 1 infection of adult outbred Swiss mice. J Virol 69:8084–8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guida JD, Fejer G, Pirofski L-A, Brosnan CF, Horwitz MS. 1995. Mouse adenovirus type 1 causes a fatal hemorrhagic encephalomyelitis in adult C57BL/6 but not BALB/c mice. J Virol 69:7674–7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spindler KR, Fang L, Moore ML, Brown CC, Hirsch GN, Kajon AK. 2001. SJL/J mice are highly susceptible to infection by mouse adenovirus type 1. J Virol 75:12039–12046. doi: 10.1128/JVI.75.24.12039-12046.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gralinski LE, Ashley SL, Dixon SD, Spindler KR. 2009. Mouse adenovirus type 1-induced breakdown of the blood-brain barrier. J Virol 83:9398–9410. doi: 10.1128/JVI.00954-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engelhardt B, Liebner S. 2014. Novel insights into the development and maintenance of the blood-brain barrier. Cell Tissue Res 355:687–699. doi: 10.1007/s00441-014-1811-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Engelhardt S, Patkar S, Ogunshola OO. 2014. Cell-specific blood-brain barrier regulation in health and disease: a focus on hypoxia. Br J Pharmacol 171:1210–1230. doi: 10.1111/bph.12489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hawkins BT, Davis TP. 2005. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev 57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 11.Agrawal S, Anderson P, Durbeej M, van Rooijen N, Ivars F, Opdenakker G, Sorokin LM. 2006. Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis. J Exp Med 203:1007–1019. doi: 10.1084/jem.20051342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Candelario-Jalil E, Yang Y, Rosenberg GA. 2009. Diverse roles of matrix metalloproteinases and tissue inhibitors of metalloproteinases in neuroinflammation and cerebral ischemia. Neuroscience 158:983–994. doi: 10.1016/j.neuroscience.2008.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yong VW. 2005. Metalloproteinases: mediators of pathology and regeneration in the CNS. Nat Rev Neurosci 6:931–944. doi: 10.1038/nrn1807. [DOI] [PubMed] [Google Scholar]
- 14.Spindler KR, Hsu T-H. 2012. Viral disruption of the blood-brain barrier. Trends Microbiol 20:282–290. doi: 10.1016/j.tim.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bojarski C, Weiske J, Schoneberg T, Schroder W, Mankertz J, Schulzke JD, Florian P, Fromm M, Tauber R, Huber O. 2004. The specific fates of tight junction proteins in apoptotic epithelial cells. J Cell Sci 117:2097–2107. doi: 10.1242/jcs.01071. [DOI] [PubMed] [Google Scholar]
- 16.Giebel SJ, Menicucci G, McGuire PG, Das A. 2005. Matrix metalloproteinases in early diabetic retinopathy and their role in alteration of the blood-retinal barrier. Lab Invest 85:597–607. doi: 10.1038/labinvest.3700251. [DOI] [PubMed] [Google Scholar]
- 17.Gurney KJ, Estrada EY, Rosenberg GA. 2006. Blood-brain barrier disruption by stromelysin-1 facilitates neutrophil infiltration in neuroinflammation. Neurobiol Dis 23:87–96. doi: 10.1016/j.nbd.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 18.Manicone AM, McGuire JK. 2008. Matrix metalloproteinases as modulators of inflammation. Semin Cell Dev Biol 19:34–41. doi: 10.1016/j.semcdb.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tirumuru N, Pretto CD, Castro Jorge L, Spindler KR. 2016. Mouse adenovirus type 1 early region 1A effects on the blood-brain barrier. mSphere 1:e00079-16. doi: 10.1128/mSphere.00079-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Welton AR, Chesler EJ, Sturkie C, Jackson AU, Hirsch GN, Spindler KR. 2005. Identification of quantitative trait loci for susceptibility to mouse adenovirus type 1. J Virol 79:11517–11522. doi: 10.1128/JVI.79.17.11517-11522.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spindler KR, Welton AR, Lim ES, Duvvuru S, Althaus IW, Imperiale JE, Daoud AI, Chesler EJ. 2010. The major locus for mouse adenovirus susceptibility maps to genes of the hematopoietic cell surface-expressed LY6 family. J Immunol 184:3055–3062. doi: 10.4049/jimmunol.0903363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsu T-H, Althaus IW, Foreman O, Spindler KR. 2012. Contribution of a single host genetic locus to mouse adenovirus type 1 infection and encephalitis. mBio 3:e00131-12. doi: 10.1128/mBio.00131-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith K, Brown CC, Spindler KR. 1998. The role of mouse adenovirus type 1 early region 1A in acute and persistent infections in mice. J Virol 72:5699–5706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang Y, Hill JW, Rosenberg GA. 2011. Multiple roles of metalloproteinases in neurological disorders. Prog Mol Biol Transl Sci 99:241–263. doi: 10.1016/B978-0-12-385504-6.00006-3. [DOI] [PubMed] [Google Scholar]
- 25.Dehouck MP, Méresse S, Delorme P, Fruchart JC, Cecchelli R. 1990. An easier, reproducible, and mass-production method to study the blood-brain barrier in vitro. J Neurochem 54:1798–1801. doi: 10.1111/j.1471-4159.1990.tb01236.x. [DOI] [PubMed] [Google Scholar]
- 26.Fischer S, Wobben M, Kleinstück J, Renz D, Schaper W. 2000. Effect of astroglial cells on hypoxia-induced permeability in PBMEC cells. Am J Physiol Cell Physiol 279:C935–C944. [DOI] [PubMed] [Google Scholar]
- 27.Rist RJ, Romero IA, Chan MW, Couraud PO, Roux F, Abbott NJ. 1997. F-actin cytoskeleton and sucrose permeability of immortalised rat brain microvascular endothelial cell monolayers: effects of cyclic AMP and astrocytic factors. Brain Res 768:10–18. doi: 10.1016/S0006-8993(97)00586-6. [DOI] [PubMed] [Google Scholar]
- 28.Al Ahmad A, Gassmann M, Ogunshola OO. 2009. Maintaining blood-brain barrier integrity: pericytes perform better than astrocytes during prolonged oxygen deprivation. J Cell Physiol 218:612–622. doi: 10.1002/jcp.21638. [DOI] [PubMed] [Google Scholar]
- 29.Rubin LL, Hall DE, Porter S, Barbu K, Cannon C, Horner HC, Janatpour M, Liaw CW, Manning K, Morales J. 1991. A cell culture model of the blood-brain barrier. J Cell Biol 115:1725–1735. doi: 10.1083/jcb.115.6.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raub TJ, Kuentzel SL, Sawada GA. 1992. Permeability of bovine brain microvessel endothelial cells in vitro: barrier tightening by a factor released from astroglioma cells. Exp Cell Res 199:330–340. doi: 10.1016/0014-4827(92)90442-B. [DOI] [PubMed] [Google Scholar]
- 31.Deli MA, Ábrahám CS, Kataoka Y, Niwa M. 2005. Permeability studies on in vitro blood-brain barrier models: physiology, pathology, and pharmacology. Cell Mol Neurobiol 25:59–127. doi: 10.1007/s10571-004-1377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spindler KR, Moore ML, Cauthen AN. 2007. Mouse adenoviruses, p 49–65, The mouse in biomedical research, 2nd ed, vol 2 Academic Press, New York, NY. [Google Scholar]
- 33.Behzadian MA, Wang XL, Windsor LJ, Ghaly N, Caldwell RB. 2001. TGF-beta increases retinal endothelial cell permeability by increasing MMP-9: possible role of glial cells in endothelial barrier function. Invest Ophthalmol Vis Sci 42:853–859. [PubMed] [Google Scholar]
- 34.Furr SR, Marriott I. 2012. Viral CNS infections: role of glial pattern recognition receptors in neuroinflammation. Front Microbiol 3:201. doi: 10.3389/fmicb.2012.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ball AO, Beard CW, Villegas P, Spindler KR. 1991. Early region 4 sequence and biological comparison of two isolates of mouse adenovirus type 1. Virology 180:257–265. doi: 10.1016/0042-6822(91)90030-F. [DOI] [PubMed] [Google Scholar]
- 36.Cauthen AN, Welton AR, Spindler KR. 2007. Construction of mouse adenovirus type 1 mutants. Methods Mol Med 130:41–59. [DOI] [PubMed] [Google Scholar]
- 37.Smith K, Ying B, Ball AO, Beard CW, Spindler KR. 1996. Interaction of mouse adenovirus type 1 early region 1A protein with cellular proteins pRb and p107. Virology 224:184–197. doi: 10.1006/viro.1996.0520. [DOI] [PubMed] [Google Scholar]
- 38.Robinson M, Li B, Ge Y, Ko D, Yendluri S, Harding T, VanRoey M, Spindler KR, Jooss K. 2009. Novel immunocompetent murine tumor model for evaluation of conditionally replication-competent (oncolytic) murine adenoviral vectors. J Virol 83:3450–3562. doi: 10.1128/JVI.02561-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cauthen AN, Brown CC, Spindler KR. 1999. In vitro and in vivo characterization of a mouse adenovirus type 1 early region 3 mutant. J Virol 73:8640–8646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McKimmie CS, Fazakerley JK. 2005. In response to pathogens, glial cells dynamically and differentially regulate Toll-like receptor gene expression. J Neuroimmunol 169:116–125. doi: 10.1016/j.jneuroim.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 41.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 42.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Opdenakker G, Van den Steen PE, Dubois B, Nelissen I, Van Coillie E, Masure S, Proost P, Van Damme J. 2001. Gelatinase B functions as regulator and effector in leukocyte biology. J Leukoc Biol 69:851–859. [PubMed] [Google Scholar]
- 44.Frankowski H, Gu YH, Heo JH, Milner R, Del Zoppo GJ. 2012. Use of gel zymography to examine matrix metalloproteinase (gelatinase) expression in brain tissue or in primary glial cultures. Methods Mol Biol 814:221–233. doi: 10.1007/978-1-61779-452-0_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nikodemova M, Watters JJ. 2012. Efficient isolation of live microglia with preserved phenotypes from adult mouse brain. J Neuroinflammation 9:147. doi: 10.1186/1742-2094-9-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beard CW, Spindler KR. 1995. Characterization of an 11K protein produced by early region 3 of mouse adenovirus type 1. Virology 208:457–466. doi: 10.1006/viro.1995.1176. [DOI] [PubMed] [Google Scholar]