

ABSTRACT
The GDP polyribonucleotidyltransferase (PRNTase) domain of the multifunctional L protein of rhabdoviruses, such as vesicular stomatitis virus (VSV) and rabies virus, catalyzes the transfer of 5′-phospho-RNA (pRNA) from 5′-triphospho-RNA (pppRNA) to GDP via a covalent enzyme-pRNA intermediate to generate a 5′-cap structure (GpppA). Here, using an improved oligo-RNA capping assay with the VSV L protein, we showed that the Michaelis constants for GDP and pppAACAG (VSV mRNA-start sequence) are 0.03 and 0.4 μM, respectively. A competition assay between GDP and GDP analogues in the GpppA formation and pRNA transfer assay using GDP analogues as pRNA acceptors indicated that the PRNTase domain recognizes the C-2-amino group, but not the C-6-oxo group, N-1-hydrogen, or N-7-nitrogen, of GDP for the cap formation. 2,6-Diaminopurine-riboside (DAP), 7-deazaguanosine (7-deaza-G), and 7-methylguanosine (m7G) diphosphates efficiently accepted pRNA, resulting in the formation of DAPpppA, 7-deaza-GpppA, and m7GpppA (cap 0), respectively. Furthermore, either the 2′- or 3′-hydroxyl group of GDP was found to be required for efficient pRNA transfer. A 5′-diphosphate form of antiviral ribavirin weakly inhibited the GpppA formation but did not act as a pRNA acceptor. These results indicate that the PRNTase domain has a unique guanosine-binding mode different from that of eukaryotic mRNA capping enzyme, guanylyltransferase.
IMPORTANCE mRNAs of nonsegmented negative-strand (NNS) RNA viruses, such as VSV, possess a fully methylated cap structure, which is required for mRNA stability, efficient translation, and evasion of antiviral innate immunity in host cells. GDP polyribonucleotidyltransferase (PRNTase) is an unconventional mRNA capping enzyme of NNS RNA viruses that is distinct from the eukaryotic mRNA capping enzyme, guanylyltransferase. In this study, we studied the pRNA acceptor specificity of VSV PRNTase using various GDP analogues and identified chemical groups of GDP as essential for the substrate activity. The findings presented here are useful not only for understanding the mechanism of the substrate recognition with PRNTase but also for designing antiviral agents targeting this enzyme.
KEYWORDS: GDP polyribonucleotidyltransferase, L protein, cap structure, mRNA capping, nonsegmented negative-strand RNA viruses, rabies virus, vesicular stomatitis virus
INTRODUCTION
The 5′-terminal cap 1 structure of viral mRNAs is required for mRNA stability, efficient translation, and escape from antiviral innate immunity in host cells (reviewed in references 1, 2, and 3). GDP polyribonucleotidyltransferase (PRNTase [EC 2.7.7.88]) is a novel mRNA capping enzyme that is present as an enzymatic domain in multifunctional large (L) proteins of nonsegmented negative-strand (NNS) RNA viruses belonging to the Rhabdoviridae family in the order Mononegavirales (reviewed in references 4 and 5). Using native and/or recombinant L proteins of three rhabdoviruses, vesicular stomatitis virus (VSV) (6–8), Chandipura virus (9), and rabies virus (RABV) (10), we have demonstrated that the molecular mechanism of mRNA capping with the PRNTase activity is strikingly different from that with the conventional mRNA capping enzyme, guanylyltransferase (GTase [EC 2.7.7.50]), of eukaryotes and nucleocytoplasmic large DNA viruses (e.g., vaccinia virus) (reviewed in references 11 and 12). Although PRNTase-like domains are found in L proteins of all other NNS RNA viruses classified into the Paramyxoviridae (e.g., measles virus), Pneumoviridae (e.g., respiratory syncytial virus), Filoviridae (e.g., Ebola virus), Bornaviridae (e.g., Borna disease virus), Nyamiviridae, Mymonaviridae, and Sunviridae families (4, 13), none of their PRNTase activities have so far been demonstrated.
The PRNTase domain in the VSV L protein (2,109 amino acids) carries out mRNA capping by covalent catalysis with a nucleophilic histidine residue at position 1227 (H1227) (8). A lone pair of electrons at the Nε2 position of H1227 nucleophilically attacks the α-phosphorus in the 5′-triphophate (ppp) group of the VSV mRNA start sequence (AACAG), resulting in the formation of a covalent enzyme-(hystidyl-Nε2)-pRNA (called L-pRNA) intermediate most probably with the concomitant release of inorganic pyrophosphate (PPi) (6, 8). A conserved arginine residue (R1228) adjacent to H1227 is also essential for the intermediate formation and can be partially replaced with histidine, but not with lysine or other neutral amino acids (8), suggesting that the secondary amine at the ε position in its positively charged guanidino group plays important roles in a catalytic step(s), such as binding to the 5′-triphosphate group of pppRNA and a proton donation to leaving PPi. To produce the cap structure, G(5′)ppp(5′)A, the pRNA moiety covalently linked to H1227 is transferred to GDP (pRNA acceptor) (6, 8), which is generated from GTP by the L protein-associated guanosine 5′-triphosphatase (GTPase) activity (6, 7).
The PRNTase domains of NNS RNA viral L proteins contain five conserved amino sequence elements, Rx(3)Wx(3–8)ΦxGxζx(P/A) (motif A; Φ, hydrophobic; ζ, hydrophilic), (Y/W)ΦGSxT (motif B), W (motif C), HR (motif D), and ζxxΦx(F/Y)QxxΦ (motif E) (13). Motif D of the VSV PRNTase domain contains the catalytic residues H1227 and R1228 (8, 13). Other key residues, G1100 in motif A, T1157 in motif B, W1188 in motif C, and F1269 and Q1270 in motif E, were identified as required for the L-pRNA intermediate formation with the VSV L protein (13). Counterparts of these key residues in the RABV L protein were recently found to be essential for RABV mRNA capping (10). In the three-dimensional structure of the VSV L protein determined by cryoelectron microscopy (14), these key residues in motifs A, B, C, and E surround motif D, forming a unique active site of the PRNTase domain (13). These conserved residues in motifs A to E are essential for VSV gene expression and growth in host cells (13, 15). So far, no amino acid residues specifically required for the pRNA transfer to GDP (e.g., GDP binding) have been identified in the VSV L protein. Although Liang et al. (14) claimed that G1154 and T1157 in motif B of the VSV PRNTase domain participate in guanosine nucleotide binding, there is no experimental evidence to support this conjecture.
We have previously reported that the PRNTase domain in the VSV L protein transfers pRNA from pppRNA to GDP, but not to other NDPs, to produce the cap structure (8). However, it remained unknown which chemical groups of GDP determine the pRNA acceptor specificity. In this study, using in vitro oligo-RNA capping assays and various GDP analogues, we identified chemical groups of GDP that are required for the pRNA acceptor activity.
RESULTS
Improvement of the in vitro oligo-RNA capping assay with the VSV L protein.
Our previous in vitro oligo-RNA capping assay with pppAACAG (VSV mRNA start sequence) and [α-32P]GDP includes many steps, such as calf intestine alkaline phosphatase (CIAP) treatment, phenol-chloroform extraction, ethanol precipitation, and nuclease P1 treatment, to analyze the GpppA cap structure by polyethyleneimine (PEI)-cellulose thin-layer liquid chromatography (TLC) (6, 8, 16). Here, we developed a simple method to measure the capping activity of the VSV L protein without the above-described laborious steps. In this system, after stopping capping reactions by the addition of a formamide solution containing EDTA, the reaction mixtures were directly resolved by 20% urea-PAGE (Fig. 1A). When [α-32P]GDP and pppAACAG were incubated with the recombinant VSV L protein, a 32P-labeled RNA product migrating slower than [α-32P]GDP was detected on the gel (lane 3). We confirmed that this RNA product comigrates with GpppAACAG (data not shown), which had been synthesized by capping of pppAACAG by the vaccinia virus capping enzyme as previously reported (6, 8). In the absence of the VSV L protein (lane 1) or pppAACAG (lane 2), no product was generated. Furthermore, as previously reported (6), 5′-diphosphorylated RNA (ppAACAG, lane 4) did not serve as a pRNA donor substrate. We also confirm that the cap-defective mutant L protein (H1227R) (8) does not synthesize the product (lane 5).
FIG 1.
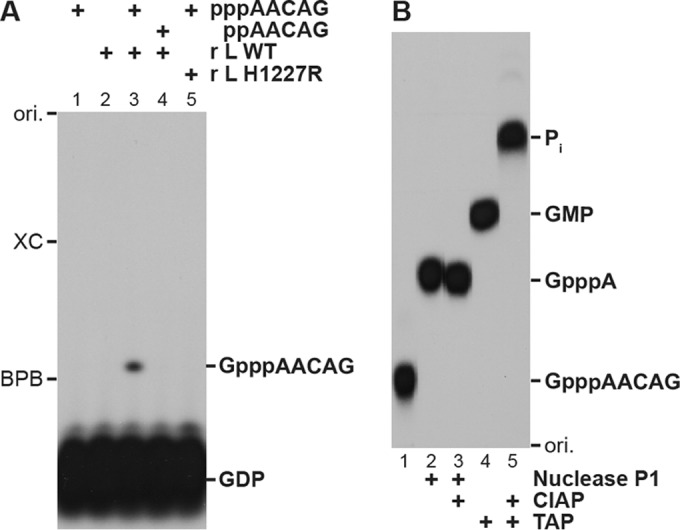
Improved oligo-RNA capping assay with the recombinant VSV L protein. (A) [α-32P]GDP (0.25 μM) was incubated in capping reaction mixtures with the indicated RNA (2.5 μM) and/or recombinant VSV L protein (0.2 μg, wild type [WT] or cap-defective mutant [H1227R]) for 20 min. The reaction mixtures were directly analyzed by 20% urea-PAGE followed by autoradiography. (B) [32P]GpppAACAG synthesized by the WT L protein was purified from a polyacrylamide gel and digested with nuclease P1, calf intestine alkaline phosphatase (CIAP), and/or tobacco acid pyrophosphatase (TAP). The digests were analyzed by PEI-cellulose TLC followed by autoradiography. ori., origin.
To confirm the presence of the cap structure on the RNA product, it was purified from the gel and then enzymatically digested (Fig. 1B). Nuclease P1 digestion of the 32P-labeled RNA product generated 32P-labeled GpppA (lane 2), which was resistant to CIAP (lane 3). Tobacco acid pyrophosphatase (TAP) hydrolyzed the RNA product into [α-32P]GMP (lane 4), which could be further digested into inorganic phosphate (Pi) with CIAP (lane 5). These results demonstrated that the capped RNA product (GpppAACAG) in the reaction mixtures could be directly analyzed by urea-PAGE.
Kinetic characteristics of the GpppA formation with the VSV L protein.
Using the improved oligo-RNA capping assay in the presence of saturating concentrations of [α-32P]GDP and pppAACAG, we showed that the amounts of the capped RNA product increase linearly with increasing amounts of the L protein, at least up to 0.3 μg, during incubation for 20 min (Fig. 2A). In the presence of 0.2 μg of the L protein, the product amounts increased linearly with time over an incubation period of 40 min (Fig. 2B). Therefore, to measure initial velocities (v0) of the capping reaction, 0.2 μg of the L protein was incubated with various concentrations of one substrate in the presence of a saturating concentration of another substrate for 20 min (Fig. 2C and D), which is within the linear range of the time course. To fit the data to the Michaelis-Menten equation, we used a curve-fitting method in nonlinear regression (17). A Michaelis-Menten plot at various concentrations of GDP estimated that the Michaelis constant (Km) and maximal velocity (Vmax) for the GpppA production are 31 ± 6 nM and 16 ± 1 pmol/min/mg protein (means ± standard deviations from three independent experiments), respectively (Fig. 2C). Km for pppAACAG and Vmax for the GpppA formation were estimated to be 0.43 ± 0.03 μM and 16 ± 2 pmol/min/mg protein, respectively, from a Michaelis-Menten plot at various concentrations of pppAACAG (Fig. 2D).
FIG 2.
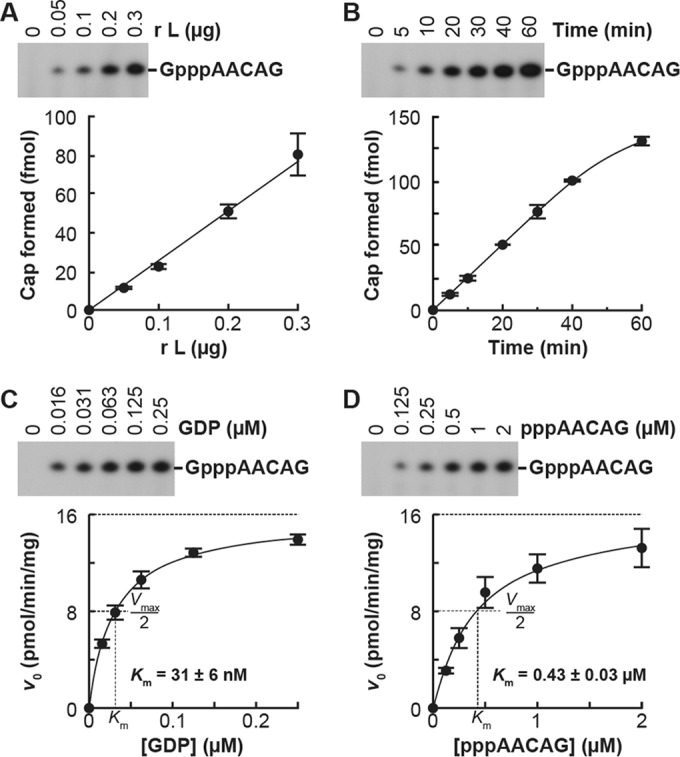
Kinetic characteristics of the GpppA formation with the VSV L protein. (A) Various amounts of the recombinant VSV L protein were incubated with 0.25 μM [α-32P]GDP and 2.5 μM pppAACGA for 20 min. The resulting GpppAACAG was analyzed as described for Fig. 1A (upper panel). The autoradiogram is a representative of three independent experiments. Amounts of GpppA formed on the RNA were determined by counting the 32P radioactivities of the indicated bands and plotted against the protein amounts (lower panel). Symbols and error bars represent the means and standard deviations, respectively. (B) The time course of the GpppA formation was determined with 0.25 μM [α-32P]GDP, 2.5 μM pppAACGA, and 0.2 μg of the recombinant VSV L protein. (C and D) The recombinant VSV L protein (0.2 μg) was incubated with various concentrations of [α-32P]GDP and 2.5 μM pppAACGA (C) or 0.25 μM [α-32P]GDP and various concentrations of pppAACGA (D) for 20 min. The initial velocity (v0) of the GpppA formation (pmol/min/mg protein) was plotted against the substrate concentrations. The curved lines represent the best fits to the Michaelis-Menten equation. Km values for respective substrates were estimated as described in Materials and Methods.
Inhibition of the GpppA formation with GDP analogues.
To find competitive inhibitors against the GpppA formation with GDP as well as to determine which moieties of GDP are required for the substrate recognition with the PRNTase domain, we performed the oligo-RNA capping assay with 0.25 μM [α-32P]GDP in the presence of various concentrations of GDP analogues depicted in Fig. 3. Representative results with selected GDP analogues are shown in Fig. 4, and absolute 50% inhibitory concentrations (IC50s) for all GDP analogues used in this study are summarized in Table 1. It should be noted that their Ki values estimated from IC50s (Table 1) are valid only if they competitively inhibit the GpppA formation against GDP. We found that the GpppA formation is strongly inhibited by GDP analogues with modifications at guanine-C-6, i.e., 2,6-diaminopurine-riboside 5′-diphosphate (DAPDP; IC50 = 0.37 μM) > 2-aminopurine-riboside 5′-diphosphate (APDP; IC50 = 1.3 μM), and at N-7, i.e., 7-deazaguanosine 5′-diphosphate (7-deaza-GDP; IC50 = 0.4 μM) and 7-methylguanosine 5′-diphosphate (m7GDP; IC50 = 0.39 μM) positions (Table 1). In contrast, GDP analogues lacking the C-2-amino group (IDP [IC50 = 6.4 μM] and XDP [IC50 = 58 μM]) as well as ADP (IC50 = 23 μM) exhibited weak inhibitory effects on GpppA formation. Furthermore, 2′-deoxyguanosine 5′-diphosphate (2′-dGDP; IC50 = 0.53 μM) and 3′-deoxyguanosine 5′-diphosphate (3′-dGDP; IC50 = 0.57 μM), but not ddGDP (IC50 = 3.5 μM), efficiently inhibited GpppA formation. Although we examined the effect of a 5′-diphosphate form of broad-spectrum antiviral compound ribavirin (RDP) on cap formation, it showed a weak inhibitory effect (IC50 = 8.6 μM).
FIG 3.

Structures of GDP analogues. The structure of GDP is shown with numbered positions of some base and ribose ring atoms. Nucleoside 5′-diphosphates with the indicated base or ribose were used in this study. Their full names and abbreviations are listed in Table 1.
FIG 4.
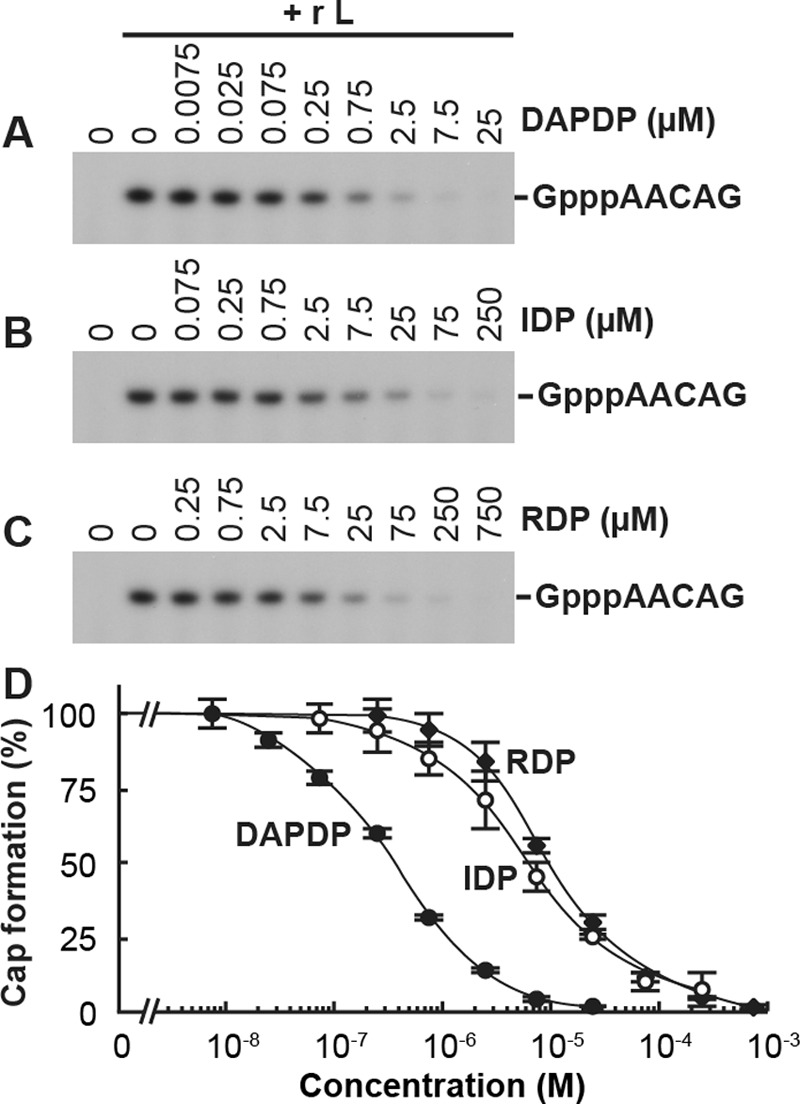
GDP analogues inhibit the GpppA formation catalyzed by the VSV L protein. The oligo-RNA capping assay was performed with 0.25 μM [α-32P]GDP and 2.5 μM pppAACGA in the presence or absence of various concentrations of 2,6-diaminopurine-riboside 5′-diphosphate (DAPDP) (A), IDP (B), or ribavirin 5′-diphosphate (RDP) (C). The resulting GpppAACAG was analyzed as described for Fig. 1A. The results are expressed as percentages of the GpppA formation activity observed in the absence of the inhibitors (set to 100%) (D). Symbols and error bars represent the means and standard deviations, respectively, for three independent experiments.
TABLE 1.
Inhibition of GpppA formation with GDP analogues
| GDP analogue (abbreviation) | IC50 (μM)a | Estimated Ki (μM)b |
|---|---|---|
| ADP | 23 ± 2 | 2.6 |
| IDP | 6.4 ± 0.2 | 0.70 |
| XDP | 58 ± 3 | 6.4 |
| 2-Aminopurine-riboside 5′-diphosphate (APDP) | 1.3 ± 0.1 | 0.14 |
| 2,6-Diaminopurine-riboside 5′-diphosphate (DAPDP) | 0.37 ± 0.02 | 0.036 |
| 7-Deazaguanosine 5′-diphosphate (7-deaza-GDP) | 0.40 ± 0.04 | 0.040 |
| 7-Methylguanosine 5′-diphosphate (m7GDP) | 0.39 ± 0.02 | 0.038 |
| 1-Methylguanosine 5′-diphosphate (m1GDP) | 1.6 ± 0.2 | 0.17 |
| 2′-Deoxyguanosine 5′-diphosphate (2′-dGDP) | 0.53 ± 0.01 | 0.053 |
| 3′-Deoxyguanosine 5′-diphosphate (3′-dGDP) | 0.57 ± 0.03 | 0.058 |
| 2′,3′-Dideoxyguanosine 5′-diphosphate (ddGDP) | 3.5 ± 0.3 | 0.38 |
| Ribavirin 5′-diphosphate (RDP) | 8.6 ± 0.4 | 0.95 |
Oligo-RNA capping was performed with 0.25 μM μM [α-32P]GDP and 2.5 μM pppAACAG in the presence or absence of various concentrations of GDP analogues. Absolute IC50s were determined as described in Materials and Methods. Data represent the means and standard deviations from three independent experiments.
IC50s were converted to Ki values against GDP as described in Materials and Methods.
Using DAPDP and IDP as representatives of strong and weak inhibitors, respectively, we analyzed their inhibition modes (Fig. 5). Lineweaver-Burk plots showed that these GDP analogues exhibit competitive and noncompetitive inhibition against GDP (Fig. 5A and B) and pppAACAG (Fig. 5C and D), respectively. Based on apparent Ki values estimated by data fitting to Morrison's equation (18, 19), Ki values for DAPDP and IDP against GDP were determined to be 0.034 ± 0.002 μM (Fig. 5E) and 0.72 ± 0.21 μM (Fig. 5F), respectively.
FIG 5.
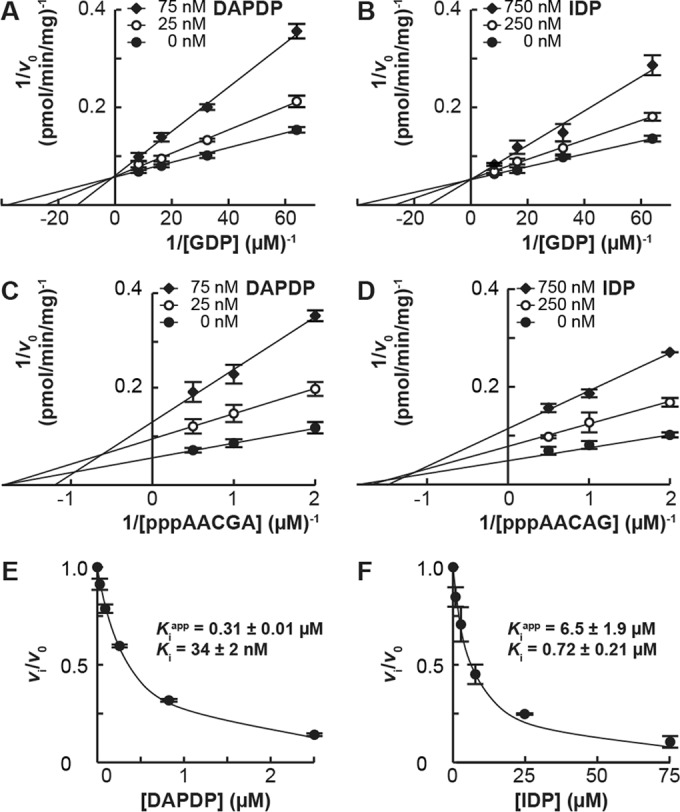
Competitive inhibition of the GpppA formation against GDP by DAPDP and IDP. (A and B) The oligo-RNA capping assay was performed with 2.5 μM pppAACGA and [α-32P]GDP (0.016, 0.031, 0.063, or 0.125 μM) in the presence or absence of various concentrations of DAPDP (A) or IDP (B). The reciprocal of the velocity (1/v0) was plotted against the reciprocal of GDP concentrations (Lineweaver-Burk plots). (C and D) The oligo-RNA capping assay was performed with 0.25 μM [α-32P]GDP and pppAACGA (0.125, 0.25, 0.5, or 1 μM) in the presence or absence of various concentrations of DAPDP (C) or IDP (D). The reciprocal of the velocity (1/v0) was plotted against the reciprocal of pppAACAG concentrations (Lineweaver-Burk plots). (E and F) The relative velocity (vi/v0) of the GpppA formation in the presence of various concentrations of DAPDP or IDP, calculated from the data shown in Fig. 4D, was plotted against inhibitor concentrations. The curved lines represent the best fits to Morrison's equation. Apparent and actual Ki values were estimated as described in Materials and Methods.
Incorporation of GDP analogues into cap structures.
The results from the competition assay between GDP and GDP analogues in the GpppA formation raised the possibility that some GDP analogues act as pRNA acceptors for the cap formation rather than as enzyme inhibitors. To explore directly this possibility, we performed the pRNA transfer assay using the GDP analogues as pRNA acceptors (Fig. 6A). The L-pRNA intermediate was generated by incubation of the L protein with pppAACAG (labeled with [α-32P]AMP) and purified as previously described (8). The purified L-pRNA intermediate was incubated with the GDP analogues, and the resulting RNAs released from the L protein were analyzed by 20% urea-PAGE.
FIG 6.
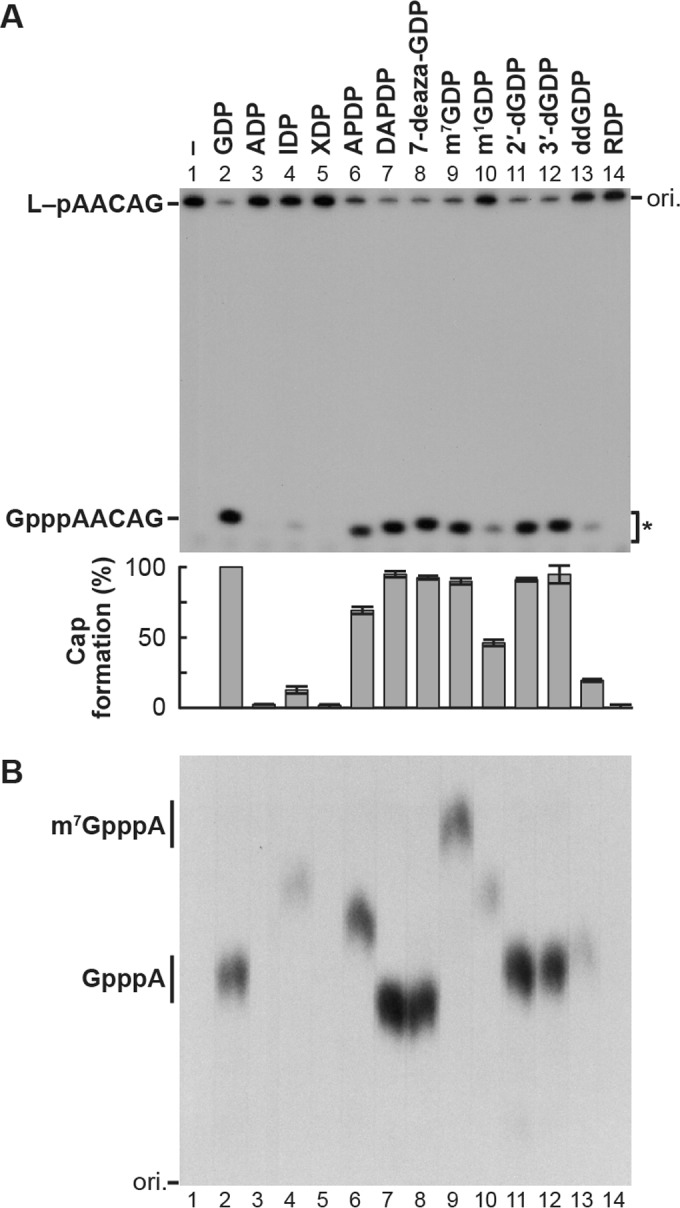
pRNA acceptor activity of GDP analogues in RNA capping with the VSV L protein. (A) The purified covalent L-pRNA intermediate (containing 32P-labeled pAACAG) was incubated with GDP analogues for 1 min. The resulting capped RNAs released from the L protein were analyzed by 20% urea-PAGE followed by autoradiography. The autoradiogram is a representative of three independent experiments. Relative cap formation activities with GDP (defined as 100%) and GDP analogues are shown in the lower panel. Columns and error bars indicate the means and standard deviations, respectively. (B) Capped RNAs were eluted from gel pieces (excised from the region marked by an asterisk in panel A) and digested with nuclease P1 and CIAP. Cap structures were purified by DEAE Sephacel and analyzed together with standard GpppA and m7GpppA by PEI-cellulose TLC followed by autoradiography. Note that band intensities of purified cap structures did not match the radioactivities of the capped RNA products before purification shown in panel A.
As reported previously (8), pRNA was transferred from the L-pRNA intermediate to GDP (lane 2) and 2′-dGDP (lane 11) but not to ADP (lane 3). When the L-pRNA intermediate was incubated with APDP (lane 6), DAPDP (lane 7), 7-deaza-GDP (lane 8), m7GDP (lane 9), or 3′-dGDP (lane 12), the RNA was efficiently released from the intermediate as a putative capped RNA. In contrast, IDP (lane 4), m1GDP (lane 10), and ddGDP (lane 13) did not act as efficient pRNA acceptors, and XDP (lane 5) and RDP (lane 14) were inert as pRNA acceptors. To demonstrate that the released RNAs have cap structures composed of the guanosine analogues, the RNAs were eluted from the gel and subsequently digested with nuclease P1 and CIAP. The cap structures resistant to nuclease P1 and CIAP were further purified by using DEAE Sephacel to remove 32Pi and analyzed by PEI-cellulose TLC. As shown in Fig. 6B, cap structures formed by the pRNA transfer to GDP and m7GDP comigrated with standard GpppA (lane 2) and m7GpppA (lane 9), respectively, on the TLC plate, whereas unique cap structures containing different guanosine analogues, i.e., IpppA (lane 4), APpppA (lane 6), DAPpppA (lane 7), 7-deaza-GpppA (lane 8), m1GpppA (lane 10), 2′-dGpppA (lane 11), 3′-dGpppA (lane 12), and ddGpppA (lane 13), migrated differently from the standard caps.
Taken together, these results indicate that GDP analogues with higher inhibitory activities against the GpppA formation (e.g., DAPDP, 7-deaza-GDP, m7GDP) act as better pRNA acceptors for VSV PRNTase than do the analogues with lower inhibitory activities. Thus, the estimated Ki values for these GDP analogues against the GpppA formation with GDP (Table 1) appear to be nearly equal to the Km values for them in the cap formation. These results indicate that the C-2-amino group, but not C-6-oxo group, N-1-hydrogen, or N-7-nitrogen, of the guanine ring and either of the hydroxyl groups at the 2′ or 3′ position of the ribose ring are essential for the efficient pRNA acceptor activity of GDP.
DISCUSSION
Rhabdoviral PRNTase is distinctly different from host mRNA capping enzymes (GTases) (6–10) and essential for viral gene expression and growth in host cells (13, 15). Importantly, the five PRNTase motifs are conserved among L proteins of NNS RNA viruses, including human pathogens (e.g., measles, Nipah, and Ebola viruses), belonging to different families (13), suggesting that NNS RNA viral L proteins synthesize the cap structure with the PRNTase activity. Thus, characterization of this unique enzyme is significantly important not only for understanding the unique enzymatic reaction but also for developing antiviral agents targeting this enzymatic domain.
We have previously developed an in vitro oligo-RNA capping assay with the VSV L protein (6, 8, 16). Here, we adapted this assay to kinetic analyses and improved its product analysis method. The major improvement was to omit the laborious steps, such as CIAP treatment, RNA extraction, RNA precipitation, and nuclease P1 treatment. We demonstrated that capping reaction mixtures can be directly analyzed by denaturing PAGE, which completely separates capped RNA products from remaining [α-32P]GDP. Using the improved oligo-RNA capping assay, we showed that the Km for GDP (pRNA acceptor, 0.03 μM) is significantly lower than that for pppAACAG (pRNA donor, 0.4 μM). In contrast, for eukaryotic GTase, the Km for ppRNA (GMP acceptor) is known to be lower than that of GTP (GMP donor) (20).
Our previous studies (6, 8) revealed that the PRNTase domain of the VSV L protein efficiently transfers pRNA from pppRNA to GDP, but not to GMP or GTP, through the covalent L-pRNA intermediate. The enzyme uses GDP and 2′-dGDP, but not ADP, UDP, or CDP, as pRNA acceptors (8), suggesting that the PRNTase domain specifically recognizes a chemical group(s) of guanine that is not present in adenine. There are structural differences between guanine and adenine: guanine has amino and oxo groups at the C-2 and C-6 positions of the purine ring, respectively, while adenine has the hydrogen atom and amino group at the C-2 and C-6 positions, respectively (see Fig. 3). Furthermore, a hydrogen atom is present at the N-1 position in guanine, but not in adenine.
In this study, we showed that GDP analogues lacking the C-2-amino group in the guanine ring (IDP and XDP) act as poor pRNA acceptors for PRNTase, suggesting that the C-2-amino group is essential for efficient pRNA transfer to GDP. In contrast, eukaryotic GTase effectively uses ITP as an IMP donor for the formation of an IpppN cap (20), indicating that the C-2-amino group of GTP is dispensable for the substrate recognition with GTase. On the other hand, GDP analogues lacking the C-6-oxo group (APDP, DAPDP) served as efficient pRNA acceptors to form cap structures with VSV PRNTase, indicating that the C-6-oxo group of GDP is not essential for the pRNA acceptor recognition with the PRNTase domain. Furthermore, since DAPDP efficiently accepted pRNA, the N-1-hydrogen as well as the C-6-oxo group is dispensable for the pRNA acceptor activity. However, the pRNA acceptor activity of 1-methyl-GDP was lower than that of GDP, suggesting that the presence of the N-1-methyl group reduces its affinity to PRNTase by a steric hindrance mechanism.
m7GDP as well as 7-deaza-GDP was efficiently utilized as a pRNA acceptor with VSV PRNTase. It is interesting that mRNA 7-methylguanylyltransferase (m7GTase) of positive-strand RNA viruses in the alphavirus-like superfamily uses m7GTP as an m7GMP donor to generate m7GpppN (21–23), whereas eukaryotic GTase does not (20). On the other hand, 8-iodo-GDP worked as an efficient pRNA acceptor for VSV PRNTase (data not shown). Thus, the PRNTase domain appears to have an ample space for the N-7-methyl or C-8-iodo group when m7GDP or 8-iodo-GDP binds to the putative GDP binding site in the PRNTase domain.
It has been known that detergent-disrupted VSV particles can synthesize mRNAs containing internal 2′-dGMP residues and 2′-dGpppA cap using 2′-dGTP as substrates for mRNA synthesis and capping (24). Furthermore, using the pRNA transfer assay with the isolated L-pRNA intermediate, we have demonstrated directly that the PRNTase domain of the VSV L protein transfers pRNA to 2′-dGDP to form 2′-dGpppA cap (8). Surprisingly, 3′-dGDP as well as 2′-dGDP, but not ddGDP, was found to serve as an efficient pRNA acceptor. These results suggest that the PRNTase domain of the VSV L protein flexibly recognizes one of the hydroxyl groups on the ribose ring. In contrast, the 2′-hydroxyl group of GTP is essential for mRNA capping with eukaryotic GTase (20).
This study also showed that the diphosphate form of antiviral ribavirin (RDP) does not act as a pRNA acceptor, although it inhibits the GpppA formation at higher concentrations. Consistent with this observation, RDP does not have a six-membered nitrogen-containing ring and a counterpart of the C-2-amino group of GDP that is critical for the efficient pRNA transfer reaction with VSV PRNTase. In contrast, vaccinia viral GTase has been reported to use ribavirin 5′-triphosphate (RTP) as a donor of ribavirin 5′-monophosphate (RMP) to generate an RpppN cap structure on RNA (25), again highlighting the difference in the guanosine recognition modes between PRNTase and GTase.
Since the estimated Ki for RDP against the cap formation with GDP by the VSV L protein is about 30-fold higher than the Km for GDP, RDP is theoretically not able to compete with submillimolar concentrations of GDP and GTP in host cells (26). However, since ribavirin was reported to render VSV mRNAs untranslatable without affecting primary transcription or incorporation into RNA chains in infected cells (27), there might be a possibility that ribavirin metabolites inhibit VSV mRNA capping, but only if concentrations of GTP and GDP are significantly reduced by inhibition of cellular IMP dehydrogenase, a target of ribavirin metabolites (28). Further studies are necessary to determine whether ribavirin metabolites inhibit VSV mRNA capping in infected cells.
Our previous (6, 8) and current studies indicate that the PRNTase domain of the VSV L protein recognizes 2-aminopurine, 2′- or 3′-hydroxyl-ribose, and 5′-diphosphate in GDP for the unique pRNA transfer reaction. However, currently, there is no information about amino acid residues of the VSV L protein required for the GDP binding. Our extensive mutagenesis analyses have shown conserved amino acid residues (G1100, T1157, W1188, H1227, R1228, F1269, and Q1270) in the PRNTase motifs A to E as essential for the L-pRNA intermediate formation (8, 13). If the putative GDP binding site is conserved in PRNTase domains of NNS RNA viral L proteins, some of these conserved amino acid residues should be involved in the GDP binding as well as the L-pRNA intermediate formation. Furthermore, hydrogen-bond donors and/or acceptors in a peptide backbone and/or side chains of non- and/or semiconserved amino acid residues close to the catalytic H1227 residue may be required for hydrogen-bonding interactions between the enzyme and GDP. We speculate that the β,γ-diphosphate of pppRNA (leaving PPi) and the α,β-diphosphate of GDP are possibly recognized with the same amino acid residue(s) (e.g., R1228). The GDP binding to the enzyme may be stabilized by a stacking interaction(s) of the purine moiety with an aromatic amino acid(s) (e.g., W1188, F1269) and/or H1227. The C-2-amino group and 2′- or 3′-hydroxyl group of GDP may contribute to hydrogen-bonding interactions with the enzyme. It is currently unknown whether the N-3-nitrogen of guanine and the 4′-oxygen of ribose have any roles in the GDP binding to the enzyme, although they are potential hydrogen bond acceptors. Further biochemical and structural analyses of the interaction of the enzyme with GDP are under way to identify the amino acid residues responsible for the GDP binding. These studies have the potential to lead to future development of antiviral agents targeting the putative GDP binding site in the PRNTase domain.
MATERIALS AND METHODS
GDP analogues.
GDP, ADP, IDP, 7-methylguanosine 5′-diphosphate (m7GDP), and 2′-deoxyguanosine 5′-diphosphate (2′-dGDP) were obtained from Sigma-Aldrich. XDP and 3′-deoxyguanosine 5′-diphosphate (3′-dGDP) were from Jena Bioscience. 2-Aminopurine-riboside 5′-diphosphate (APDP), 2,6-diaminopurine-riboside 5′-diphosphate (DAPDP), 7-deazaguanosine 5′-diphosphate (7-deaza-GDP), 1-methylguanosine 5′-diphosphate (m1GDP), and 2′,3′-dideoxyguanosine 5′-diphosphate (ddGDP) were synthesized by Trilink Biotechnologies. Ribavirin 5′-diphosphate (RDP) was from TLC PharmaChem.
In vitro oligo-RNA capping assay.
The recombinant VSV L protein (0.2 μg) was incubated in 10-μl reaction mixtures containing 0.25 μM [α-32P]GDP (150 to 250 cpm/fmol) and 2.5 μM pppAACAG for 20 min at 30°C under the previously reported conditions (6, 16). The reactions were stopped by adding 10 μl of a 95% formamide solution containing 10 mM EDTA, bromophenol blue (BPB), and xylene cyanol FF (XC). The resulting capped RNA (GpppAACAG) was directly analyzed by electrophoresis in 20% polyacrylamide gels containing 7 M urea (urea-PAGE) followed by autoradiography (6, 16).
To confirm the presence of the cap structure on the RNA product, it was purified from a urea-polyacrylamide gel and digested with nuclease P1 (Sigma-Aldrich), tobacco acid pyrophosphatase (TAP; Epicentre), and/or calf intestine alkaline phosphatase (CIAP; Roche) as described previously (8). The digests were analyzed by polyethyleneimine (PEI)-cellulose thin-layer liquid chromatography (TLC) with 0.4 M ammonium sulfate (8).
To determine Michaelis constants (Km) for the substrates and maximal velocities (Vmax) of VSV PRNTase, the oligo-RNA capping assay was carried out with a saturating concentration of a substrate and various concentrations of another substrate. Nonlinear least-squares regression was performed by fitting experimental data to the Michaelis-Menten equation, v0 = Vmax [S]/(Km + [S]), where v0 is the initial velocity and [S] is the concentration of substrate, using the Microsoft Excel software with the Solver add-in (17).
Inhibition of the GpppA cap formation with GDP analogues.
The oligo-RNA capping assay was performed in the presence or absence of various concentrations of GDP analogues as described above. Concentrations of GDP analogues that inhibit the RNA capping activity by 50% were determined as absolute IC50s by fitting experimental data, including values from two points closest to the midpoint of dose-response curves, to the four-parameter logistic equation (29) using the Solver tool of Microsoft Excel. Inhibition constants (Ki) were estimated from IC50s, a Km value for GDP (Km = 0.031 μM [Fig. 2C]), and concentrations of GDP ([S] = 0.25 μM) and the L protein ([E] = 0.08 μM) using the equation Ki = (IC50 − [E]/2)/(1 + [S]/Km) as described by Copeland et al. (30).
Apparent Ki values (Kiapp) for DAPDP and IDP were determined by fitting experimental data to Morrison's equation (18, 19) using the Solver tool of Microsoft Excel, and actual Ki values were estimated using the equation for competitive inhibition against GDP, Ki = Kiapp/(1 + [S]/Km), as described by Copeland (19).
The oligo-RNA capping assay was carried out with a saturating concentration of a substrate and various concentrations of another substrate in the presence or absence of various concentrations of DAPDP or IDP as described above. Their modes of inhibition were analyzed by using the Lineweaver-Burk plot (31).
pRNA transfer assay.
The L-pRNA intermediate containing [α-32P]AMP-labeled AACAG was isolated and incubated with 0.25 μM GDP or GDP analogues for 1 min as previously described (8). The resulting capped RNAs were analyzed by 20% urea-PAGE followed by autoradiography. To analyze cap structures, 32P-labeled capped RNAs were eluted from gel pieces, precipitated with ethanol, and digested with nuclease P1 and CIAP as described previously (8). After incubation of the digests with DEAE Sephacel resins (GE Healthcare) in 0.1 M triethylamine-bicarbonate buffer (TEA-HCO3, pH 7.5), the resins were washed with the same buffer to remove 32Pi. Cap structures were eluted from the resins with 1 M TEA-HCO3. After lyophilizing the eluates, the cap structures were dissolved in H2O and analyzed by PEI-cellulose TLC with 0.4 M ammonium sulfate (8).
ACKNOWLEDGMENTS
We thank Timothy W. Nilsen (Case Western Reserve University) for helpful discussions and critical comments on the manuscript.
The HHS NIH National Institute of Allergy and Infectious Diseases (NIAID) provided funding to Tomoaki Ogino under grant number R01AI093569.
REFERENCES
- 1.Topisirovic I, Svitkin YV, Sonenberg N, Shatkin AJ. 2011. Cap and cap-binding proteins in the control of gene expression. Wiley Interdiscip Rev RNA 2:277–298. doi: 10.1002/wrna.52. [DOI] [PubMed] [Google Scholar]
- 2.Hyde JL, Diamond MS. 2015. Innate immune restriction and antagonism of viral RNA lacking 2′-O methylation. Virology 479–480:66–74. doi: 10.1016/j.virol.2015.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramanathan A, Robb GB, Chan SH. 2016. mRNA capping: biological functions and applications. Nucleic Acids Res 44:7511–7526. doi: 10.1093/nar/gkw551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ogino T, Banerjee AK. 2011. An unconventional pathway of mRNA cap formation by vesiculoviruses. Virus Res 162:100–109. doi: 10.1016/j.virusres.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ogino T, Banerjee AK. 2011. mRNA capping by vesicular stomatitis virus and other related viruses, p 79–94. In Luo M. (ed), Negative strand RNA virus. World Scientific, Singapore. [Google Scholar]
- 6.Ogino T, Banerjee AK. 2007. Unconventional mechanism of mRNA capping by the RNA-dependent RNA polymerase of vesicular stomatitis virus. Mol Cell 25:85–97. doi: 10.1016/j.molcel.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 7.Ogino T, Banerjee AK. 2008. Formation of guanosine(5′)tetraphospho(5′)adenosine cap structure by an unconventional mRNA capping enzyme of vesicular stomatitis virus. J Virol 82:7729–7734. doi: 10.1128/JVI.00326-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ogino T, Yadav SP, Banerjee AK. 2010. Histidine-mediated RNA transfer to GDP for unique mRNA capping by vesicular stomatitis virus RNA polymerase. Proc Natl Acad Sci U S A 107:3463–3468. doi: 10.1073/pnas.0913083107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ogino T, Banerjee AK. 2010. The HR motif in the RNA-dependent RNA polymerase L protein of Chandipura virus is required for unconventional mRNA-capping activity. J Gen Virol 91:1311–1314. doi: 10.1099/vir.0.019307-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ogino M, Ito N, Sugiyama M, Ogino T. 2016. The rabies virus L protein catalyzes mRNA capping with GDP polyribonucleotidyltransferase activity. Viruses 8:144. doi: 10.3390/v8050144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shuman S. 2001. Structure, mechanism, and evolution of the mRNA capping apparatus. Prog Nucleic Acid Res Mol Biol 66:1–40. [DOI] [PubMed] [Google Scholar]
- 12.Ghosh A, Lima CD. 2010. Enzymology of RNA cap synthesis. Wiley Interdiscip Rev RNA 1:152–172. doi: 10.1002/wrna.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neubauer J, Ogino M, Green TJ, Ogino T. 2016. Signature motifs of GDP polyribonucleotidyltransferase, a non-segmented negative strand RNA viral mRNA capping enzyme, domain in the L protein are required for covalent enzyme–pRNA intermediate formation. Nucleic Acids Res 44:330–341. doi: 10.1093/nar/gkv1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liang B, Li Z, Jenni S, Rahmeh AA, Morin BM, Grant T, Grigorieff N, Harrison SC, Whelan SP. 2015. Structure of the L protein of vesicular stomatitis virus from electron cryomicroscopy. Cell 162:314–327. doi: 10.1016/j.cell.2015.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ogino T. 2014. Capping of vesicular stomatitis virus pre-mRNA is required for accurate selection of transcription stop-start sites and virus propagation. Nucleic Acids Res 42:12112–12125. doi: 10.1093/nar/gku901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ogino T. 2013. In vitro capping and transcription of rhabdoviruses. Methods 59:188–198. doi: 10.1016/j.ymeth.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kemmer G, Keller S. 2010. Nonlinear least-squares data fitting in Excel spreadsheets. Nat Protoc 5:267–281. doi: 10.1038/nprot.2009.182. [DOI] [PubMed] [Google Scholar]
- 18.Morrison JF. 1969. Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors. Biochim Biophys Acta 185:269–286. doi: 10.1016/0005-2744(69)90420-3. [DOI] [PubMed] [Google Scholar]
- 19.Copeland RA. 2013. Evaluation of enzyme inhibitors in drug discovery: a guide for medicinal chemists and pharmacologists, 2nd ed, p 245–285. John Wiley & Sons, Inc., Hoboken, NJ. [PubMed] [Google Scholar]
- 20.Venkatesan S, Moss B. 1980. Donor and acceptor specificities of HeLa cell mRNA guanylyltransferase. J Biol Chem 255:2835–2842. [PubMed] [Google Scholar]
- 21.Ahola T, Kääriäinen L. 1995. Reaction in alphavirus mRNA capping: formation of a covalent complex of nonstructural protein nsP1 with 7-methyl-GMP. Proc Natl Acad Sci U S A 92:507–511. doi: 10.1073/pnas.92.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang YL, Hsu YH, Han YT, Meng M. 2005. mRNA guanylation catalyzed by the S-adenosylmethionine-dependent guanylyltransferase of bamboo mosaic virus. J Biol Chem 280:13153–13162. doi: 10.1074/jbc.M412619200. [DOI] [PubMed] [Google Scholar]
- 23.Li C, Guillen J, Rabah N, Blanjoie A, Debart F, Vasseur JJ, Canard B, Decroly E, Coutard B. 2015. mRNA capping by Venezuelan equine encephalitis virus nsP1: functional characterization and implications for antiviral research. J Virol 89:8292–8303. doi: 10.1128/JVI.00599-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schubert M, Lazzarini RA. 1982. In vitro transcription of vesicular stomatitis virus. Incorporation of deoxyguanosine and deoxycytidine, and formation of deoxyguanosine caps. J Biol Chem 257:2968–2973. [PubMed] [Google Scholar]
- 25.Bougie I, Bisaillon M. 2004. The broad spectrum antiviral nucleoside ribavirin as a substrate for a viral RNA capping enzyme. J Biol Chem 279:22124–22130. doi: 10.1074/jbc.M400908200. [DOI] [PubMed] [Google Scholar]
- 26.Traut TW. 1994. Physiological concentrations of purines and pyrimidines. Mol Cell Biochem 140:1–22. doi: 10.1007/BF00928361. [DOI] [PubMed] [Google Scholar]
- 27.Toltzis P, Huang AS. 1986. Effect of ribavirin on macromolecular synthesis in vesicular stomatitis virus-infected cells. Antimicrob Agents Chemother 29:1010–1016. doi: 10.1128/AAC.29.6.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Streeter DG, Witkowski JT, Khare GP, Sidwell RW, Bauer RJ, Robins RK, Simon LN. 1973. Mechanism of action of 1-β-D-ribofuranosyl-1,2,4-triazole-3-carboxamide (Virazole), a new broad-spectrum antiviral agent. Proc Natl Acad Sci U S A 70:1174–1178. doi: 10.1073/pnas.70.4.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sebaugh JL. 2011. Guidelines for accurate EC50/IC50 estimation. Pharm Stat 10:128–134. doi: 10.1002/pst.426. [DOI] [PubMed] [Google Scholar]
- 30.Copeland RA, Lombardo D, Giannaras J, Decicco CP. 1995. Estimating Ki values for tight binding inhibitors from dose-response plots. Bioorg Med Chem Lett 5:1947–1952. doi: 10.1016/0960-894X(95)00330-V. [DOI] [Google Scholar]
- 31.Lineweaver H, Burk D. 1934. The determination of enzyme dissociation constants. J Am Chem Soc 56:658–666. doi: 10.1021/ja01318a036. [DOI] [Google Scholar]