

ABSTRACT
As human cytomegalovirus (HCMV) is the most common infectious cause of fetal anomalies during pregnancy, development of a vaccine that prevents HCMV infection is considered a global health priority. Although HCMV immune correlates of protection are only poorly defined, neutralizing antibodies (NAb) targeting the envelope pentamer complex (PC) composed of the subunits gH, gL, UL128, UL130, and UL131A are thought to contribute to the prevention of HCMV infection. Here, we describe a continuous target sequence within UL128 that is recognized by a previously isolated potent PC-specific NAb termed 13B5. By using peptide-based scanning procedures, we identified a 13-amino-acid-long target sequence at the UL128 C terminus that binds the 13B5 antibody with an affinity similar to that of the purified PC. In addition, the 13B5 binding site is universally conserved in HCMV, contains a previously described UL128/gL interaction site, and interferes with the 13B5 neutralizing function, indicating that the 13B5 epitope sequence is located within the PC at a site of critical importance for HCMV neutralization. Vaccination of mice with peptides containing the 13B5 target sequence resulted in the robust stimulation of binding antibodies and, in a subset of immunized animals, in the induction of detectable NAb, supporting that the identified 13B5 target sequence constitutes a PC-specific neutralizing epitope. These findings provide evidence for the discovery of a continuous neutralizing epitope within the UL128 subunit of the PC that could be an important target of humoral immune responses that are involved in protection against congenital HCMV infection.
IMPORTANCE Neutralizing antibodies (NAb) targeting the human cytomegalovirus (HCMV) envelope pentamer complex (PC) are thought to be important for preventing HCMV transmission from the mother to the fetus, thereby mitigating severe developmental disabilities in newborns. However, the epitope sequences within the PC that are recognized by these potentially protective antibody responses are only poorly defined. Here, we provide evidence for the existence of a highly conserved, continuous, PC-specific epitope sequence that appears to be located within the PC at a subunit interaction site of critical importance for HCMV neutralization. These discoveries provide insights into a continuous PC-specific neutralizing epitope, which could be an important target for a vaccine formulation to interfere with congenital HCMV infection.
KEYWORDS: HCMV, UL128, pentamer complex, peptide, epitope
INTRODUCTION
Human cytomegalovirus (HCMV) is a widely distributed herpesvirus that persists latently or chronically in the human host following primary infection (1–3). Although HCMV only rarely causes disease in healthy individuals, primary and recurrent infections (i.e., reactivation and reinfection) by HCMV are often associated with morbidity and mortality in immunologically vulnerable groups such as transplant recipients, AIDS patients, and pregnant women with their developing fetuses (4, 5). Congenital HCMV infection is a leading cause of fetal abnormalities, which most commonly results in microcephaly, intellectual impairment, sensorineural hearing loss, and, in rare cases, even death and multiorgan failure (2, 6). Permanent birth defects are associated more frequently with congenital HCMV infection than with other well-known childhood diseases, including trisomy 21, spina bifida, or fetal alcohol syndrome (2, 5, 7–9). Given the medical, societal, and economical benefits for the human population that would result from an effective HCMV vaccine, the Institute of Medicine assessed HCMV vaccine development as a major public health priority. Despite this assessment and research efforts that span more than 4 decades, a vaccine candidate that effectively prevents HCMV infection remains elusive.
Although the immune responses that correlate with protection against HCMV infection are only poorly defined, the induction of neutralizing antibodies (NAb) that block virus entry into host cells is thought to contribute to the prevention of HCMV infection (10–15). Most subunit vaccine strategies have focused on envelope glycoprotein B (gB) as an immunogen based on its essential role in HCMV entry, abundance in the virion envelope, and immunodominance in eliciting fibroblast (FB)-specific NAb in HCMV-seropositive (HCMV+) individuals (16, 17). These efforts culminated in the clinical findings obtained with gB admixed in the adjuvant MF59, showing efficacy rates of 50% and 43% in preventing primary infection in HCMV-seronegative (HCMV−) women and adolescent girls, respectively and an ability to significantly reduce viremia in transplant patients (18–20). These encouraging results with gB/MF59 indicate that other antigen compositions or vaccine formulations with an improved ability to stimulate humoral or cellular immune responses could confer levels of protection against HCMV that are considered sufficient for having a significant health impact (21).
Many findings obtained over the past years suggest that NAb targeting the HCMV envelope pentamer complex (PC) composed of gH, gL, UL128, UL130, and UL131A play a critical role in preventing congenital HCMV infection (12, 13). While the glycoprotein complexes gB and gH/gL/gO appear necessary for HCMV entry into all susceptible cell types, the PC is dispensable for HCMV entry into FBs but is required for virus entry into many other important cell types, such as epithelial cells (EC), endothelial cells, monocytes/macrophages, and dendritic cells (22–27). Most importantly from the vaccine point of view, NAb that block PC-mediated steps of HCMV entry are substantially more potent than are NAb that interfere with the entry function of gB or gH/gL/gO (28–31). In addition, we recently demonstrated that PC-specific antibodies have a potent ability to neutralize infection of primary cytotrophoblasts, which are considered the key placental cells that HCMV utilizes to cross the fetal-maternal interface (32). In line with these in vitro data, there is evidence for a correlation of NAb to the PC and reduced virus spread in vivo as well as a reduced risk of intrauterine virus transmission (12, 13). As a consequence of these observations, many innovative vaccine strategies based on the PC have been preclinically evaluated, which consistently showed that the PC is a potent immunogen to elicit NAb that block HCMV entry into EC and other cells (29, 30, 33, 34). Collectively, these results suggest that the stimulation of PC-specific NAb could be an important immune component for a vaccine candidate to prevent or control congenital HCMV infection.
However, the epitope sequences that are recognized by PC-specific NAb are only poorly defined. Here, we describe a continuous binding site within UL128 that is recognized by a potent NAb previously raised in mice by a modified vaccinia virus Ankara (MVA) vector coexpressing all five PC subunits (30, 32). The identified 13B5 target site comprises at minimum a 13-amino-acid-long sequence at the C terminus of UL128 and has a similar affinity for binding of the 13B5 antibody compared to that of the purified PC. In addition, this binding site is universally conserved in HCMV and localized at a previously described putative critical interaction site of UL128 and gL (35). Furthermore, peptides based on this target sequence have the ability to interfere with HCMV neutralization and to elicit HCMV-specific NAb in mice. These observations indicate the discovery of a continuous neutralizing epitope within the UL128 subunit of the PC that could be an important target of potentially protective HCMV-specific humoral immune responses.
RESULTS
NAb 13B5 binds a linear sequence within UL128.
We recently isolated a panel of NAb from MVA-PC-immunized mice that showed antigen recognition and neutralization potency that were similar to those of human PC-specific NAb previously isolated from HCMV+ individuals (32). Intracellular flow cytometry staining of the PC subunits expressed either alone or in different combinations with one another revealed that most of the isolated NAb recognized conformational epitopes requiring the assembly of two or more PC subunits (UL130/UL131A or UL128/UL130/UL131A). However, similarly to NAb isolated from HCMV+ individuals, one potent NAb, termed 13B5, that we isolated showed binding to UL128 when expressed individually or in any combination with the other PC subunits, but it did not show binding in the absence of UL128, indicating that it recognizes an epitope constituted by UL128 alone (32). To determine whether NAb 13B5 recognizes a continuous or discontinuous sequence within UL128, we performed Western blot analysis with the 13B5 antibody to detect UL128 under denaturing and reducing conditions following its expression as a single subunit or together with all other PC subunits from adenovirus (Ad) vectors in ARPE-19 EC. For comparison, we used identical concentrations of a well-characterized anti-UL128 antibody, termed Z9G11, that lacks neutralization activity and that we routinely use to confirm UL128 expression from our MVA constructs (31). As shown in Fig. 1, antibodies 13B5 and Z9G11 demonstrated similar antigen recognition of UL128 under denaturing/reducing conditions via Western blotting. Slightly lower recognition signals were observed with 13B5 than with Z9G11. These results suggest that the potent NAb 13B5 that was isolated from MVA-PC-immunized mice targets a continuous binding site within UL128, and this interaction presumably does not require disulfide bonding between UL128 and gL.
FIG 1.

Recognition of denatured UL128 by 13B5 and Z9G11. Shown is immunoblot detection of UL128 expressed from Ad vectors either alone (128) or in combination with the other four PC subunits (PC) in infected ARPE-19 EC using nonneutralizing Ab Z9G11 (top) or PC-specific NAb 13B5 (bottom). Cells infected with Ad-tet only (tet) were analyzed as a control. Mass markers (in kilodaltons) are shown next to each panel.
NAb 13B5 targets a binding site that is localized at the UL128 C terminus.
Based on data from the Western blot analyses that suggested 13B5 recognition of a continuous sequence within UL128, we proceeded to precisely map the 13B5 binding site using peptide-based scanning methods. Therefore, we synthesized a UL128 peptide library consisting of 15-mers with an offset of 4 for a total of 41 peptides (Table 1) and evaluated 13B5 binding to the synthesized peptides via an enzyme-linked immunosorbent assay (ELISA). For comparison, we determined the binding of the UL128-specific nonneutralizing Ab Z9G11. As shown in Fig. 2, NAb 13B5 reacted with three peptides, peptides 39, 40, and 41, that span the extreme C-terminal part of UL128. The strongest binding was observed with peptide 40, which was composed of the sequence 157-KRLDVCRAKMGYMLQ-171, indicating that this peptide contains the major amino acid residues that are required for 13B5 binding. Note that 13B5 did not show reactivity with any other peptides besides peptides 39, 40, and 41, supporting that only the C-terminal part of UL128 contains amino acid sequences that constitute the 13B5 target site. In contrast to NAb 13B5, nonneutralizing Ab Z9G11 demonstrated strong binding to three peptides at the UL128 N terminus: peptides 7, 8, and 9. Notably, not all peptides bind to plastic with the same efficiency. Hence, the ELISA data presented in Fig. 2 (and Fig. 3) may be biased by certain peptides failing to bind efficiently to the ELISA plate wells considering that the UL128 library peptides vary significantly. Nonetheless, these results indicated that the target epitope of NAb 13B5 is located at the C terminus of UL128, while the target site of nonneutralizing Ab Z9G11 is located at the N terminus of the protein.
TABLE 1.
UL128 peptide library
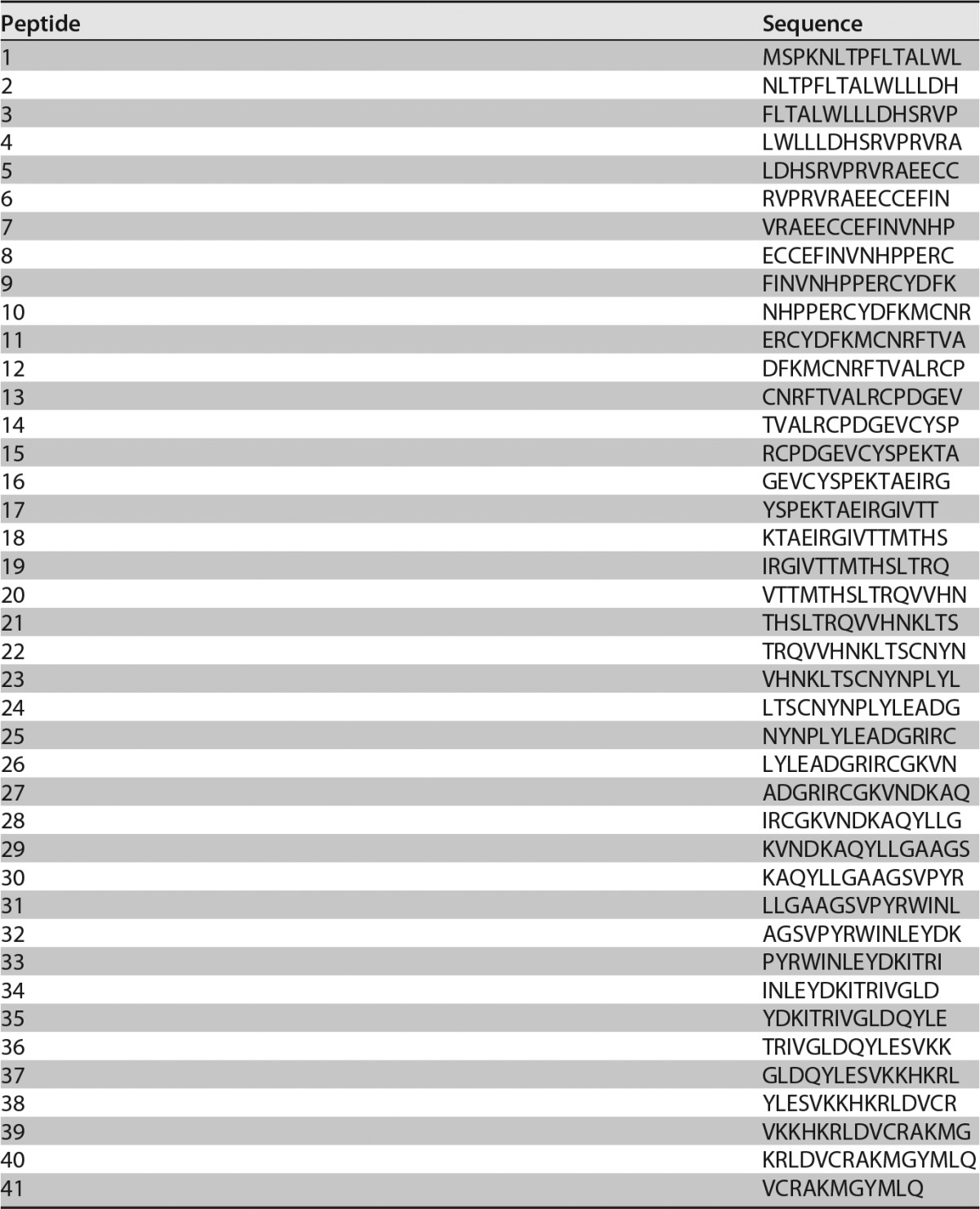
FIG 2.

UL128 peptide library binding of 13B5 and Z9G11. An ELISA was used to detect the binding of PC-specific NAb 13B5 and nonneutralizing Ab Z9G11 to a UL128 peptide library consisting of 41 overlapping 15-mers with an offset of 4. OD, optical density.
FIG 3.

Binding of human serum antibodies to the UL128 peptide library. An ELISA was performed to determine the reactivity of the UL128 peptide library with commercial serum products from HCMV+ (S4360, S4234, and S4371; SeraCare) or HCMV− (SNeg; SeraCare) individuals or CMV-HIG. Human serum was diluted 1:100, and CMV-HIG was used at a concentration of 1 μg/ml.
The UL128 binding site of NAb 13B5 is only minimally reactive with antibodies from HCMV+ individuals.
Since NAb 13B5 was isolated from mice immunized with MVA-PC, we investigated whether HCMV+ individuals develop antibodies to the UL128 binding site of NAb 13B5 following natural HCMV infection. To address this, commercially available antisera from three HCMV+ individuals and CMV hyperimmune globulins that represent an antibody pool of over 1,000 HCMV+ individuals (36) were evaluated by an ELISA to assess whether the human antisera contained antibodies that bound to the peptides of the UL128 peptide library. We identified two regions in UL128 that showed strong binding of human antibodies, corresponding to peptides 6 to 9 at the N terminus and peptides 37 to 39 at the C terminus of the UL128 amino acid sequence (Fig. 3). Interestingly, C-terminal peptide 40 of UL128 containing the 13B5 binding site reacted only minimally with human antibodies, while peptides comprising the target site of nonneutralizing Ab Z9G11 (peptides 7, 8, and 9) showed very strong binding with the antibody preparations (Fig. 3). Hence, these data may suggest that the 13B5 target site within UL128 is not a dominant HCMV PC epitope sequence, or it is only poorly recognized by antibodies induced by HCMV during natural infection. In contrast, the UL128 target site of nonneutralizing Ab Z9G11 appears to be located at a region that is highly reactive with antibodies found in HCMV-infected individuals.
The UL128 binding site of NAb 13B5 is universally conserved in HCMV.
UL128, UL130, and UL131A are known to be highly conserved in HCMV and hence are considered excellent targets for the development of broadly effective prophylactic or therapeutic antiviral strategies (37). Of these three proteins, UL128 has the lowest variability, with a reported amino acid identity score of 97.0% (37). In order to evaluate the conservation of the 13B5 binding site within UL128, we aligned 124 UL128 amino acid sequences available in GenBank (data not shown) using Clustal Omega. This alignment included UL128 protein sequences from highly passaged laboratory strains (e.g., AD169 and Towne), common clinical-like strains (e.g., TB40/E and TR), clinical isolates, and HCMV strain Merlin, which is considered the HCMV reference strain. Consistent with previously reported findings, our alignment confirmed that UL128 is highly conserved over almost the entire sequence. We found the highest amino acid variability at the UL128 N terminus. In contrast, the UL128 C terminus, ranging from amino acids 148 to 171, which includes the 13B5 binding site, was universally conserved in the analyzed HCMV strains. These data show that the 13B5 binding site is highly conserved in HCMV.
The NAb 13B5 binding site within UL128 is at minimum 13 amino acids long.
Linear B cell epitopes vary greatly in length and range from 5 to 22 amino acids (38, 39), with an average of 15 amino acids (40). In order to define the amino acid sequence within UL128 that constitutes the 13B5 epitope, we evaluated the binding of 13B5 to N-terminally and C-terminally truncated sequences of peptide 40 of the UL128 peptide library via an ELISA. As mentioned above, peptide 40, composed of the amino acids 157-KRLDVCRAKMGYMLQ-171 of the UL128 protein, demonstrated the strongest binding to the 13B5 antibody and hence was predicted to contain the minimal 13B5 binding sequence (Fig. 2). The removal of the N-terminal amino acid K and the sequential removal of the four following amino acids (RLDV) from the N terminus of peptide 40 resulted in dramatically reduced 13B5 binding. A complete loss of 13B5 binding was observed by removing six or more N-terminal amino acids from peptide 40 (Fig. 4A). Truncation of the two C-terminal amino acids LQ from peptide 40 did not show a reduction in 13B5 binding, although slightly decreased 13B5 binding was observed by additionally removing the M residue from the peptide 40 C terminus (Fig. 4B). 13B5 binding was substantially decreased by the removal of the four C-terminal amino acids YMLQ from peptide 40 and was completely lost when five or more C-terminal amino acids of peptide 40 were removed (Fig. 4B). These results indicated that a 13-amino-acid-long sequence ranging from K157 to M169 (KRLDVCRAKMGYM) at the C terminus of the UL128 protein is necessary and sufficient for efficient 13B5 binding. In addition, surface plasmon resonance (SPR) analysis showed a comparable and high affinity of the 13B5 antibody for the binding peptides based on the identified minimal 13B5 binding sequence (K13M) (KD [equilibrium dissociation constant] = 2.1 × 10−8 M−1) and purified PC (KD = 1.0 × 10−8 M−1), supporting that the mapped binding site represents a continuous epitope. To investigate further whether additional amino acid residues localized at the N terminus of K157 of UL128 are critical for 13B5 recognition, we compared 13B5 binding to peptides composed of the defined minimal 13B5 binding site with peptides comprising the 13B5 binding site and one or two additional amino acids (K155 and H156) of UL128 added to the N terminus. As shown in Fig. 4C, the addition of the N-terminal amino acids (KH) to the 13B5 target sequence improved the binding of the 13B5 antibody only minimally, suggesting that these amino acids are not critical for 13B5 binding, but they may slightly improve the interaction of the 13B5 antibody and its target site. As mentioned above, different peptides may bind to plastic with various efficiencies. Thus, minor variations in 13B5 binding of the different peptides may be difficult to interpret, as they may be a result of different efficiencies of peptides for binding to the ELISA plate wells. In sum, these results indicate that NAb 13B5 targets a 13-amino-acid-long continuous epitope sequence at the C terminus of UL128 that is composed of residues 157-KRLDVCRAKMGYM-169 of the protein.
FIG 4.

Mapping of the NAb 13B5 minimal binding site. (A and B) C- and N-terminally truncated peptides based on library peptide 40 were used in an ELISA to identify the shortest amino acid sequence needed for binding of NAb 13B5. (C) ELISA to compare 13B5 binding to K13M, comprising the minimal 13B5 epitope sequence, and peptides based on K13M with one (H14M) or two (K15M) additional amino acid residues of UL128 added to the N terminus. (D) Alanine scanning based on peptide K13M to identify amino acid residues involved in 13B5 binding. Bars represent standard deviations of data from triplicate wells. Vertical axes represent the optical density at 450 nm.
Most residues of the 13B5 target site within UL128 are critical for antibody binding.
In order to define the amino acid residues of the 13B5 target site within UL128 that are critical for antibody binding, we serially replaced each residue of the defined 13-amino-acid-long 13B5 binding site with alanine residues and evaluated the influence of these changes on 13B5 binding. Figure 4D shows the individual sequences of the mutated peptides as well as the results of testing of 13B5 binding to these peptides via an ELISA. Note that peptide 8 of the alanine scanning library is identical to the original sequence of the 13B5 binding site within UL128 (157-KRLDVCRAKMGYM-169 [K13M]). This peptide comprises an internal alanine residue and hence was used as a control in the ELISA for testing 13B5 binding. Consistent with our results obtained with the truncation libraries, alanine substitution of the N-terminal K residue and the C-terminal Y or M residue of K13M significantly reduced the binding of the 13B5 antibody. Similarly, replacement of most of the internal amino acids (C, R, K, M, and G) of K13M with alanine residues resulted in a dramatically reduced or complete loss of 13B5 binding. In contrast, 13B5 binding was not impaired when one of the internal residues L, D, and V of K13M was replaced. In addition, the substitution of L or D within K13M slightly increased 13B5 binding compared to that of the original K13M sequence, suggesting that these amino acid substitutions slightly improved the interaction of 13B5 and its target sequence. Interestingly, the internal cysteine residue within K13M that appeared essential for 13B5 binding corresponds to amino acid C162 of UL128, which was previously shown to form a disulfide bridge with gL in the PC (35). This suggests that NAb 13B5 targets a sequence within UL128 that is critical for interactions of the PC subunits. In sum, these results indicate that most of the amino acids of the 13B5 target sequence within UL128 are necessary for 13B5 binding, while three residues within the target site appear not to be critical for 13B5 binding.
Peptides based on the 13B5 target site interfere with HCMV neutralization.
For obtaining functional evidence that the identified 13B5 target sequence within UL128 represents a neutralizing epitope, we sought to evaluate whether peptides based on the 13B5 binding site can interfere with 13B5 neutralizing activity. Serial dilutions of the peptides were preincubated with a constant concentration of 13B5 antibody at which the antibody is known to neutralize over 95% of HCMV infection of ARPE-19 EC. The antibody-peptide mixture was then tested via a standard assay on ARPE-19 EC for HCMV neutralization to determine the functional inhibition of the 13B5 antibody. Another potent PC-specific NAb (54E11) that was previously shown to recognize a conformational epitope constituted by UL130/UL131A was used as a control (32). As shown in Fig. 5, a strong dose-dependent interference with 13B5-mediated neutralization with over 87% inhibition of 13B5 neutralizing activity at the highest peptide concentration tested was observed only with peptides containing the entire minimal 13B5 target sequence (library peptide 40, K13M, and K14L). A significantly lower inhibition of 13B5 function was observed with library peptide 41 that was missing four N-terminal residues of the 13B5 binding site (Table 1). In contrast, neutralization by 13B5 was not or was only minimally inhibited by peptides in which all (library peptides 37 and 38), most (library peptide 39), or one or two (K12Y and K11G) C-terminal residues of the 13B5 binding site sequence were absent. As expected, none of the tested peptides interfered with the neutralizing function of anti-PC antibody 54E11. These results show that peptides containing the identified 13B5 binding site specifically interfere with the 13B5 neutralizing function, providing functional evidence that the 13B5 target sequence is critically involved in HCMV neutralization.
FIG 5.
13B5 neutralization interference by 13B5 binding peptides. Serial dilutions of peptides 37 to 41 of the UL128 peptide library (A) and peptides K14L to K11G of the C-terminal truncation library (B) were evaluated for interference with 13B5 neutralizing activity to block TB40/E entry into ARPE-19 EC. Peptides were preincubated with 150 ng/ml 13B5 or 50 ng/ml 54E11 as a control and then incubated for 2 h with 9,000 PFU of HCMV TB40/E before the mixture was transferred to the cells.
Peptide construction based on the 13B5 binding site to test antibody induction.
To further support whether the identified 13B5 target site represents a neutralizing epitope, we evaluated keyhole limpet hemocyanin (KLH)-coupled peptides based on the 13B5 binding site for immunogenicity to elicit NAb in mice. For this, we generated three different KLH-coupled peptide constructs based on the 13B5 target sequence. In one construct, termed KLH-K15M, KLH was coupled to the minimal 13B5 target sequence (K13M) via the existing internal C162 residue, and two additional residues of UL128 were added to the peptide N terminus, which appeared to slightly increase the binding of the 13B5 antibody (Fig. 4C). The second construct, named KLH-K14CS, was generated by the coupling of KLH via a C-terminally added C residue to K13M in which the internal C162 residue of the minimal 13B5 target sequence was replaced with a serine. The third construct, termed KLH-K16CS, was generated in a way similar to that used for the second construct (KLH-K14CS), except that it included two additional amino acid residues of UL128 at the N terminus, similarly to the first construct (KLH-K15M). C-to-S amino acid substitutions in the second and third constructs (KLH-K14CS and KLH-K16CS) were chosen because of the similarity in steric occupancy between these two residues. As shown in Fig. 6A, all KLH constructs showed strong binding of the 13B5 antibody, indicating that KLH coupling and amino substitutions did not alter the interaction of the 13B5 target sequence with the 13B5 antibody. As a control, we also generated a peptide construct consisting of library peptide 38 (KLH-38), which only partially overlapped the 13B5 target site (Table 1). This peptide was similar to a recently tested UL128 peptide that failed to stimulate NAb in rabbits (41). As expected, KLH-38 was unable to bind the 13B5 antibody (Fig. 6A). In sum, these results indicated that all KLH-coupled peptide constructs based on the 13B5 epitope sequence presented intact 13B5 binding sites.
FIG 6.

Induction of binding and neutralizing antibodies by peptides based on the 13B5 epitope sequence. (A) Peptides based on the 13B5 binding site (K15M, K14CS, and K16CS) and peptides containing only partial sequences of the 13B5 binding site (UL128 library peptide 38) were coupled to KLH and tested for binding to 13B5 antibody by an ELISA. KLH alone was used as a control. Bars represent standard deviations of data from triplicate wells. (B) Pooled sera from mice immunized with the peptide constructs KLH-K15M, KLH-K14CS, and KLH-K16CS were tested by Western blot analysis for the detection of UL128 expressed from Ad vectors in ARPE-19 EC. EC infected with Ad-tet (tet) were analyzed as a control. Mass markers (in kilodaltons) are shown at the right. (C and D) BALB/c mice (5 animals per group) were immunized three times 4 weeks apart with KLH-coupled peptides admixed in Freund's adjuvant. (C) Levels of peptide-specific binding antibodies in sera of immunized mice were measured via an ELISA 1 week before (−1wp1st) and 3 weeks after the first, second, and third immunizations (3wp1st, 3wp2nd, and 3wp3rd, respectively) by using the peptides (not conjugated to KLH) that were used for immunization as coating antigens. (D) Serum neutralizing antibody titers (NT50) from immunized mice against HCMV TB40/E were measured on ARPE-19 EC by using a standard microneutralization assay. Lines in panels C and D indicate the group means.
Peptides based on the 13B5 target site have the ability to elicit NAb in mice.
To test whether the generated KLH-coupled peptide constructs based on the 13B5 target site (KLH-K15M, KLH-K14CS, and KLH-K16CS) have the ability to elicit NAb, BALB/c mice were intraperitoneally immunized three times 4 weeks apart with the peptide constructs admixed in Freund's adjuvant. Levels of serum binding antibodies and NAb were determined 1 week before and 3 weeks after each immunization. Levels of binding antibodies of the individual groups were determined via an ELISA using peptides that were used for immunization as target antigens (Fig. 6C). The NAb titer at which infection was inhibited by 50% (NT50) was determined by a microneutralization assay using ARPE-19 EC as the cell substrate and HCMV strain TB40/E for infection (Fig. 6D). As a control, mice were immunized with KLH only or KLH-38 with the C-terminal UL128 peptide sequence that only partially overlapped the 13B5 target site (Table 1). As shown in Fig. 6C, all mice immunized with the peptide constructs developed binding antibodies against the peptide sequence with which they were vaccinated after the first immunization. These responses were not or were only minimally boosted by second and third immunizations. In addition, Western blot analysis of UL128 expressed from Ad vectors showed that mice immunized with KLH-K15M, KLH-K14CS, and KLH-K16CS developed antibodies that recognized denatured UL128 (Fig. 6B). NAb responses with NT50 values ranging from 200 to 300 were detectable after the first immunization in only two out of five animals in each vaccine group immunized with KLH-K14CS or KLH-K16CS (Fig. 6D). These responses were boosted in only one animal each in the KLH-K14CS and KLH-K16CS vaccine groups, reaching NT50 values of 800 to 1,200. These titers remained stable after the third immunization. In the two other animals that developed NAb after the first immunization, NAb levels declined and were undetectable after the booster immunizations. None of the animals immunized with the peptide construct KLH-K15M, the control construct KLH-38, or KLH only developed NAb. These results indicated that peptides based on the 13B5 target site in which the internal cysteine residue was replaced by a serine residue and KLH was coupled via an additional cysteine residue to the C terminus had the ability to elicit NAb in mice. In contrast, peptides composed of the 13B5 target site that were coupled to KLH via the existing internal C162 residue did not show immunogenicity for NAb induction. These results provide evidence that the identified 13B5 target sequence has the potential to elicit NAb, supporting that it constitutes a neutralizing epitope within UL128.
DISCUSSION
In this study, we provide evidence for the discovery of a linear neutralizing epitope within the HCMV PC, which is thought to be an important target of humoral immune responses preventing congenital HCMV infection. Previous studies demonstrated that the PC comprises predominantly quaternary conformational epitopes that are formed by two or more subunits and in only rare cases epitopes that are constituted by single subunits of the PC (28, 29). In addition, Ciferri et al. identified amino acid residues that are either in close proximity to or part of conformational neutralizing epitopes within the PC (42). However, amino acid residues that constitute the minimal target sequence of a potent PC-specific NAb have not been described. By taking advantage of an existing NAb with specificity for UL128 (NAb 13B5) that was previously raised in mice by immunization with the entire PC (32), we defined a continuous NAb target sequence at the C terminus of UL128. This discovery represents the first detailed characterization of a putative neutralizing epitope within the HCMV PC and hence provides a first glimpse into the epitope sequences within the PC that may serve as key targets of protective humoral immunity.
Several observations support that the minimal 13B5 target sequence is located at the C terminus of UL128. Our previous study using intracellular flow cytometry to evaluate 13B5 subunit specificity demonstrated that UL128 is necessary and sufficient for efficient 13B5 binding, indicating that UL128 alone comprises the major amino acid residues that constitute the 13B5 epitope (32). In addition, the 13B5 target site does not overlap target sites of a panel of PC-specific NAb to conformational epitopes that we isolated previously (32). Here, we have used Western blot analysis and peptide-based scanning methods to show that a continuous sequence composed of the 13 C-terminal residues 157-KRLDVCRAKMGYM-169 of UL128 is minimally required for 13B5 binding. Without the full complement of these residues, 13B5 binding is dramatically decreased or completely abrogated. Moreover, truncation peptides of UL128 that only partially overlap the 13B5 target site or that correspond to other regions within UL128 are not reactive with the 13B5 antibody, indicating that no other residues within UL128 besides the 13 C-terminal residues contribute to the formation of the 13B5 epitope. This conclusion is further supported by our SPR analysis showing that the 13B5 antibody has a similar and high affinity for binding of peptides based on the 13B5 minimal epitope sequence and purified PC. In addition, our neutralization interference assays and immunization studies provide functional evidence that the identified 13B5 target sequence within UL128 represents a PC-specific neutralizing epitope. Whether additional amino acids or peptide sequences of UL128 or the other PC subunits besides the 13 C-terminal residues of UL128 may minimally contribute to the formation of the 13B5 epitope or the binding of 13B5 to its target sequence within UL128 in the assembled PC can be determined only by detailed structural analysis. Nonetheless, these findings provide the first evidence for the existence of a continuous neutralizing epitope within the UL128 subunit of the HCMV PC.
Considering the strong neutralizing potency of the 13B5 antibody (50% inhibitory concentration [IC50] of 15 ng/ml) (32), which is only slightly lower than those of most other anti-PC NAb (IC50 of 1 to 10 ng/ml) but exceeds those of most gB- and gH/gL-specific NAb (IC50 of 100 to 1,000 ng/ml) (28, 29, 32), the low immunogenicity of the 13B5 peptides for the induction of NAb was unexpected. Whereas all of the 13B5 peptides stimulated robust binding antibodies, only 1 out of 5 mice in each of the vaccine groups immunized with K14CS or K16CS developed long-lasting NAb, and these responses were only modest compared to those observed previously by immunization with the PC. While the reasons for this discrepancy in 13B5 NAb potency and 13B5 peptide immunogenicity for stimulating NAb remain speculative, it is possible that the 13B5 epitope sequence in its native state within the PC forms a helical, bended, or “kinked” three-dimensional structure that is required for NAb induction (43). Consequently, without structural constraint by PC subunits, the 13B5 epitope structure is only inefficiently or rarely formed. This may account for the inconsistent ability of the KLH-coupled 13B5 peptides to stimulate NAb despite their property of eliciting robust peptide-specific binding antibodies. Therefore, the formation of the three-dimensional 13B5 epitope structure is likely promoted in the presence of the 13B5 antibody; otherwise, we would not expect to observe strong binding of the 13B5 NAb and 13B5-based peptides.
Alternative explanations may account for the low immunogenicity of the 13B5 peptides to stimulate NAb. For example, additional amino acid residues within UL128 or other PC subunits may minimally contribute to the 13B5 epitope structure, rendering the identified 13B5 epitope sequence insufficient for the effective stimulation of NAb (44). Another possibility is that the 13B5 epitope sequence is simply not optimal for EC-specific NAb induction because the neutralization potency of the 13B5 antibody is lower than those of most other anti-PC NAb. Alternatively, the chosen KLH-coupling strategy, despite the fact that it did not abrogate 13B5 binding, may have interfered with NAb induction by impairing the ability of the epitope sequence to form a properly folded three-dimensional structure. Note that all ELISA binding experiments were performed at a single, near-saturating antibody concentration, which may not have allowed the capture of significant differences in the ELISA absorbance values. This might explain why serum antibodies of mice in all vaccine groups immunized with the 13B5 peptides appear to bind their immunogen peptide efficiently but fail to neutralize the virus in most cases or why KLH-K15M appears to bind the 13B5 antibody with an efficiency similar to those of KLH-K14CS and KLH-K16CS but, unlike KLH-K14CS and KLH-K16CS, is unable to elicit neutralizing responses. Nonetheless, although we observed only limited NAb induction by peptide variants based on the 13B5 epitope, this observation provides further evidence that the identified target sequence represents a PC-specific neutralizing epitope.
Our observations may suggest that the 13B5 target sequence is located within the PC at an antigenic site that is largely protected from immune recognition during natural HCMV infection. The finding that the 13B5 target sequence is universally conserved in a minimum of 124 known HCMV isolates suggests that this invariant sequence plays a critical role in HCMV host cell entry. In line with this, C162 of UL128, which is centrally positioned within the 13B5 target sequence, was previously shown to form a disulfide bond with a cysteine residue in gL (35), highlighting that the 13B5 binding sequence is located at a critical interaction site of the PC subunits. In addition, the observation that antibodies in HCMV+ individuals react only minimally with peptides containing the 13B5 target sequence may suggest that this sequence is only minimally immunogenic or only rarely recognized by the immune system during natural HCMV infection, which is consistent with characteristics of an immunologically “cryptic” epitope (45). Epitope crypticity is a common pattern of immune evasion and has been described for many other pathogens such as HIV, influenza virus, dengue virus, Plasmodium falciparum, and Bacillus anthracis (45–49). While it appears contradictory how the 13B5 epitope can be immunologically cryptic and at the same time a target of potent NAb, this could be explained by differences in the accessibility of the 13B5 binding site during natural HCMV infection. The 13B5 binding site may be largely inaccessible in mature HCMV virions, but it may temporarily unfold, for instance, upon PC receptor binding during HCMV entry (50). Alternatively, the discrepancy in 13B5 epitope crypticity and 13B5 epitope recognition may be explained by the difference in the definition and meaning of epitope immunogenicity and antibody function. For example, highly conserved neutralizing epitopes of the HIV envelope protein are in many cases poorly immunogenic and induce NAb in only about 1% of the HIV-infected population, but these antibodies are extremely potent in neutralizing HIV (51, 52). While the details may be different, a parallel to the HIV case can be made in the comparison of the putative low immunogenicity of the 13B5 epitope and the potent ability of the 13B5 antibody to neutralize infection. The 13B5 epitope could be of low immunogenicity because it competes for antibody access with immunodominant nonneutralizing epitopes that are localized in close proximity to the 13B5 NAb binding site (45). However, only a very small amount of the 13B5 antibody may be required for potent neutralization, which could be associated with either the high affinity of the 13B5 antibody or the small amounts of the PC in HCMV virions (53).
However, as peptide-based scanning methods apply only for characterizing continuous epitope sequences, which represent only a minority of the epitopes within the HCMV PC, our findings do not allow us to make general conclusions for PC-specific antibodies. Most of the antibodies targeting the PC are specific for more complex epitope structures, and hence, it may well be that many other antibodies bind to an area within the PC that is similar to that of 13B5, although they recognize conformational epitopes rather than continuous sequences. Competition experiments with the 13B5 antibody and antibodies from HCMV+ individuals to recognize the PC could clarify whether the 13B5 epitope sequence is located at a common recognition site for PC-specific humoral immune responses. In this context, it may also be possible that the 13B5 epitope within the PC forms a three-dimensional rather than a linear structure and that the 13B5 peptide folds into this structure in the presence of the 13B5 antibody but is not well recognized by lower-affinity serum antibodies of HCMV+ individuals. Hence, our conclusion could apply only to continuous epitopes, which occur only very infrequently within the HCMV PC.
In light of the observations made with the 13B5 target sequence, this sequence may have value for a peptide-based vaccine strategy to stimulate or enhance NAb responses in HCMV− or HCMV+ individuals. As naturally acquired immunity as a consequence of primary HCMV infection provides only incomplete protection against reinfection (7), it is hypothesized that vaccine-mediated immunity needs to have improved properties over that stimulated by HCMV itself to alter the outcome of congenital infection. In addition, congenital HCMV infection is more often a result of nonprimary infection (reinfection or reactivation) than of primary infection, suggesting than HCMV+ individuals, in addition to HCMV− individuals, may benefit from a vaccine that is able to augment naturally induced immune responses (7, 54). How such a vaccine candidate can be realized remains elusive, particularly because of the limited knowledge about the immune correlates of protection. However, based on the universal conservation and the putative crypticity of the 13B5 epitope sequence, it can be surmised that a vaccine strategy based on the 13B5 target sequence would allow the elicitation of broadly effective NAb that are focused on potentially protective antigenic sites without inducing antibodies to less protective or nonprotective epitopes. Such a vaccine approach based either on only the 13B5 epitope or on multiple nonoverlapping linear epitope sequences within the PC could induce antibody responses with improved quality compared to those induced during natural infection and hence provide higher protection efficacy than naturally acquired immunity.
In sum, we provide evidence for the identification of a continuous, 13-amino-acid-long neutralizing epitope at the UL128 C terminus within the HCMV PC that is universally conserved and located at an antigenic site of critical importance for HCMV entry and neutralization. These observations enhance our understating of PC-specific neutralizing epitopes, which represent major targets of NAb responses that are considered essential for protection against congenital HCMV infection.
MATERIALS AND METHODS
Cells.
ARPE-19 (American Type Culture Collection [ATCC]) cells were maintained in Dulbecco minimal essential medium (DMEM; Corning, NY). Medium was supplemented with 10% fetal bovine serum (FBS; HyClone, Logan, UT). HEK 293-6E cells (NRC-BRI, Montreal, Canada) were maintained in Freestyle F17 medium (Thermo-Fisher Scientific [Thermo], Carlsbad, CA).
Viruses.
HCMV TB40/E expressing green fluorescent protein (GFP) was derived from TB40/Ewt-GFP bacterial artificial chromosome (BAC) DNA, a kind gift from T. A. Shenk and E. A. Murphy (Princeton University, NJ) (55). HCMV stocks were prepared as previously described (30). Briefly, ARPE-19 cells were infected with HCMV TB40/E and periodically reseeded until 70 to 80% of the cells expressed GFP. Clarified medium was ultracentrifuged (70,000 × g for 1 h) over 20% (wt/vol) sucrose in Tris-buffered saline (0.1 M Tris-Cl [pH 7.4], 0.1 M NaCl), and concentrated virus was resuspended in Tris-buffered saline and stored at −80°C until use.
Antibodies.
Purified 13B5 and 54E11 were obtained from hybridoma cells as previously described (32). Briefly, hybridoma cells were cultured in Hybridoma-SFM (Thermo). The hybridoma supernatant was collected, and NAb was purified by using a HiTrap Protein G HP column (GE Healthcare, Chicago, IL) according to the manufacturer's instructions. NAb quantification was performed by using Bradford-Coomassie brilliant blue dye (Thermo). The Z9G11 unpurified hybridoma supernatant was a kind gift of G. Gerna and E. Percivalle (31).
Immunoblotting.
Immunoblotting to determine 13B5 or mouse serum binding to denatured UL128 was performed by using lysates from cells infected with a UL128-expressing adenoviral vector (kindly provided by David Johnson, Oregon Health & Sciences University, Portland, OR, USA) (56) as previously described (30). Briefly, ARPE-19 cells were coinfected with 20 multiplicities of infection (MOI) of an adenoviral vector expressing the tetracycline transactivator (Ad tet-trans) alone; UL128 and Ad tet-trans; and UL128, UL130, UL131, gH, gL, and Ad tet-trans. Infected cells were harvested at 4 days postinfection, centrifuged at 300 × g, resuspended in Tris-HCl buffer (10 mM Tris-HCl [pH 7.9], 1.0 mM EDTA, and 1× cOmplete Mini protease inhibitor cocktail tablets [Roche, Basel, Switzerland]), and sonicated. Samples were lysed in loading buffer (2× Laemmli sample buffer, 200 mM dithiothreitol), boiled, and used for SDS-PAGE. Primary antibodies 13B5 and Z9G11 were diluted to 100 ng/ml in phosphate-buffered saline (PBS)–0.1% Tween (PBS-T)–3% bovine serum albumin (BSA). Mouse serum was diluted 1:10,000 in PBS-T–3% BSA. A secondary anti-mouse horseradish peroxidase (HRP) conjugate (Sigma) was employed at a dilution of 1:2,000 in PBS-T–2.5% BSA. Chemiluminescence detection was performed by using the ECL WB substrate (Thermo).
Peptide synthesis.
Peptides were synthesized by using the Symphony peptide synthesizer (Protein Technologies, Inc., Tucson, AZ) or by manual synthesis utilizing 9-fluorenylmethoxy carbonyl (Fmoc)-based solid-phase peptide synthesis. Fmoc-protected amino acids, HOBT (N-hydroxybenzotriazole), and HBTU [2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate] were purchased from Anaspec (Fremont, CA). DMF (N,N-dimethylformamide) and TFA (trifluoroacetic acid) were purchased from Protein Technologies, Inc. (Tucson, AZ). Preloaded Wang resins were purchased from EMD Millipore (Temecula, CA). Piperidine, DIPEA (N,N-diisopropylethylamine), DICI (N,N′-diisopropylcarbodiimide), EDT (1,2-ethanedithiol), TIS (triisopropylsilane), and NMM (4-methylmorpholine) were purchased from Sigma-Aldrich (St. Louis, MO). Peptides were purified and characterized by using an Agilent 1200 preparative liquid chromatography/mass spectrometry (LC/MS) system and an Agilent 1260 analytical high-performance liquid chromatography (HPLC) instrument (Agilent Technologies, Santa Clara, CA). A typical synthesis reaction starts with mixing 25 μmol of Wang resin with a 20% piperidine–DMF solution to remove the Fmoc-protecting group for 30 min. In the next step, 3 eq of Fmoc-protected amino acid, HBTU, and NMM are added to the resin in a DMF solution. The reaction is mixed under nitrogen for 1 to 2 h. After draining, the resin is washed with DMF, the Fmoc group is removed with 20% piperidine, and the next amino acid in the sequence is added. Once the sequence is complete, the peptide is cleaved from the resin with a cleavage cocktail containing TFA, EDT, water, and TIS overnight. The peptide is collected and precipitated with cold ether. All peptides were purified and characterized individually to at least 80% purity. All peptides were lyophilized to powder and reconstituted in stock solutions for easier development of library pools as well as individual use.
Surface plasmon resonance.
Analysis of the affinity of 13B5 for the purified PC and K13M was performed by using a Biacore T100 SPR spectrometer according to the manufacturer's instructions. The Biacore CM5 sensor chip was bound by amine coupling with 13B5. Affinity constants were calculated by using the Biacore T100 software provided by the manufacturer. The HCMV PC was produced and purified as described by Chiuppesi et al. (F. Chiuppesi, F. Wussow, L. Sharf, H. Contreras, H. Gao, Z. Meng, J. Nguyen, P. A. Barry, P. J. Bjorkman, and D. J. Diamond, unpublished data). Briefly, HEK 293-6E cells (NRC-BRI) were cotransfected with plasmids (pTT5; NRC-BRI) encoding HCMV PC subunit genes from TB40/E by using polyethylenimine HCl Max (Polysciences). The supernatant was collected at 10 days posttransfection and purified by using an affinity chromatography column (Hi-Trap NHS-activated HP column; GE Healthcare Bio-Sciences, Pittsburgh, PA) activated with UL128/UL130/UL131A-specific monoclonal antibody 12E2 (32). Size exclusion chromatography (S200; GE Healthcare Bio-Sciences) was performed on the eluate, and fractions were analyzed by SDS-PAGE followed by Coomassie gel staining.
Keyhole limpet hemocyanin conjugation.
Peptides were conjugated to KLH (Thermo) according to the manufacturer's instructions. Briefly, 20 mg of KLH was reacted with 10 mg sulfo-SMCC [sulfosuccinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate] for 1 h at room temperature. After the removal of the excess cross-linker by using a desalting column, 20 mg of the peptide was added, and the mixture was incubated for 2 h at room temperature. To remove the unconjugated peptide, the conjugate was purified by desalting and subsequently aliquoted and stored at −20°C until use. Conjugate quantification was performed by reading the absorbance at 280 nm and by performing a quantitative ELISA using the peptides as a standard.
Immunizations.
Groups of five BALB/c mice (Jackson Laboratory, Bar Harbor, ME) were prime vaccinated with 100 μg of a peptide-KLH conjugate (quantified based on the peptide concentration) emulsified with complete Freund's adjuvant (CFA) and boosted two times with 50 μg of the peptide-KLH conjugate admixed in incomplete Freund's adjuvant (IFA). Mice were immunized via the intraperitoneal route every 4 weeks. Blood samples were collected by eye bleed 1 week before prime immunization as well as 3 weeks after prime and booster immunizations.
ELISA.
For analysis of 13B5 binding to UL128 peptides or to KLH-peptide conjugates, ELISA plates (Thermo) were coated overnight with peptides or KLH-peptide constructs at a dilution of 10 μg/ml in PBS. Negative-control wells were coated with PBS alone. After 2 h of blocking with PBS–1% BSA, 13B5 was added at a concentration of 3 μg/ml for 2 h. After washing, anti-mouse HRP (Thermo) was added at a dilution of 1:2,000. Plates were developed with the Ultra TMB (3,3′,5,5′-tetramethylbenzidine)-ELISA substrate (Thermo), and the reaction was blocked with 1 M H2SO4. An ELISA to evaluate the recognition of the UL128 library peptides by human serum was performed by diluting three commercially available sera from HCMV+ individuals [lots BM204234 (S4234), BM204360 (S4360), and BM204371 (S4371); SeraCare], HCMV IgG-negative human serum [lot BM216642 (SNeg); SeraCare] at a 1:100 dilution in PBS, and an IgG preparation (CMV-hyperimmune globulin [CMV-HIG]) (Cytogam; Baxter-Healthcare Corp., Irvine, CA) to 1 μg/ml. HRP-conjugated goat anti-human IgG secondary antibody (Promega) was diluted 1:2,000 in PBS. Sera from immunized mice were analyzed as described above for human sera but with the differences that the mouse sera were diluted 1:100 in PBS and ELISA plate wells were coated with 10 μg/ml of the peptide used to immunize the animals. Sera from mice immunized with only KLH were tested by an ELISA using uncoated wells.
Neutralization assay.
ARPE-19 cells were seeded at 1.5 × 104 cells/well in a clear-bottom 96-well plate (Corning). Approximately 24 h later, the medium in every plate was replaced with 50 μl per well of fresh growth medium. A neutralization assay to analyze serum NAb from immunized mice was performed as previously described (30, 32). Briefly, serial 2-fold dilutions of the mouse sera were prepared in complete growth medium in a final volume of 75 μl. NAb dilutions were mixed with 75 μl of complete growth medium containing ∼9,000 PFU of HCMV TB40/E and incubated for 2 h at 37°C. The mixture was transferred to the cells (50 μl each, in duplicate wells). After 48 h, cells were fixed, and IE-1 immunostaining was performed as previously described (30). The NAb concentration inhibiting 50% of virus infectivity (IC50) was also calculated as previously described (30). A neutralization assay to quantify peptide-mediated inhibition of antibody neutralizing activity was performed as described above, with a few changes. Serial 2-fold dilutions of the peptides starting from 100 μg/ml were incubated with 10 NT50s of NAb 13B5 (150 ng/ml) or 54E11 (50 ng/ml) (32). After 2 h of incubation, ∼9,000 PFU of HCMV TB40/E were added to each well, and the wells were incubated for an additional 2 h. The mixture was transferred to the cells (50 μl each, in duplicate wells), and cells were analyzed after 48 h as described above.
Software.
Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo/) was used to align 124 UL128 amino acid sequences randomly selected from the available complete HCMV genomes in GenBank (http://www.ncbi.nlm.nih.gov/GenBank/).
ACKNOWLEDGMENTS
We are grateful to David Johnson (Oregon Health & Science University, Portland, OR, USA) for providing replication-deficient Ad vectors and to Giuseppe Gerna and Elena Percivalle (Fondazione IRCCS Policlinico San Matteo, Pavia, Italy) for providing UL128 antibody Z9G11. We thank the personnel of the X-Ray Crystallography Core of the Beckman Research Institute of the City of Hope for their professional help with the SPR study. We also thank Heidi Contreras (City of Hope) for critically reading the manuscript.
This research was funded by USPHS grant AI103960 to D.J.D. and P.A.B. D.J.D. was partially supported by grants CA077544 and CA181045. The City of Hope Cancer Center is supported by the National Cancer Institute of the National Institutes of Health under award number CA33572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1.Boeckh M, Geballe AP. 2011. Cytomegalovirus: pathogen, paradigm, and puzzle. J Clin Invest 121:1673–1680. doi: 10.1172/JCI45449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manicklal S, Emery VC, Lazzarotto T, Boppana SB, Gupta RK. 2013. The “silent” global burden of congenital cytomegalovirus. Clin Microbiol Rev 26:86–102. doi: 10.1128/CMR.00062-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Britt W. 2008. Manifestations of human cytomegalovirus infection: proposed mechanisms of acute and chronic disease. Curr Top Microbiol Immunol 325:417–470. [DOI] [PubMed] [Google Scholar]
- 4.Landolfo S, Gariglio M, Gribaudo G, Lembo D. 2003. The human cytomegalovirus. Pharmacol Ther 98:269–297. doi: 10.1016/S0163-7258(03)00034-2. [DOI] [PubMed] [Google Scholar]
- 5.Griffiths P, Plotkin S, Mocarski E, Pass R, Schleiss M, Krause P, Bialek S. 2013. Desirability and feasibility of a vaccine against cytomegalovirus. Vaccine 31(Suppl 2):B197–B203. doi: 10.1016/j.vaccine.2012.10.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goderis J, De Leenheer E, Smets K, Van Hoecke H, Keymeulen A, Dhooge I. 2014. Hearing loss and congenital CMV infection: a systematic review. Pediatrics 134:972–982. doi: 10.1542/peds.2014-1173. [DOI] [PubMed] [Google Scholar]
- 7.Britt W. 13 March 2015. Controversies in the natural history of congenital human cytomegalovirus infection: the paradox of infection and disease in offspring of women with immunity prior to pregnancy. Med Microbiol Immunol doi: 10.1007/s00430-015-0399-9. [DOI] [PubMed] [Google Scholar]
- 8.Kenneson A, Cannon MJ. 2007. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev Med Virol 17:253–276. doi: 10.1002/rmv.535. [DOI] [PubMed] [Google Scholar]
- 9.Lanzieri TM, Dollard SC, Bialek SR, Grosse SD. 2014. Systematic review of the birth prevalence of congenital cytomegalovirus infection in developing countries. Int J Infect Dis 22:44–48. doi: 10.1016/j.ijid.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Plotkin SA. 2013. Complex correlates of protection after vaccination. Clin Infect Dis 56:1458–1465. doi: 10.1093/cid/cit048. [DOI] [PubMed] [Google Scholar]
- 11.Heineman TC. 2007. Human cytomegalovirus vaccines, p 1274–1291. In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom. [PubMed] [Google Scholar]
- 12.Lilleri D, Kabanova A, Lanzavecchia A, Gerna G. 2012. Antibodies against neutralization epitopes of human cytomegalovirus gH/gL/pUL128-130-131 complex and virus spreading may correlate with virus control in vivo. J Clin Immunol 32:1324–1331. doi: 10.1007/s10875-012-9739-3. [DOI] [PubMed] [Google Scholar]
- 13.Lilleri D, Kabanova A, Revello MG, Percivalle E, Sarasini A, Genini E, Sallusto F, Lanzavecchia A, Corti D, Gerna G. 2013. Fetal human cytomegalovirus transmission correlates with delayed maternal antibodies to gH/gL/pUL128-130-131 complex during primary infection. PLoS One 8:e59863. doi: 10.1371/journal.pone.0059863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McVoy MA. 2013. Cytomegalovirus vaccines. Clin Infect Dis 57(Suppl 4):S196–S199. doi: 10.1093/cid/cit587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boppana SB, Britt WJ. 1995. Antiviral antibody responses and intrauterine transmission after primary maternal cytomegalovirus infection. J Infect Dis 171:1115–1121. doi: 10.1093/infdis/171.5.1115. [DOI] [PubMed] [Google Scholar]
- 16.Manghera A, McLean GR. 2016. Human cytomegalovirus vaccination: progress and perspectives of recombinant gB. Future Virol 11:439–449. doi: 10.2217/fvl-2016-0039. [DOI] [Google Scholar]
- 17.Lilja AE, Mason PW. 2012. The next generation recombinant human cytomegalovirus vaccine candidates—beyond gB. Vaccine 30:6980–6990. doi: 10.1016/j.vaccine.2012.09.056. [DOI] [PubMed] [Google Scholar]
- 18.Bernstein DI, Munoz FM, Callahan ST, Rupp R, Wootton SH, Edwards KM, Turley CB, Stanberry LR, Patel SM, McNeal MM, Pichon S, Amegashie C, Bellamy AR. 2016. Safety and efficacy of a cytomegalovirus glycoprotein B (gB) vaccine in adolescent girls: a randomized clinical trial. Vaccine 34:313–319. doi: 10.1016/j.vaccine.2015.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pass RF. 2009. Development and evidence for efficacy of CMV glycoprotein B vaccine with MF59 adjuvant. J Clin Virol 46(Suppl 4):S73–S76. doi: 10.1016/j.jcv.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Griffiths PD, Stanton A, McCarrell E, Smith C, Osman M, Harber M, Davenport A, Jones G, Wheeler DC, O'Beirne J, Thorburn D, Patch D, Atkinson CE, Pichon S, Sweny P, Lanzman M, Woodford E, Rothwell E, Old N, Kinyanjui R, Haque T, Atabani S, Luck S, Prideaux S, Milne RS, Emery VC, Burroughs AK. 2011. Cytomegalovirus glycoprotein-B vaccine with MF59 adjuvant in transplant recipients: a phase 2 randomised placebo-controlled trial. Lancet 377:1256–1263. doi: 10.1016/S0140-6736(11)60136-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dempsey AF, Pangborn HM, Prosser LA. 2012. Cost-effectiveness of routine vaccination of adolescent females against cytomegalovirus. Vaccine 30:4060–4066. doi: 10.1016/j.vaccine.2012.04.011. [DOI] [PubMed] [Google Scholar]
- 22.Wille PT, Knoche AJ, Nelson JA, Jarvis MA, Johnson DC. 2010. A human cytomegalovirus gO-null mutant fails to incorporate gH/gL into the virion envelope and is unable to enter fibroblasts and epithelial and endothelial cells. J Virol 84:2585–2596. doi: 10.1128/JVI.02249-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hahn G, Revello MG, Patrone M, Percivalle E, Campanini G, Sarasini A, Wagner M, Gallina A, Milanesi G, Koszinowski U, Baldanti F, Gerna G. 2004. Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J Virol 78:10023–10033. doi: 10.1128/JVI.78.18.10023-10033.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang D, Shenk T. 2005. Human cytomegalovirus UL131 open reading frame is required for epithelial cell tropism. J Virol 79:10330–10338. doi: 10.1128/JVI.79.16.10330-10338.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou M, Lanchy JM, Ryckman BJ. 17 June 2015. Human cytomegalovirus gH/gL/gO promotes the fusion step of entry into all cell types whereas gH/gL/UL128-131 broadens virus tropism through a distinct mechanism. J Virol doi: 10.1128/JVI.01325-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang XJ, Adler B, Sampaio KL, Digel M, Jahn G, Ettischer N, Stierhof YD, Scrivano L, Koszinowski U, Mach M, Sinzger C. 2008. UL74 of human cytomegalovirus contributes to virus release by promoting secondary envelopment of virions. J Virol 82:2802–2812. doi: 10.1128/JVI.01550-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adler B, Scrivano L, Ruzcics Z, Rupp B, Sinzger C, Koszinowski U. 2006. Role of human cytomegalovirus UL131A in cell type-specific virus entry and release. J Gen Virol 87:2451–2460. doi: 10.1099/vir.0.81921-0. [DOI] [PubMed] [Google Scholar]
- 28.Macagno A, Bernasconi NL, Vanzetta F, Dander E, Sarasini A, Revello MG, Gerna G, Sallusto F, Lanzavecchia A. 2010. Isolation of human monoclonal antibodies that potently neutralize human cytomegalovirus infection by targeting different epitopes on the gH/gL/UL128-131A complex. J Virol 84:1005–1013. doi: 10.1128/JVI.01809-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kabanova A, Perez L, Lilleri D, Marcandalli J, Agatic G, Becattini S, Preite S, Fuschillo D, Percivalle E, Sallusto F, Gerna G, Corti D, Lanzavecchia A. 2014. Antibody-driven design of a human cytomegalovirus gHgLpUL128L subunit vaccine that selectively elicits potent neutralizing antibodies. Proc Natl Acad Sci U S A 111:17965–17970. doi: 10.1073/pnas.1415310111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wussow F, Chiuppesi F, Martinez J, Campo J, Johnson E, Flechsig C, Newell M, Tran E, Ortiz J, La Rosa C, Herrmann A, Longmate J, Chakraborty R, Barry PA, Diamond DJ. 2014. Human cytomegalovirus vaccine based on the envelope gH/gL pentamer complex. PLoS Pathog 10:e1004524. doi: 10.1371/journal.ppat.1004524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gerna G, Sarasini A, Patrone M, Percivalle E, Fiorina L, Campanini G, Gallina A, Baldanti F, Revello MG. 2008. Human cytomegalovirus serum neutralizing antibodies block virus infection of endothelial/epithelial cells, but not fibroblasts, early during primary infection. J Gen Virol 89:853–865. doi: 10.1099/vir.0.83523-0. [DOI] [PubMed] [Google Scholar]
- 32.Chiuppesi F, Wussow F, Johnson E, Bian C, Zhuo M, Rajakumar A, Barry PA, Britt WJ, Chakraborty R, Diamond DJ. 2015. Vaccine-derived neutralizing antibodies to the human cytomegalovirus gH/gL pentamer potently block primary cytotrophoblast infection. J Virol 89:11884–11898. doi: 10.1128/JVI.01701-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wen Y, Monroe J, Linton C, Archer J, Beard CW, Barnett SW, Palladino G, Mason PW, Carfi A, Lilja AE. 2014. Human cytomegalovirus gH/gL/UL128/UL130/UL131A complex elicits potently neutralizing antibodies in mice. Vaccine 32:3796–3804. doi: 10.1016/j.vaccine.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 34.Freed DC, Tang Q, Tang A, Li F, He X, Huang Z, Meng W, Xia L, Finnefrock AC, Durr E, Espeseth AS, Casimiro DR, Zhang N, Shiver JW, Wang D, An Z, Fu TM. 2013. Pentameric complex of viral glycoprotein H is the primary target for potent neutralization by a human cytomegalovirus vaccine. Proc Natl Acad Sci U S A 110:E4997–E5005. doi: 10.1073/pnas.1316517110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ciferri C, Chandramouli S, Donnarumma D, Nikitin PA, Cianfrocco MA, Gerrein R, Feire AL, Barnett SW, Lilja AE, Rappuoli R, Norais N, Settembre EC, Carfi A. 2015. Structural and biochemical studies of HCMV gH/gL/gO and pentamer reveal mutually exclusive cell entry complexes. Proc Natl Acad Sci U S A 112:1767–1772. doi: 10.1073/pnas.1424818112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fouts AE, Chan P, Stephan JP, Vandlen R, Feierbach B. 2012. Antibodies against the gH/gL/UL128/UL130/UL131 complex comprise the majority of the anti-cytomegalovirus (anti-CMV) neutralizing antibody response in CMV hyperimmune globulin. J Virol 86:7444–7447. doi: 10.1128/JVI.00467-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baldanti F, Paolucci S, Campanini G, Sarasini A, Percivalle E, Revello MG, Gerna G. 2006. Human cytomegalovirus UL131A, UL130 and UL128 genes are highly conserved among field isolates. Arch Virol 151:1225–1233. doi: 10.1007/s00705-005-0696-5. [DOI] [PubMed] [Google Scholar]
- 38.Van Regenmortel MH. 2006. Immunoinformatics may lead to a reappraisal of the nature of B cell epitopes and of the feasibility of synthetic peptide vaccines. J Mol Recognit 19:183–187. doi: 10.1002/jmr.768. [DOI] [PubMed] [Google Scholar]
- 39.Singh H, Ansari HR, Raghava GP. 2013. Improved method for linear B-cell epitope prediction using antigen's primary sequence. PLoS One 8:e62216. doi: 10.1371/journal.pone.0062216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kringelum JV, Nielsen M, Padkjaer SB, Lund O. 2013. Structural analysis of B-cell epitopes in antibody:protein complexes. Mol Immunol 53:24–34. doi: 10.1016/j.molimm.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saccoccio FM, Sauer AL, Cui X, Armstrong AE, Habib E-SE, Johnson DC, Ryckman BJ, Klingelhutz AJ, Adler SP, McVoy MA. 2011. Peptides from cytomegalovirus UL130 and UL131 proteins induce high titer antibodies that block viral entry into mucosal epithelial cells. Vaccine 29:2705–2711. doi: 10.1016/j.vaccine.2011.01.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ciferri C, Chandramouli S, Leitner A, Donnarumma D, Cianfrocco MA, Gerrein R, Friedrich K, Aggarwal Y, Palladino G, Aebersold R, Norais N, Settembre EC, Carfi A. 2015. Antigenic characterization of the HCMV gH/gL/gO and pentamer cell entry complexes reveals binding sites for potently neutralizing human antibodies. PLoS Pathog 11:e1005230. doi: 10.1371/journal.ppat.1005230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Serrano S, Araujo A, Apellaniz B, Bryson S, Carravilla P, de la Arada I, Huarte N, Rujas E, Pai EF, Arrondo JL, Domene C, Jimenez MA, Nieva JL. 2014. Structure and immunogenicity of a peptide vaccine, including the complete HIV-1 gp41 2F5 epitope: implications for antibody recognition mechanism and immunogen design. J Biol Chem 289:6565–6580. doi: 10.1074/jbc.M113.527747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Malito E, Faleri A, Lo Surdo P, Veggi D, Maruggi G, Grassi E, Cartocci E, Bertoldi I, Genovese A, Santini L, Romagnoli G, Borgogni E, Brier S, Lo Passo C, Domina M, Castellino F, Felici F, van der Veen S, Johnson S, Lea SM, Tang CM, Pizza M, Savino S, Norais N, Rappuoli R, Bottomley MJ, Masignani V. 2013. Defining a protective epitope on factor H binding protein, a key meningococcal virulence factor and vaccine antigen. Proc Natl Acad Sci U S A 110:3304–3309. doi: 10.1073/pnas.1222845110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Good MF, Yanow SK. 21 March 2016. Cryptic epitopes should not be forgotten in vaccine design. Expert Rev Vaccines doi: 10.1586/14760584.2016.1154791. [DOI] [PubMed] [Google Scholar]
- 46.Shen Y, Wang J, Huang Y, Liang J, Liu X, Wu D, Jiang H, Zhao Y, Li Y. 30 January 2016. Analysis of the immune response of a new malaria vaccine based on the modification of cryptic epitopes. Parasitol Res doi: 10.1007/s00436-016-4931-7. [DOI] [PubMed] [Google Scholar]
- 47.Ascough S, Ingram RJ, Chu KK, Musson JA, Moore SJ, Gallagher T, Baillie L, Williamson ED, Robinson JH, Maillere B, Boyton RJ, Altmann DM. 2015. CD4+ T cells targeting dominant and cryptic epitopes from Bacillus anthracis lethal factor. Front Microbiol 6:1506. doi: 10.3389/fmicb.2015.01506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rathore D, Nagarkatti R, Jani D, Chattopadhyay R, de la Vega P, Kumar S, McCutchan TF. 2005. An immunologically cryptic epitope of Plasmodium falciparum circumsporozoite protein facilitates liver cell recognition and induces protective antibodies that block liver cell invasion. J Biol Chem 280:20524–20529. doi: 10.1074/jbc.M414254200. [DOI] [PubMed] [Google Scholar]
- 49.Oscherwitz J, Yu F, Jacobs JL, Liu TH, Johnson PR, Cease KB. 2009. Synthetic peptide vaccine targeting a cryptic neutralizing epitope in domain 2 of Bacillus anthracis protective antigen. Infect Immun 77:3380–3388. doi: 10.1128/IAI.00358-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rieder FJ, Biebl J, Kastner MT, Schneider M, Jungbauer C, Redlberger-Fritz M, Britt WJ, Kundi M, Steininger C. 2016. Microbial cryptotopes are prominent targets of B-cell immunity. Sci Rep 6:31657. doi: 10.1038/srep31657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mascola JR, Haynes BF. 2013. HIV-1 neutralizing antibodies: understanding nature's pathways. Immunol Rev 254:225–244. doi: 10.1111/imr.12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rusert P, Kouyos RD, Kadelka C, Ebner H, Schanz M, Huber M, Braun DL, Hoze N, Scherrer A, Magnus C, Weber J, Uhr T, Cippa V, Thorball CW, Kuster H, Cavassini M, Bernasconi E, Hoffmann M, Calmy A, Battegay M, Rauch A, Yerly S, Aubert V, Klimkait T, Boni J, Fellay J, Regoes RR, Gunthard HF, Trkola A, Swiss HIV Cohort Study . 2016. Determinants of HIV-1 broadly neutralizing antibody induction. Nat Med 22:1260–1267. doi: 10.1038/nm.4187. [DOI] [PubMed] [Google Scholar]
- 53.Buscher N, Paulus C, Nevels M, Tenzer S, Plachter B. 2015. The proteome of human cytomegalovirus virions and dense bodies is conserved across different strains. Med Microbiol Immunol 204:285–293. doi: 10.1007/s00430-015-0397-y. [DOI] [PubMed] [Google Scholar]
- 54.Wang C, Zhang X, Bialek S, Cannon MJ. 2011. Attribution of congenital cytomegalovirus infection to primary versus non-primary maternal infection. Clin Infect Dis 52:e11–e13. doi: 10.1093/cid/ciq085. [DOI] [PubMed] [Google Scholar]
- 55.O'Connor CM, Murphy EA. 2012. A myeloid progenitor cell line capable of supporting human cytomegalovirus latency and reactivation, resulting in infectious progeny. J Virol 86:9854–9865. doi: 10.1128/JVI.01278-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ryckman BJ, Rainish BL, Chase MC, Borton JA, Nelson JA, Jarvis MA, Johnson DC. 2008. Characterization of the human cytomegalovirus gH/gL/UL128-131 complex that mediates entry into epithelial and endothelial cells. J Virol 82:60–70. doi: 10.1128/JVI.01910-07. [DOI] [PMC free article] [PubMed] [Google Scholar]