

ABSTRACT
African swine fever is an acute hemorrhagic disease of pigs. Extensive recent spread in the Russian Federation and Eastern Europe has increased the risk to global pig production. The virus is a large DNA virus and is the only member of the Asfarviridae family. In pigs, the virus replicates predominantly in macrophages. We review how the virus overcomes the barriers to replication in the macrophage and the virus mechanism to inhibit key host defense pathways.
KEYWORDS: immune evasion, African swine fever, interferon, protein synthesis, apoptosis
INTRODUCTION
Virulent isolates of African swine fever (ASF) virus cause an acutely lethal hemorrhagic disease in domestic pigs and wild boar. Following emergence from the sylvatic cycle between warthogs and Ornithodoros species of soft ticks in the early 1900s, the virus spread to most sub-Saharan African countries. In 2007, ASF appeared in Europe for the second time after it was introduced into Georgia, from where it has spread through the Russian Federation into eastern countries of the European Union (EU).
As there is no vaccine against ASF, the disease has a very high socioeconomic impact and threatens the livelihoods of resource-poor farmers. In the Baltic States, the cost of ASF was estimated to be around $55 million during 2014 and 2015 and the net benefit of preventing ASF in the United States has been estimated to be US$4500 million (1). Further spread of the disease throughout the world might have disastrous consequences for global supplies of pork, an increasingly important source of relatively cheap protein.
ASF virus (ASFV) is the only known DNA virus that replicates in both mammalian and arthropod hosts. Since ASFV replicates predominantly in the cell cytoplasm, the 170- to 190-kbp genome encodes the enzymes and factors for genome replication and transcription, but the virus devotes considerable coding capacity to genes that help the virus survive and evade the host's defenses (Table 1).
TABLE 1.
Host cell pathways known to be modulated by African swine fever virus infectiona

Column A shows host antiviral pathways known to be modulated by ASFV. Column B shows the effect of ASFV infection on these pathways, and column C shows ASFV proteins known to modulate the pathways. Column D indicates whether the gene is known to be essential or nonessential and also includes any known phenotypic effect of deletion.
MACROPHAGE AS TARGET CELLS FOR VIRUS REPLICATION
ASFV replicates predominantly in macrophages. Since these cells play a key role in activating and orchestrating the host's innate and adaptive responses, this provides the virus with a critical advantage to block or manipulate these responses (Fig. 1). However, this strategy comes at a cost since macrophages are at the frontline in the sensing and destruction of pathogens and activation of the host's defense. Strategies to overcome these barriers are critical. Thus, ASFV provides an excellent model to better understand how viruses replicate in and manipulate macrophage functions.
FIG 1.
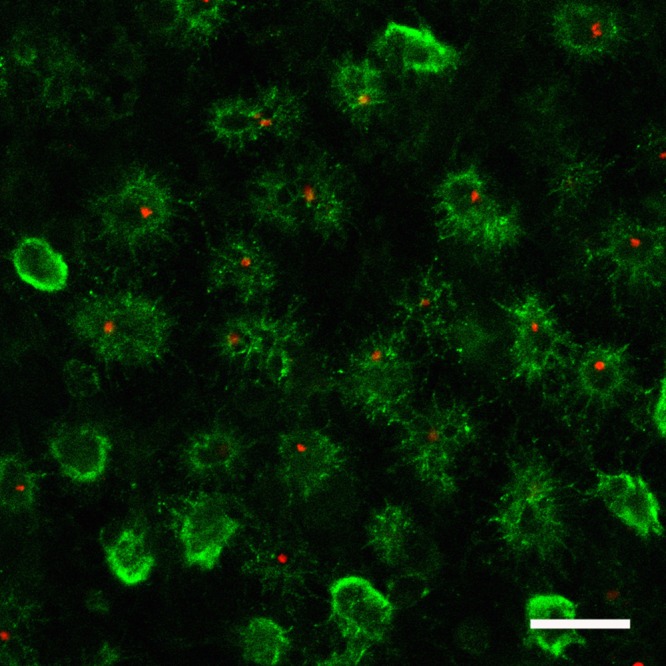
African swine fever virus in spleen samples from pigs infected with virulent virus. Pigs were infected with the virulent ASFV Benin 97/1 isolate, and after 3 days pigs were euthanized and tissue samples were collected. A section of infected spleen is shown with the myeloid-specific cell surface marker SWC3 (CD172a) labeled in green and the ASFV p54 protein in red. Bar, 30 μm. (Image courtesy of Pippa Hawes, Pirbright Institute, reproduced with permission.)
To enter swine macrophages, ASFV can use either clathrin-mediated endocytosis, which involves virus interaction with an as yet unknown receptor, or macropinocytosis, a nonspecific uptake mechanism (2). The virus core enters the cell cytoplasm following fusion of the viral internal membrane with that of the host late endosome compartment in a pH-dependent step to initiate the replication cycle.
Challenges for virus replication in the macrophage include the oxidizing environment, which might induce DNA damage, and the accumulation of mutations, which might inhibit virus replication. A specific adaptation of ASFV to replicate in this environment includes the acquisition of a base excision DNA repair system comprising a DNA polymerase X, a type I DNA ligase, and an apurinic/apyrimidinic (AP) endonuclease. Deletion of the genes encoding either the DNA polymerase X or AP endonuclease results in impaired virus growth and an increased mutation frequency of virus DNA in macrophages compared to that in Vero cells. These data confirm a role for the repair enzymes in the preservation of viral genetic information in macrophages (3). ASFV also encodes a protein, A238L, that inhibits host transcription factors involved in activating transcription of host defense proteins, including inducible nitric oxide synthase (4).
ASFV MANIPULATION OF THE HOST'S DEFENSES TO ENABLE VIRUS REPLICATION AND INHIBIT HOST INNATE AND ADAPTIVE IMMUNE RESPONSES
ASFV replication requires subversion of several host pathways which can lead to rapid cell death through programmed pathways, including apoptosis, pyroptosis, and necrosis. The virus subverts these to keep the host cell alive long enough such that it can replicate its progeny. After replication, induction of such pathways may be beneficial to induce host cell lysis, particularly as an ordered induction of apoptosis will recruit phagocytic cells, the principal target cells for ASFV.
Inhibition of cell death by ASFV.
ASFV induces apoptosis both in vitro and in vivo, and in cultured cells, apoptosis is triggered in the absence of virus replication. Apoptotic signatures in infected cells are detected after DNA replication has commenced, and the virus encodes at least four proteins that interact with the programmed cell death pathway: A179L, A224L, and EP153R can inhibit apoptosis, whereas E183L can induce it.
The A179L gene of ASFV encodes a bcl-2 homologue that can inhibit the induction of apoptosis by different stimuli in a BH1 domain-dependent manner (5). More recently, A179L was shown to directly interact with a broad range of BH3-only cellular proteins, including Bid, and can inhibit apoptotic signaling induced by overexpression of such proteins. A179L can also interact with beclin-1, a critical regulator of autophagy, suggesting that this viral protein can modulate a wider range of potential host cell responses to infection (21).
A224L is a nonessential homologue of the cellular inhibitor of the apoptosis protein (IAP) family of genes that can protect cells from external induction of apoptosis through an interaction with caspase-3 (6). Deletion of A224L does not affect the virulence of ASFV in pigs but may play a more important role in the wildlife hosts, such as the tick or warthog.
Recombinant ASFV lacking the C-type lectin EP153R induced increased apoptosis after infection of macrophages (7), suggesting that this protein may also play a role in regulating programmed cell death. Conversely, the E183L gene, which encodes the structural protein p54, induces caspase-3 activity and apoptosis through a mechanism that requires a 13-amino-acid dynein binding motif (8) that is similar to a dynein light chain-8 binding motif in the BH3-only proapoptotic protein Bim. Bim is recruited from the cytoskeleton to mitochondria in infected cells at a time similar to the time for the increase in other indicators of apoptosis. p54 may be involved in the transport of virions along microtubules after entry as well as retaining membranes involved in assembly in the virus factories. The interaction between p54 and the microtubular network may contribute to the induction of apoptosis that is triggered shortly after ASFV infection.
Cell death resulting from pyroptosis and programmed necrosis signals through inflammasomes and involves caspase-1 and possibly caspase-8. Research on the interaction between ASFV and the different pathways of programmed cell death in macrophages is likely to be fruitful. For example, caspase-3 activity has been reported by many authors in Vero cells infected with tissue culture-adapted ASFV. However, experiments in macrophages suggest little caspase-3 activity after infection with virulent field isolates, although low-virulence isolates do induce caspase-3 activation (9).
Avoiding shutoff of protein synthesis.
ASFV inhibits the global shutoff of protein synthesis triggered by the phosphorylation of translation initiation factor α subunit of eukaryotic initiation factor 2 (eIF2α). eIF2α is phosphorylated by the double-stranded RNA-activated protein kinase PKR and a number of other stress-inducible cellular kinases. The ASFV DP71L protein recruits host protein phosphatase 1 (PP1) to dephosphorylate eIF2α, thus restoring global protein synthesis. However, deletion of the DP71L gene from ASFV did not result in increased phosphorylation of eIF2α in mammalian cells, indicating that the virus has other complementary functions to reduce eIF2α phosphorylation (10). PP1 has many different cellular functions controlled by the interaction of the protein with different regulatory subunits; as a consequence, PP1 is relatively conserved. Other viruses inhibit phosphorylation of eIF2α by blocking the activation of PKR. This may have led to the relatively high divergence of PKR due to pathogen pressure to evade this critical antiviral defense. Targeting of conserved points of critical pathways, for example, PP1 rather than PKR, may contribute to the successful replication of ASFV in diverse mammalian and arthropod hosts.
INHIBITION OF HOST INNATE AND ADAPTIVE RESPONSES
Type I interferon responses.
ASFV encodes a suite of proteins that inhibit transcription of type I interferon (IFN), cytokines, chemokines, adhesion molecules, and other immunomodulatory genes. Secreted IFNs are recognized by cell surface receptors in both infected and neighboring cells, resulting in expression of interferon-stimulated genes (ISGs). These have roles in controlling viral replication, in the maturation of dendritic cells, thus contributing to the cross-presentation of viral antigens to CD8+ cells, and in the activation of natural killer cells.
Virulent isolates of ASFV suppress expression of type I IFN and ISGs in infected macrophages. Experiments with deletion mutant viruses lacking multiple members of the multigene family 360 (MGF360) and MGF530 show that these genes inhibit the induction of IFN and may also have an impact on the STAT signaling cascade and/or components of the antiviral state (11–13). Importantly, recombinant viruses with similar deletions of MGF360 and MGF530 that were derived from three different genotypes are attenuated in pigs (11, 13, 14). This indicates that modulation of the IFN response plays a crucial role in viral pathogenesis. Interestingly, the deletion of genes from MGF360 and MGF530 severely reduces viral replication in ticks (15). The recent discovery that ticks may have an IFN-like system dependent on STAT signaling raises the hypothesis that these genes may have evolved to modulate parallel pathways in both the mammalian and the arthropod hosts. Despite the importance of these genes in the inhibition of the IFN response, their mechanism(s) of action remains elusive.
ASFV encodes several other proteins for modulation by the host's innate immunity with some redundancy in their mechanisms of action. For example, both I329L, a viral Toll-like receptor (TLR) homologue, and A528R inhibit IFN induction by poly(I·C) upstream of IRF3 and NF-κB activation (16). The MGF360-15R gene inhibits the same pathway, but at the level of IRF3. The A528R gene was also shown to inhibit cellular responses to type I and type II IFN, a good example of a viral multifunctional protein.
Virulent isolates are resistant to pretreatment of macrophages with IFN (12), showing that ASFV also inhibits the antiviral state. Two host ISG proteins have emerged as potential targets for ASFV. These are the interferon-induced transmembrane (IFITM) protein family members, which affect viral entry/uncoating, and MxA, which affects viral replication (17, 18). Several hundred ISGs are known, and ASFV may also modulate the functions of a number of these.
Virus adhesion protein CD2v.
Infection of swine macrophages with virulent ASFV isolates causes binding of red blood cells (RBCs) as a result of the interaction between a virus CD2-like protein (CD2v) and its ligand expressed on these cells. The CD2v protein is also incorporated into the virus particles, mediating attachment of extracellular virions to RBCs. This facilitates virus spreading in the host since deletion of the CD2v gene caused a significant delay in the onset of both viremia and the development of clinical signs. CD2v was also shown to be immunosuppressive by inhibiting lymphocyte proliferation in vitro (19). In addition to its role in the viral pathogenesis in the mammalian host, deletion of CD2v was also shown to reduce viral replication in the tick vector. Restoration of CD2v expression resulted in a significant increase in virus titers in ticks, possibly through enhancement of virus uptake across the gut wall (20). This may be particularly important for the maintenance of ASFV in the sylvatic cycle, in which the tick vector is thought to play an essential role.
FUTURE DIRECTIONS
Good progress has been made in understanding the “armory” employed by ASFV to replicate in the hostile environment of the macrophage and avoid detection by and activation of the host's defenses. Research priorities going forward will include understanding the relative importance of the different entry mechanisms in different virus hosts and identification of the host cell receptor(s) and the virus proteins which interact with these. This information would lead to improved understanding of the cellular tropism and pathogenesis of the virus and to new targets for vaccine development.
The failure of the host's intrinsic and innate immune systems to control virus replication results in rapid ASFV replication and induction of hemorrhagic pathology in infected pigs. Evidence suggests that the inhibition of type I IFN induction and responses by virulent ASFV isolates is of critical importance in facilitating rapid virus replication and that deletion of genes that inhibit this induction can attenuate the virus. The host pattern recognition receptors (PRRs) involved in sensing ASFV infection and the mechanism of virus IFN inhibitory proteins are largely unknown. Several multigene families (MGFs) have been amplified on the ASFV genome, and an attractive hypothesis is that these have been selected to evade the host's innate immune responses. A key challenge is to understand the targets and functions of these MGFs.
The delayed onset of cell death in infected macrophages and induction of apoptosis in bystander lymphocytes are key factors in enabling virus replication and immune evasion. The roles of other cell death pathways, pyroptosis and necrosis, in ASFV infection are unknown and are particularly relevant since the pathways signal through inflammasomes. Mechanisms by which apoptosis is induced in uninfected bystander lymphocytes are also unclear. Additional proteins involved in inhibiting or modulating translation remain to be characterized. The impact of ASFV on critical host pathways, including autophagy and inflammasome activation, has been little studied. Recent analysis of the pig genome has shown differences in the repertoire of host response genes, including those for inflammasome components and the pyhin domain DNA sensors between pigs and other mammals. This indicates that there may be important differences in how these pathways function. Extension of genomic analysis to the warthog may reveal the basis for their resistance to ASFV.
ACKNOWLEDGMENTS
We acknowledge helpful discussions with many colleagues and apologize for not citing all relevant work due to space constraints. We thank colleagues at The Pirbright Institute, including Pippa Hawes for providing the confocal image, and Dave Chapman, Pedro Sanchez-Cordon, Maria Montoya, Tamara Jabbar, Lynnette Goatley, and Geraldine Taylor for support and discussion.
This work was supported by BBSRC grants BB/L004267/1 and BB/L026562/1.
Footnotes
For a companion article on this topic, see https://doi.org/10.1128/JVI.02228-16.
REFERENCES
- 1.Costard S, Wieland B, de Glanville W, Jori F, Rowlands R, Vosloo W, Roger F, Pfeiffer DU, Dixon LK. 2009. African swine fever: how can global spread be prevented? Philos Trans R Soc Lond B Biol Sci 364:2683−2696. doi: 10.1098/rstb.2009.0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hernáez B, Guerra M, Salas ML, Andres G. 2016. African swine fever virus undergoes outer envelope disruption, capsid disassembly and inner envelope fusion before core release from multivesicular endosomes. PLoS Pathog 12:e1005595. doi: 10.1371/journal.ppat.1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Redrejo-Rodríguez M, Salas ML. 2014. Repair of base damage and genome maintenance in the nucleo-cytoplasmic large DNA viruses. Virus Res 179:12–25. doi: 10.1016/j.virusres.2013.10.017. [DOI] [PubMed] [Google Scholar]
- 4.Granja AG, Sabina P, Salas ML, Fresno M, Revilla Y. 2006. Regulation of inducible nitric oxide synthase expression by viral A238L-mediated inhibition of p65/RelA acetylation and p300 transactivation. J Virol 80:10487–10496. doi: 10.1128/JVI.00862-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Revilla Y, Cebrian A, Baixeras E, Martinez C, Vinuela E, Salas ML. 1997. Inhibition of apoptosis by the African swine fever virus bcl-2 homologue: role of the BH1 domain. Virology 228:400–404. doi: 10.1006/viro.1996.8395. [DOI] [PubMed] [Google Scholar]
- 6.Nogal ML, González de Buitrago G, Rodriguez C, Cubelos B, Carrascosa AL, Salas ML, Revilla Y. 2001. African swine fever virus IAP homologue inhibits caspase activation and promotes cell survival in mammalian cells. J Virol 75:2535–2543. doi: 10.1128/JVI.75.6.2535-2543.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hurtado C, Granja AG, Bustos MJ, Nogal ML, de Buitrago GG, de Yebenes VG, Salas ML, Revilla Y, Carrascosa AL. 2004. The C-type lectin homologue gene (EP153R) of African swine fever virus inhibits apoptosis both in virus infection and in heterologous expression. Virology 326:160–170. doi: 10.1016/j.virol.2004.05.019. [DOI] [PubMed] [Google Scholar]
- 8.Hernaez B, Diaz-Gil G, Garcia-Gallo M, Quetglas JI, Rodriguez-Crespo I, Dixon L, Escribano JM, Alonso C. 2004. The African swine fever virus dynein-binding protein p54 induces infected cell apoptosis. FEBS Lett 569:224–228. doi: 10.1016/j.febslet.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 9.Portugal R, Leitao A, Martins C. 2009. Apoptosis in porcine macrophages infected in vitro with African swine fever virus (ASFV) strains with different virulence. Arch Virol 1441–1450. doi: 10.1007/s00705-009-0466-x. [DOI] [PubMed] [Google Scholar]
- 10.Zhang FQ, Moon A, Childs K, Goodbourn S, Dixon LK. 2010. The African swine fever virus DP71L protein recruits the protein phosphatase 1 catalytic subunit to dephosphorylate eIF2 alpha and inhibits CHOP induction but is dispensable for these activities during virus infection. J Virol 84:10681–10689. doi: 10.1128/JVI.01027-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Afonso CL, Piccone ME, Zaffuto KM, Neilan J, Kutish GF, Lu Z, Lu Z, Balinsky CA, Gibb TR, Bean TJ, Zsak L, Rock DL. 2004. African swine fever virus multigene family 360 and 530 genes affect host interferon response J Virol 78:1858–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Golding JP, Goatley L, Goodbourn S, Dixon LK, Taylor G, Netherton CL. 2016. Sensitivity of African swine fever virus to type I interferon is linked to genes within multigene families 360 and 505. Virology 493:154−161. doi: 10.1016/j.virol.2016.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reis AL, Abrams CC, Goatley LC, Netherton C, Chapman DG, Sanchez-Cordon P, Dixon LK. 2016. Deletion of African swine fever virus interferon inhibitors from the genome of a virulent isolate reduces virulence in domestic pigs and induces a protective response. Vaccine 34:4698–4705. doi: 10.1016/j.vaccine.2016.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O'Donnell V, Holinka LG, Gladue DP, Sanford B, Krug PW, Lu X, Arzt J, Reese B, Carrillo C, Risatti GR, Borca MV. 2015. African swine fever virus Georgia isolate harboring deletions of MGF360 and MGF505 genes is attenuated in swine and confers protection against challenge with virulent parental virus. J Virol 89:6048–6056. doi: 10.1128/JVI.00554-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burrage TG, Lu Z, Neilan JG, Rock DL, Zsak L. 2004. African swine fever virus multigene family 360 genes affect virus replication and generalization of infection in Ornithodoros porcinus ticks. J Virol 78:2445–2453. doi: 10.1128/JVI.78.5.2445-2453.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Correia S, Ventura S, Parkhouse RM. 2013. Identification and utility of innate immune system evasion mechanisms of ASFV. Virus Res 173:87–100. doi: 10.1016/j.virusres.2012.10.013. [DOI] [PubMed] [Google Scholar]
- 17.Muñoz-Moreno R, Cuesta-Geijo MA, Martinez-Romero C, Barrado-Gil L, Galindo I, Garcia-Sastre A, Alonso C. 2016. Antiviral role of IFITM proteins in African swine fever virus infection. PLoS One 11:e0154366. doi: 10.1371/journal.pone.0154366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Netherton CL, Simpson J, Haller O, Wileman TE, Takamatsu HH, Monaghan P, Taylor G. 2009. Inhibition of a large double-stranded DNA virus by MxA protein. J Virol 83:2310–2320. doi: 10.1128/JVI.00781-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borca MV, Carrillo C, Zsak L, Laegreid WW, Kutish GF, Neilan JG, Burrage TG, Rock DL. 1998. Deletion of a CD2-like gene, 8-DR, from African swine fever virus affects viral infection in domestic swine. J Virol 72:2881–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rowlands RJ, Duarte MM, Boinas F, Hutchings G, Dixon LK. 2009. The CD2v protein enhances African swine fever virus replication in the tick vector, Ornithodoros erraticus. Virology 393:319–328. doi: 10.1016/j.virol.2009.07.040. [DOI] [PubMed] [Google Scholar]
- 21.Banjara S, Sofia Caria S, Dixon LK, Hinds MG, Kvansakul M. 2017. Structural insight into African swine fever virus A179L-mediated inhibition of apoptosis. J Virol 91:e02228-16. doi: 10.1128/JVI.02228-16. [DOI] [PMC free article] [PubMed] [Google Scholar]