

ABSTRACT
The assembly of hepatitis C virus (HCV), a complicated process in which many viral and cellular factors are involved, has not been thoroughly deciphered. NS3 is a multifunctional protein that contains an N-terminal amphipathic α helix (designated helix α0), which is crucial for the membrane association and stability of NS3 protein, followed by a serine protease domain and a C-terminal helicase/NTPase domain. NS3 participates in HCV assembly likely through its C-terminal helicase domain, in which all reported adaptive mutations enhancing virion assembly reside. In this study, we determined that the N-terminal helix α0 of NS3 may contribute to HCV assembly. We identified a single mutation from methionine to threonine at amino acid position 21 (M21T) in helix α0, which significantly promoted viral production while having no apparent effect on the membrane association and protease activity of NS3. Subsequent analyses demonstrated that the M21T mutation did not affect HCV genome replication but rather promoted virion assembly. Further study revealed a shift in the subcellular localization of core protein from lipid droplets (LD) to the endoplasmic reticulum (ER). Finally, we showed that the M21T mutation increased the colocalization of core proteins and viral envelope proteins, leading to a more efficient envelopment of viral nucleocapsids. Collectively, the results of our study revealed a new function of NS3 helix α0 and aid understanding of the role of NS3 in HCV virion morphogenesis.
IMPORTANCE HCV NS3 protein possesses the protease activity in its N-terminal domain and the helicase activity in its C-terminal domain. The role of NS3 in virus assembly has been mainly attributed to its helicase domain, because all adaptive mutations enhancing progeny virus production are found to be within this domain. Our study identified, for the first time to our knowledge, an adaptive mutation within the N-terminal helix α0 domain of NS3 that significantly enhanced virus assembly while having no effect on viral genome replication. The mechanistic studies suggested that this mutation promoted the relocation of core proteins from LD to the ER, leading to a more efficient envelopment of viral nucleocapsids. Our results revealed a possible new function of helix α0 in the HCV life cycle and provided new clues to understanding the molecular mechanisms for the action of NS3 in HCV assembly.
KEYWORDS: HCV, NS3, core, helix α0, assembly, hepatitis C virus
INTRODUCTION
Hepatitis C virus (HCV) is an enveloped positive-stranded RNA virus that belongs to the Flaviviridae family. It causes chronic infections in over 170 million people all over the world who are at a risk of developing into liver cirrhosis and hepatocellular carcinoma (1). Despite advances in recently approved potent direct antiviral agents, the worldwide application of these therapies remains limited due to the expense and potential drug resistance (2). In addition, no HCV vaccine is available, and new transmissions still occur, especially among the high-risk individuals (3). Continuous investigation of the HCV life cycle is important not only for better understanding of the biology and pathogenesis of the virus but also for eventual eradication of the pathogen.
The 9.6-kb HCV RNA genome encodes a single polyprotein precursor that is cleaved by viral and host proteases into 10 distinct proteins: the structural protein core, E1 and E2, the viral porin protein p7, and the nonstructural proteins NS2, NS3, NS4A, NS4B, NS5A, and NS5B. Core proteins oligomerize to form a nucleocapsid that packages the viral RNA genome, while E1 and E2 envelope proteins form heterodimers on the endoplasmic reticulum (ER) membrane, where viral nucleocapsids bud and acquire lipid envelope. The C-terminal nonstructural proteins play essential roles in both viral genome replication and virion assembly. The viral replicase, consisting of NS5B, the RNA-dependent RNA polymerase, and NS3, NS4A, NS4B, and NS5A, carries out viral genome replication in ER-associated double membrane vesicles. With the help of NS5A and possibly other nonstructural proteins, the produced progeny viral genomes are transferred to core proteins (4). Core precursor is processed by signal peptidase and signal peptide peptidase into a 19-kDa mature form that targets the surface of lipid droplets (LD) (5) or remains on the ER membrane in a manner critically relying on NS2 and p7 (6). The oligomerized core proteins encapsidate viral RNA genomes to form nucleocapsids (7, 8) that bud into the ER lumen, during which they acquire the phospholipid envelope and heterodimeric E1/E2 glycoproteins. The final step of HCV morphogenesis involves host very-low-density lipoprotein (VLDL) secretory pathway during which HCV virions become associated with VLDL or its components to form lipoviral particles (LVPs) prior to release (9, 10).
NS3 is a multifunctional protein with a serine protease domain at the N terminus, which together with its cofactor NS4A cleaves HCV polyprotein from NS3 to NS5B, and a helicase/NTPase domain at its C terminus, which binds and unwinds RNA duplexes during viral genome replication (11–14). An conserved amphiphathic α-helix (designated helix α0), located in front of the protease domain at amino acids 12 to 23 of NS3, was previously reported to mediate the membrane association of NS3/4A complex (15). Our previous work also suggested that helix α0 is indispensable for the subcellular localization as well as stability of NS3 and NS3/4A complex (16). In this study, we identified a single mutation in the helix α0 of NS3 that promotes HCV assembly, suggesting a previously unreported putative role of helix α0 in the process of HCV assembly.
RESULTS
M21T promoted HCV viral particle production.
In our previous study, a point mutation from methionine to threonine at amino acid residue position 21 (M21T) in NS3 protein was identified in a tissue-culture adaptive HCV (HCVcc) that gains enhanced infectivity (17). The M21T mutation lies in the N-terminal amphipathic helix α0 (Fig. 1A), which is conserved among all HCV genotypes and was reported to play important roles in anchoring NS3 and NS3/4A to the intracellular membrane and thus preventing NS3 from degradation (15, 16, 18). To determine whether the M21T mutation contributes to HCV infection, we engineered this mutation into the JFH1 genome. The in vitro-transcribed full-length JFH1 RNAs containing the wild type (WT), the M21T mutant, or the NS5B polymerase defective GND mutant were electroporated into Huh7.5.1 cells. HCV E2 immunofluorescence and intracellular RNA quantification at day 2 postelectroporation were conducted to monitor electroporation and virus replication efficiency, and the supernatant viral infectivity titers at days 1, 3, 5, and 7 postelectroporation were measured to monitor virus production efficiency. As shown in Fig. 1B and C, there were similar percentages of E2-positive cells and intracellular HCV RNA levels at day 2 postelectroporation, indicating the comparable electroporation and virus replication efficiencies between the WT and M21T. However, the supernatant infectivity titers of the M21T mutant were significantly higher than those of the WT (Fig. 1D). These results suggested that the M21T mutation in the NS3 helix α0 domain could promote the production of HCV progeny viruses.
FIG 1.

The M21T mutant promoted HCV virus production. (A) Schematic representation of JFH1-WT and JFH1-M21T mutant in helix α0 of NS3 protein. Amino acids 1 to 632 are numbered according to the numbering used for NS3 protein. Amino acids 1040 and 1050, indicated in the parentheses, are numbered according to the HCV polyprotein. The point mutation at position 21 is indicated by an asterisk. The M21 side chain is presented as blue sticks in the model of helix α0. (B to D) Huh7.5.1 cells were electroporated with in vitro-transcribed JFH1 RNA of the WT, M21T mutant, or GND mutant. The cells at day 2 postelectroporation were analyzed by immunofluorescence for HCV E2 and nuclei (red and blue channels, respectively) (B) and intracellular HCV RNA levels (C), and the supernatant infectivity titers were determined at different time points after electroporation (D). Error bars indicate standard deviations calculated from 3 independent experiments. ns, not significant.
The M21T mutation did not affect NS3 membrane association, protease activity, polyprotein processing, or MAVS cleavage activity.
Helix α0 was originally characterized as a structural determinant of membrane association of NS3, and we previously showed that a point mutation from methionine to proline at position 21 (M21P) of helix α0 disrupted the membrane association of NS3 and led to the degradation of NS3 (16). Therefore, we first examined whether the NS3 membrane association was affected by the M21T mutation. Green fluorescent protein (GFP) fused with WT, M21T mutant, or M21P mutant helix α0 (the first 10 to 24 amino acids of NS3) was expressed in Huh7 cells. As shown in Fig. 2A, while GFP alone and GFP fused with the M21P mutant helix α0 displayed a diffused subcellular localization pattern, GFP fused with the WT or M21T mutant helix α0 displayed a comparable membrane-associated pattern, as previously reported (15, 16). Next, we examined the subcellular localization of wild-type and M21T NS3 together with its cofactor NS4A. Plasmids expressing the WT or M21T mutant NS3/4A complex were transfected into Huh7.5.1 cells, and immunostaining of NS3 and calnexin (ER marker) was performed. As shown in Fig. 2B, the WT and M21T mutant NS3 showed similar subcellular localizations at the ER.
FIG 2.

M21T did not impair NS3 membrane association or protease activity. (A) Subcellular localization of GFP fused with WT or M21T mutant helix α0. Plasmids GFP-N1 (Vector), NS310-24wt-GFP [Helix α0(WT)-GFP)], NS310-24M21T-GFP [Helix α0(M21T)-GFP], or NS310-24M21P-GFP [Helix α0(M21P)-GFP] were transfected into Huh7 cells, and GFP expression was examined by fluorescence microscopy at 48 h posttransfection. (B) Confocal imaging of NS3 and calnexin (CNX) colocalization. Huh7.5.1 cells were transfected with plasmids expressing WT or M21T NS3/4A for 72 h and subjected to immunofluorescence staining of NS3 (red) and calnexin (green). (C) NS3 protease activity quantified by FRET assay. WT, M21T mutant, or protease-defective H57A mutant recombinant NS3 proteins with or without NS4A were subjected to protease activity assay. The fluorescent signals, activated by NS3 protease activity, were recorded 1 h after the addition of the FRET substrate. Error bars indicate standard deviations calculated from 3 independent experiments. (D) Western blot analysis of HCV nonstructural protein cleavage by NS3. Plasmids expressing HCV NS3-NS5B containing WT, M21T, or H57A NS3 were transfected into Huh7.5.1 cells for 48 h, and cell lysates were analyzed by Western blotting using antibodies against NS3, Flag, and actin. (E) Western blot analysis of MAVS cleavage by NS3. Plasmids expressing Flag-tagged MAVS and NS3-4A proteins (WT, M21T mutant, or empty vector control) were cotransfected into Huh7.5.1 cells for 48 h, and cell lysates were analyzed by Western blotting using antibodies against Flag, NS3, and actin. (F) IFN-β luciferase assay. Poly(I·C) was transfected into Huh7 cells that had been initially transfected with plasmids encoding the IFN-β promoter luciferase reporter and plasmids expressing NS3 (WT, M21T mutant, or vector control) for 36 h. Luciferase activities were measured at 16 h posttransfection. Error bars indicate standard deviations calculated from 3 independent experiments. (G) Western blot analysis of NS2-NS3 cleavage. Plasmids expressing N-terminally HA-tagged NS2 and NS3 (WT, M21T mutant, or the NS2-3 cleavage-defective C184A mutant) were transfected into Huh7.5.1 cells. At 48 h posttransfection, cell lysates were analyzed by Western blotting using antibodies against HA and NS3.
Next, we sought to determine whether the M21T mutation had any effects on the protease activity of NS3. We employed a previously described fluorescent resonance energy transfer (FRET)-based NS3 protease assay in which the fluorescence signals were activated by NS3 cleaving a peptide substrate containing the fluorescent dye 5-[(2′-aminoethyl)amino]naphthalene sulfonic acid (EDANS) and the quencher dye 4-([4′-(dimethylamino)phenyl]azo)benzoic acid (DABCYL) on its two ends (19). As shown in Fig. 2C, while the protease-defective H57A mutant (11, 20) did not cleave the substrate, the WT and M21T mutant NS3 proteins, with the help of their cofactor NS4A, displayed similar cleavage efficiencies, suggesting that the M21T mutation did not affect the protease activity of NS3.
We then assessed the impact of the M21T mutation on the ability of NS3 to cleave HCV nonstructural proteins, the viral protease's natural substrates. Plasmids that express NS3-NS5B containing the wild-type or the M21T or H57A mutant form of NS3 were transfected into Huh7.5.1 cells, and the mature NS3 and NS5B protein levels, which reflect the NS3 protease activity, were determined at day 2 posttransfection. As shown in Fig. 2D, there was no significant difference in mature NS3 and NS5B protein levels between the WT and M21T mutant, while no mature NS3 and NS5B protein was detected in the protease-deficient H57A mutant. The low level of NS3-NS5B in H57A was likely due to the instability of the uncleaved precursor.
It is known that HCV NS3/4A protease can shut down host interferon (IFN) signaling by cleaving MAVS, a mitochondrion-associated host protein essential for the antiviral interferon response (21), and helix α0 of NS3 could control its ability to suppress host innate immune signaling as well (18). To assess the possibility that the M21T mutation may enhance NS3's capability to cleave MAVS to suppress interferon production for viral production, we examined MAVS cleavage and interferon suppression by the wild-type and M21T mutant NS3. For the MAVS cleavage assay, plasmids expressing Flag-tagged MAVS and NS3-4A proteins (WT and M21T mutant) were cotransfected into Huh7.5.1 cells, and MVAS cleavage at 48 h posttransfection was analyzed by Western blotting. As shown in Fig. 2E, NS3 protein and the cleaved MAVS protein levels were comparable between the WT and M21T mutant. For the interferon signaling assay, poly(I·C) was transfected into Huh7 cells that had been transfected with an IFN-β promoter luciferase reporter plasmid and plasmids expressing NS3 (WT, M21T mutant, or vector control). As shown in Fig. 2F, WT and M21T mutant NS3 displayed similar capabilities in inhibiting the poly(I·C)-induced interferon response. Taken together, our results described above clearly demonstrated that M21T mutation did not affect the capability of NS3 to cleave MAVS or to suppress interferon signaling.
As the N terminus of NS3 is a cofactor of the NS2 autoprotease, which carries out the autocleavage of NS2 and NS3, we next assessed the impact of M21T mutation on the cleavage between NS2 and NS3. Plasmids expressing hemagglutinin (HA)-tagged NS2 fused with NS3 of the WT, the M21T mutant, or an NS2-3 cleavage-defective C184A mutant (13, 22) were transfected into Huh7.5.1 cells, and the mature NS2 and NS3 protein levels were analyzed by Western blotting. As shown in Fig. 2G, there was no significant difference of mature NS2 and NS3 protein levels between the WT and M21T mutant, suggesting that the M21T mutation did not affect the cleavage of NS2 and NS3.
The M21T mutation had no apparent effects on HCV genome replication.
Next, we examined which step of the HCV life cycle the M21T mutation would affect. We first determined whether the M21T mutation altered HCV RNA replication using the JFH1 subgenomic replicon, a bicistronic subgenomic replicon that expresses firefly luciferase (16). In vitro-transcribed WT, M21T mutant, or GND mutant subgenomic replicon RNAs were electroporated into Huh7 cells, and the luciferase activity was measured at 4, 24, 48, and 96 h postelectroporation. As shown in Fig. 3A, the luciferase activities of both the WT and M21T mutant increased about 26-fold, reaching the peak at 48 h postelectroporation, indicating similar replication capacities between WT and M21T mutant replicons.
FIG 3.
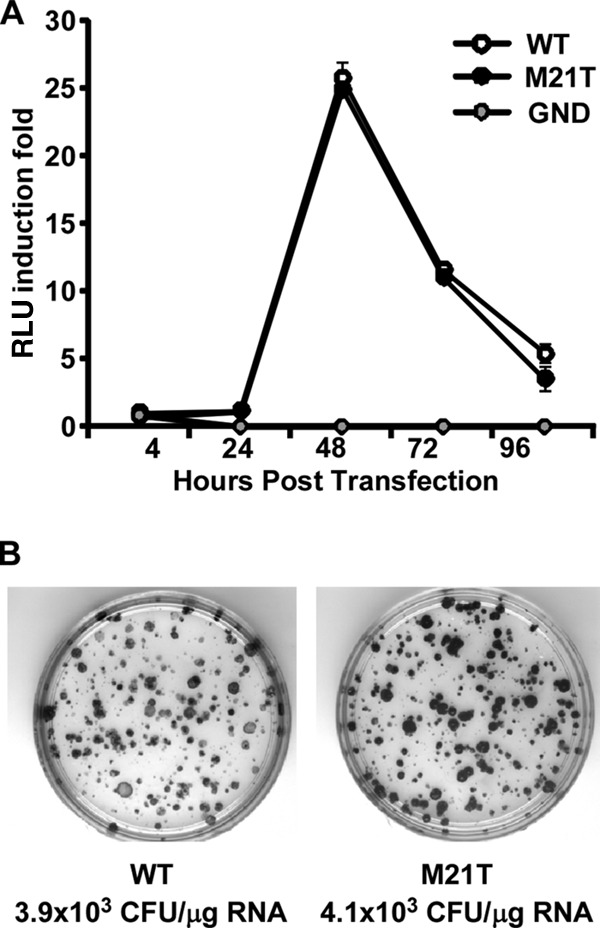
The M21T mutation had no effects on viral genome replication. (A) HCV RNA levels in a transient-transfection assay. HCV subgenomic replicon RNA (WT, M21T mutant, or GND mutant) were electroporated into Huh7 cells and luciferase activities were measured at the indicated time points. Luciferase activities are expressed as the fold change of relative light unit (RLU) values at different time points relative to the RLU value at 4 h posttransfection. Error bars indicate standard deviations calculated from 3 independent experiments. (B) HCV replicon colony formation assay. HCV subgenomic replicon RNAs containing WT or M21T NS3 were electroporated into Huh7 cells. After 3 weeks of G418 selection, the surviving cells were stained with crystal violet.
In addition, we examined the effect of the M21T mutation on viral genome replication using a replicon colony formation assay. The in vitro-transcribed WT or M21T mutant subgenomic replicon RNAs were electroporated into Huh7 cells, followed by 3-week G418 selection. As shown in Fig. 3B, consistent with the transient-transfection assay, the colony forming efficiencies of the WT and M21T mutant were also comparable. All these results demonstrated that the M21T mutation did not affect HCV genome replication.
The M21T mutation significantly enhanced viral assembly.
Next, we examined the effect of the M21T mutation on HCV virion assembly. In vitro-transcribed full-length JFH1 RNAs containing WT or M21T mutant NS3 sequences were electroporated into Huh7.5.1 cells, and the intracellular HCV RNA levels and extracellular and intracellular infectivity titers were measured. As shown in Fig. 4A, while the intracellular HCV RNA levels were comparable between the WT and M21T mutant at day 2 postelectroporation, the M21T mutant had a higher HCV RNA level than the WT at day 3, likely due to reinfection of produced progeny viruses. Consistently, the extracellular viral titers of the M21T mutant were more than 1 log higher than those of the WT at days 2 and 3 postelectroporation (Fig. 4B). Importantly, the intracellular viral titers of the M21T mutant were also significantly higher than those of the WT (Fig. 4C), suggesting that the increased virus production of the M21T mutant was likely due to the enhanced virion assembly in the cells.
FIG 4.
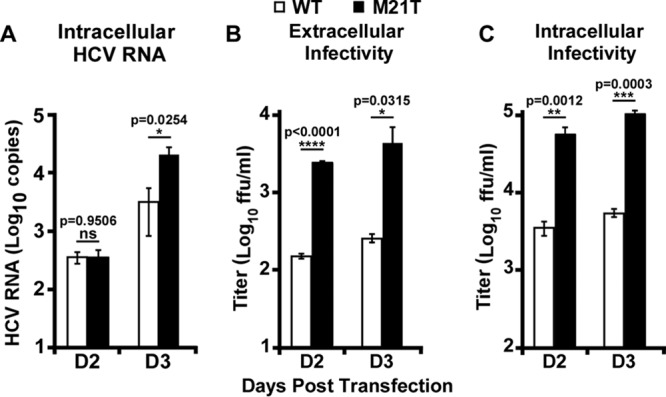
M21T significantly enhanced viral assembly efficiency. Full-length JFH1 RNAs containing WT or M21T NS3 were electroporated into Huh7.5.1 cells, and the cells were harvested at day 2 and day 3 postelectroporation. (A) Intracellular HCV RNA levels were analyzed by quantitative reverse transcription-PCR (RT-qPCR). Extracellular (B) and intracellular (C) infectivities were determined by titration assay. Error bars indicate standard deviations calculated from 3 independent experiments.
The M21T mutation altered the localization of core proteins from LD to the ER.
The nascent HCV RNA genomes are encapsidated by core proteins on lipid droplets (LD), and the resulting nucleocapsids are then translocated to the ER for budding. Previous work showed that HCV virion assembly rate was linked to the subcellular localization of the core (6, 23, 40). The Jc1 strain, whose core proteins are located on the ER, has more efficient virion assembly than the JFH1 strain, whose core proteins are mostly located on LD. We speculated that the M21T mutation may accelerate virus assembly by altering the core subcellular localization. In order to test this hypothesis, we analyzed the subcellular localization of core proteins in Huh7.5 cells infected with the JFH1 virus with WT or M21T mutant NS3. Huh7.5 cells infected with the Jc1 virus were also analyzed as a control. The results of colocalization of core and LD as well as core and ER are presented in Fig. 5A and B, respectively. The percentages of the cells with different core subcellular localization patterns (exclusively on either LD or the ER, or found on both organelles) are summarized in Fig. 5C. Consistent with previous observations (6, 23, 40), the core proteins of wild-type JFH1 were largely coating LD (75% on LD, 5% on the ER, and 20% on LD and the ER), whereas the majority of the cells infected with the M21T mutant had core proteins either on the ER or on both the ER and LD (5% on LD, 30% on the ER, and 65% on LD and the ER), which was similar to the case with Jc1 (3% on LD, 71% on the ER, and 26% on LD and the ER). These results demonstrated that the M21T mutation altered the subcellular localization of core from LD to the ER for successive virion assembly processes.
FIG 5.

The M21T mutation altered the subcellular localization of HCV core protein from LD to the ER. Confocal imaging of core and LD colocalization. Huh7.5 cells were infected with WT-JFH1, M21T-JFH1, or WT-Jc1 virus at a multiplicity of infection (MOI) of 0.2 for 72 h and subjected to immunofluorescence staining of core (red) and lipid droplets (LD) (green) (A) and immunofluorescence staining of core (red) and calnexin (CNX) (green) (B). (C) Core localization pattern analysis. The LD localization was scored when core proteins were found only at LD surface without any residual reticular pattern, while the ER localization was scored when core staining was found as a reticular pattern with less than 10 LD fully covered by core. The ER/LD localization was scored when core was present as a reticular pattern and with at least 10 LD fully covered by core. For each virus, at least 30 HCV-positive cells were analyzed in 3 different experiments.
The M21T mutation promoted envelopment of viral nucleocapsids.
HCV nucleocapsids bud on the ER membrane, during which viral envelope glycoproteins E1 and E2 are acquired and viral envelopes are formed. Therefore, we determined whether the M21T mutation promoted the budding of HCV nucleocapsids. To do so, we first examined the colocalization of core and envelope proteins by immunofluorescence analysis. Representative images of core and E2 immunofluorescence in the cells transfected with the full-length JFH1 RNA containing WT or M21T mutant NS3 are shown in Fig. 6A, and the merged images as well as the pixel dot plots with x and y axes representing E2 (red)- and core (green)-positive signals are also included (right images in Fig. 6A). Pearson's coefficient was used to reflect the extent of dot regression (24), which represents the correlation of the core- and E2-positive signals and thus colocalization of the two proteins. Statistical analysis of 25 randomly selected images is presented in Fig. 6B. Our results showed that the M21T mutant had a significantly higher Pearson's coefficient, suggesting a better colocalization of core and E2.
FIG 6.
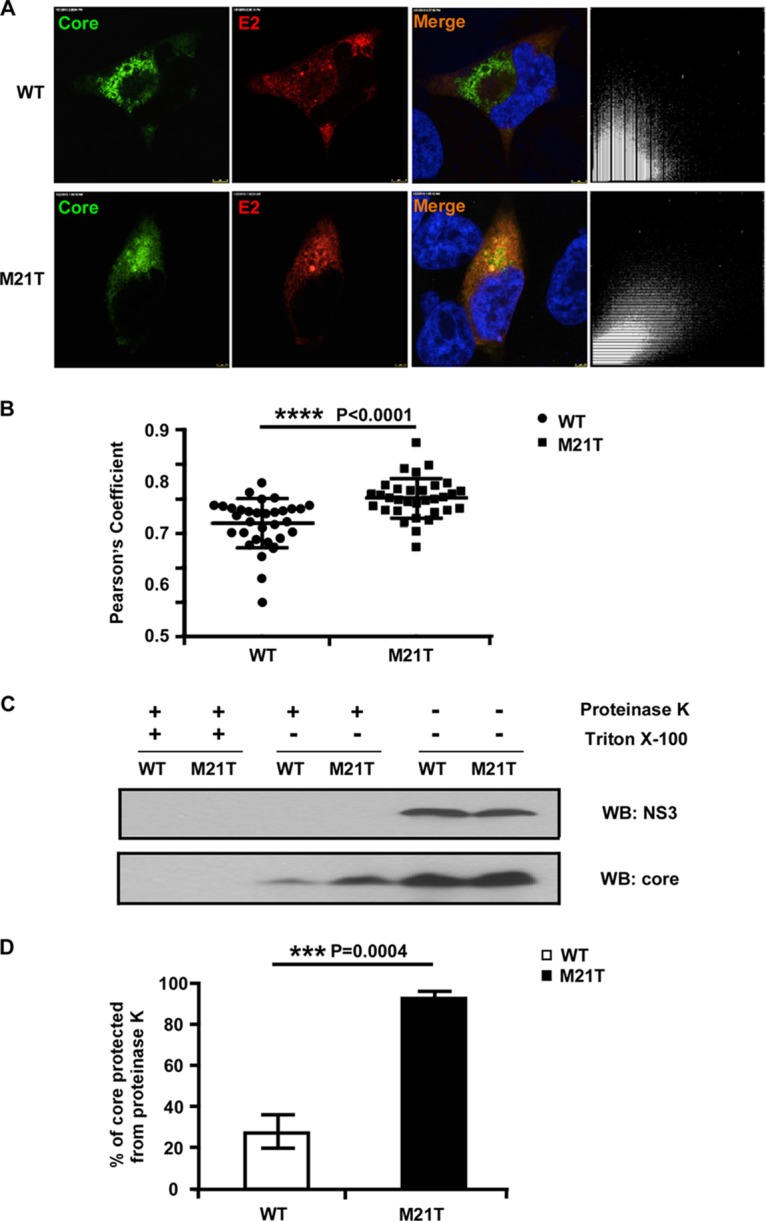
The M21T mutation promoted envelopment of viral nucleocapsid. (A) Confocal immunofluorescence analysis of core and E2 colocalization. Huh7.5.1 cells were electroporated with full-length JFH1 RNA containing WT or M21T NS3 for 3 days and subjected to immunofluorescence staining of core (green) and E2 (red). The colocalization of core and E2 was analyzed in pixel dot plots on the right, with the x axis representing red signal intensity (E2) and the y axis representing green signal (core). (B) Statistical analysis of core and E2 colocalization. Pearson's coefficient, the indicator of colocalization, was determined by regression of the pixel dot plots. Twenty-five individual images were analyzed for each group. (C) Proteinase K digestion protection assay. Huh7.5.1 cells were electroporated with full-length JFH1 RNA containing WT or M21T NS3 for 2 days. Cell lysates were treated with or without detergent (Triton X-100) followed by proteinase K digestion. Digested cell lysates were then analyzed by Western blotting to determine NS3 and core protein levels. (D) Statistical analysis of the percentage of core protected from the proteinase K digestion. Core signals were quantified by ImageJ, and the percentages of protected core were calculated as the ratio of core in the digested group and the undigested control. Error bars indicate standard deviations calculated from 3 independent experiments.
Next, a proteinase K digestion assay was employed to evaluate the envelopment of viral nucleocapsids. The intracellular virions of the WT and M21T mutant were subjected to proteinase K digestion in the presence or absence of detergent treatment that removes the lipid envelope and exposes the naked nucleocapsids to the proteinase K digestion. As shown in Fig. 6C, the mock-treated WT and M21T mutant had comparable levels of core and NS3 proteins, indicating similar transfection and viral replication efficiencies between the WT and M21T mutant. The proteinase K digestion without the detergent pretreatment completely degraded NS3 but not core proteins. However, the residual core proteins were completely degraded if the detergent pretreatment was applied before the proteinase K digestion. These results were consistent with the notion that the core proteins in enveloped nucleocapsids, but not NS3, are protected from the proteinase digestion. Interestingly, significantly more core proteins were protected from the proteinase digestion in the M21T mutant than in the WT (Fig. 6D), suggesting that more nucleocapsids acquired lipid envelope. Together, these results suggested that the M21T mutation altered core localization from LD to the ER and then promoted nucleocapsid budding and envelopment.
The M21T mutation did not enhance the production of Jc1 virus.
Next, we examined the effect of the M21T mutation on virus production of the Jc1 genome. In vitro-transcribed full-length JFH1 and Jc1 RNAs containing the WT, the M21T NS3 mutant, or the NS5B polymerase-defective GND mutant were electroporated into Huh7.5.1 cells. HCV E2 immunofluorescence and intracellular RNA quantification at day 2 postelectroporation were conducted to monitor the electroporation and virus replication efficiency, and the supernatant viral infectivity titers at day 2, 4, 6, and 8 postelectroporation were measured to monitor the virus production efficiency. As shown in Fig. 7A and B, there were similar levels of virus replication between the WT and M21T mutant for both JFH1 and Jc1. Interestingly, while the M21T mutant significantly increased the virus production of JFH1, it had no effect on the virus production of Jc1 (Fig. 7C).
FIG 7.
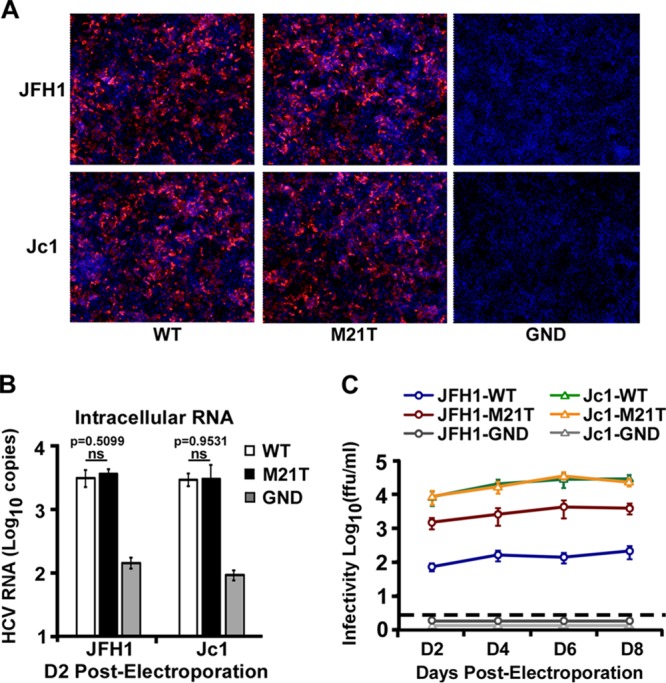
The M21T mutation did not enhance production of Jc1 virus. Huh7.5.1 cells were electroporated with in vitro-transcribed WT, M21T mutant, or GND mutant JFH1 or Jc1 RNA. Transfected cells were analyzed for HCV E2 immunofluorescence at day 2 postelectroporation (A) and intracellular HCV RNA levels (B), and supernatant infectivity titers were determined at different time points after electroporation (C). Error bars indicate standard deviations calculated from 3 independent experiments.
DISCUSSION
HCV assembly is a complicated process that has not been thoroughly deciphered. Several viral and cellular factors have been defined to be involved in the process of assembly. NS3 participates in HCV assembly, but the precise molecular mechanism remains elusive. In this study, we identified a single amino acid mutation, M21T, located in helix α0 of NS3 that could promote viral production (Fig. 1). Much to our surprise, despite the fact that this mutation resides in helix α0, the structural determinant of NS3's membrane anchorage and protein stability, it did not affect the membrane association of NS3 (Fig. 2A). Since the M21T mutation is in close proximity to NS3's protease domain, we also examined its potential effect on the protease activity of NS3. By using artificial and natural protease substrates, we found that this mutation had no effect on the protease activity (Fig. 2B to F). Finally, we ruled out the potential cis effect of this mutation on the NS2-NS3 cleavage (Fig. 2G). Consistent with these observations, the M21T mutation did not promote viral genome replication (Fig. 3). Instead, it increased the intracellular infectivity titers (Fig. 4), suggesting that this mutation enhanced virus assembly. Immunofluorescence and biochemical analyses demonstrated that the M21T mutation promoted the relocation of core proteins from LD to the ER in the cells (Fig. 5) and ultimately enhanced virion envelopment and budding (Fig. 6). The mutation had no effect on the virus production of Jc1 (Fig. 7), likely because Jc1 virus has a high percentage of ER-associated core proteins (Fig. 5), rendering its highly efficient virion assembly not being further increased by the M21T mutation.
The amino acid sequence at the 10 to 24 first residues of NS3 is highly conserved among different genotypes. Analysis of about 5,000 HCV sequences from the European Hepatitis C Virus Database (http://euhcvdb.ibcp.fr) (25) demonstrated that the vast majority of HCV sequences have leucine or methionine at this residue, and only 5 prolines, 2 phenylalanines, and 1 histidine, but no threonine, were identified at this position of NS3. The failure to find threonine at residue 21 of NS3 in natural HCV isolates could possibly be explained by the highly strain-specific effect of the M21T mutation on HCV production, or by potential additional adverse effects of this mutation on overall virus fitness in vivo which do not affect virus propagation in vitro, for example, induction of host immune responses by shedding virus.
Several other adaptive mutations in NS3 have also been reported to enhance HCV assembly. A single mutation, Q221L, within subdomain I of the NS3 helicase could rescue a defect in infectious particle production of HJ3, a chimeric HCV genome comprised of H77 core-NS2 and JFH1 NS2-NS5B (26). The Q221L mutation had no effect on viral RNA replication or NS3-associated enzymatic activities but enhanced virus assembly. Furthermore, subdomains I and II of the NS3 helicase seemed to work together to promote HCV assembly because combination of an adaptive mutation, Q221N or I286V, in subdomain I and a second mutation, I399V, in subdomain II rescued virus production (26). Another study found that an adaptive mutation, K272R, in NS3 helicase could rescue virus production of defective HCVcc that has mutations in core protein (27). Recently, Kohlway and colleagues suggested that amino acids 171 to 194, located in a linker between the helicase and protease domains, also participate in HCV assembly (28). Interestingly, our study identified an adaptive mutation within the N-terminal helix α0 domain of NS3 that significantly enhances virus assembly. To our knowledge, it is the first NS3 mutation outside the helicase domain that enhances HCV assembly.
Despite the fact that many adaptive mutations in NS3 have been identified to enhance virus assembly, the underlying molecular mechanisms are largely unknown. It has been proposed that NS3 may work with other viral proteins, for example, core and NS2, to promote nucelocapsid formation. Genetic interactions of NS3 and NS2 (8, 29, 30) or NS3 and core (27) were implied to contribute to HCV assembly. Besides, the physical protein-protein interactions of NS3 with core or NS3 with NS2 have been demonstrated to be critical for HCV production (31, 32). However, our results showed that the M21T mutation did not alter NS3's interactions with NS2 or core compared to those of the WT (data not shown). This raises a possibility that the M21T mutation may exert its impact on HCV assembly in a manner independent of the interaction between NS3 and NS2 or core. Although the mutation did not change the ability of helix α0 to anchor NS3 to intracellular membrane, it may still cause a subtle or transient alteration in the subcellular localization of NS3 during the process of HCV assembly, and this change may somehow contribute to more efficient virus assembly. More investigations in this direction are certainly warranted.
MATERIALS AND METHODS
Plasmids and antibodies.
pUC-JFH1-WT, pUC-Jc1-WT, pSGR-JFH1-WT, pET21b-JFH1-NS3-WT, and pCMV-NS310-24-GFP-WT were constructed as previously described (16). Plasmid pNS3-5B-3Flag-WT was constructed by PCR amplification of an NS3-5B fragment from pUC-JFH1 and cloned into the EcoRI and XbaI sites of p3XFLAG-CMV-14 (Sigma, St. Louis, MO). Plasmid pNS3-4A-WT was constructed by PCR amplification of the NS3-4A fragment from pUC-JFH1 and cloned into the HindIII and XbaI sites of pcDNA3.1. All point mutations were engineered using the QuikChange site-directed mutagenesis kit (Stratagene, Santa Clara, CA) and verified by sequencing. Mouse monoclonal antibodies against HCV E2 (1C9 and 1B4), core, and NS3 were raised in our lab. Other antibodies used in this study include mouse monoclonal anti-Flag (M2; Sigma), anti-HA (F7; Santa Cruz Biotechnology Inc., Santa Cruz, CA), anti-actin (A5441; Sigma), anti-ADRP (AP125; Progen Biotechnik, Heidelberg, Germany), rabbit polyclonal anti-calnexin (C4731; Sigma), and human monoclonal anti-E2 antibody (AR3A, provided by Denis Burton, The Scripps Research Institute, USA) (33).
Cell culture and plasmid transfection.
The cell culture conditions were described previously (17, 34). Plasmid DNA transfection into Huh7 or Huh7.5.1 cells was performed with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions.
In vitro transcription, HCV RNA transfection, indirect immunofluorescence, titration analysis, HCV transfection kinetics assay, and Western blotting.
In vitro transcription, HCV RNA transfection, indirect immunofluorescence, titration analysis, HCV transfection kinetics assay, and Western blotting were described previously (17, 35).
NS3 protease activity assay.
The NS3 protease activity was measured by fluorescent resonance energy transfer (FRET) assay using the SensoLyte490 HCV protease assay kit (AnaSpec, San Jose, CA). Briefly, 5-pmol quantities of purified recombinant NS3 proteins (WT and the M21T and H57A mutants) (16), with or without a 10-fold excess of NS4A cofactor peptide, were mixed with the FRET substrate in 1× assay buffer for 1 h. The fluorescent signals were measured using a SpectraMax M2 microplate reader (Molecular Devices, Sunnyvale, CA).
IFN-β luciferase reporter assay.
The protocol for the IFN-β luciferase reporter assay was described previously (36–38). Briefly, Huh7 cells seeded in 48-well plates were transfected with 15 ng of IFN-β promoter luciferase reporter plasmid pIFΔ(−125)lucter, 15 ng of Renilla luciferase-expressing plasmid pRL-Tk (Promega, Madison, WI) used to normalize transfection, and 50 ng of pNS3-4A-WT/M21T or the empty vector control using the Exgen 500 transfection reagent (Fermentas, Hanover, MD) for 36 h, followed by another transfection with 0.3 μg of poly(I·C) using the Lipofectamine 2000 transfection reagent (Invitrogen) for additional 16 h. The cell lysates were collected, and luciferase activities were measured using the dual-luciferase reporter assay system (Promega) according to the manufacturer's protocol.
HCV replicon transient-transfection and colony formation assay.
Five micrograms of HCV subgenomic replicon RNA (WT or M21T mutant) was electroporated into Huh7 cells, and the luciferase activities were measured at the desired time points. For the colony formation assay, Huh7 cells electroporated with the subgenomic RNA were seeded into 10-cm dishes at a density of 4 × 106 per dish. The cells were maintained in culture medium containing 3 μg/ml of G418 at day 3 postelectroporation and thereafter. After 3 weeks of antibiotic selection, the cells were stained with 1.25% crystal violet in 20% ethanol for 5 min for visualizing the surviving cell colonies.
Immunostaining and confocal microscopy.
Immunostaining and confocal microscopy were performed as previously described (6). Cells were seeded on 14-mm-diameter glass coverslips prior to infection. The cells were washed with phosphate-buffered saline (PBS), fixed with 3% of paraformaldehyde in PBS for 15 min, quenched with 50 mM NH4Cl, and permeabilized with 0.1% Triton X-100 for 7 min. Subsequently, the cells were incubated with the primary antibody in 1% bovine serum albumin (BSA)–PBS for 1 h, washed, and stained for 1 h with the secondary antibodies conjugated with ATTO-488 (Rockland Immunochemicals Inc., Gilbertsville, PA) or Alexa 555 (Molecular Probes Europe BV, Leiden, The Netherlands). The LD staining was performed using a specific cellular tracer of neutral lipids, Bodipy 493/503 (Molecular Probes Europe BV). The cells were washed with PBS and mounted in Mowiol 40-88 (Fluka, Buchs, Switzerland) prior to image acquisition with an LSM 510 confocal microscope (Carl Zeiss Inc., Thornwood, NY). The images were analyzed using ImageJ software. The core subcellular localizations were quantified as previously described (6). Briefly, the LD localization was scored when core proteins were found only at the LD surface without any residual reticular pattern, while the ER localization was scored when core staining was found as a reticular pattern with less than 10 LD fully covered by core. The ER/LD localization was scored when core was present as a reticular pattern and with at least 10 LD fully covered by core.
Intracellular viral particle preparation.
The infected or transfected cells were harvested and resuspended in Dulbecco modified Eagle medium (DMEM). The cells were then subjected to 5 quick freeze-thaw cycles, and the cell debris was removed by centrifugation at 10,000 rpm for 10 min.
Proteinase K digestion protection assay.
The proteinase K digestion protection assay was performed as previously described (39). The full-length JFH1 RNA containing the WT or M21T NS3 was electroporated into Huh7.5.1 cells. The cells were collected at day 2 postelectroporation and subjected to 5 freeze-thaw cycles. The cell lysates were obtained after centrifugation and divided into three groups: untreated, treated with 50 μg/μl of proteinase K (Sangon, Shanghai, China) on ice for 1 h, and pretreated with 5% (vol/vol) Triton X-100 before proteinase K treatment. Proteinase K was then inactivated with 5 mM phenylmethylsulfonyl fluoride (PMSF) on ice for 10 min. The amount of core protein was determined by Western blotting.
ACKNOWLEDGMENTS
We thank Wanyin Tao, Zhenliang He, and Qingcao Li (Institut Pasteur of Shanghai, CAS) for technical assistance.
We declare no conflict of interest.
This study was supported by grants from the National Natural Science Foundation of China (31270203) and the Chinese National 973 Program (2015CB554300) to J.Z. and grants from the French Agence Nationale de la Recherche sur le SIDA et les hépatites virales (ANRS), the European Research Council (ERC-2008-AdG-233130-HEPCENT), and the LabEx Ecofect (ANR-11-LABX-0048) to B.B. and F.-L.C.
REFERENCES
- 1.Simmonds P, Bukh J, Combet C, Deleage G, Enomoto N, Feinstone S, Halfon P, Inchauspe G, Kuiken C, Maertens G, Mizokami M, Murphy DG, Okamoto H, Pawlotsky JM, Penin F, Sablon E, Shin IT, Stuyver LJ, Thiel HJ, Viazov S, Weiner AJ, Widell A. 2005. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology 42:962–973. doi: 10.1002/hep.20819. [DOI] [PubMed] [Google Scholar]
- 2.Liang TJ, Ghany MG. 2013. Current and future therapies for hepatitis C virus infection. N Engl J Med 368:1907–1917. doi: 10.1056/NEJMra1213651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li DP, Huang Z, Zhong J. 2015. Hepatitis C virus vaccine development: old challenges and new opportunities. Nat Sci Rev 2:285–295. doi: 10.1093/nsr/nwv040. [DOI] [Google Scholar]
- 4.Masaki T, Suzuki R, Murakami K, Aizaki H, Ishii K, Murayama A, Date T, Matsuura Y, Miyamura T, Wakita T, Suzuki T. 2008. Interaction of hepatitis C virus nonstructural protein 5A with core protein is critical for the production of infectious virus particles. J Virol 82:7964–7976. doi: 10.1128/JVI.00826-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McLauchlan J, Lemberg MK, Hope G, Martoglio B. 2002. Intramembrane proteolysis promotes trafficking of hepatitis C virus core protein to lipid droplets. EMBO J 21:3980–3988. doi: 10.1093/emboj/cdf414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boson B, Granio O, Bartenschlager R, Cosset FL. 2011. A concerted action of hepatitis C virus p7 and nonstructural protein 2 regulates core localization at the endoplasmic reticulum and virus assembly. PLoS Pathog 7:e1002144. doi: 10.1371/journal.ppat.1002144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jirasko V, Montserret R, Lee JY, Gouttenoire J, Moradpour D, Penin F, Bartenschlager R. 2010. Structural and functional studies of nonstructural protein 2 of the hepatitis C virus reveal its key role as organizer of virion assembly. PLoS Pathog 6:e1001233. doi: 10.1371/journal.ppat.1001233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma Y, Anantpadma M, Timpe JM, Shanmugam S, Singh SM, Lemon SM, Yi M. 2011. Hepatitis C virus NS2 protein serves as a scaffold for virus assembly by interacting with both structural and nonstructural proteins. J Virol 85:86–97. doi: 10.1128/JVI.01070-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.André P, Perlemuter G, Budkowska A, Brechot C, Lotteau V. 2005. Hepatitis C virus particles and lipoprotein metabolism. Semin Liver Dis 25:93–104. doi: 10.1055/s-2005-864785. [DOI] [PubMed] [Google Scholar]
- 10.Nielsen SU, Bassendine MF, Burt AD, Martin C, Pumeechockchai W, Toms GL. 2006. Association between hepatitis C virus and very-low-density lipoprotein (VLDL)/LDL analyzed in iodixanol density gradients. J Virol 80:2418–2428. doi: 10.1128/JVI.80.5.2418-2428.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bartenschlager R, Ahlborn-Laake L, Mous J, Jacobsen H. 1993. Nonstructural protein 3 of the hepatitis C virus encodes a serine-type proteinase required for cleavage at the NS3/4 and NS4/5 junctions. J Virol 67:3835–3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eckart MR, Selby M, Masiarz F, Lee C, Berger K, Crawford K, Kuo C, Kuo G, Houghton M, Choo QL. 1993. The hepatitis C virus encodes a serine protease involved in processing of the putative nonstructural proteins from the viral polyprotein precursor. Biochem Biophys Res Commun 192:399–406. doi: 10.1006/bbrc.1993.1429. [DOI] [PubMed] [Google Scholar]
- 13.Grakoui A, McCourt DW, Wychowski C, Feinstone SM, Rice CM. 1993. A second hepatitis C virus-encoded proteinase. Proc Natl Acad Sci U S A 90:10583–10587. doi: 10.1073/pnas.90.22.10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim DW, Gwack Y, Han JH, Choe J. 1995. C-terminal domain of the hepatitis C virus NS3 protein contains an RNA helicase activity. Biochem Biophys Res Commun 215:160–166. doi: 10.1006/bbrc.1995.2447. [DOI] [PubMed] [Google Scholar]
- 15.Brass V, Berke JM, Montserret R, Blum HE, Penin F, Moradpour D. 2008. Structural determinants for membrane association and dynamic organization of the hepatitis C virus NS3-4A complex. Proc Natl Acad Sci U S A 105:14545–14550. doi: 10.1073/pnas.0807298105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He Y, Weng L, Li R, Li L, Toyoda T, Zhong J. 2012. The N-terminal helix alpha(0) of hepatitis C virus NS3 protein dictates the subcellular localization and stability of NS3/NS4A complex. Virology 422:214–223. doi: 10.1016/j.virol.2011.10.021. [DOI] [PubMed] [Google Scholar]
- 17.Zhong J, Gastaminza P, Chung J, Stamataki Z, Isogawa M, Cheng G, McKeating JA, Chisari FV. 2006. Persistent hepatitis C virus infection in vitro: coevolution of virus and host. J Virol 80:11082–11093. doi: 10.1128/JVI.01307-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horner SM, Park HS, Gale M Jr. 2012. Control of innate immune signaling and membrane targeting by the hepatitis C virus NS3/4A protease are governed by the NS3 helix alpha0. J Virol 86:3112–3120. doi: 10.1128/JVI.06727-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yon C, Viswanathan P, Rossignol JF, Korba B. 2011. Mutations in HCV non-structural genes do not contribute to resistance to nitazoxanide in replicon-containing cells. Antiviral Res 91:233–240. doi: 10.1016/j.antiviral.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grakoui A, McCourt DW, Wychowski C, Feinstone SM, Rice CM. 1993. Characterization of the hepatitis C virus-encoded serine proteinase: determination of proteinase-dependent polyprotein cleavage sites. J Virol 67:2832–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li XD, Sun L, Seth RB, Pineda G, Chen ZJ. 2005. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci U S A 102:17717–17722. doi: 10.1073/pnas.0508531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hijikata M, Mizushima H, Akagi T, Mori S, Kakiuchi N, Kato N, Tanaka T, Kimura K, Shimotohno K. 1993. Two distinct proteinase activities required for the processing of a putative nonstructural precursor protein of hepatitis C virus. J Virol 67:4665–4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shavinskaya A, Boulant S, Penin F, McLauchlan J, Bartenschlager R. 2007. The lipid droplet binding domain of hepatitis C virus core protein is a major determinant for efficient virus assembly. J Biol Chem 282:37158–37169. doi: 10.1074/jbc.M707329200. [DOI] [PubMed] [Google Scholar]
- 24.Bolte S, Cordelieres FP. 2006. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 25.Combet C, Garnier N, Charavay C, Grando D, Crisan D, Lopez J, Dehne-Garcia A, Geourjon C, Bettler E, Hulo C, Le Mercier P, Bartenschlager R, Diepolder H, Moradpour D, Pawlotsky JM, Rice CM, Trepo C, Penin F, Deleage G. 2007. euHCVdb: the European hepatitis C virus database. Nucleic Acids Res 35:D363–D366. doi: 10.1093/nar/gkl970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma Y, Yates J, Liang Y, Lemon SM, Yi M. 2008. NS3 helicase domains involved in infectious intracellular hepatitis C virus particle assembly. J Virol 82:7624–7639. doi: 10.1128/JVI.00724-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones DM, Atoom AM, Zhang X, Kottilil S, Russell RS. 2011. A genetic interaction between the core and NS3 proteins of hepatitis C virus is essential for production of infectious virus. J Virol 85:12351–12361. doi: 10.1128/JVI.05313-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kohlway A, Pirakitikulr N, Ding SC, Yang F, Luo D, Lindenbach BD, Pyle AM. 2014. The linker region of NS3 plays a critical role in the replication and infectivity of hepatitis C virus. J Virol 88:10970–10974. doi: 10.1128/JVI.00745-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Counihan NA, Rawlinson SM, Lindenbach BD. 2011. Trafficking of hepatitis C virus core protein during virus particle assembly. PLoS Pathog 7:e1002302. doi: 10.1371/journal.ppat.1002302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Phan T, Beran RK, Peters C, Lorenz IC, Lindenbach BD. 2009. Hepatitis C virus NS2 protein contributes to virus particle assembly via opposing epistatic interactions with the E1-E2 glycoprotein and NS3-NS4A enzyme complexes. J Virol 83:8379–8395. doi: 10.1128/JVI.00891-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang J, Luo G. 2012. Cell culture-adaptive mutations promote viral protein-protein interactions and morphogenesis of infectious hepatitis C virus. J Virol 86:8987–8997. doi: 10.1128/JVI.00004-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stapleford KA, Lindenbach BD. 2011. Hepatitis C virus NS2 coordinates virus particle assembly through physical interactions with the E1-E2 glycoprotein and NS3-NS4A enzyme complexes. J Virol 85:1706–1717. doi: 10.1128/JVI.02268-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gastaminza P, Kapadia SB, Chisari FV. 2006. Differential biophysical properties of infectious intracellular and secreted hepatitis C virus particles. J Virol 80:11074–11081. doi: 10.1128/JVI.01150-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A 102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li R, Qin Y, He Y, Tao W, Zhang N, Tsai C, Zhou P, Zhong J. 2011. Production of hepatitis C virus lacking the envelope-encoding genes for single-cycle infection by providing homologous envelope proteins or vesicular stomatitis virus glycoproteins in trans. J Virol 85:2138–2147. doi: 10.1128/JVI.02313-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ding Q, Cao XZ, Lu J, Huang B, Liu YJ, Kato N, Shu HB, Zhong J. 2013. Hepatitis C virus NS4B blocks the interaction of STING and TBK1 to evade host innate immunity. J Hepatol 59:52–58. doi: 10.1016/j.jhep.2013.03.019. [DOI] [PubMed] [Google Scholar]
- 37.Cheng G, Zhong J, Chisari FV. 2006. Inhibition of dsRNA-induced signaling in hepatitis C virus-infected cells by NS3 protease-dependent and -independent mechanisms. Proc Natl Acad Sci U S A 103:8499–8504. doi: 10.1073/pnas.0602957103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cao XZ, Ding Q, Lu J, Tao WY, Huang B, Zhao YN, Niu JQ, Liu YJ, Zhong J. 2015. MDA5 plays a critical role in interferon response during hepatitis C virus infection. J Hepatol 62:771–778. doi: 10.1016/j.jhep.2014.11.007. [DOI] [PubMed] [Google Scholar]
- 39.Gentzsch J, Brohm C, Steinmann E, Friesland M, Menzel N, Vieyres G, Perin PM, Frentzen A, Kaderali L, Pietschmann T. 2013. Hepatitis C virus p7 is critical for capsid assembly and envelopment. PLoS Pathog 9:e1003355. doi: 10.1371/journal.ppat.1003355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boson B, Denolly S, Turlure F, Chamot C, Dreux M, Cosset FL. 2016. Daclatasvir prevents hepatitis C virus by blocking transfer of viral genome to assembly sites. Gastroenterology doi: 10.1053/j.gastro.2016.11.047. [DOI] [PubMed] [Google Scholar]