

ABSTRACT
In five experimentally characterized arterivirus species, the 5′-end genome coding region encodes the most divergent nonstructural proteins (nsp's), nsp1 and nsp2, which include papain-like proteases (PLPs) and other poorly characterized domains. These are involved in regulation of transcription, polyprotein processing, and virus-host interaction. Here we present results of a bioinformatics analysis of this region of 14 arterivirus species, including that of the most distantly related virus, wobbly possum disease virus (WPDV), determined by a modified 5′ rapid amplification of cDNA ends (RACE) protocol. By combining profile-profile comparisons and phylogeny reconstruction, we identified an association of the four distinct domain layouts of nsp1-nsp2 with major phylogenetic lineages, implicating domain gain, including duplication, and loss in the early nsp1 evolution. Specifically, WPDV encodes highly divergent homologs of PLP1a, PLP1b, PLP1c, and PLP2, with PLP1a lacking the catalytic Cys residue, but does not encode nsp1 Zn finger (ZnF) and “nuclease” domains, which are conserved in other arteriviruses. Unexpectedly, our analysis revealed that the only catalytically active nsp1 PLP of equine arteritis virus (EAV), known as PLP1b, is most similar to PLP1c and thus is likely to be a PLP1b paralog. In all non-WPDV arteriviruses, PLP1b/c and PLP1a show contrasting patterns of conservation, with the N- and C-terminal subdomains, respectively, being enriched with conserved residues, which is indicative of different functional specializations. The least conserved domain of nsp2, the hypervariable region (HVR), has its size varied 5-fold and includes up to four copies of a novel PxPxPR motif that is potentially recognized by SH3 domain-containing proteins. Apparently, only EAV lacks the signal that directs −2 ribosomal frameshifting in the nsp2 coding region.
IMPORTANCE Arteriviruses comprise a family of mammalian enveloped positive-strand RNA viruses that include some of the most economically important pathogens of swine. Most of our knowledge about this family has been obtained through characterization of viruses from five species: Equine arteritis virus, Simian hemorrhagic fever virus, Lactate dehydrogenase-elevating virus, Porcine respiratory and reproductive syndrome virus 1, and Porcine respiratory and reproductive syndrome virus 2. Here we present the results of comparative genomics analyses of viruses from all known 14 arterivirus species, including the most distantly related virus, WPDV, whose genome sequence was completed in this study. Our analysis focused on the multifunctional 5′-end genome coding region that encodes multidomain nonstructural proteins 1 and 2. Using diverse bioinformatics techniques, we identified many patterns of evolutionary conservation that are specific to members of distinct arterivirus species, both characterized and novel, or their groups. They are likely associated with structural and functional determinants important for virus replication and virus-host interaction.
KEYWORDS: arterivirus, bioinformatics, comparative genomics, duplication, evolution, nidovirus, papain-like proteases, phylogenetic analysis, ribosome frameshifting, wobbly possum disease
INTRODUCTION
Arteriviruses are a family of enveloped nonsegmented positive-strand RNA viruses of mammals that belongs to the order Nidovirales (1, 2). The arterivirus genetic diversity was recently classified into 14 species (3), five of which include relatively well-characterized viruses, i.e., equine arteritis virus (EAV) (4, 5), lactate dehydrogenase-elevating virus (LDV) (6, 7), simian hemorrhagic fever virus (SHFV) (8), porcine respiratory and reproductive syndrome virus 1 (PRRSV-1) (9), and PRRSV-2 (10). Among the newly identified viruses is wobbly possum disease virus (WPDV), a marsupial virus that is most distantly related to the current members of the family Arteriviridae (11, 12). Infection with WPDV has been linked to a fatal neurological disease of the Australian brushtail possum (Trichosurus vulpecula) (13). The disease has been identified in both captive (14) and free-living (15) possum populations in New Zealand. It is currently unknown if the virus is present in other parts of the world. Experimental research on arteriviruses was driven by the need to develop robust control measures against PRRSV infection, which causes considerable losses to the swine industry (16), and aimed to reveal molecular mechanisms of replication and virus-host interactions of this family, which are often characterized using the EAV model. Comparative sequence analyses involving arteriviruses contributed to these goals by informing experimental research about natural constraints imposed on the structure and function of different genome regions and encoded products (5, 7, 17–25).
The arterivirus genome includes multiple open reading frames (ORFs), most of which overlap and are flanked by short noncoding regions at the 5′ and 3′ termini. Protein machineries controlling genome expression and replication are encoded in the first two ORFs, ORF1a and ORF1b, while capsid proteins controlling virus dissemination are encoded in the downstream ORFs, whose number varies among arteriviruses. ORF1a directs the synthesis of replicase polyprotein 1a (pp1a) and, together with ORF1b, also pp1ab. The latter involves a −1 programmed ribosomal frameshift (PRF) at the ORF1a/b overlap. pp1a and pp1ab are co- and posttranslationally processed by viral proteases to mature products (and their intermediates) that are designated nonstructural proteins (nsp's) 1 to 12 (1). The release of the nsp1-to-nsp3 and nsp3-to-nsp12 proteins from pp1a/ab is mediated by papain-like proteases (PLPs) and chymotrypsin-like protease (3CLpro), respectively (nsp3 release is controlled by two proteases) (20). No domain variation was reported for the nsp3-to-nsp12 region, with the size of ORF1b-encoded nsp9 to nsp12 being found to be under particularly strong constraint (21). According to the characterization of mostly EAV and PRRSVs, these proteins include four RNA processing enzymes (residing in nsp9 to nsp11), diverse enigmatic cofactors (nsp6 to -8 and nsp12) of replication, 3CLpro (nsp4), and two transmembrane domains, TM2 and TM3, anchoring the replication-transcription complex (RTC) (nsp3 and nsp5) (1, 22, 23, 26).
In contrast, the domain organization and size of the nsp1-nsp2 region vary considerably among the five characterized arterivirus species. For other arteriviruses with fully sequenced genomes, this region has not been studied, while for the most distantly related virus, WPDV, the respective region of the genome was not available (11). nsp2 is one of the two largest arterivirus proteins. Its size varies >2-fold, from 572 amino acids (aa) (EAV) to 1,232 aa (PRRSV-2). It invariably includes a multifunctional Zn-binding PLP2 domain at the N terminus and adjacent transmembrane (TM1) and Cys-rich (CR) domains at the C terminus (1, 27). These conserved domains are separated by a poorly conserved domain known as the hypervariable region (HVR) (1), which is notable for its high content of proline in PRRSV (28, 29), while another poorly conserved domain of unknown function (hinge) is found upstream of PLP2 in some arteriviruses (27). PLP2 mediates the processing of the nsp2-nsp3 junction and removes ubiquitin and ubiquitin-like moieties from target proteins, thus regulating both genome expression and virus-host interaction (30). The HVR contains antigenic sites (31), and the TM1 domain seems to contribute to anchoring the RTC, but the function of the CR domain remains totally obscure (1). In addition, EAV nsp2 is a cofactor essential for cleavage of the nsp4-nsp5 junction by 3CLpro (32). Recently, two truncated and modified derivatives of nsp2 of unknown function, and possibly different localizations, were identified in PRRSVs (33, 34). Their generation involves −1 and −2 PRFs in the nsp2 genome region encoding the HVR-TM1 junction and is directed by a slippery sequence and a downstream C-rich element conserved in several arteriviruses but not EAV (33, 34). PRFs are transactivated by a complex of viral nsp1b and host poly(C) binding protein (PCBP) that binds to the downstream C-rich element (34, 35).
The scale of variation in the nsp1 coding region is even larger, since it may encode either one (nsp1; EAV), two (nsp1a and nsp1b; LDV and PRRSV-1 and -2), or three (nsp1a, nsp1b, and nsp1c; SHFV) proteins. Each of these nsp1 variants includes an enzymatically active PLP domain that liberates the respective nsp from pp1a/ab by cleavage at the C terminus and, for SHFV PLP1c, possibly also the N terminus (17, 36–41). The name of each PLP includes a suffix matching that in the name of the nsp in which it resides. The only exception is EAV nsp1, which uniquely includes an enzymatically silent PLP (PLP1a) upstream of the active PLP, accordingly named PLP1b (36, 37). The variable number of adjacent PLPs present in the nsp1 region is likely to have emerged by duplication, implying that they are paralogs. Yet similarity between paralogous PLPs is (extremely) low at both the primary and tertiary structure (available for PLP1a and PLP1b of PRRSV-2 [18, 42]) levels, which suggests that this region has been under diversifying positive selection and/or that considerable time has passed since the duplication. nsp1/nsp1a of the characterized arteriviruses also includes an N-terminal Zn finger (ZnF) domain (37). All three domains of nsp1 are important for template-specific complex regulation of subgenomic mRNA production (transcription) and virion biogenesis in EAV (19, 43, 44); nsp1a of PRRSV-1 seems to play a similar role in transcriptional regulation (45). Analysis of tertiary structure and biochemical characterization of nsp1b revealed a small domain with weak nuclease activity residing upstream of PLP1b in PRRSV-2 (42) (hereinafter called nuclease domain). This protein was also shown to facilitate −1 and −2 PRFs in the nsp2 genome region of PRRSV-1 and -2 (34). All individual nsp1 subunits of PRRSV-2, LDV, SHFV, and EAV were found to have anti-innate-immunity activities (46, 47).
In the present study, we sought to gain insight into the structure and function of the nsp1-nsp2 genomic region by characterizing the evolution of this region in all known arteriviruses, including seven newly identified arteriviruses of monkeys and one arterivirus of the forest giant pouched rat (12, 87–91). To increase the resolution of this analysis, we also extended it to the most distantly related arterivirus, WPDV, whose sequence in this region is reported for the first time, thus completing its full genome sequence. We were interested in mapping the already identified domains to the nsp1-nsp2 region for arteriviruses that have not been characterized in this respect, reconstructing the evolutionary history of PLP duplications and other nsp1 domains, identifying molecular markers of PLP paralogs, and searching for new sites under constraint. Below we describe the obtained results along with the challenges of analysis of distant relations, summarize our conclusions, and outline directions for future studies.
RESULTS
Completion of genome sequencing of WPDV by a modified RACE method.
The available WPDV sequence, obtained using a classic 5′ rapid amplification of cDNA ends (RACE) protocol, comprised 10,087 nucleotides (nt), which included the published partial sequence of 9,509 nt [excluding the poly(A) tail] (11). However, analysis of this sequence suggested that it may still lack the 5′-terminal region which encodes nsp1-nsp2 in other arteriviruses. Several attempts to further extend this sequence by use of a commercially available kit utilizing a modification of the classic approach to 5′ RACE (5′ RNA ligase-mediated [RLM] RACE) according to the manufacturer's instructions were unsuccessful. To address this challenge, we modified the 5′ RLM RACE protocol by addition of a target enrichment step (Fig. 1). Three rounds of RACE reactions (RACE 1–3; see Materials and Methods) were performed using different capture probes and different target-specific primers (Table S1). Using the modified protocol, three bands of different sizes (∼0.4 kbp, 1.5 kbp, and 2.5 kbp) were obtained by primary PCR with the RACE.outer/WPD.S5.R primer pair (RACE 1) (Fig. 2). Each band was reamplified separately with the RACE.inner primer in combination with either the WPD.S5.R, WPD.S7.R, or WPD.S8.R primer. Sequencing of the nested bands indicated that the 0.4-kbp band represented nonspecific amplification, while the 1.5-kbp and 2.5-kbp bands assembled into one sequence, which extended the available sequence of WPDV by another 2,006 nt. The longest PCR product obtained in RACE 2 extended the RACE 1 assembly by 808 nt. Small bands of about 250 bp were amplified following seminested PCRs with the RACE.outer/inner and WPD.S16.R primer pairs and either HOT FIREPol or Kappa LongRange enzyme mix (RACE 3). Both bands contained the same sequence, which aligned with the existing WPDV sequence but did not extend it. The RACE adapter was ligated at nt 343 of the WPDV sequence obtained in RACE 2.
FIG 1.
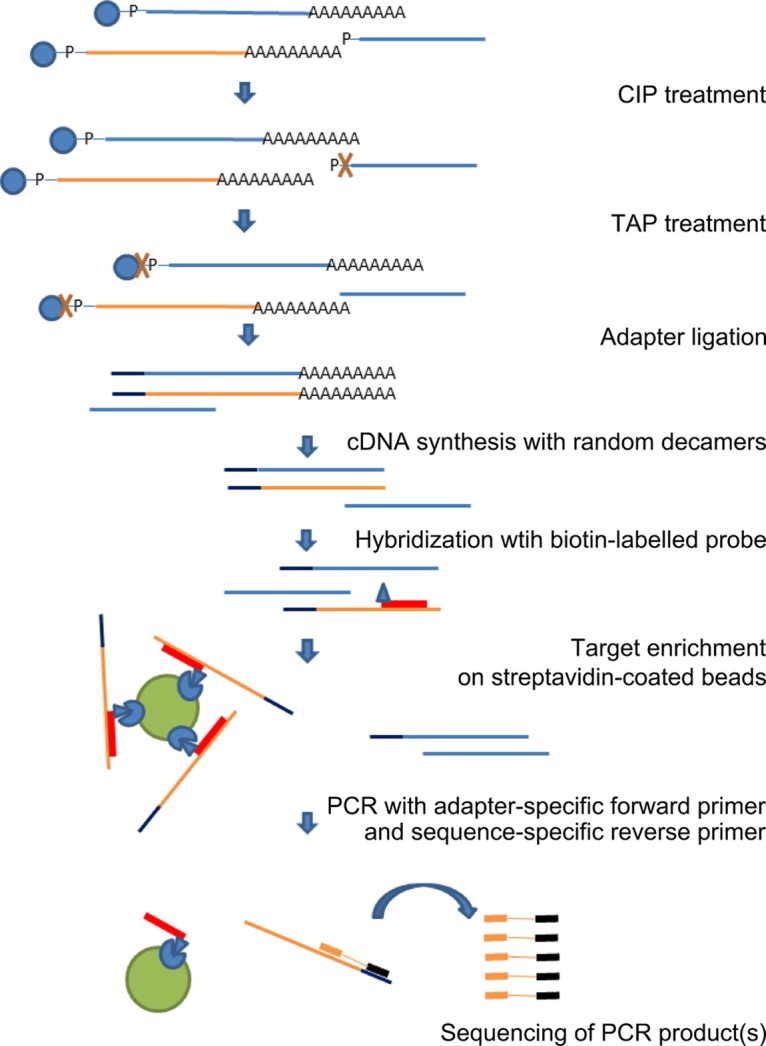
Overview of the target-enriched 5′ RLM RACE protocol. Total RNA extracted from WPD-affected tissues was treated with calf intestine alkaline phosphatase (CIP) to remove free 5′ phosphates from all noncapped nucleic acids, followed by treatment with tobacco acid pyrophosphatase (TAP) to remove the cap structure from full-length mRNA (including capped positive-sense viral RNA), ligation of the RACE adapter to decapped mRNA containing 5′ phosphates, and reverse transcription of the ligated mRNA to cDNA by use of random decamers. These steps were performed according to the manufacturer's instructions (RLM RACE; Invitrogen). The ligated cDNA was then hybridized to biotinylated virus-specific probes. The viral sequences captured on streptavidin-coated magnetic beads were used in the PCR step of the 5′ RLM RACE protocol. The unknown 5′ end was amplified with a selection of virus-specific reverse primers and adapter-specific RACE primers. The target (WPDV) and nontarget (host) nucleic acids are depicted in orange and blue, respectively.
FIG 2.
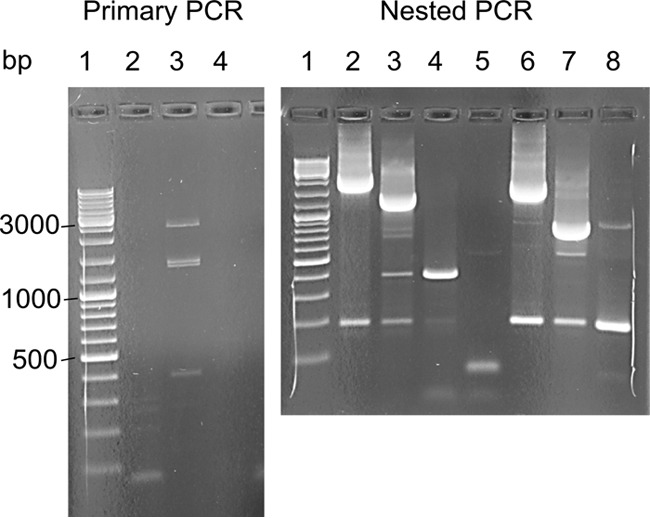
Example of the results obtained using modified 5′ RLM RACE as described in the text. (Left) One of the primary PCRs (lane 3) using the RACE.outer/WPD.S5.R primer pair (see Table S1 in the supplemental material) and target-enriched cDNA captured on streptavidin-coated magnetic beads as the template produced three bands, with approximate sizes of 2,500 bp (band 1), 1,500 bp (band 2), and 400 bp (band 3), with no bands in the no-template control (lane 4). Lane 2 represents an unsuccessful 5′ RLM RACE reaction with a different source of starting material. (Right) Nested PCR with the RACE.inner primer and either the WPD.S5.R (lanes 2 to 5) or WPD.S7.R (lanes 6 to 8) reverse primer. DNA extracted from primary band 1 (lanes 2 and 6), band 2 (lanes 3 and 7), band 3 (lanes 4 and 8), or water (lane 5) was used as the template. No bands were visible in the no-template control with the RLM-RACE inner/WPD.S7.R primer pair (not shown in the picture). A DNA ladder (GeneRuler DNA ladder mix; Fermentas) was included in lane 1 of both gels.
Based on our inability to amplify any further sequences in the 5′-end direction by using a variety of primers and amplification conditions, combined with the bioinformatics analysis of the newly identified sequence (see below), we concluded that we most likely amplified the full genomic sequence of WPDV. If so, then the length of the WPDV genome is 12,901 nt, excluding the poly(A) tail, and the length of predicted polyprotein 1ab (pp1ab) of WPDV is 3,402 aa, whose coding sequence is flanked by a 245-nt 5′ untranslated region (5′-UTR) and a 97-nt 3′-UTR (Table 1).
TABLE 1.
Predicted ORFs in the genome of WPDV and their corresponding protein products
| Name | Start position (nt) | Stop position (nt) | Length (nt) | Protein product | Protein size (aa) |
|---|---|---|---|---|---|
| ORF1a | 246 | 6,242 | 5,997 | Polyprotein 1a | 1,998 |
| ORF1b | 6,218 | 10,453 | 4,236 | Polyprotein 1aba | 3,402 |
| ORF2 | 10,309 | 10,932 | 624 | Glycoprotein 2 (GP2) | 207 |
| ORF2a | 10,553 | 10,690 | 138 | Envelope protein (E) | 45 |
| ORF3 | 10,794 | 11,447 | 654 | Glycoprotein 3 (GP3) | 217 |
| ORF4 | 11,330 | 11,581 | 252 | Glycoprotein 4 (GP4) | 83 |
| ORF5 | 11,578 | 12,114 | 537 | Glycoprotein 5 (GP5) | 178 |
| ORF5a | 11,603 | 11,839 | 237 | Glycoprotein 5a (GP5a) | 78 |
| ORF6 | 12,051 | 12,593 | 543 | Membrane protein (M) | 180 |
| ORF7 | 12,424 | 12,804 | 381 | Nucleocapsid protein (N) | 126 |
Polyprotein 1ab is predicted to be expressed from ORF1a and -b via a −1 ribosomal frameshift.
Phylogeny of arteriviruses.
To facilitate analysis of the nsp1-nsp2 genomic region, we reconstructed the phylogeny of arteriviruses by using multiple-sequence alignment (MSA) of seven conserved nsp domains (see Materials and Methods; see Table S2 in the supplemental material) of 14 viruses, including WPDV (Fig. 3A; also see below). The total size of the analyzed MSA was 2,055 columns, which accounted for 43% of the pp1ab sites (bottom panel in Fig. 3A). The viruses represented all species defined by DEmARC (Table 2) (see Materials and Methods), whose sampling varied by 2 orders of magnitude (Fig. 3C). Based on the tree topology and the distance-based results of DEmARC, we recognized five clades (Table 2), including three represented by single virus species (WPDV, EAV, and African pouched rat arterivirus [APRAV]), one represented by eight virus species (named the simian clade), and one represented by three virus species (named the LDV-PRRSV clade). A large Bayesian sample of rooted trees revealed well-resolved branches for all viruses except APRAV and, to a lesser extent, simian hemorrhagic encephalitis virus (SHEV) (Fig. 3A). APRAV formed the basal branch to either the LDV-PRRSV (less favored; 33.66% of trees) or LDV-PRRSV and simian (most favored; 66.30% of trees) clades. Also, in a small fraction of trees (4.43%), SHEV formed a basal branch to the Kibale red colobus virus 1 (KRCV-1) and KRCV-2 clade, while in the majority of trees (95.50%) SHEV was basal to all simian arteriviruses other than SHEV.
FIG 3.

Phylogeny and nsp1-nsp2 domain organization of arteriviruses. (A) The phylogeny is presented by a posterior sample of phylogenetic trees, reconstructed by BEAST software. The trees are colored blue, red, or green, in descending order of prevalent topology. The genome organization, polyprotein processing scheme, and polyprotein domains used for phylogeny reconstruction (shaded in gray) are detailed in the bottom left corner for PRRSV-2 (accession number NC_001961.1). (B) The domain organization of nsp1-nsp2 is shown for each arterivirus species. Protein domains are represented by colored bars. The bar representing PLP1b of EAV has dark green stripes to emphasize its affinity with PLP1c. Bars representing the PLP1 domains of WPDV have white stripes to show their weak sequence similarity with the PLP1 domains of other arteriviruses. The positions of nsp2 PRF-related motifs are indicated by orange triangles, those of experimentally established cleavage sites by black triangles, and those of PxPxPR motifs by cyan diamonds. (C) Number of genomes sequenced for each of the characterized species (with sampling size and bias).
TABLE 2.
Representative set of arteriviruses whose genome sequences were used in the present study
| Acronyma | Virus name | Accession no. | Cluster nameb |
|---|---|---|---|
| PRRSV-2 | Porcine reproductive and respiratory syndrome virus 2 | EU624117.1 | LDV-PRRSV |
| PRRSV-1 | Porcine reproductive and respiratory syndrome virus 1 | DQ489311.1 | LDV-PRRSV |
| LDV | Lactate dehydrogenase-elevating virus | U15146.1 | LDV-PRRSV |
| KRCV-2 | Kibale red colobus virus 2 | KC787658.1 | Simian |
| PBJV | Pebjah virus | KR139839.1 | Simian |
| SHFV | Simian hemorrhagic fever virus | AF180391.1 | Simian |
| DeMAV | De Brazza's monkey arterivirus | KP126831.1 | Simian |
| KRTGV | Kibale red-tailed guenon virus 1 | JX473849.1 | Simian |
| KRCV-1 | Kibale red colobus virus 1 | KC787630.1 | Simian |
| SHEV | Simian hemorrhagic encephalitis virus | KM677927.1 | Simian |
| MYBV-1 | Mikumi yellow baboon virus 1 | KM110938.1 | Simian |
| EAV | Equine arteritis virus | X53459.3 | EAV |
| APRAV | African pouched rat arterivirus | KP026921.1 | APRAV |
| WPDV | Wobbly possum disease virus | JN116253.3 | WPDV |
Sequences of all full-length arterivirus genomes retrieved from GenBank and RefSeq, as well as that of the full-length WPDV genome, were grouped into 14 species by DEmARC. One sequence from each species was selected as a representative for further analysis.
Delineated species were further grouped into five clusters.
Domain organization of the nsp1-nsp2 genomic region of arteriviruses.
We then analyzed the domain organization of the nsp1-nsp2 genomic region in different arteriviruses. Using arterivirus-wide MSA, we found that all poorly characterized viruses of the simian clade adopted the domain organization described for SHFV (Fig. 3B; Table S3). The sequence affinity of viruses of the LDV-PRRSV clade in this region, except for the highly divergent hinge and HVR domains, was also previously documented and confirmed by our analysis. Quality MSAs of the nsp1-nsp2 region for both the simian and LDV-PRRSV clades and three other single-species clades were converted into the respective HMM profiles that were used for all-versus-all profile-profile comparisons by HHalign. The most informative comparisons were profile comparisons of the most diverse simian clade with itself and other clades, whose results are visualized as two-dimensional plots in Fig. 4. Based on the number of high-scoring domains and the level of confidence (measured by both probability and E values), sequence affinity between the simian clade and other clades was ranked in the descending order LDV-PRRSV > APRAV > EAV > WPDV. Notably, this ranking was in agreement with the phylogenetic relationships of the clades (Fig. 3A). This analysis also enabled nsp1-nsp2 domain assignment for APRAV (Fig. 3B; Table S3; see below). At the domain level, support was obtained for the conservation of ZnF and “nuclease” domains in all viruses except for EAV and WPDV, and for PLP2 of all non-WPDV arteriviruses (Fig. 4 and 5). Since EAV also encodes the ZnF domain (37), this result showed that the conducted profile-profile comparisons were not sufficiently sensitive to reveal the most remote relationships. We then inspected residues conserved in the respective MSAs of the ZnF and nuclease domains (Fig. 5A and B). As expected, four Zn-binding residues were conserved in the ZnF MSA of non-WPDV arteriviruses (Fig. 5A). In contrast, the Lys and Glu residues implicated in the nuclease activity of PPRSV-2 (42) were found to be among the least conserved residues in the MSA of the nuclease domain (Fig. 5B). Accordingly, the “nuclease” domain included only two residues (Trp and Pro) that were invariant in arteriviruses, further indicating that this domain is unlikely to have any enzymatic activity that can broadly be conserved in arteriviruses; its name is thus retained purely for historical reasons.
FIG 4.
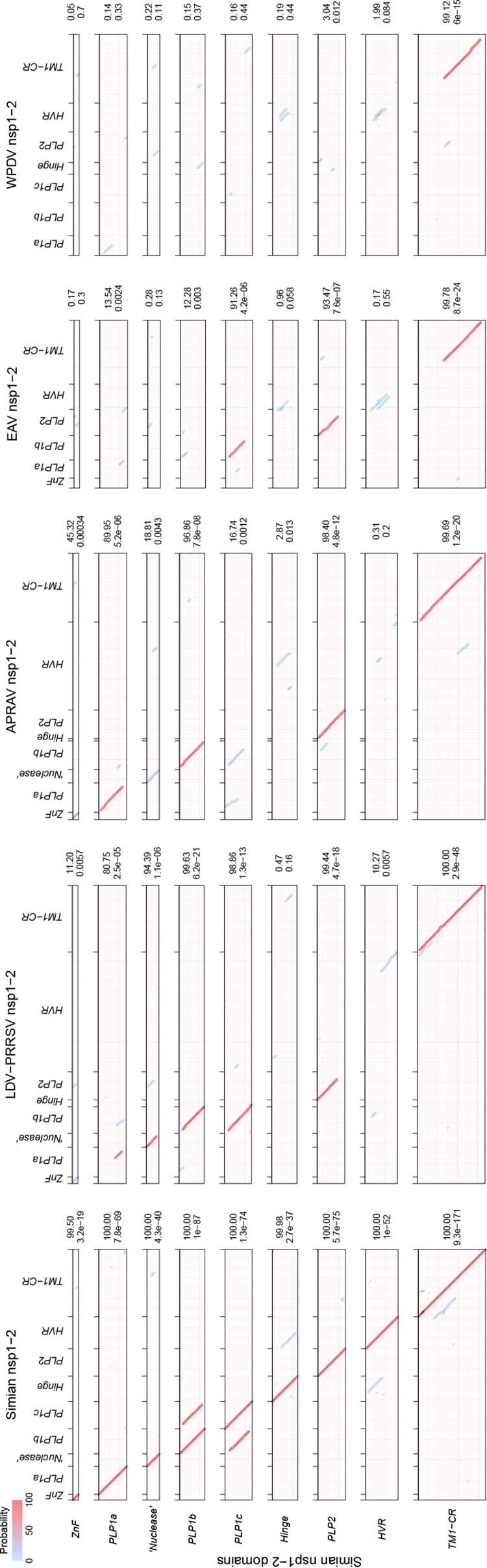
Profile-profile comparisons of nsp1-nsp2 domains of the simian lineage and five other arterivirus lineages. The plots shown are HHalign dot plots, with domains and viruses indicated on the respective axes and alignment paths of the two top-scoring hits drawn with transparent lines. The color of each line indicates the probability of the hit. On the right side of each dot plot, the probability and E value of the top-scoring hit are depicted.
FIG 5.

Multiple-sequence alignments of selected nsp1-nsp2 domains of arteriviruses. (A) MSA of ZnF domains. Zinc-binding residues are marked with black triangles. (B) MSA of “nuclease” domains. Columns of the MSA that contain PRRSV-2 nsp1b residues whose mutation to alanine led to abolishment of PRRSV-2 nsp1b nuclease activity (42) are marked with black triangles. (C) MSA of PLP2 domains. Catalytic residues are marked with black triangles. MSAs were visualized with the help of Espript 2.1 (82). Secondary structures were derived from PDB entries.
Relationships between paralogous and orthologous PLPs of all non-WPDV arteriviruses.
Before proceeding to analyze WPDV further, we first clarified the relationships between paralogous and orthologous PLPs by using profile-profile plots. In agreement with prior observations, no significant similarity between PLP1a and either PLP1b or PLP1c was found for PLPs of any origin. In contrast, similarity between PLP1b and PLP1c variants of different origins was significant, although it varied considerably depending on the pair (Fig. 6A). The most significant similarity was that between PLP1b variants of the simian and LDV-PRRSV clades, which was supported much more strongly than the next most significant hit between either of these PLP1b variants and simian PLP1c (3.9e−26 versus 1.1e−15). Likewise, PLP1b of APRAV showed much higher sequence similarity to PLP1b of the LDV-PRRSV or simian clade than to PLP1c of the simian cluster (8.2e−11 and 6.1e−09 versus 0.0001). In contrast, the enzymatically active PLP of EAV (known as PLP1b) was most similar to simian PLP1c rather than to PLP1b variants of different origins (7.28e−08 versus 0.00091 to 0.00013). In all comparisons of PLP1b and PLP1c, the result depended on the inclusion of EAV PLP1b: almost entire domains were similar without EAV PLP1b being involved, while the similarity was limited to the N-terminal half of the domain when EAV PLP1b was compared. To extend these observations further, MSA of combined PLP1b/PLP1c was used to infer the Bayesian phylogeny of these domains (Fig. 6B). While the obtained sample of rooted trees lacked a prevalent topology, PLPs were clearly partitioned into two major PLP1b- and PLP1c-based clades according to the sequence affinities revealed in the profile analysis. For each clade, considerable uncertainty of the branching was observed for most viruses, likely due to the extremely large scale of divergence of the entire tree (more than four times that of the nsp-based tree of non-WPDV arteriviruses) (compare Fig. 6B with Fig. 3A), confounded by the small size of the PLP1 domains. The only notable exception to the domain-clade association was EAV PLP1b, which was basal to either PLP1c (70.26% of trees) or PLP1b and PLP1c (23.36% of trees), and this was also sustained in the comparable tree including WPDV (Fig. S1; see below). These results combined strongly suggested an orthologous relationship between PLP1b variants of the simian, LDV-PRRSV, and APRAV clades but not that of EAV, which is most likely either an ortholog of PLP1c enzymes or a direct descendant of the ancestral enzyme for PLP1b and PLP1c (see Discussion).
FIG 6.

Sequence similarity and evolutionary relationships of PLP1b and PLP1c. (A) HHalign comparisons between PLP1b and PLP1c domains of different arteriviruses. For each comparison, a dot plot is shown. On the dot plot, the alignment path of the top-scoring hit is drawn with a transparent line. The color of the line indicates the probability of the hit. Below the dot plot, the probability and E value of the top-scoring hit are given. (B) Posterior sample of phylogenetic trees generated by BEAST, based on MSA of PLP1b and PLP1c. For other designations, see the legend to Fig. 3A.
Domain organization of the nsp1-nsp2 region in WPDV.
To improve the limited resolution of the domains mapping in the nsp1-nsp2 region of WPDV by profile-profile comparison (Fig. 4), we combined four clade-specific MSAs of domains of this region. These MSAs were then compared with the N-terminal 1,096 aa of WPDV pp1a/pp1ab in the profile-profile mode by using HHalign (Fig. 7). Significant similarities were observed for the PLP2 (8.4e−06) and TM1-CR (1.7e−23) domains, which were much stronger than those observed using simian-based profiles only (Fig. 4), facilitating mapping of these domains in the WPDV polyprotein. In line with the considerable divergence of WPDV, its PLP2 domain included a 16-aa insertion between catalytic Cys and His residues in the otherwise uniformly compact PLP2 sequences of different origins (Fig. 5C). Although no other domain showed statistically significant similarity, the sizes of regions upstream of PLP2 and between the TM1-CR and PLP2 domains in the WPDV pp1ab protein were sufficiently large to accommodate other canonical domains.
FIG 7.
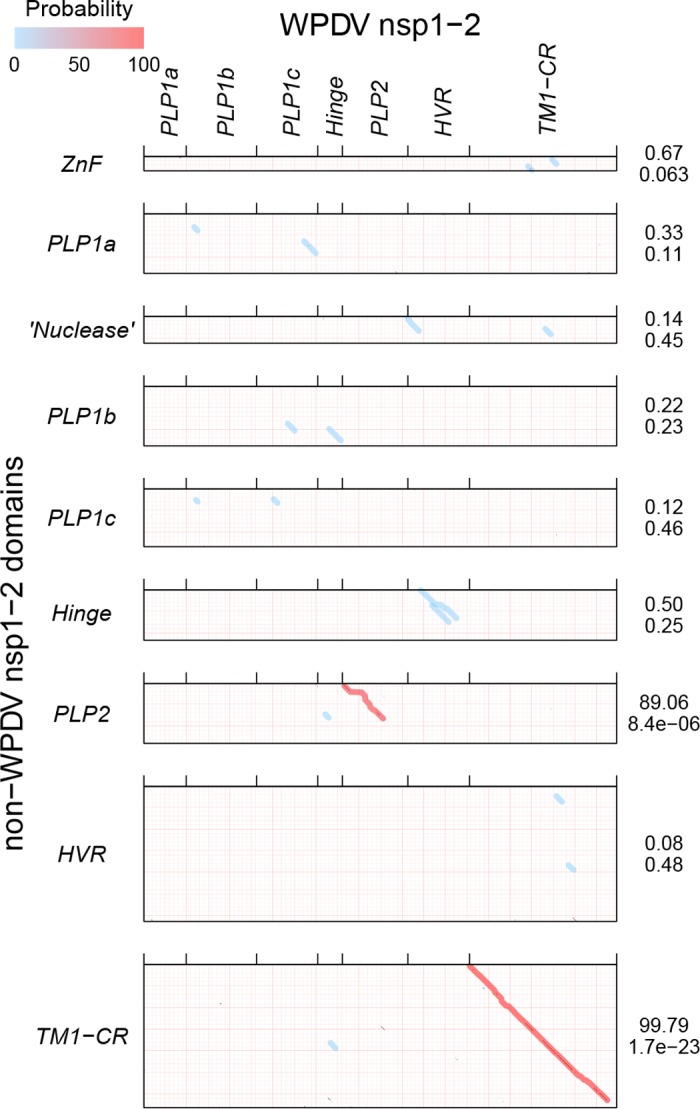
HHalign profile-profile comparisons of nsp1-nsp2 domains of WPDV and non-WPDV arteriviruses. EAV PLP1b was regarded as PLP1c for this figure. For details, see the legend to Fig. 4.
To learn whether WPDV could indeed encode highly divergent homologs of arterivirus PLP1, we scanned WPDV pp1ab with HMM profiles of short regions around the catalytic cysteine and histidine residues of nsp1 PLPs of other arteriviruses by using HHalign; since enzymatically silent PLP1a of EAV lacks the catalytic cysteine, it was not included in the corresponding HMM profile. Hit probability distributions (Fig. 8) revealed that two top-scoring hits for the cysteine motif had considerably higher probabilities (1.62% and 0.47%, respectively) than those of other hits (≤0.04% and ≤0.06% for Cys and His motifs, respectively), indicating that they may be genuine. These top-scoring hits were mapped upstream of the putative PLP2 domain, to aa 121 to 125 and 301 to 311 of the WPDV polyprotein, positions compatible with belonging to two PLP1 varieties. Accordingly, these hits included CysTrp and CysTyr dipeptides, respectively, which either matched or closely resembled the CysTrp dipeptide with a catalytic Cys residue of PLP1b and PLP1c, besides conservation at other, less prominent positions (Fig. 9 and 10A). These observations were used to guide MSAs between WPDV and arteriviruses for PLP1a, PLP1b, and PLP1c, including putative catalytic His residues (Fig. 9), and to delineate the hinge domain in WPDV (Fig. 3B; Table S3). Like its EAV counterpart, the delineated PLP1a domain of WPDV lacks the catalytic cysteine and is expected to be proteolytically silent. However, like PLP1a enzymes of all arteriviruses, it did include the most characteristic HXXXXXF motif (Fig. 10A), which is the core of the Ha conservation peak in Fig. 10B. Secondary structure predictions (Fig. 9) and the modest impact of the WPDV inclusion in the respective MSAs on their mean conservation (Fig. 10B) further supported the identification of these most divergent PLP1s (Fig. S1). No ZnF or nuclease domains were evident in WPDV.
FIG 8.
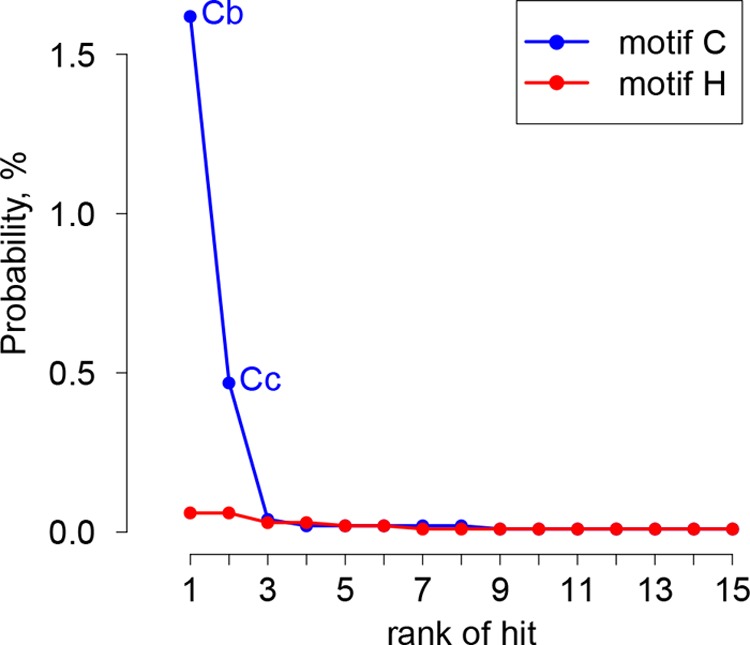
Rank distribution of top HHalign hits between PLP1 active site motifs of arteriviruses and WPDV pp1ab. HMM profiles representing cysteine and histidine motifs of PLP1s of all non-WPDV arterivirus species, with the EAV PLP1a cysteine motif excluded, were compared with WPDV pp1ab. The 15 top hits were ranked in descending order of probability (indicated on the y axis). Hits potentially including the catalytic cysteines of WPDV PLP1b and PLP1c are designated Cb and Cc, respectively.
FIG 9.

Multiple-sequence alignment of arterivirus nsp1 PLPs. The top two secondary structures were derived from PDB entries. All other secondary structures were predicted by Jpred4 (74). Red triangles indicate columns of the PLP1a and PLP1b/PLP1c MSAs that have conservation scores above 0.75 for non-WPDV arteriviruses and were mapped on PDB structures (see Fig. 11). Columns containing the first residues of the PRRSV-2 PLP1a and PLP1b C-terminal subdomains are indicated by ochre bars. Catalytic motifs of nsp1 PLPs are underlined in cyan. The MSAs were visualized with Espript 2.1 (82).
FIG 10.

Distribution of sequence conservation in the N-terminal region of pp1ab of arteriviruses. (A) MSAs of nsp1 PLP motifs of all non-WPDV arteriviruses are depicted as logos, with the homologous WPDV sequence specified below each logo. PLP motifs, including the catalytic residues Cys (C) and His (H) and putative RNA-binding residues (R), are labeled with domain-specific suffixes. Logos were prepared with the R package RWebLogo 1.0.3 (83). (B) The conservation profile, calculated based on the MSA of sequences from non-WPDV clusters, is shown for each domain of nsp1 and the N-terminal domains of nsp2. Areas above and below the mean conservation lines are shaded in black and gray, respectively. Dotted red lines indicate the mean conservation of the domains after the addition of the WPDV sequence to the MSA. EAV PLP1b was regarded as PLP1c for this figure.
N- and C-terminal subdomains of PLP1 are enriched with sites that are conserved in paralogous PLP1b/c and PLP1a, respectively.
Sequence similarity between PLP1a and PLP1b/PLP1c is limited to very few residues (Fig. 9). This profound divergence was also evident upon comparison of the resolved crystal structures of PRRSV-2 nsp1a and nsp1b (Protein Data Bank [PDB] entries 3IFU [18] and 3MTV [42]) by use of DALI (48), which revealed the similarity between PLP1a and PLP1b to be below the Z-score cutoff (Z-scores of 4.2 and 3.6 and root mean square deviations [RMSD] of 4.0 and 4.6 with PLP1b and PLP1a as queries, respectively) (see Materials and Methods). To gain insight into the selection that drove the divergence of these enzymes, we mapped residues conserved in PLP1a and PLP1b/c of non-WPDV arteriviruses on the structure of PRRSV-2 PLP1a (Fig. 11A) and PLP1b (Fig. 11B), respectively. Six of 9 residues conserved in PLP1a were found in the right subdomain of the papain fold, while 9 of 13 residues conserved in PLP1b/c were located in the left subdomain of the papain fold. This contrasting pattern suggests that the divergence of PLP1a and PLP1b/c has been constrained and/or promoted in a subdomain-specific fashion, which thus explains the exceptionally low similarity between these paralogs.
FIG 11.
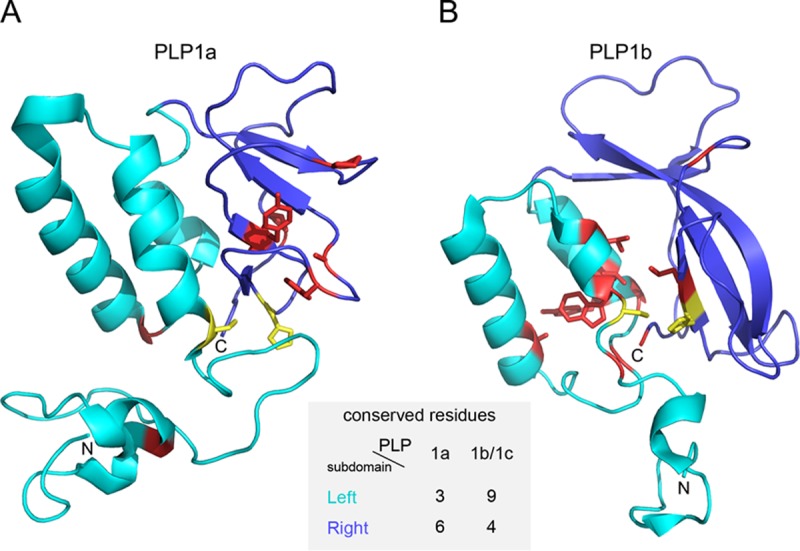
Subdomain-specific distribution of residues conserved in PLP1a and PLP1b/c. The structures shown are tertiary structures of PRRSV-2 PLP1a (A) and PLP1b (B) with residues conserved in all non-WPDV arteriviral PLP1a and PLP1b/PLP1c domains, respectively. The N-terminal subdomain, formed by α-helices, is shown in cyan; and the C-terminal subdomain, consisting of antiparallel β-strands, is shown in blue. Conserved residues are shown in yellow (catalytic dyad) and red (all the rest). The following residues were conserved in the PLP1a alignment and mapped on PRRSV-2 (accession number EU624117.1) nsp1a: left subdomain, Gly45, Cys76, and Gly109; and right subdomain, Pro134, Tyr141, His146, Phe152, Ala155, and Pro175. The following residues were conserved in the PLP1b/c alignment and mapped on PRRSV-2 (accession number EU624117.1) nsp1b: left subdomain, Gly88, Cys90, Trp91, Leu94, Ala110, Gly120, Gly123, Tyr125, and Leu126; and right subdomain, Gly143, His159, Leu160, and Gly203. The figure was prepared with PyMOL (85).
Conservation of a novel proline-rich motif in the nsp2 HVR of WPDV and other arteriviruses.
One of the two most divergent regions of nsp2 is the HVR, located between PLP2 and the TM1-CR domains (Fig. 3B). We found that the size of this domain varied >5-fold, from 125 aa (EAV) to 716 aa (PRRSV-2). Since the size difference might have emerged as a result of duplications, we searched for repeats in this domain. The presence of tandem repeats was initially detected in the WPDV HVR when it was compared to itself by use of HHalign and was subsequently corroborated by RADAR (49). The MSA of WPDV HVR tandem repeats was converted into an HMM profile and compared with the nsp2 HVRs of different arteriviruses by using HHalign, resulting in multiple significant hits that conformed to the pattern PxPxPR or a close derivative (Fig. 12A and C). Similar results were obtained using MEME (50), which identified extended versions of this motif (E value = 9.3e−9) in representatives of eight species (Fig. 12B and C). A subsequent search for strict matches to the PxPxPR motif in the pp1ab proteins of all sequenced arteriviruses identified at least one copy of the motif in the HVR for most viruses of 10 arterivirus species, with the number of motif copies varying in some species (Table 3). Two of the remaining four species, SHEV and KRCV-1, were found to contain a PxPxPR motif(s) in the hinge domain, while none of the pp1ab domains of two other species, Pebjah virus (PBJV) and De Brazza's monkey arterivirus (DeMAV), contain this motif. Overall, PxPxPR motifs were found predominantly in the HVR and much less frequently in the hinge domain, with the only exception being two isolates of Kibale red-tailed guenon virus 1 (KRTGV) that contain one copy of the motif in the PLP1a domain.
FIG 12.

Conservation of PxPxPR motifs in the HVR of arteriviruses. (A) Rank distribution of the top 30 hits obtained during HHalign comparison between WPDV HVR tandem repeats and individual HVR domain sequences of arteriviruses. The red line depicts the 5% probability threshold. WPDV HVR tandem repeats identified by RADAR are shown in the top right corner. (B) Locations of motifs identified by MEME in the HVR of arterivirus species. Extended PxPxPR motifs are shown in green, and conserved C-terminal motifs corresponding to the nsp2 PRF site are shown in red. (C) MSA of the PxPxPR motif and its derivatives in the HVR of viruses representing arterivirus species. Coordinates in the names of motifs refer to their domain position. Numbers to the right of the MSA show support for the identification of each motif by three methods. The first column shows probability values assigned to hits containing PxPxPR motifs by HHalign in analyses comparing HVR sequences of the respective arteriviruses to the MSA of tandem repeats of the WPDV HVR. The second column shows P values assigned to motifs by MEME. The third column shows matches (+) and mismatches (−) of the PxPxPR pattern.
TABLE 3.
Intraspecies variation in the number of PxPxPR motifs in the HVR and elsewhere in pp1ab
| Species | Total no. of genomes | Motifs in HVR |
|
|---|---|---|---|
| No. of motifs per domain | No. of genomes | ||
| WPDV | 1 | 3 | 1 |
| EAV | 27 | 3 | 27 |
| SHEVa | 1 | 0 | 1 |
| MYBV-1 | 13 | 1 | 13 |
| PBJV | 3 | 0 | 3 |
| SHFV | 1 | 1 | 1 |
| KRTGVb | 4 | 1 | 4 |
| DeMAV | 1 | 0 | 1 |
| KRCV-2 | 29 | 2 | 1 |
| 1 | 26 | ||
| 0 | 2 | ||
| KRCV-1c | 15 | 0 | 15 |
| LDV | 2 | 1 | 1 |
| 0 | 1 | ||
| PRRSV-2 | 368 | 4 | 1 |
| 3 | 310 | ||
| 2 | 53 | ||
| 1 | 4 | ||
| PRRSV-1 | 36 | 1 | 34 |
| 0 | 2 | ||
| APRAV | 1 | 2 | 1 |
One motif is present in the hinge domain.
One motif is present in the PLP1a domain of 2 out of 4 isolates.
One or two motifs are present in the hinge domain of 11 or 4 out of 15 isolates, respectively.
EAV may be the only arterivirus that has no PRF motifs in the nsp2 region.
The above-described MEME analysis also identified residue conservation in all arteriviruses except the most divergent ones, EAV and WPDV, at the very C terminus of the HVR (Fig. 12B, red boxes), which is adjacent to the TM1-CR domains conserved in all arteriviruses (Fig. 13). Upon conversion of the arterivirus-wide MSA of the HVR domain C terminus (Fig. 14B) into the nucleotide MSA (Fig. 14C), it became evident that amino acid conservation identified by MEME corresponds to nucleotide PRF motif conservation, slippery sequence RG_GUU_UUU (R = G or A) and downstream element CCCANCUCC (33). These motifs were shown to guide translation of the genome region encoding HVR/TM1-CR junction in two alternative open reading frames, −1TF and −2TF, in PRRSV-1 and -2 (33, 34). These two ORFs are expressed via −1 and −2 PRF with the production of nsp2N and nsp2TF, respectively (Fig. 14A). Previously, it was suggested that during nsp2 PRF in arteriviruses, complete codon-anticodon repairing is required at the closely monitored ribosomal A site, while mismatches are tolerated at the P site (33). Accordingly, slippery sequences observed in our analysis conformed to the patterns NN_NUU_UUU, NN_NUU_UUC, and NN_NUC_UCU (with the exception of PRRSV-1 EU076704.1 slippery sequence GG_GUU_UGU), which allow the integrity of the A-site duplex to be maintained after the −1/−2 shift or, in the case of the latter pattern, only the −2 shift (51). Deviations in the downstream element were rare and did not involve more than one nucleotide, while observed sizes of −2TF domains were comparable (with the exception of LDV L13298.1 47 aa -2TF domain) to the experimentally verified size of the −2TF domain of PRRSV (Table 4). These results suggest that the observed variations in PRF motifs in our large virus data set (Table 4) may be compatible with their function despite the detrimental effects of some of these variations artificially introduced into PRRSV-2 (33, 35).
FIG 13.

Multiple-sequence alignment of the nsp2 C termini of arteriviruses. Columns containing amino acids whose tRNAs are expected to be present in the ribosomal P and A sites prior to −1/−2 frameshifting are marked with orange triangles. The first column of the TM1-CR domains is marked with a black box. Amino acid residues predicted by TMHMM 2.0 (75) to form transmembrane regions are colored blue. The MSA was visualized with Espript 2.1 (82).
FIG 14.

Arteriviral nsp2 PRF. (A) Schematic representation of the expression of nsp2 moieties (based on LDV; accession number U15146.1). (B) Fragment of the pp1ab alignment corresponding to the site of nsp2 PRF. Columns containing amino acids whose tRNAs are present in the ribosomal P and A sites prior to frameshifting are highlighted with orange triangles. (C) Nucleotide alignment corresponding to the protein alignment presented in panel B. The slippery sequence is shown in orange and the C-rich element in cyan. Deviations from the canonical motifs, i.e., RG_GUU_UUU (R = G or A) and CCCANCUCC, are highlighted in red. For each sequence, the genome coordinate of the first nucleotide in the alignment is specified. If the frameshift site allows complete A-site duplex repairing in the −1 or −2 frame, then the length of the corresponding hypothetical protein product is specified. Otherwise, it is marked with a dash. Alignment columns containing the first nucleotides of −1TF and −2TF are highlighted with pink and blue bars, respectively.
TABLE 4.
Intraspecies variation of nsp2 PRF-related elements
| Species | Total no. of genomes | Slippery sequence |
C-rich region |
−1TF |
−2TF |
||||
|---|---|---|---|---|---|---|---|---|---|
| Sequencea | No. of genomes | Sequencea | No. of genomes | Length (aa) | No. of genomes | Length (aa) | No. of genomes | ||
| SHEV | 1 | Au_uUc_UcU | 1 | CCuANCUCC | 1 | 25 | 1 | 219 | 1 |
| MYBV-1 | 13 | cG_GUc_UcU | 10 | CCCANCUCC | 13 | 10 | 4 | 220 | 13 |
| uG_GUc_UcU | 3 | 0 | 4 | ||||||
| 21 | 3 | ||||||||
| 15 | 1 | ||||||||
| 13 | 1 | ||||||||
| PBJV | 3 | Gu_GUU_UUU | 3 | CCCANCUCC | 3 | 73 | 3 | 225 | 3 |
| SHFV | 1 | GG_GUU_UUU | 1 | CCCANCUCC | 1 | 77 | 1 | 225 | 1 |
| KRTGV | 4 | Gu_GUU_UUU | 2 | CCCANaUCC | 4 | 60 | 2 | 230 | 4 |
| GG_GUU_UUU | 2 | 55 | 2 | ||||||
| DeMAV | 1 | Gu_GUU_UUU | 1 | CCCANCUCC | 1 | 73 | 1 | 225 | 1 |
| KRCV-2 | 29 | AG_GUc_UcU | 29 | CCCANCUCC | 29 | 24 | 29 | 220 | 29 |
| KRCV-1 | 15 | AG_GUc_UcU | 15 | CCCANCUCC | 15 | 13 | 15 | 219 | 15 |
| LDV | 2 | AG_GUU_UUU | 2 | CCCANCUCC | 2 | 23 | 1 | 47 | 1 |
| 20 | 1 | 169 | 1 | ||||||
| PRRSV-2 | 368 | AG_GUU_UUU | 298 | CCCANCUCC | 366 | 0 | 314 | 169 | 365 |
| GG_GUU_UUU | 40 | CCCgNCUCC | 2 | 23 | 33 | 168 | 1 | ||
| GG_GUU_UUc | 17 | 16 | 16 | 128 | 1 | ||||
| AG_GUU_UUc | 6 | 18 | 5 | 115 | 1 | ||||
| AG_aUU_UUU | 5 | ||||||||
| uG_GUU_UUU | 1 | ||||||||
| Au_GUU_UUU | 1 | ||||||||
| PRRSV-1 | 36 | GG_GUU_UUU | 35 | CCCANCUCC | 36 | 0 | 36 | 169 | 35 |
| GG_GUU_UgU | 1 | 170 | 1 | ||||||
| APRAV | 1 | cG_GUU_UUc | 1 | CCCgNCUCC | 1 | 55 | 1 | 173 | 1 |
Deviations from the canonical motifs, i.e., RG_GUU_UUU (R = G or A) and CCCANCUCC, are shown by lowercase bold letters.
To learn about WPDV in this respect, we compared HMM profiles of nsp2 PRF-related motifs of arteriviruses and the WPDV nsp2 nucleotide sequence by using NHMMER. The two motifs were found in close proximity and canonical order in the expected region of the WPDV nsp2 locus, with the third best hit to each of the two queries. Remarkably, the hit to the slippery sequence profile was the only one observed that allowed complete A-site duplex repairing in the −2 frame. While each of these hits was statistically insignificant (with E values of 4.6 and 2.3), the probability of observing their combination in this place by chance may be approximately 2 orders of magnitude smaller than that of observing each hit separately, given the size of the nsp2 locus. Importantly, no comparably located proximal hits were found upon scanning of the EAV nsp2 locus, which served as a negative control. Accordingly, WPDV compared to EAV deviated from PRRSV much less in both motifs, but these were separated by 18 rather than the canonical 10 nucleotides (Fig. 14C). In WPDV, the −1 frameshift is expected to lead to immediate termination of translation (as observed in PRRSV) (Fig. 15A), while translation in the −2 frame may result in the product being extended with a domain as in other arteriviruses, with the following caveats: the size of this domain is much smaller than those of arteriviruses (32 versus 169 to 230 aa) (but see Table 4), and it lacks a TM module (Fig. 15B). The −1/−2 PRF is stimulated by a complex of PCBP and nsp1b in PRRSV (34, 35). Its effector region, located in PLP1b, is most conserved in arteriviruses (peak and logo Rb in Fig. 10) and, further, has a conserved counterpart in PLP1c (Rc). WPDV deviates considerably from arteriviruses in this region, in both PLP1b and PLP1c, which may be due to either coevolution with the PRF motifs or the lack of involvement of these domains in PRF regulation in WPDV.
FIG 15.

Multiple-sequence alignments of alternative nsp2 C termini. (A) C terminus of nsp2N, translated as a result of −1 PRF. (B) C terminus of nsp2TF, translated as a result of −2 PRF. MSAs were guided by the MSA presented in Fig. 13. For other details, see the legend to Fig. 13.
DISCUSSION
In this report, we present the current state of the art for domain characterization of the nsp1-nsp2 genome region of arteriviruses by comparative sequence analysis. This work has confirmed and considerably extended the results of prior analyses of this region (5, 7, 17–20, 25, 39, 41). Below, we briefly discuss the limitations and implications of the obtained results as well as the challenges of the conducted analyses.
We analyzed the genomes of all arteriviruses available on 11 June 2015 plus the genome sequence of WPDV, the most distantly related arterivirus, reported in full here for the first time. We extended the available WPDV genome sequence of 10,087 nt by 2,814 nt in the 5′ direction. This was accomplished with a modified 5′ RLM RACE protocol in two steps, RACE 1 (2,006-nt extension) and RACE 2 (808-nt extension), and benefited from bioinformatics analyses. No further extension was observed with an additional step, RACE 3, and no major genomic element was missing in this sequence according to our bioinformatics analysis. Collectively, these results attest to the completion of the genome sequence of WPDV, although we acknowledge that the exact terminal nucleotide(s) remains to be verified. Even with the target enrichment step, the full 5′ end was obtained in only one PCR, with several shorter PCR fragments amplified in various PCR runs. The difficulties encountered in amplification of the 5′ end of the WPDV sequence may be related to the presence of complex secondary structures at the 5′ end of the viral genomic RNA. Such structures play a role in virus replication and have been described for the genomes of other nidoviruses (52).
The traditional 5′ RACE protocol relies on homopolymer tailing of cDNA. The tail is then used to attach a linker sequence in the first rounds of PCR with a sequence-specific reverse primer and a linker-specific forward primer (53). The main drawbacks of the technique are that it does not provide the ability to select full-length cDNA transcripts of interest and that it introduces bias for amplification of shorter sequences. As a result, a range of heterogeneous amplicons are produced, often including nonspecific products (54, 55). To overcome these difficulties, 5′ RLM RACE was used in the current study, which supports amplification of only capped, full-length mRNA. However, in all 5′ RLM RACE reactions, only some of the amplified bands extended the sequence in the 5′ direction despite the presence of a ligated adapter at the 5′ ends of several other PCR products. This may have occurred due to one or more different factors of a biological and technical nature, including the presence of defective genomes and/or low efficiencies of the calf intestine alkaline phosphatase (CIP) and tobacco acid pyrophosphatase (TAP) treatments for preventing ligation of the adapter to noncapped RNA.
WPDV is the most distant of the arteriviruses based on conservation of the nonstructural proteins, and it infects the most distantly related mammalian host, a marsupial. In the arterivirus tree, its basal single-virus lineage could be contrasted with the sister lineage represented by a dozen arterivirus species which infect different placental hosts and which are separated by considerably shorter evolutionary distances. The divergence of WPDV from other arteriviruses is so profound that neither of the arterivirus-specific domains was delineated upstream of PLP2 in the putative nsp1 region by application of the most powerful profile-profile comparison techniques using conventional criteria. Only after the established homology of WPDV and arteriviruses in other nsp's was accounted for in the analysis did the profile-profile comparison identify three putative and highly divergent PLP1 domains in pp1ab. While this delineation will guide further experimental characterization of these domains, the mere presence of three paralogous PLP1 domains in WPDV is already significant for understanding the evolution of PLP1 domains in the sister lineage.
Given the presence of two or three PLP1 domains in all arteriviruses, the most recent common ancestor (MRCA) of all known arteriviruses most likely already encoded at least two PLP1 domains, one of which is expected to be the ancestor of the ubiquitous PLP1a domain (Fig. 3). This consideration also implies that a duplication of PLP1 must have happened before the emergence of this MRCA; it remains to be established whether descendants of the ancestral arterivirus lineage with a single PLP1 domain have yet to be discovered or already went extinct. Gene duplication often results in subfunctionalization or neofunctionalization, driven by positive selection to improve fitness that is facilitated by an increased evolvability of duplicates, each of which is less constrained than their ancestor (56, 92). This framework may explain the observed subdomain-specific association of most conserved residues in PLP1a and PLP1b/c of all non-WPDV arteriviruses. Given the large evolutionary distance involved (Fig. 3), these residues must be under the strong purifying selection that is commonly associated with a conserved function(s). Besides the Cys and His residues involved in catalysis, the functions of other residues have yet to be established.
Since PLP1b and PLP1c of the non-WPDV arteriviruses are much more closely related to each other than to PLP1a, they must have emerged through a second duplication and subsequent diversification. This duplication must have happened before the emergence of the MRCA of the simian group, all of whose members encode three PLP1 domains. The type- and lineage-specific evolutionary dynamics of PLP1 domains might have involved both divergent and convergent evolution and/or parallel duplication. These dynamics remain untested computationally due to poor sampling of three long-branch lineages that include just a single species each (WPDV, EAV, and APRAV), compounded by the observed variation in the number of PLP1 domains and the complexity of their similarities. For instance, similarity between PLP1b and PLP1c varies from very strong in viruses of the simian group to extremely weak in WPDV, while sequence affinity of the second PLP1 domain for PLP1b and PLP1c differs for EAV and LDV/PRRSV/APRAV, respectively. In the case of EAV, the observed affinity of the second PLP1 domain for PLP1c is both compatible with the published experimental research (33, 34) and incompatible with the current designation of this domain, PLP1b, which reflects its order in the pp1ab polyprotein. Consequently, our results predict EAV PLP1b to be closer functionally to PLP1c, which has not yet been characterized beyond its proteolytic activity in SHFV (39, 41, 46).
One of the recently identified functions of PLP1b is transactivation of nsp2 PRF, which was demonstrated for PRRSV-2 and shown to be lacking in EAV (33, 34), in line with PLP1b of the latter being similar to PLP1c (see above). Results of comparative sequence analyses by the discoverers of this phenomenon (33, 34) and those presented in this paper support the conservation of nsp2 PRF in all non-EAV arteriviruses. According to our analysis, the most divergent version of nsp2 PRF may be employed by WPDV, which deviates from other non-EAV arteriviruses in the sizes of the −2TF domain (smaller) and the region separating two PRF-related nucleotide motifs (larger). Both deviations may have functional implications. The −2TF domain of WPDV does not have hydrophobic regions predicted for other non-EAV arteriviruses, which may imply different localizations of nsp2TF proteins. The production of nsp2N and nsp2TF in PRRSV was highly sensitive to mutations that changed the size of the spacer between PRF motifs (35). Consequently, during evolution, the unusually large size of this region in WPDV must have been associated with changes elsewhere in the genome and/or been host specific. In this respect, PLP1b is a prime candidate to consider due to its role as the major domain of nsp1b in the transactivation of nsp2 PRF (34). Compared to its orthologs, PLP1b of WPDV has a unique large insertion in the left subdomain and accepted many mutations to the putative equivalent of the positively charged α-helix that was implicated in the interaction with the PRF motifs in PRRSV. While further experimental research could address a possible connection between sequence specifics of the PLP1b and PRF motifs in WPDV, our results indicate that the −2 PRF in nsp2 may be a universal feature of non-EAV arteriviruses.
The apparent production of several molecular forms of nsp2 in arteriviruses may be linked to multifunctionality of this large nsp, which remains poorly characterized. We described here the large (5-fold) variation of the size of the most divergent domain of nsp2, the HVR, among arterivirus species, which is indicative of this domain being involved in arterivirus adaptation to hosts. Duplication was likely one of the mechanisms used to increase the size of this domain, as could be deduced from the presence of three tandem repeats with the formula PxPxPR in WPDV. These repeats may mediate a conserved function, since various numbers of their counterparts were identified in many but not all arteriviruses. Their interacting partners may be host proteins containing an SH3 domain(s), which have been shown to recognize PxPxPR motifs (57). A similar suggestion was first made in a previous study (28), based on the presence of canonical SH3-binding PxxP motifs in nsp2 of PRRSV-1. However, PxxP motifs were not detected in our MEME analysis, suggesting that their presence in the HVR may be due to a disproportionally high Pro content in this domain.
In conclusion, our comparative genomic analysis of the most divergent region of replicative polyproteins revealed evolutionarily conserved patterns that are either specific to distinct species or common for different groups of arteriviruses. While the obtained insights were often the first ones for recently identified arteriviruses (12, 87–91), this analysis is also expected to promote further characterization of prototype arteriviruses, thus connecting the exploration of genetic diversity with experimental research on arteriviruses.
MATERIALS AND METHODS
Modified 5′ RLM RACE.
The protocol supplied with a commercial kit (FirstChoice RLM RACE; Invitrogen) was modified by addition of a target enrichment step. Briefly, total RNA was isolated from a standard inoculum (SI) that had been used in the previous WPD transmission studies (58) and from which the previously described partial viral sequence was obtained (11). Extracted total RNA was used as a template for the initial steps of 5′ RML RACE, performed according to the manufacturer's instructions (Fig. 1). The steps comprised treatment of total RNA with calf intestine alkaline phosphatase (CIP) to remove free 5′ phosphates from all noncapped nucleic acids, treatment with tobacco acid pyrophosphatase (TAP) to remove the cap structure from full-length mRNA (including capped positive-sense viral RNA), ligation of the provided RACE adapter to decapped mRNA containing 5′ phosphates, and reverse transcription of the ligated mRNA to cDNA by use of random decamers.
In the first round of 5′ RLM RACE (RACE 1), the cDNA was enriched for the target sequence before proceeding with the PCR step of the protocol. The enrichment step was performed using the magnetic bead, sequence capture, nested PCR method according to principles described by others (59–61). Briefly, 0.24 pmol of a biotinylated capture probe that matched the available 5′ sequence of viral RNA (biotin-WPD.S5.F) (see Table S1 in the supplemental material) was added to a reaction mix that comprised 5 μl of cDNA, 1 μl of 10× buffer O (containing 50 mM Tris-HCl, 10 mM MgCl2, 100 mM NaCl, and 0.1 mg/ml bovine serum albumin [BSA] at a 1× dilution; Fermentas), and water in a final volume of 10 μl. The nucleic acids were denatured at 95°C for 5 min and hybridized at 60°C for 23 h. An equal volume of 2× wash buffer (10 mM Tris-HCl, pH 7.5, 1 mM EDTA, 2 M NaCl) and 1 μl (5 μg) of streptavidin-coated magnetic beads (Dynabeads M280; Invitrogen) were then added to the hybridization reaction mixture, and the mixture was incubated for 3 h at 43°C with gentle shaking. The viral sequences captured on streptavidin-coated magnetic beads were then washed 3 times in 1× wash buffer and resuspended in 8 μl of water.
An aliquot (2 μl) of bead suspension was used in the PCR step of the 5′ RLM RACE protocol. Primary PCRs were performed using a 0.2 μM final concentration of each primer (RACE.outer forward primer and a virus-specific reverse primer) in 1× HOT FIREPol PCR master mix (Solis Biodyne) with 2 mM (final concentration) MgCl2. The amplification conditions included 15 min of initial denaturation at 95°C followed by 35 cycles of denaturation (95°C for 10 s), annealing (60°C for 10 s), and elongation (72°C for 1 to 3 min), followed by a final extension step (72°C for 7 min). Nested PCRs were performed as primary reactions, but a nested adapter-specific primer (RACE.inner) was used in combination with each of several virus-specific reverse primers (Table S1). The template used for nested PCR was either the primary PCR product (1 μl) or a gel-purified band from the primary PCR (1 μl). Since the lengths of the expected PCR fragments were unknown, primary PCRs were also performed using an Expand long-range PCR kit (Roche) according to the manufacturer's instructions, with an initial elongation step of 4 min at 68°C.
In order to determine whether the longest PCR product represented the 5′ end of the full-length genomic RNA, the RLM RACE protocol was repeated using another capture probe (Biotyn_S12.F) targeting a region within the newly determined 5′ end of the sequence, in combination with virus-specific primers WPD.S10.R, WPD.S13.R, WPD.S14.R, and WPD.S15.R (RACE 2) (Table S1).
The final round of 5′ RLM RACE reactions (RACE 3) was performed using virus-specific primers (WPD.S16.R and WPD.S18.R) (Table S1) located close to the 5′ end identified in RACE 2. The RACE 3 reactions were performed according to the manufacturer's instructions, without the target enrichment step. Primary and nested PCR amplifications were performed with either HOT FIREPol PCR mix or Kappa LongRange HotStart ReadyMix (Kappa Biosystems). The long-range mix was used as recommended by the manufacturer to support amplification of fragments of up to 15 kbp. The non-TAP control was included in the RACE reaction mixtures to further assess whether any of the RACE-amplified bands originated from capped RNA sequences.
The final assembly of the newly identified 5′ end with the previously published sequence (11) was confirmed by amplification of a set of overlapping PCR fragments by use of virus-specific primers and SI cDNA as the template.
The previous GenBank record (accession number JN116253) was updated to include the 5′ end of the viral sequence.
Designation of nsp1 and PLP domains.
In the literature, nsp1 PLPs and corresponding cleavage products are labeled with either the Latin letters a, b, and c or the Greek letters α, β, and γ (for example, PLP1a or PLP1α and nsp1a or nsp1α). In this report, we use Latin letters as labels.
Arterivirus genomes and classification.
Full-length genomes of arteriviruses available on 11 June 2015 were retrieved from GenBank (62) and RefSeq (63) by using the homology-annotation hybrid retrieval of genetic sequences (HAYGENS) tool (http://veb.lumc.nl/HAYGENS). The sequence of the WPDV genome, including the newly sequenced 5′ terminus, whose annotation was updated accordingly (Table 1), was added to the set. With the help of DEmARC 1.3 (64; https://talk.ictvonline.org/files/ictv_official_taxonomy_updates_since_the_8th_report/m/animal-ssrna-viruses/5890), genomes of a total of 502 viruses were clustered into 14 species (3) that were grouped into five clusters. One virus representative was selected to represent each arterivirus species in further analyses (Table 2).
MSAs.
Multiple-sequence alignments (MSAs) of pp1ab domains were generated using the Viralis platform (65) and assisted by use of the HMMER 3.1 (66), Muscle 3.8.31 (67), and ClustalW 2.012 (68) programs in default modes, with subsequent manual local refinement of MSAs of most divergent domains. Domain borders in nsp1-nsp2 proteins were tentatively identified (Table S3) through limited similarity with protein domains and cleavage cites that were studied experimentally (27, 36–39). They may differ from the ones defined elsewhere. The MSA of nsp1 PLP paralogs was prepared using the profile mode of ClustalW in a stepwise manner: first, the PLP1b and PLP1c domain alignments were combined, and then the PLP1a MSA was added. MAFFT v7.123b (69) was used to align tandem repeats (see below). All presented protein MSAs were deposited at https://github.com/aag1/Arteriviridae_nsp1-2 in FASTA format.
Quantification of MSA conservation.
To quantify residue conservation at each position of the MSA, we used the R package Bio3D 1.1.-5 (70), the “conserv” command, the “similarity” conservation assessment method, and the substitution matrix BLOSUM62 (71). Individual columns of arteriviral PLP1a and PLP1b/PLP1c alignments (WPDV sequences excluded) were considered to be conserved if their conservation score exceeded 0.75. To transform conservation scores of individual columns in the arteriviral nsp1-nsp2 MSA into a conservation profile for plotting, a sliding window of 11 MSA columns was used to calculate mean conservation score values.
Secondary structure retrieval and prediction.
Information about the PRRSV-2 nsp1a and nsp1b and EAV PLP2 secondary structures was retrieved from PDB structures 3IFU (18), 3MTV (42), and 4IUM (30), respectively, using the DSSP database (72) via the MRS system (73). Secondary structure predictions were made for individual nsp1 PLP sequences of different origins by use of Jpred4 (74) in MSA mode.
Transmembrane region prediction.
Transmembrane regions of proteins were predicted with the help of TMHMM 2.0 (75).
Profile-profile comparisons.
We employed HHmake 2.0.16 to convert protein MSAs into HMM profiles and an in-house version of HHalign 2.0.16 (76) (deposited at https://github.com/dvs/hhsuite) to conduct profile-profile comparisons. The in-house version of HHalign enables the user control over the SMIN score threshold, otherwise hard coded to be 20. The SMIN score threshold is utilized by the HHalign algorithm to decide which hits will be reported, based on their raw Viterbi scores. By lowering the SMIN score threshold, the user can increase the number of alternative hits reported, which may be informative for analyzing extremely remote relationships.
HHalign comparisons were performed with the following parameters: SMIN score threshold of 5, local alignment mode, and realignment by the MAC algorithm not applied. To visualize profile-profile comparisons in default mode, dot plots were generated.
Repeat and motif identification.
We used a multistep procedure to characterize sequence repeats and associated motifs. First, the protein sequence of a virus was compared to itself by use of HHalign. In the produced diagonal plot, overlapping off-diagonal hits with high statistical support were indicative of tandem repeats. Subsequently, the protein sequence was submitted to the RADAR Web server (49) to verify the presence of tandem repeats and to delineate their exact positions. To study if an identified repeat motif was present in sequences representing other arterivirus species, the sequences were scanned with a RADAR-produced MSA of repeats by use of HHalign (probability threshold, 5%). At the next stage, the obtained results were verified and extended by use of MEME 4.11.2 (50), which was applied to the selected protein domain of representatives of all arterivirus species. In the MEME analysis, the number of unique motifs to be found was set to 10, the expected distribution of the unique motifs' occurrences in a sequence was defined as “any number of repetitions,” the lengths of motifs were allowed to range from 4 to 50 aa, and other parameters were set to their defaults.
Nucleotide sequence profile comparisons.
We used NHMMER 3.1b1 (77) with the parameters rna-toponly-max-nonull2 to scan the EAV and WPDV genome regions encoding nsp2 for similarity to nucleotide MSAs of nsp2 PRF-related motifs from genomes representing 12 other arterivirus species.
Phylogeny reconstruction.
The phylogeny of arteriviruses was reconstructed using a concatenated MSA of most conserved nsp domains (Table S2). To select a model of evolution that best fits the data, ProtTest 3.4 (78) was used. All models offered by ProtTest were tested. When a discrete gamma distribution was employed to model various rates of mutation among sites (+G), four rate categories were used. Maximum likelihood (ML) tree topology optimization strategy, employing a subtree pruning and regrafting (SPR) algorithm, was used. Two model selection criteria, the Akaike information criterion (AIC) and the Bayesian information criterion (BIC), were employed. According to both criteria, the LG+I+G+F model is the best. Subsequently, the phylogeny was reconstructed using the BEAST 1.8.2 package (79) and the LG+I+G4+F model. Two models, a strict clock and a relaxed clock with an uncorrelated lognormal rate distribution, were tested. The latter was found to be superior (log10 Bayes factor of 5.48). Markov chain Monte Carlo (MCMC) chains were run for 10 million steps and sampled every 1,000 steps; the first 10% were discarded as burn-in. Mixing and convergence were verified with the help of Tracer (http://beast.bio.ed.ac.uk/Tracer).
A similar procedure was used to reconstruct the phylogeny of PLP1 domains by using MSA of PLP1b and PLP1c domains. Among the models available in BEAUti 1.8.2, ProtTest favored the LG+I+G4+F model, which was employed for BEAST phylogeny reconstruction. A relaxed clock with an uncorrelated lognormal rate distribution was favored over a strict clock (log10 Bayes factors of 4.88 and 3.80 for data sets with and without WPDV PLP1 domains, respectively). MCMC chains were run for 5 million steps and sampled every 500 steps; the first 10% were discarded as burn-in. The R package APE 3.5 was used to calculate the percentage of trees in the sample that differed in terms of the phylogenetic positions of major clades (80).
Tertiary protein structure comparison.
We used the DALI server (48) for comparison of PLP tertiary structures. Conventionally, two folds are considered to be similar if their similarity Z-score is above 2. However, to be considered strongly supported, the similarity Z-score must be above the cutoff, defined as n/10 − 4, where n is the number of residues in the query structure (81). For PRRSV-2 PLP1a and PLP1b queries, Z-score cutoffs were calculated to be 10.7 and 9.4, respectively.
Visualization of results of bioinformatics analyses.
Protein MSAs with highlighted conservation and assigned secondary structure were visualized with Espript 2.1 (82), using the BLOSUM62 similarity coloring scheme and a similarity global score of 0.2. MSA conservation was also presented in the logo format with the help of the R package RWebLogo 1.0.3 (83). To visualize the posterior sample of trees, DensiTree.v2.2.1 was used (84). Protein tertiary structures were processed for presentation by use of PyMOL 1.7.6.6 (85). R was used extensively for other data plotting (86).
Supplementary Material
ACKNOWLEDGMENTS
We thank Igor Sidorov for helpful discussions and for help with Viralis.
This study was partially funded by an Agreement about Cooperation in Bioinformatics between LUMC and MSU (MoBiLe Program) and by EU project EVAg 653316, the Leids Universiteits Fonds, the Massey University Research Fund, and Lewis Fitch and McGeorge Veterinary Research grants.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.02096-16.
REFERENCES
- 1.Snijder EJ, Kikkert M, Fang Y. 2013. Arterivirus molecular biology and pathogenesis. J Gen Virol 94:2141–2163. doi: 10.1099/vir.0.056341-0. [DOI] [PubMed] [Google Scholar]
- 2.Faaberg KS, Balasuriya UB, Brinton MA, Gorbalenya AE, Leung FC-C, Nauwynck H, Snijder EJ, Stadejek T, Yang H, Yoo D. 2012. Family Arteriviridae, p 796–805. In King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (ed), Virus taxonomy: classification and nomenclature of viruses. Ninth report of the International Committee on Taxonomy of Viruses. Academic Press, London, United Kingdom. [Google Scholar]
- 3.Adams MJ, Lefkowitz EJ, King AM, Harrach B, Harrison RL, Knowles NJ, Kropinski AM, Krupovic M, Kuhn JH, Mushegian AR, Nibert M, Sabanadzovic S, Sanfacon H, Siddell SG, Simmonds P, Varsani A, Zerbini FM, Gorbalenya AE, Davison AJ. 2016. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2016). Arch Virol 161:2921–2949. doi: 10.1007/s00705-016-2977-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balasuriya UB, Snijder EJ, Heidner HW, Zhang J, Zevenhoven-Dobbe JC, Boone JD, McCollum WH, Timoney PJ, MacLachlan NJ. 2007. Development and characterization of an infectious cDNA clone of the virulent Bucyrus strain of equine arteritis virus. J Gen Virol 88:918–924. doi: 10.1099/vir.0.82415-0. [DOI] [PubMed] [Google Scholar]
- 5.den Boon JA, Snijder EJ, Chirnside ED, de Vries AA, Horzinek MC, Spaan WJ. 1991. Equine arteritis virus is not a togavirus but belongs to the coronaviruslike superfamily. J Virol 65:2910–2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palmer GA, Kuo L, Chen Z, Faaberg KS, Plagemann PG. 1995. Sequence of the genome of lactate dehydrogenase-elevating virus: heterogenicity between strains P and C. Virology 209:637–642. doi: 10.1006/viro.1995.1296. [DOI] [PubMed] [Google Scholar]
- 7.Godeny EK, Chen L, Kumar SN, Methven SL, Koonin EV, Brinton MA. 1993. Complete genomic sequence and phylogenetic analysis of the lactate dehydrogenase-elevating virus (LDV). Virology 194:585–596. doi: 10.1006/viro.1993.1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zeng L, Godeny EK, Methven SL, Brinton MA. 1995. Analysis of simian hemorrhagic fever virus (SHFV) subgenomic RNAs, junction sequences, and 5′ leader. Virology 207:543–548. doi: 10.1006/viro.1995.1114. [DOI] [PubMed] [Google Scholar]
- 9.Meulenberg JJ, Hulst MM, de Meijer EJ, Moonen PL, den Besten A, de Kluyver EP, Wensvoort G, Moormann RJ. 1993. Lelystad virus, the causative agent of porcine epidemic abortion and respiratory syndrome (PEARS), is related to LDV and EAV. Virology 192:62–72. doi: 10.1006/viro.1993.1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nelsen CJ, Murtaugh MP, Faaberg KS. 1999. Porcine reproductive and respiratory syndrome virus comparison: divergent evolution on two continents. J Virol 73:270–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dunowska M, Biggs PJ, Zheng T, Perrott MR. 2012. Identification of a novel nidovirus associated with a neurological disease of the Australian brushtail possum (Trichosurus vulpecula). Vet Microbiol 156:418–424. doi: 10.1016/j.vetmic.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuhn JH, Lauck M, Bailey AL, Shchetinin AM, Vishnevskaya TV, Bao Y, Ng TF, LeBreton M, Schneider BS, Gillis A, Tamoufe U, Diffo Jle D, Takuo JM, Kondov NO, Coffey LL, Wolfe ND, Delwart E, Clawson AN, Postnikova E, Bollinger L, Lackemeyer MG, Radoshitzky SR, Palacios G, Wada J, Shevtsova ZV, Jahrling PB, Lapin BA, Deriabin PG, Dunowska M, Alkhovsky SV, Rogers J, Friedrich TC, O'Connor DH, Goldberg TL. 2016. Reorganization and expansion of the nidoviral family Arteriviridae. Arch Virol 161:755–768. doi: 10.1007/s00705-015-2672-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giles J, Perrott M, Roe W, Dunowska M. 2016. The aetiology of wobbly possum disease: reproduction of the disease with purified nidovirus. Virology 491:20–26. doi: 10.1016/j.virol.2016.01.005. [DOI] [PubMed] [Google Scholar]
- 14.Mackintosh C, Crawford J, Thompson E, McLeod B, Gill J, O'Keefe J. 1995. A newly discovered disease of the brushtail possum: wobbly possum syndrome. N Z Vet J 43:126. doi: 10.1080/00480169.1995.35869. [DOI] [PubMed] [Google Scholar]
- 15.Perrott M, Meers J, Cooke M, Wilks C. 2000. A neurological syndrome in a free-living population of possums (Trichosurus vulpecula). N Z Vet J 48:9–15. doi: 10.1080/00480169.2000.36150. [DOI] [PubMed] [Google Scholar]
- 16.Holtkamp DJ, Kliebenstein JB, Neumann EJ, Zimmerman JJ, Rotto HF, Yoder TK, Wang C, Yeske PE, Mowrer CL, Haley CA. 2013. Assessment of the economic impact of porcine reproductive and respiratory syndrome virus on United States pork producers. J Swine Health Prod 21:72–84. [Google Scholar]
- 17.Snijder EJ, Wassenaar AL, Spaan WJ. 1992. The 5′ end of the equine arteritis virus replicase gene encodes a papainlike cysteine protease. J Virol 66:7040–7048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun Y, Xue F, Guo Y, Ma M, Hao N, Zhang XC, Lou Z, Li X, Rao Z. 2009. Crystal structure of porcine reproductive and respiratory syndrome virus leader protease Nsp1alpha. J Virol 83:10931–10940. doi: 10.1128/JVI.02579-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nedialkova DD, Gorbalenya AE, Snijder EJ. 2010. Arterivirus Nsp1 modulates the accumulation of minus-strand templates to control the relative abundance of viral mRNAs. PLoS Pathog 6:e1000772. doi: 10.1371/journal.ppat.1000772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ziebuhr J, Snijder EJ, Gorbalenya AE. 2000. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J Gen Virol 81:853–879. doi: 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]
- 21.Lauber C, Goeman JJ, Parquet MC, Nga PT, Snijder EJ, Morita K, Gorbalenya AE. 2013. The footprint of genome architecture in the largest genome expansion in RNA viruses. PLoS Pathog 9:e1003500. doi: 10.1371/journal.ppat.1003500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lehmann KC, Gulyaeva A, Zevenhoven-Dobbe JC, Janssen GM, Ruben M, Overkleeft HS, van Veelen PA, Samborskiy DV, Kravchenko AA, Leontovich AM, Sidorov IA, Snijder EJ, Posthuma CC, Gorbalenya AE. 2015. Discovery of an essential nucleotidylating activity associated with a newly delineated conserved domain in the RNA polymerase-containing protein of all nidoviruses. Nucleic Acids Res 43:8416–8434. doi: 10.1093/nar/gkv838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lehmann KC, Hooghiemstra L, Gulyaeva A, Samborskiy DV, Zevenhoven-Dobbe JC, Snijder EJ, Gorbalenya AE, Posthuma CC. 2015. Arterivirus nsp12 versus the coronavirus nsp16 2′-O-methyltransferase: comparison of the C-terminal cleavage products of two nidovirus pp1ab polyproteins. J Gen Virol 96:2643–2655. doi: 10.1099/vir.0.000209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Snijder EJ, Bredenbeek PJ, Dobbe JC, Thiel V, Ziebuhr J, Poon LLM, Guan Y, Rozanov M, Spaan WJM, Gorbalenya AE. 2003. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J Mol Biol 331:991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Snijder EJ, Wassenaar ALM, Spaan WJM, Gorbalenya AE. 1995. The arterivirus nsp2 protease—an unusual cysteine protease with primary structure similarities to both papain-like and chymotrypsin-like proteases. J Biol Chem 270:16671–16676. doi: 10.1074/jbc.270.28.16671. [DOI] [PubMed] [Google Scholar]
- 26.Manolaridis I, Gaudin C, Posthuma CC, Zevenhoven-Dobbe JC, Imbert I, Canard B, Kelly G, Tucker PA, Conte MR, Snijder EJ. 2011. Structure and genetic analysis of the arterivirus nonstructural protein 7alpha. J Virol 85:7449–7453. doi: 10.1128/JVI.00255-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kikkert M, Snijder EJ, Gorbalenya AE. 2013. Arterivirus nsp2 cysteine proteinase, p 2210–2215. In Rawlings ND, Salvesen GS (ed), Handbook of proteolytic enzymes, 3rd ed, vol 2 Academic Press, London, United Kingdom. [Google Scholar]
- 28.Ropp SL, Wees CE, Fang Y, Nelson EA, Rossow KD, Bien M, Arndt B, Preszler S, Steen P, Christopher-Hennings J, Collins JE, Benfield DA, Faaberg KS. 2004. Characterization of emerging European-like porcine reproductive and respiratory syndrome virus isolates in the United States. J Virol 78:3684–3703. doi: 10.1128/JVI.78.7.3684-3703.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Faaberg KS, Kehrli ME Jr, Lager KM, Guo B, Han J. 2010. In vivo growth of porcine reproductive and respiratory syndrome virus engineered nsp2 deletion mutants. Virus Res 154:77–85. doi: 10.1016/j.virusres.2010.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Kasteren PB, Bailey-Elkin BA, James TW, Ninaber DK, Beugeling C, Khajehpour M, Snijder EJ, Mark BL, Kikkert M. 2013. Deubiquitinase function of arterivirus papain-like protease 2 suppresses the innate immune response in infected host cells. Proc Natl Acad Sci U S A 110:E838–E847. doi: 10.1073/pnas.1218464110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fang Y, Snijder EJ. 2010. The PRRSV replicase: exploring the multifunctionality of an intriguing set of nonstructural proteins. Virus Res 154:61–76. doi: 10.1016/j.virusres.2010.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wassenaar AL, Spaan WJ, Gorbalenya AE, Snijder EJ. 1997. Alternative proteolytic processing of the arterivirus replicase ORF1a polyprotein: evidence that NSP2 acts as a cofactor for the NSP4 serine protease. J Virol 71:9313–9322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fang Y, Treffers EE, Li Y, Tas A, Sun Z, van der Meer Y, de Ru AH, van Veelen PA, Atkins JF, Snijder EJ, Firth AE. 2012. Efficient −2 frameshifting by mammalian ribosomes to synthesize an additional arterivirus protein. Proc Natl Acad Sci U S A 109:E2920–E2928. doi: 10.1073/pnas.1211145109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Y, Treffers EE, Napthine S, Tas A, Zhu L, Sun Z, Bell S, Mark BL, van Veelen PA, van Hemert MJ, Firth AE, Brierley I, Snijder EJ, Fang Y. 2014. Transactivation of programmed ribosomal frameshifting by a viral protein. Proc Natl Acad Sci U S A 111:E2172–E2181. doi: 10.1073/pnas.1321930111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Napthine S, Treffers EE, Bell S, Goodfellow I, Fang Y, Firth AE, Snijder EJ, Brierley I. 2016. A novel role for poly(C) binding proteins in programmed ribosomal frameshifting. Nucleic Acids Res 44:5491–5503. doi: 10.1093/nar/gkw480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nedialkova DD, Gorbalenya AE, Snijder EJ. 2013. Arterivirus papain-like proteinase 1β, p 2205–2210. In Rawlings ND, Salvesen GS (ed), Handbook of proteolytic enzymes, 3rd ed, vol 2 Academic Press, London, United Kingdom. [Google Scholar]
- 37.Nedialkova DD, Gorbalenya AE, Snijder EJ. 2013. Arterivirus papain-like proteinase 1α, p 2199–2204. In Rawlings ND, Salvesen GS (ed), Handbook of proteolytic enzymes, 3rd ed, vol 2 Academic Press, London, United Kingdom. [Google Scholar]
- 38.Li Y, Tas A, Sun Z, Snijder EJ, Fang Y. 2015. Proteolytic processing of the porcine reproductive and respiratory syndrome virus replicase. Virus Res 202:48–59. doi: 10.1016/j.virusres.2014.12.027. [DOI] [PubMed] [Google Scholar]
- 39.Vatter HA, Di H, Donaldson EF, Radu GU, Maines TR, Brinton MA. 2014. Functional analyses of the three simian hemorrhagic fever virus nonstructural protein 1 papain-like proteases. J Virol 88:9129–9140. doi: 10.1128/JVI.01020-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.den Boon JA, Faaberg KS, Meulenberg JJ, Wassenaar AL, Plagemann PG, Gorbalenya AE, Snijder EJ. 1995. Processing and evolution of the N-terminal region of the arterivirus replicase ORF1a protein: identification of two papainlike cysteine proteases. J Virol 69:4500–4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Han M, Kim CY, Rowland RR, Fang Y, Kim D, Yoo D. 2014. Biogenesis of non-structural protein 1 (nsp1) and nsp1-mediated type I interferon modulation in arteriviruses. Virology 458–459:136–150. doi: 10.1016/j.virol.2014.04.028. [DOI] [PubMed] [Google Scholar]
- 42.Xue F, Sun Y, Yan L, Zhao C, Chen J, Bartlam M, Li X, Lou Z, Rao Z. 2010. The crystal structure of porcine reproductive and respiratory syndrome virus nonstructural protein Nsp1beta reveals a novel metal-dependent nuclease. J Virol 84:6461–6471. doi: 10.1128/JVI.00301-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tijms MA, van Dinten LC, Gorbalenya AE, Snijder EJ. 2001. A zinc finger-containing papain-like protease couples subgenomic mRNA synthesis to genome translation in a positive-stranded RNA virus. Proc Natl Acad Sci U S A 98:1889–1894. doi: 10.1073/pnas.98.4.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tijms MA, Nedialkova DD, Zevenhoven-Dobbe JC, Gorbalenya AE, Snijder EJ. 2007. Arterivirus subgenomic mRNA synthesis and virion biogenesis depend on the multifunctional nsp1 autoprotease. J Virol 81:10496–10505. doi: 10.1128/JVI.00683-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kroese MV, Zevenhoven-Dobbe JC, Bos-de Ruijter JN, Peeters BP, Meulenberg JJ, Cornelissen LA, Snijder EJ. 2008. The nsp1alpha and nsp1 papain-like autoproteinases are essential for porcine reproductive and respiratory syndrome virus RNA synthesis. J Gen Virol 89:494–499. doi: 10.1099/vir.0.83253-0. [DOI] [PubMed] [Google Scholar]
- 46.Han M, Yoo D. 2014. Modulation of innate immune signaling by nonstructural protein 1 (nsp1) in the family Arteriviridae. Virus Res 194:100–109. doi: 10.1016/j.virusres.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Go YY, Li YH, Chen ZH, Han MY, Yoo DW, Fang Y, Balasuriya UBR. 2014. Equine arteritis virus does not induce interferon production in equine endothelial cells: identification of nonstructural protein 1 as a main interferon antagonist. Biomed Res Int 2014:420658. doi: 10.1155/2014/420658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Holm L, Rosenstrom P. 2010. Dali server: conservation mapping in 3D. Nucleic Acids Res 38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heger A, Holm L. 2000. Rapid automatic detection and alignment of repeats in protein sequences. Proteins 41:224–237. doi: 10.1002/1097-0134(20001101)41:2<224::AID-PROT70>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 50.Bailey TL, Elkan C. 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2:28–36. [PubMed] [Google Scholar]
- 51.Percudani R. 2001. Restricted wobble rules for eukaryotic genomes. Trends Genet 17:133–135. [DOI] [PubMed] [Google Scholar]
- 52.van den Born E, Posthuma CC, Gultyaev AP, Snijder EJ. 2005. Discontinuous subgenomic RNA synthesis in arteriviruses is guided by an RNA hairpin structure located in the genomic leader region. J Virol 79:6312–6324. doi: 10.1128/JVI.79.10.6312-6324.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frohman MA. 1994. On beyond classic RACE (rapid amplification of cDNA ends). PCR Methods Appl 4:S40–S58. doi: 10.1101/gr.4.1.S40. [DOI] [PubMed] [Google Scholar]
- 54.Schaefer BC. 1995. Revolutions in rapid amplification of cDNA ends: new strategies for polymerase chain reaction cloning of full-length cDNA ends. Anal Biochem 227:255–273. doi: 10.1006/abio.1995.1279. [DOI] [PubMed] [Google Scholar]
- 55.Schramm G, Bruchhaus I, Roeder T. 2000. A simple and reliable 5′-RACE approach. Nucleic Acids Res 28:E96. doi: 10.1093/nar/28.22.e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Innan H, Kondrashov F. 2010. The evolution of gene duplications: classifying and distinguishing between models. Nat Rev Genet 11:97–108. doi: 10.1038/nrg2689. [DOI] [PubMed] [Google Scholar]
- 57.Kowanetz K, Szymkiewicz I, Haglund K, Kowanetz M, Husnjak K, Taylor JD, Soubeyran P, Engstrom U, Ladbury JE, Dikic I. 2003. Identification of a novel proline-arginine motif involved in CIN85-dependent clustering of Cbl and down-regulation of epidermal growth factor receptors. J Biol Chem 278:39735–39746. doi: 10.1074/jbc.M304541200. [DOI] [PubMed] [Google Scholar]
- 58.Perrott MR, Wilks CR, Meers J. 2000. Routes of transmission of wobbly possum disease. N Z Vet J 48:3–8. doi: 10.1080/00480169.2000.36149. [DOI] [PubMed] [Google Scholar]
- 59.Allen GP. 2006. Antemortem detection of latent infection with neuropathogenic strains of equine herpesvirus-1 in horses. Am J Vet Res 67:1401–1405. doi: 10.2460/ajvr.67.8.1401. [DOI] [PubMed] [Google Scholar]
- 60.Tagle DA, Swaroop M, Lovett M, Collins FS. 1993. Magnetic bead capture of expressed sequences encoded within large genomic segments. Nature 361:751–753. doi: 10.1038/361751a0. [DOI] [PubMed] [Google Scholar]
- 61.Morgan JG, Dolganov GM, Robbins SE, Hinton LM, Lovett M. 1992. The selective isolation of novel cDNAs encoded by the regions surrounding the human interleukin 4 and 5 genes. Nucleic Acids Res 20:5173–5179. doi: 10.1093/nar/20.19.5173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. 2016. GenBank. Nucleic Acids Res 44:D67–D72. doi: 10.1093/nar/gkv1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.O'Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, Astashyn A, Badretdin A, Bao Y, Blinkova O, Brover V, Chetvernin V, Choi J, Cox E, Ermolaeva O, Farrell CM, Goldfarb T, Gupta T, Haft D, Hatcher E, Hlavina W, Joardar VS, Kodali VK, Li W, Maglott D, Masterson P, McGarvey KM, Murphy MR, O'Neill K, Pujar S, Rangwala SH, Rausch D, Riddick LD, Schoch C, Shkeda A, Storz SS, Sun H, Thibaud-Nissen F, Tolstoy I, Tully RE, Vatsan AR, Wallin C, Webb D, Wu W, Landrum MJ, Kimchi A, Tatusova T, DiCuccio M, Kitts P, Murphy TD, Pruitt KD. 2016. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 44:D733–D745. doi: 10.1093/nar/gkv1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lauber C, Gorbalenya AE. 2012. Partitioning the genetic diversity of a virus family: approach and evaluation through a case study of picornaviruses. J Virol 86:3890–3904. doi: 10.1128/JVI.07173-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gorbalenya AE, Lieutaud P, Harris MR, Coutard B, Canard B, Kleywegt GJ, Kravchenko AA, Samborskiy DV, Sidorov IA, Leontovich AM, Jones TA. 2010. Practical application of bioinformatics by the multidisciplinary VIZIER consortium. Antiviral Res 87:95–110. doi: 10.1016/j.antiviral.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Eddy SR. 2009. A new generation of homology search tools based on probabilistic inference. Genome Inform 23:205–211. [PubMed] [Google Scholar]
- 67.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 69.Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Grant BJ, Rodrigues AP, ElSawy KM, McCammon JA, Caves LS. 2006. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics 22:2695–2696. doi: 10.1093/bioinformatics/btl461. [DOI] [PubMed] [Google Scholar]
- 71.Henikoff S, Henikoff JG. 1992. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A 89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Touw WG, Baakman C, Black J, te Beek TA, Krieger E, Joosten RP, Vriend G. 2015. A series of PDB-related databanks for everyday needs. Nucleic Acids Res 43:D364–D368. doi: 10.1093/nar/gku1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hekkelman ML, Vriend G. 2005. MRS: a fast and compact retrieval system for biological data. Nucleic Acids Res 33:W766–W769. doi: 10.1093/nar/gki422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Drozdetskiy A, Cole C, Procter J, Barton GJ. 2015. JPred4: a protein secondary structure prediction server. Nucleic Acids Res 43:W389–W394. doi: 10.1093/nar/gkv332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sonnhammer EL, von Heijne G, Krogh A. 1998. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc Int Conf Intell Syst Mol Biol 6:175–182. [PubMed] [Google Scholar]
- 76.Remmert M, Biegert A, Hauser A, Soding J. 2011. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods 9:173–175. doi: 10.1038/nmeth.1818. [DOI] [PubMed] [Google Scholar]
- 77.Wheeler TJ, Eddy SR. 2013. nhmmer: DNA homology search with profile HMMs. Bioinformatics 29:2487–2489. doi: 10.1093/bioinformatics/btt403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Darriba D, Taboada GL, Doallo R, Posada D. 2011. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27:1164–1165. doi: 10.1093/bioinformatics/btr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Drummond AJ, Suchard MA, Xie D, Rambaut A. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Paradis E, Claude J, Strimmer K. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- 81.Holm L, Kaariainen S, Rosenstrom P, Schenkel A. 2008. Searching protein structure databases with DaliLite v.3. Bioinformatics 24:2780–2781. doi: 10.1093/bioinformatics/btn507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gouet P, Robert X, Courcelle E. 2003. ESPript/ENDscript: extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res 31:3320–3323. doi: 10.1093/nar/gkg556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res 14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Heled J, Bouckaert RR. 2013. Looking for trees in the forest: summary tree from posterior samples. BMC Evol Biol 13:221. doi: 10.1186/1471-2148-13-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schrödinger LLC. 2015. The PyMOL molecular graphics system, version 1.7.6.6. https://www.pymol.org/. [Google Scholar]
- 86.R Core Team. 2013. R: a language and environment for statistical computing. Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org. [Google Scholar]
- 87.Lauck M, Hyeroba D, Tumukunde A, Weny G, Lank SM, Chapman CA, O'Connor DH, Friedrich TC, Goldberg TL. 2011. Novel, divergent simian hemorrhagic fever viruses in a wild Ugandan red colobus monkey discovered using direct pyrosequencing. PLoS One 6:e19056. doi: 10.1371/journal.pone.0019056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bailey AL, Lauck M, Weiler A, Sibley SD, Dinis JM, Bergman Z, Nelson CW, Correll M, Gleicher M, Hyeroba D, Tumukunde A, Weny G, Chapman C, Kuhn JH, Hughes AL, Friedrich TC, Goldberg TL, O'Connor DH. 2014. High genetic diversity and adaptive potential of two simian hemorrhagic fever viruses in a wild primate population. PLoS One 9:e90714. doi: 10.1371/journal.pone.0090714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lauck M, Sibley SD, Hyeroba D, Tumukunde A, Weny G, Chapman CA, Ting N, Switzer WM, Kuhn JH, Friedrich TC, O'Connor DH, Goldberg TL. 2013. Exceptional simian hemorrhagic fever virus diversity in a wild African primate community. J Virol 87:688–691. doi: 10.1128/JVI.02433-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bailey AL, Lauck M, Sibley SD, Pecotte J, Rice K, Weny G, Tumukunde A, Hyeroba D, Greene J, Correll M, Gleicher M, Friedrich TC, Jahrling PB, Kuhn JH, Goldberg TL, Rogers J, O'Connor DH. 2014. Two novel simian arteriviruses in captive and wild baboons (Papio spp.). J Virol 88:13231–13239. doi: 10.1128/JVI.02203-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lauck M, Alkhovsky SV, Bao Y, Bailey AL, Shevtsova ZV, Shchetinin AM, Vishnevskaya TV, Lackemeyer MG, Postnikova E, Mazur S, Wada J, Radoshitzky SR, Friedrich TC, Lapin BA, Deriabin PG, Jahrling PB, Goldberg TL, O'Connor DH, Kuhn JH. 2015. Historical outbreaks of simian hemorrhagic fever in captive macaques were caused by distinct arteriviruses. J Virol 89:8082–8087. doi: 10.1128/JVI.01046-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang J. 2003. Evolution by gene duplication: an update. Trends Ecol Evol 18:292–298. doi: 10.1016/S0169-5347(03)00033-8. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.