

Supplemental Digital Content is available in the text.
Keywords: cor triatriatum, developmental biology, endocardium, extracellular matrix, live birth
Abstract
Background—
Hyaluronan (HA) is required for endothelial-to-mesenchymal transition and normal heart development in the mouse. Heart abnormalities in hyaluronidase 2 (HYAL2)–deficient (Hyal2−/−) mice and humans suggested removal of HA is also important for normal heart development. We have performed longitudinal studies of heart structure and function in Hyal2−/− mice to determine when, and how, HYAL2 deficiency leads to these abnormalities.
Methods and Results—
Echocardiography revealed atrial enlargement, atrial tissue masses, and valvular thickening at 4 weeks of age, as well as diastolic dysfunction that progressed with age, in Hyal2−/− mice. These abnormalities were associated with increased HA, vimentin-positive cells, and fibrosis in Hyal2−/− compared with control mice. Based on the severity of heart dysfunction, acute and chronic groups of Hyal2−/− mice that died at an average of 12 and 25 weeks respectively, were defined. Increased HA levels and mesenchymal cells, but not vascular endothelial growth factor in Hyal2−/− embryonic hearts, suggest that HYAL2 is important to inhibit endothelial-to-mesenchymal transition. Consistent with this, in wild-type embryos, HYAL2 and HA were readily detected, and HA levels decreased with age.
Conclusions—
These data demonstrate that disruption of normal HA catabolism in Hyal2−/− mice causes increased HA, which may promote endothelial-to-mesenchymal transition and proliferation of mesenchymal cells. Excess endothelial-to-mesenchymal transition, resulting in increased mesenchymal cells, is the likely cause of morphological heart abnormalities in both humans and mice. In mice, these abnormalities result in progressive and severe diastolic dysfunction, culminating in heart failure.
Congenital heart disease affects ≈50 of every 1000 live births.1 The genetic causes of congenital heart disease are highly diverse, reflecting the complexity of normal heart development. Mutations affecting components of the extracellular matrix (ECM) are associated with a range of congenital heart diseases, including Marfan syndrome and dilated cardiomyopathy.2 A deficiency in the synthesis of the ECM glycosaminoglycan, hyaluronan (HA), causes early embryonic death in mice because of defective heart development.3 Recently, defective degradation of HA because of hyaluronidase 2 (HYAL2) deficiency was identified as a cause of cor triatriatum sinister and other heart abnormalities in both humans and mice.4–6
During embryogenesis, the heart develops as a straight tube consisting of an outer layer of myocardium and an inner layer of endocardium. At embryonic day (E) 9.5 in the mouse, the ECM between these layers expands to form the cardiac jelly. Extensive remodeling of the cardiac ECM and looping of the tube forms the atrioventricular canal, outflow tract, and cardiac cushion.7 Endothelial-to-mesenchymal transition (EMT) within the cushions establishes the primordium that will develop into the valves and ventricular septum.8 This primordium grows into thin fibrous valve leaflets/cups that are matured through ECM deposition and remodeling,9 even after birth, to form the mature heart.
HA is an abundant component of the provisional matrix in the developing heart and the mature matrix of adult heart valves.3,10 A critical role for HA during heart development has been demonstrated in HA synthase 2 (HAS2)–deficient mouse embryos. These embryos died at E9.5 because of a failure to form the HA-rich cardiac jelly needed to support EMT to form the heart valves and septa.3 Studies of mouse cardiac explants revealed that exogenous high-molecular-mass HA promoted EMT, whereas HA fragments inhibited EMT and activated the vascular endothelial growth factor (VEGF) pathway to promote differentiation.11 Until now, in vivo studies of the role of hyaluronidase in normal EMT and heart development have not been done.
Degradation of HA is presumed to be accomplished in somatic cells by HYAL1 and HYAL2.12 HYAL2 is a glycophosphatidylinositol-linked protein13 with weak activity toward high-molecular-mass extracellular HA to produce fragments of ≈ 20 kDa.12 These fragments are thought to bind a cell surface receptor for endocytosis and be transported to the lysosome for degradation by HYAL1 and the exoglycosidases.14 An important role for HYAL2 in HA degradation is demonstrated by the craniofacial abnormalities, preweaning lethality (only 33% survived at weaning), atrial enlargement (50%), cor triatriatum (50%), and valve thickening (100%) present in Hyal2−/− mice.4,5,15 Further, histological analyses showed significant accumulation of HA in Hyal2−/− mice that was absent in control mice.16 However, the developmental basis of these changes, their effect on function, and whether the phenotypes progress with age was unknown.
In this study, we have analyzed heart structure and function in Hyal2−/− and control mice using echocardiography. Severe atrial dilation accompanied by diastolic dysfunction was found as early as 4 weeks, and progressed with age, in Hyal2−/− mice. Histological analyses revealed that atrial dilation resulted from excess tissue and did not correlate with the presence of cor triatriatum. Hyal2−/− mice had increased numbers of mesenchymal cells during development, suggesting increased EMT or mesenchymal cell proliferation and decreased differentiation, presumably because of the presence of excess HA. These findings suggest that HA degradation by HYAL2 is required to attenuate EMT and mesenchymal cell proliferation and that excess mesenchymal cells formed in its absence cause fibrosis that leads to diastolic dysfunction.
Materials and Methods
Mice
Mice that are null for HYAL2, Hyal2−/− mice, were generated previously.15 These mice were maintained on an outbred (C57BL6; C129; CD1) background on which 9% of Hyal2−/− mice survive.5 Hyal2−/− and control mice (Hyal2+/+) were generated through heterozygous intercrosses. Embryos for analysis of heart development were collected from timed-pregnant females at E18.5 and E14.5. Polymerase chain reaction–based genotyping of DNA from tissue samples collected from offspring or embryos was performed as described previously.5 All studies were performed using protocols approved by the University of Manitoba Animal Care Committee in accordance with the Canadian Council on Animal Care.
Ultrasound Analyses of Heart Function
Cardiac imaging of Hyal2−/− and control mice was performed using high-frequency ultrasound with the Vevo 2100 system (Visual Sonics, Toronto, Canada) equipped with a 40 MHz transducer.17 Mice were imaged at 4 weeks of age, and then every 4 weeks until 6 months of age, unless an earlier humane end point requiring euthanasia was reached. Euthanasia was performed by isoflurane overdose. During imaging, the body temperature of the mice was maintained at 37±0.5°C under mild anesthesia (sedated with 3% isoflurane and 1.0 L/min oxygen and maintained at 1–1.5% isoflurane and 1.0 L/min oxygen).
Structural and functional cardiac parameters were assessed using 3 imaging formats: brightness mode, motion mode, and Doppler imaging. Brightness mode was used to generate 2-dimensional views of cardiac and associated vasculatures, whereas the motion mode was used to characterize ventricular functional parameters, and pulsed-wave Doppler was used to determine velocity and direction of the blood flow. Measurements were averaged over 4 cardiac cycles. Each parameter used in the calculations for the study was measured in triplicate for each mouse at each time point. The data were analyzed by a trained and blinded research animal echocardiographer using the Cardiovascular Package from VisualSonics following a published approach.17
Micro-Computed Tomography
Hearts were fixed overnight in alcoholic Bouin solution containing 1% phosphotungstic acid as described previously18 and imaged at 9 μm using the Skyscan 1176 micro-computed tomographic scanner. The scanning parameters were set at 0.5-mm-thick aluminum filter, x-ray source voltage 50 kV, and current 500 μA. The reconstructed 3-dimensional images were space-filled, rotated, and colorized using Bruker-Micro-CT CT-Analyser Version 1.13.
Histology
Hearts were harvested from adult Hyal2−/− mice at 6 months of age unless a humane end point was reached earlier. Each control heart was collected at the same time as an experimental (Hyal2−/−) heart. Embryonic hearts were collected at E14.5 or E18.5. Wild-type embryos at E8.5, 11.5, and 12.5 were obtained as part of a previous study.19 Morphology was examined by hematoxylin and eosin staining, and HA was detected using the HABP (HA-binding protein) following established procedures.5 ECM components were visualized with Masson trichrome (Sigma) following the manufacturer’s instructions.
Immunochemistry and Immunoblots
Immunohistochemistry was performed as described previously,5 except that antigen retrieval was in 0.1% sodium acetate pH 6.0 for 20 minutes. Polyclonal antibodies to vimentin (Abcam; 1:700), VEGFA (Proteintech; 1:1000), and HYAL2 (Abcam; 1:200) were detected with biotinylated goat antirabbit antibody (Vector Laboratories; 1:500). Cell counting per unit area was with Image J Fiji 1.46 software.20 Immunoblots using antivimentin (1/1000) or anti-β-actin antibody (1/5000) to detect protein in heart extracts prepared by sonication in PBS were as described previously.21
Statistical Analysis
Data are presented as mean±SEM. Student t tests were used to compare groups, and values of P<0.05 were considered significant. For each cardiac phenotype with repeated measures, comparisons were conducted using PROC MIXED from SAS v9.3 (SAS Institute Inc, Cary, NC).
Results
Atrial Enlargement in Hyal2−/− Mice
To understand the developmental origins of the cardiac abnormalities in Hyal2−/− mice, we conducted a longitudinal analysis of cardiac structure and function at 4-week intervals beginning at 4 weeks of age using high-frequency ultrasound. At 4 weeks of age, all Hyal2−/− mice exhibited significant atrial enlargement (Figure 1A through 1C, dashed lines). The most severely affected mice reached a humane end point at an average of 9 weeks, 15 weeks earlier than their less severely affected Hyal2−/− littermates. The severe atrial enlargement in 50% of mice was consistent with our previous study in which 54% of mice were found to have severe atrial enlargement at death.5 On the basis of these findings, severely affected Hyal2−/− mice were deemed acute, and less severely affected Hyal2−/− mice were deemed chronic; these 2 groups were analyzed independently throughout the study. Quantification of the atrial enlargement by measuring the diameter of the atrium from 2-dimensional ultrasound images revealed a 1.8-fold increase in the acute Hyal2−/− mice at 4 weeks of age compared with controls and a 1.3-fold increase in acute compared to chronic Hyal2−/− mice (Figure 1G).
Figure 1.
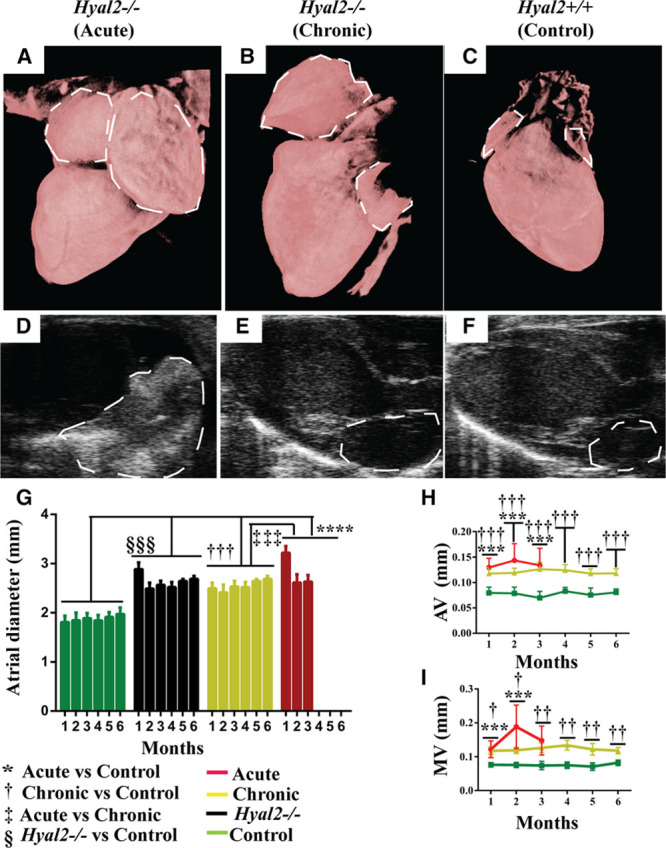
Structural abnormalities in hyaluronidase 2 (HYAL2)–deficient (Hyal2−/−) mouse hearts. A–C, High-resolution micro-computed tomographic images of Hyal2−/− and control hearts. Micro-computed tomographic images were reconstructed in 3-dimensional and colorized to enhance structural visualization of the atria (dashed lines). Hyal2−/− mice with a grossly enlarged atrium (A) were deemed acute, whereas those with a mildly enlarged atrium (B) were deemed as chronic. A heart from a control mouse is shown in (C). D–F, Ultrasound images of Hyal2−/− and control hearts. Bright-mode images of the heart showed an enlarged and dense left atrium (dashed lines) in the acute Hyal2−/− mice compared with chronic Hyal2−/− and control mice. The increased density is indicated in the image by the stronger white signal. G, Atrium diameter in Hyal2−/− and control mice. The diameter of the atrium was significantly larger in both acute and chronic Hyal2−/− mice compared with controls. H, Atrial valve (AV) and (I) mitral valve (MV) thickness was significantly increased in acute and chronic Hyal2−/− mice compared with controls. *, †, ‡P<0.05; **, ††P<0.001; ***, †††, ‡‡‡, §§§P<0.0001. The number of animals used were for atrial enlargement control and chronic (n=6); acute (at 1 mo n=7; 2 mo n=4; and 3 mo n=3), for AV thickness control and chronic (n=6); acute (at 1 mo n=7; 2 mo n=4, and 3 mo n=3), for MV thickness control and chronic (n=6), acute (at 1 mo n=6; 2 mo n=4, and 3 mo n=3).
Ultrasound imaging revealed increased tissue density in the atria of Hyal2−/− mice (Figure 1D through 1F), which in the acute group blocked the view of the atrium (Figure 1D) that was normally clearly visible (Figure 1E and 1F). No progressive change in the size of the atrium was detected in the Hyal2−/− mice (Figure 1G). We were unable to measure the ventricular diameter because the apex of the heart could not be reproducibly visualized in the ultrasound images. However, there were no instances where the ventricles were grossly distended like the atria of Hyal2−/− mice.
Valve Thickening in Hyal2−/− Mice
We previously showed that all heart valves were thickened in adult Hyal2−/− mice.5 However, whether this thickening occurred before or after birth was unknown. Brightness-mode images revealed significantly thickened valves were already present at 4 weeks of age in Hyal2−/− mice and did not change significantly with age (Figure 1H and 1I). No significant difference was found in the valve thickness of acute and chronic Hyal2−/− mice. Brightness-mode imaging allowed the measurement of only the aortic and mitral valves, which we used as a proxy for valve thickness in general (Figure I in the Data Supplement). Interestingly, histological studies of the heart valves at postnatal day 1 (P1) revealed only minimal valve thickening in Hyal2−/− mice, suggesting that thickening occurred during early postnatal valve remodeling.
Heart Function in Hyal2−/− Mice
To determine how the structural abnormalities affected cardiac function in Hyal2−/− mice,5 we analyzed several parameters using echocardiography. The peak velocities of early to late atrial filling of the left ventricle were inverted in Hyal2−/− mice (Figure II in the Data Supplement) and resulted in a significantly reduced early to late atrial filling ratio in all Hyal2−/− mice compared with controls (Figure 2A). In the acute Hyal2−/− mice, the early to late atrial filling ratio was significantly lower at 3 months than that in the chronic Hyal2−/− mice. Another measure of left ventricular (LV) diastolic function, the isovolumic relaxation time, was significantly increased in all Hyal2−/− mice at all time points (Figure 2B). The reduced early to late atrial filling ratio and prolonged isovolumic relaxation time show there is an increased interval between mitral valve closure and aortic valve opening, indicating severe diastolic dysfunction in both acute and chronic Hyal2−/− mice compared with controls, although the acute Hyal2−/− mice were severely affected at earlier ages.
Figure 2.
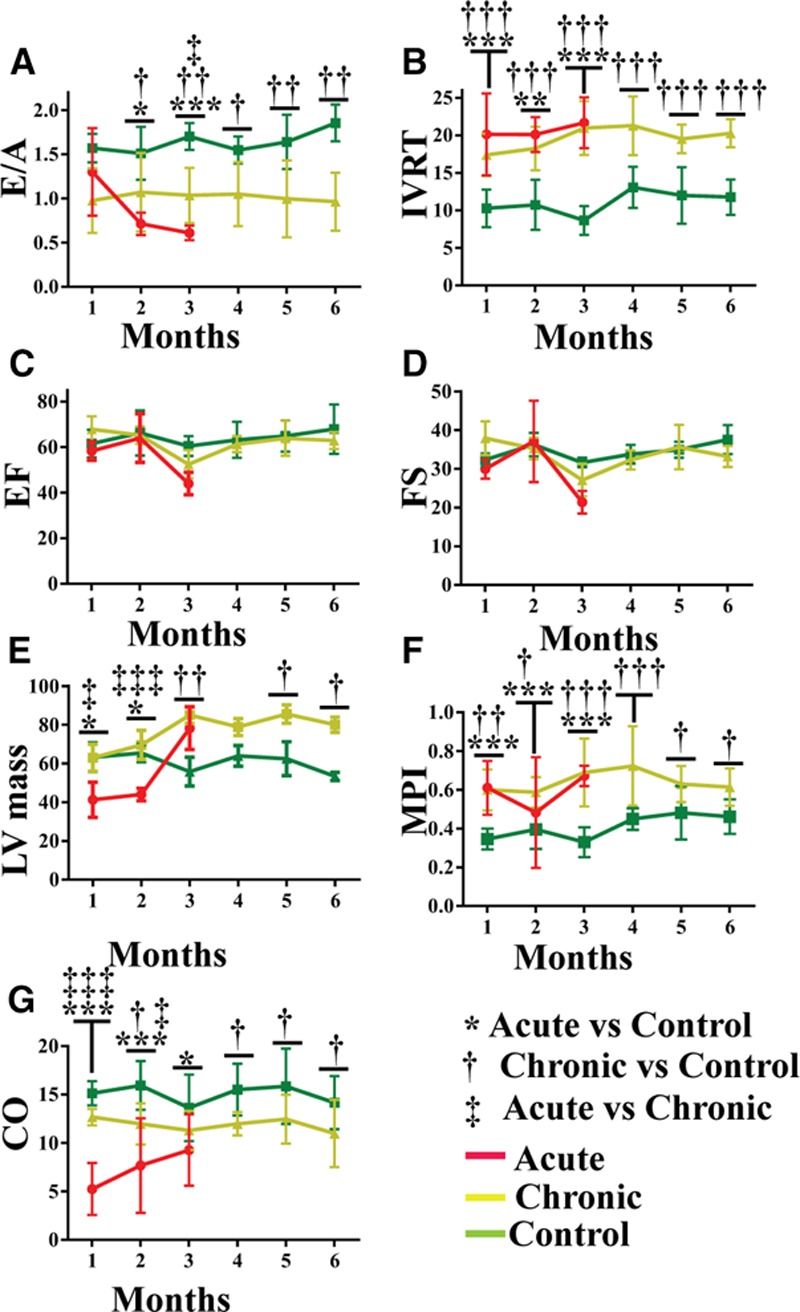
Cardiac function in hyaluronidase 2 (HYAL2)–deficient (Hyal2−/−) and control mice. Pulse-wave Doppler images were used to measure early to late atrial filling (E/A; A) and isovolumic relaxation time (IVRT; B). A and B, Acute and chronic Hyal2−/− mice showed significant impairment at all ages compared with controls. There was no significant difference between acute and chronic Hyal2−/− mice except at 4 wk where the IVRT of the acute group was significantly prolonged compared with the chronic group. C and D, Motion-mode images were used to measure systolic parameters including ejection fraction (EF) and fractional shortening (FS). No significant difference in the EF or FS was found between Hyal2−/− and control mice, although the function was trending downward in the acute Hyal2−/− mice at the last measurement before a humane end point was reached. E, Left ventricular (LV) mass. LV mass increased progressively in both the acute and chronic groups of Hyal2−/− mice compared with controls. F, The myocardial performance index (MPI)=(IVRT+IVCT)/AET was increased significantly at all ages in acute and chronic groups of Hyal2−/− mice compared with controls (AET indicates aortic ejection time; and IVCT, isovolumic contraction time). G, Cardiac output (CO) was significantly impaired in the acute group of Hyal2−/− mice compared with controls, although the chronic group of Hyal2−/− mice also showed decreased CO at most ages. *, †, ‡P<0.05; **, ††P<0.001; ***, †††, ‡‡‡P<0.0001, n=3 to 7 per group at all ages.
To evaluate systolic function, the ejection fraction and fractional shortening of acute and chronic groups of Hyal2−/− mice and controls were compared. No significant difference was observed among the groups (Figure 2C and 2D). LV corrected mass was assessed to determine whether hypertrophy detected by histological studies in Hyal2−/− mice5 was present at the level of the whole heart. Consistent with these earlier findings, both acute and chronic groups of Hyal2−/− mice showed progressively increased LV mass compared with control mice (Figure 2E). In the first 12 weeks, acute Hyal2−/− mice had reduced LV mass compared with control and chronic Hyal2−/− mice (Figure 2E).
Global cardiac function, represented by the myocardial performance index, was impaired in all Hyal2−/− mice. This value was significantly increased at all time points, reflecting the reduced overall cardiac performance (Figure 2F). Reduced cardiac output was also evident in the acute Hyal2−/− mice compared with chronic Hyal2−/− mice and controls (Figure 2G). Together, our data suggest that severe diastolic dysfunction accompanied by reduced cardiac output contributes to the development of heart failure in the acute Hyal2−/− mice within the first 3 months of life. In the chronic Hyal2−/− mice, progressive diastolic dysfunction without reduced cardiac output developed over time, leading to heart failure at an average age of 6 months.
Increased Tissue Density in Hyal2−/− Mice
Histological analyses of transverse sections of hearts from Hyal2−/− and control mice were used to investigate the basis of the increased tissue density. Hematoxylin and eosin staining revealed enlarged atria in all Hyal2−/− mice compared with controls, consistent with the ultrasound findings (Figure 3A through 3C, n=7 pairs). Further, in atria from the acute Hyal2−/− mice, tissue masses (*) were present that were absent in chronic Hyal2−/− and control mice (Figure 3A, n=4 pairs). HABP staining to detect HA revealed abundant HA in the atrium and ventricle of acute and chronic groups of Hyal2−/− mice compared with controls (Figure 3D through 3F). However, increased HA was only detected in the periphery of the atrial masses and not in the central region, which seemed to be composed of cardiomyocytes (Figure 3D, open arrow). Additionally, HABP staining revealed valve-like tissue in other regions of the heart of both acute and chronic Hyal2−/− mice compared with controls (arrow in Figure 3E). Masson trichrome staining demonstrated fibrosis in the atrium and ventricle of Hyal2−/− mice (acute and chronic, green arrows) compared with controls (Figure 3G through 3I; Figure III in the Data Supplement).
Figure 3.
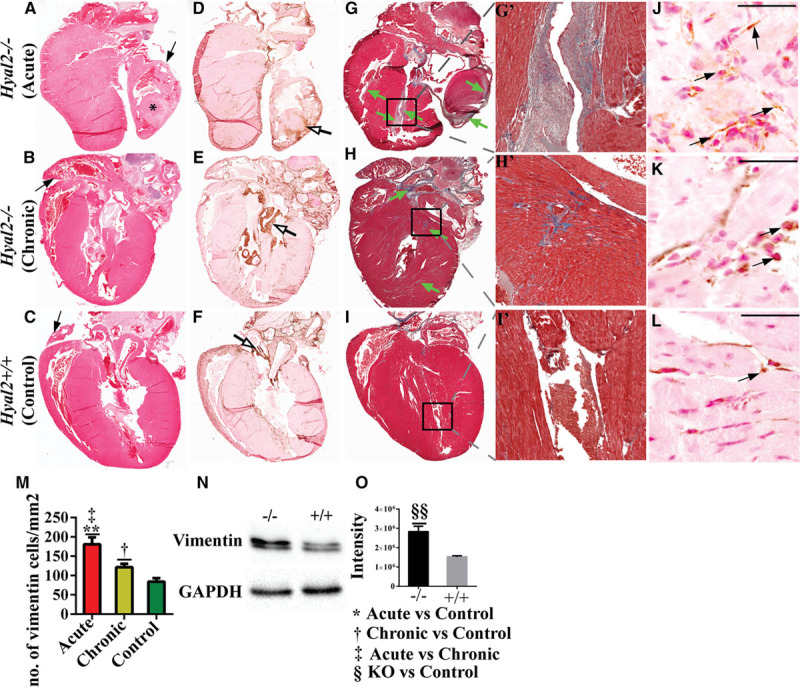
Histological analysis of hyaluronidase 2 (HYAL2)–deficient (Hyal2−/−) and control hearts. Transverse sections of hearts from Hyal2−/− (acute and chronic) and control mice were compared for differences in morphology and structure. A–C, Images of hematoxylin and eosin–stained sections revealed an enlarged atrium (arrow) in both the acute (A) and chronic (B) groups of Hyal2−/− mice compared with control mice (C). *A tissue mass in the atrium of the acute Hyal2−/− mouse. D–F, Images of hyaluronan (HA) distribution in the Hyal2−/− and control hearts. HA was detected as a brown precipitate using the HABP (HA-binding protein). There is intense brown staining in several regions of the Hyal2−/− hearts (open arrows in D and E), whereas the intense brown staining is limited to the valves in the control hearts (open arrow in F). G–I, Masson trichrome staining of Hyal2−/− and control hearts. Masson trichrome stains the extracellular matrix (ECM) components collagen and elastin as blue, and glycosaminoglycans remain unstained. Excess ECM indicating fibrosis (green arrows) is widespread in the Hyal2−/− hearts compared with the control heart (I). G’–I’, Enlarged view of the area in the box in (G)–(I). J–L, Detection of mesenchymal cells in Hyal2−/− and control hearts. Anti-vimentin (brown) indicates the presence of mesenchymal cells. There are increased numbers of vimentin-positive cells (arrow) in both the acute and chronic Hyal2−/− atria (J and K) compared with the control atrium (L). M, Semi-quantitative analysis of vimentin-positive cells in Hyal2−/− and control atria. Significantly increased numbers of vimentin-positive cells are present in Hyal2−/− atria compared with control atria and significantly more in the acute Hyal2−/− than in control Hyal2−/− atria. N and O, Vimentin protein levels in Hyal2−/− and controls hearts (atrium and ventricle). N, Western blot analysis showed increased expression of vimentin in the Hyal2−/− heart compared with controls. GAPDH was used as protein-loading control. O, Quantification of vimentin levels in (N). The chemiluminescent images from Western blot analysis of vimentin from Hyal2−/− and control hearts (n=4) were quantified using a BioRad ChemiDoc. The columns represent the average level of vimentin±SEM (n=4). Significance was determined using the Student t test. Bar=50 μm. The images in this figure are representative of those from 7 pairs of Hyal2−/− and control mice.
The excess fibrous tissue suggested there may be large numbers of fibroblasts secreting ECM. Indeed, an abundance of cells positive for the mesenchymal marker, vimentin, were detected. In a representative image of the atrium, excess vimentin-positive cells were obvious in the Hyal2−/− mice compared with controls, although the number of mesenchymal cells was significantly higher in the acute compared with the chronic Hyal2−/− mice (Figure 3J through 3M). Similarly, mesenchymal cells in the ventricle were also higher in the Hyal2−/− mice compared with controls (Figure III in the Data Supplement), although in this case, the number of vimentin-positive cells was higher in the chronic than in the acute Hyal2−/− and control mice (Figure IIIM in the Data Supplement). The increase in the level of vimentin in Hyal2−/− mice was also detected using Western blot analysis of whole hearts (Figure 3N and 3O).
Morphological Analysis of Embryonic Heart in Hyal2−/− and Control Mice
To determine whether the structural abnormalities in the adult heart of the Hyal2−/− mice originated during development, we analyzed Hyal2−/− and control hearts at E18.5, after the 4-chambered heart had formed. Hematoxylin and eosin staining revealed an enlarged atrium (Figure 4A, open arrow) and excess fibrous tissues in Hyal2−/− mice (arrow in Figure 4A, n=8) compared with controls (Figure 4B, n=5). Further, HABP confirmed the presence of excess HA in the Hyal2−/− atrium compared with controls (Figure 4C and 4D, n=3). The excess fibrous tissue was also accompanied by significantly increased numbers of vimentin-positive cells in the atria and ventricles of Hyal2−/− embryos compared with controls (Figure 4E through 4H, n=3 and 4).
Figure 4.
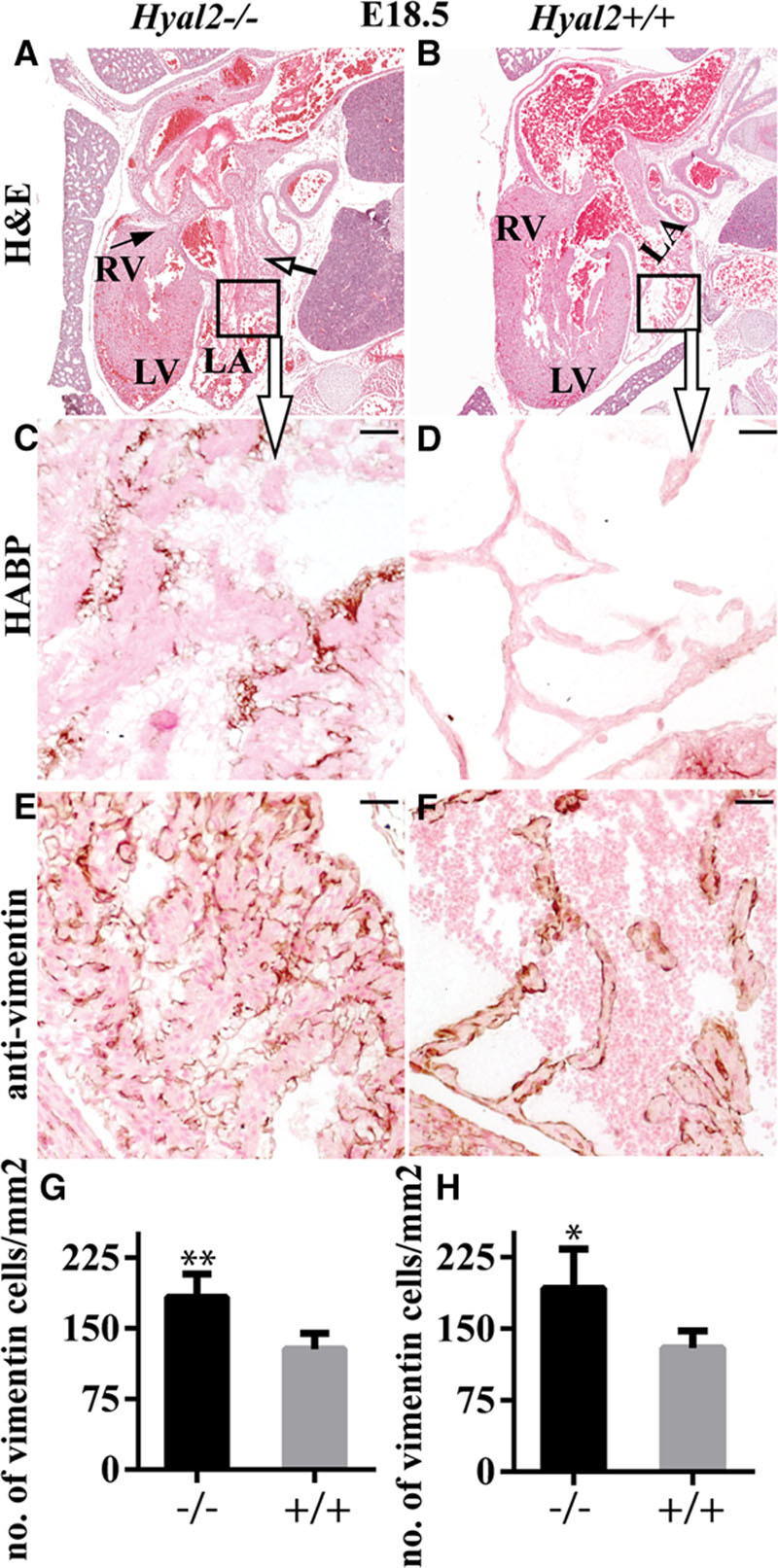
Histological analysis of Hyal2−/− and control hearts at embryonic day (E) 18.5. Paraffin sections from E18.5 embryos were stained or used for immunohistochemistry. A and B, Images of hematoxylin and eosin–stained embryo sections revealed an enlarged atrium (open arrow) and the presence of fibrosis (arrow) in the Hyal2−/− heart (A) compared with the control (B) heart. (Hyal2−/−, n=8; control, n=6). C–F, Enlarged view of the area in the box in (A) and (B) stained for hyaluronan (HA). C and D, Images showing increased HA (brown) in the Hyal2−/− atrium (C) compared with the control atrium (D; n=3). E and F, Detection of vimentin-positive cells (brown) revealed an excess in the Hyal2−/− atrium (E) compared with the control (F; n=3). G–H, Semi-quantitative analysis of vimentin-positive cells in the atrium (G; n=3) and ventricle (H; n=4) of Hyal2−/− and control hearts. There were significantly increased numbers of mesenchymal cells in the Hyal2−/− hearts compared with controls. Bar=50 μm. LA, left atrium; LV, left ventricle; and RV; right ventricle. *P<0.05; **P<0.001.
Abnormal EMT in Hyal2−/− Mice
Previous ex vivo studies at E9.5 showed that high-molecular-mass HA promoted EMT, whereas HA fragments inhibited EMT and activated the VEGF pathway.11 To study this in vivo, we first analyzed the distribution of HYAL2 and HA in embryonic tissues of wild-type mice at E8.5, 11.5, and 12.5. HYAL2 was detected primarily in the endocardial lining of the developing E8.5 heart (Figure 5A through 5F), the bulbus cordis (open arrow), endocardial cushion, and wall of the ventricular chamber (arrow) of the developing E11.5 and E12.5 heart (Figure IV in the Data Supplement). The specificity of the HYAL2 signal was verified by comparing the pattern in wild-type and Hyal2−/− E14.5 and E18.5 embryos (Figure 5G through 5J). HA was most abundant in the E8.5 developing heart, and progressively decreased during development (Figure IV in the Data Supplement). These findings are consistent with HYAL2 having a role in the degradation of HA during development.11
Figure 5.

Hyaluronidase 2 (HYAL2) distribution in embryonic hearts. Sections of the embryonic heart at embryonic day (E) 8.5, from a previous study,19 were used for the detection of HYAL2 using immunofluorescent and immunohistochemical approaches. A–C, Detection of HYAL2 (red) in the endocardial lining of the blood vessels of the E8.5 heart. Nuclei are stained blue with Hoechst 33342. D–F, Enlarged view of the image in (A)–(C). G–J, HYAL2 distribution in E14.5 and E18.5 embryos. The brown staining indicates the presence of HYAL2 in the endothelial cells of blood vessels and heart valves of wild-type heart at E14.5 (H; n=3) and E18.5 (J; n=3), respectively. As expected, this signal is absent in the Hyal2−/− hearts (G and I; n=3). Bar=50 μm.
Examination of the hearts at E14.5 showed the presence of fibrous tissues and HA in the atrium and ventricle in Hyal2−/− mice (n=3) compared with controls (n=3; Figure 6A through 6D). Consistent with increased EMT in the Hyal2−/− heart, there were significantly increased numbers of vimentin-positive cells (Figure 6E, 6F, 6I, and 6J) and decreased levels of VEGF (Figure 6G and 6H). Therefore, our data suggest that disruption of normal HA catabolism in the heart results in increased EMT. However, it is possible that the increase in vimentin-positive cells is because of increased mesenchymal cell proliferation alone or in combination with increased EMT.
Figure 6.
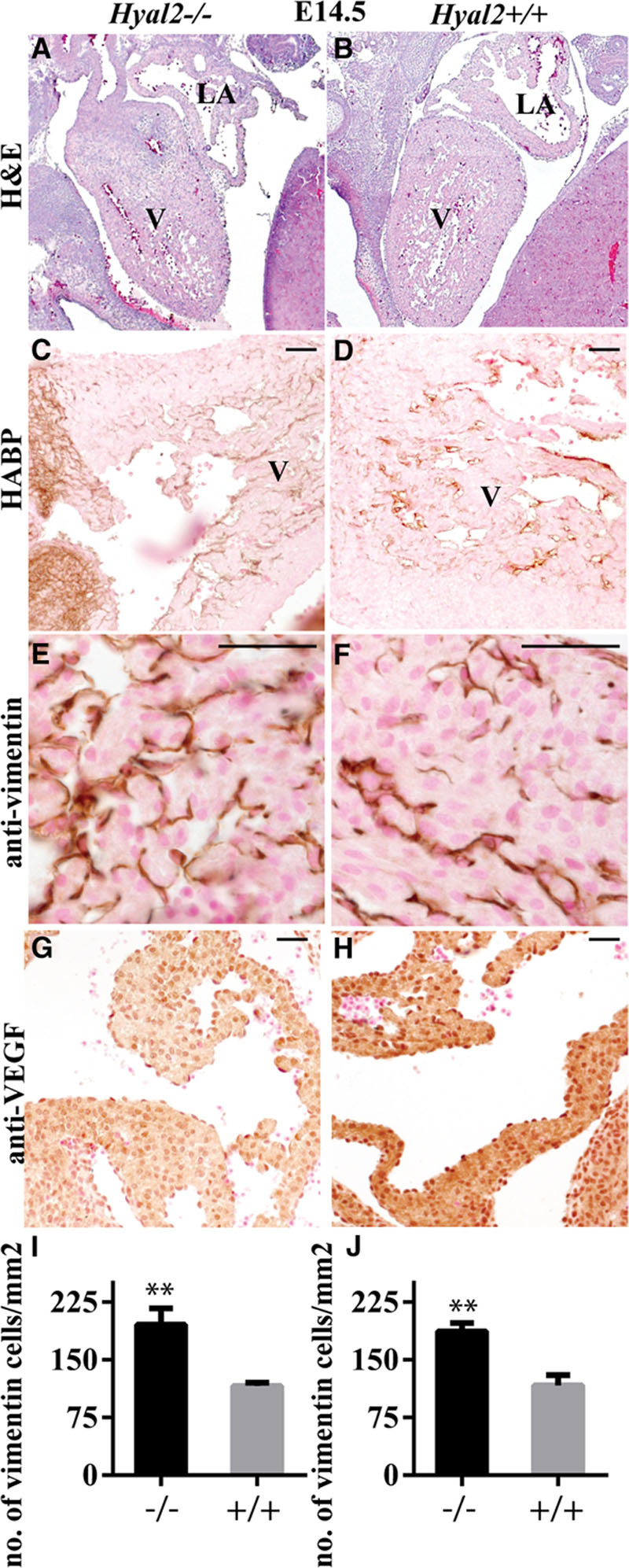
Histological analysis of Hyal2−/− and control hearts at embryonic day (E) 14.5. A–B, Images of hematoxylin and eosin–stained sections show excess tissue in the ventricle in hyaluronidase 2 (HYAL2)–deficient (Hyal2−/−; A) compared with control (B) hearts. C and D, Detection of hyaluronan (HA; brown) showed increased HA in the Hyal2−/− ventricle (C) compared with the control (D). E and F, Vimentin-positive cells were found to be more abundant in the Hyal2−/− heart (E) compared with the control (F). G and H, VEGF (brown) seemed to be more abundant in the Hyal2−/− (G) than in control (H) heart. I and J, Semi-quantitative analysis of vimentin-positive cells in the atrium (I) and ventricle (J) of Hyal2−/− and control hearts at E14.5. The number of mesenchymal cells in the Hyal2−/− hearts was significantly increased compared with controls. Bar=50 μm. **P<0.001, n=3; LA indicates left atrium; and V, ventricle.
Discussion
HA is abundant in the provisional matrix of the developing embryo.22 Its importance in heart development was clearly demonstrated by the early embryonic death of mice deficient in HA synthesis (HAS2 deficient).3 Without HA, EMT in the cardiac cushions of Has2−/− embryos was not supported to form the heart valves and septum. Previously, we have demonstrated that a failure to degrade HA also resulted in cardiac abnormalities in Hyal2−/− mice and HYAL2-deficient humans.5,6 Herein, we show that these abnormalities, and others, are present by 4 weeks of age and result in progressive and severe diastolic dysfunction. The persistence of HA in the absence of HYAL2 presumably promotes EMT and mesenchymal cell proliferation, resulting in excess mesenchymal cells in all Hyal2−/− mice, providing a molecular explanation for the fibrosis, and abnormal heart structures including thickened valves and atrial masses.
A specific role for hyaluronidase in development has been proposed in studies of the cardiac cushion and muscles of the embryonic chick23 and by in vitro studies showing opposing roles for high- and low-molecular-mass HA in EMT using cardiac cushion explants.11 Our studies provide in vivo evidence that normal cardiac development in the mouse requires the hyaluronidase, HYAL2. The absence of HYAL2 results in the accumulation of high-molecular-mass HA4 and excess mesenchymal cells. Taken together, this suggests that HYAL2 is normally required to remove HA to inhibit EMT and mesenchymal cell proliferation and promote differentiation. Although we focused on the characterization of the cardiovascular defect in Hyal2−/− mice, craniofacial abnormalities and a missing kidney also affect a proportion of Hyal2−/− mice.5 HA may also be important in the development of these organs because HA levels are elevated during embryonic development of these tissues.22 The early lethality of HAS2-deficient embryos prevented determination of whether HA was required for the development of organs other than the heart.
Taken together with previous studies of HAS2 deficiency, it is clear that HA levels must be regulated for normal heart development. Heart defects in HYAL2-deficient mice and humans show that increased HA poses a risk for abnormal heart development. Similarly, embryonic lethality because of abnormal heart development in HAS2 deficiency indicates that too little HA also disturbs development.3 In humans, a single case of partial HAS2 deficiency was associated with a ventricular septal defect24; a complete HAS2 deficiency is unlikely to be compatible with life. We have recently described humans with HYAL2 deficiency and demonstrated that the cardiac phenotypes, which included cor triatriatum, atrial enlargement, valvular thickening and accessory tissue, and dilated coronary sinus,6 were similar to those in the Hyal2−/− mice. In addition, both the humans and mice shared palate abnormalities and hearing loss. Taken together, the Hyal2−/− mice provide an excellent model for further study of HYAL2 function and potentially for the development of therapies for the human disorder.
The cardiac abnormalities found in Hyal2−/− mice are uncommon in humans, although there are examples of excess EMT leading to valve thickening. For example, mutations in PTPN11 encoding the protein tyrosine phosphatase SHP2 result in valve thickening in Noonan syndrome.25 In mice deficient in ephrin-A1, aortic and mitral valves are thickened, and there are increased numbers of mesenchymal cells, consistent with increased EMT.26 More studies are needed to understand how HA accumulation, specifically HYAL2 deficiency, impacts these pathways.
In this and a previous study,5 Hyal2−/− mice clearly fell into 2 groups differing in the severity of their heart phenotype. In the previous study, the acute group exhibited severe atrial dilation leading to death at ≈ 3.2 months of age, whereas the chronic (nonacute) group died at ≈ 5.8 months of age.5 In this study, the acutely affected mice died earlier than in our previous study, and often this occurred soon after an ultrasound evaluation, suggesting that the anesthesia may have worsened the cardiac function. Ultrasound evaluation revealed that atrial dilation, valve hypertrophy, and diastolic dysfunction were already present at 4 weeks of age. Although overall cardiac function decreased until death, the structural parameters did not change significantly. The excess tissue growth found in the hearts of the acute mice was consistent with the finding of increased numbers of mesenchymal cells and ECM that results in rapid-onset diastolic dysfunction. Our findings are consistent with several studies that showed that in the presence of preserved systolic function the increased isovolumic relaxation time and atrial size27 are indicators of diastolic dysfunction, that are independent of atrial pressure,28 heart failure,29 or heart rate.30 The basis for the differences in the severity of the phenotype in acute and chronic groups of Hyal2−/− mice is probably because of a genetic determinant segregating in the outbred background. Modifying genes that influence the severity of a cardiac phenotype are extremely common,31 and future studies are required to determine the modifying genes that are involved in the acute and chronic phenotypes of the Hyal2−/− mice.
Thickening of the heart valves and walls and restricted blood flow and regurgitation through the affected valves (data not shown) were present in all Hyal2−/− mice. Together with abnormally placed valve tissues, these phenotypes are the probable cause of diastolic dysfunction. Several studies show that LV hypertrophy,32 interstitial fibrosis,33 and thickened valves34 are the principle causes of diastolic dysfunction of the heart. In the acute Hyal2−/− mice, the presence of tissue masses in the atria further disrupted cardiac function, resulting in earlier and more severe diastolic dysfunction and heart failure. In the chronic Hyal2−/− mice, the increased fibrosis in the ventricles may have resulted from compensatory changes for the ongoing diastolic dysfunction in the heart. In both cases, the eventual outcome was cardiac failure, although it is interesting that the ejection fraction was preserved in the chronic Hyal2−/− mice as models of this type are rare.
The impact of interstitial fibrosis on cardiac function is seen in other disorders of ECM molecules. Normally, the ECM provides support for the contractile forces produced by the cardiac myocytes, and disruption of ECM homeostasis can result in impaired force transmission, causing dilation or hypertrophy. For example, in ADAMTS 9- or 5-deficient mice, accumulation of versican in the heart disrupts ECM homeostasis and causes cardiac disease pathology.35,36 In addition, accumulation of glycosaminoglycans in the heart valves resulted in valve thickening that changed the atrial and ventricular volume overload and contributed to atrial dilation, ventricular hypertrophy, and ultimately diastolic dysfunction in many mucopolysaccharidoses.37 The cardiac disease pathology appears early in the life of patients with defects in glycosaminoglycan degradation and progresses rapidly to cause heart failure and sudden death. Almost 60% to 90% of patients with mucopolysaccharidoses have valvular disease.38 Surgical replacement of heart valves and continuous monitoring of cardiac function through echocardiogram are the common practice in the treatment of mucopolysaccharidoses patients with cardiovascular disease. Given that the valve thickening in our model occurs postnatally, generating a model with a postnatal deletion of HYAL2 might be beneficial for the study of valve disease.
These studies of the cardiac phenotype in Hyal2−/− mice clearly demonstrate an important role for HYAL2 and HA degradation in heart development. The presence of increased numbers of mesenchymal cells and decreased VEGF expression in Hyal2−/− embryos strongly suggests that HYAL2 is needed to inhibit EMT and that in its absence excess EMT leads to congenital malformations. Further studies are needed to clearly differentiate the effects of HYAL2 deficiency on EMT and mesenchymal cell proliferation and to determine whether it is the presence of excess high-molecular-mass HA or the absence of low-molecular-mass HA that results in the phenotypic changes.
Acknowledgments
We thank the University of Manitoba Small Animal and Materials Imaging Facility for assistance.
Sources of Funding
This work was supported by the Canadian Glycomics Network (GlycoNet), a member of the Networks of Centres of Excellence Canada program, the Manitoba Centres of Excellence Fund through Research Manitoba, the Canadian Cancer Society (grant no 702828), the Mizutani Foundation for Glycoscience, the Canadian Foundation for Innovation (projects 23290 and 31284), and a joint Research Manitoba and Manitoba Institute of Child Health studentship (Dr Chowdhury).
Disclosures
None.
Supplementary Material
Footnotes
The Data Supplement is available at http://circgenetics.ahajournals.org/lookup/suppl/doi:10.1161/CIRCGENETICS.116.001598/-/DC1.
CLINICAL PERSPECTIVE
Hyaluronan is an abundant component of extracellular matrix that has been shown in mice to be required for normal cardiac development. As well, human cardiac pathologies such as cardiac fibrosis and myxomatous valve degeneration are associated with increased levels of hyaluronan. Recently, we have identified mutations in the hyaluronan-degrading enzyme, hyaluronidase 2 (HYAL2), as the cause of an autosomal recessive syndrome characterized by severe cardiovascular and palatal abnormalities. Comparison of humans and mice with HYAL2 deficiency revealed that several of the cardiovascular abnormalities, including thickened heart valves, enlarged atria, and cor triatriatum sinister, were shared. In the current article, we examine the origin and functional impact of the cardiovascular abnormalities in HYAL2-deficient mice. We found abnormally distributed valve-like tissue in the atria and ventricles, cell masses in the atria, and increased levels of hyaluronan and mesenchymal cells throughout the heart of HYAL2-deficient mice. These findings were associated with early-onset diastolic dysfunction with preserved ejection fraction that ultimately progressed to systolic heart failure in the mice. These studies clearly demonstrate an important role for hyaluronan degradation in normal heart development and function. Individuals with HYAL2 deficiency typically have cardiac anomalies and may be at risk for the development of heart dysfunction with age. HYAL2-deficient mice could be a valuable tool to determine the risk for further cardiovascular complications because of HYAL2 deficiency and for testing therapeutic interventions for these conditions.
References
- 1.Pierpont ME, Basson CT, Benson DW, Jr, Gelb BD, Giglia TM, Goldmuntz E, et al. American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young. Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation. 2007;115:3015–3038. doi: 10.1161/CIRCULATIONAHA.106.183056. doi: 10.1161/CIRCULATIONAHA.106.183056. [DOI] [PubMed] [Google Scholar]
- 2.Payne RM, Johnson MC, Grant JW, Strauss AW. Toward a molecular understanding of congenital heart disease. Circulation. 1995;91:494–504. doi: 10.1161/01.cir.91.2.494. [DOI] [PubMed] [Google Scholar]
- 3.Camenisch TD, Spicer AP, Brehm-Gibson T, Biesterfeldt J, Augustine ML, Calabro A, Jr, et al. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J Clin Invest. 2000;106:349–360. doi: 10.1172/JCI10272. doi: 10.1172/JCI10272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chowdhury B, Xiang B, Muggenthaler M, Dolinsky VW, Triggs-Raine B. Hyaluronidase 2 deficiency is a molecular cause of cor triatriatum sinister in mice. Int J Cardiol. 2016;209:281–283. doi: 10.1016/j.ijcard.2016.02.072. doi: 10.1016/j.ijcard.2016.02.072. [DOI] [PubMed] [Google Scholar]
- 5.Chowdhury B, Hemming R, Hombach-Klonisch S, Flamion B, Triggs-Raine B. Murine hyaluronidase 2 deficiency results in extracellular hyaluronan accumulation and severe cardiopulmonary dysfunction. J Biol Chem. 2013;288:520–528. doi: 10.1074/jbc.M112.393629. doi: 10.1074/jbc.M112.393629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muggenthaler MM, Chowdhury B, Hasan SN, Cross HE, Mark B, Harlalka GV, et al. Mutations in HYAL2, encoding hyaluronidase 2, cause a syndrome of orofacial clefting and cor triatriatum sinister in humans and mice. PLoS Genet. 2017;13:e1006470. doi: 10.1371/journal.pgen.1006470. doi: 10.1371/journal.pgen.1006470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin CJ, Lin CY, Chen CH, Zhou B, Chang CP. Partitioning the heart: mechanisms of cardiac septation and valve development. Development. 2012;139:3277–3299. doi: 10.1242/dev.063495. doi: 10.1242/dev.063495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hinton RB, Yutzey KE. Heart valve structure and function in development and disease. Annu Rev Physiol. 2011;73:29–46. doi: 10.1146/annurev-physiol-012110-142145. doi: 10.1146/annurev-physiol-012110-142145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hinton RB, Jr, Lincoln J, Deutsch GH, Osinska H, Manning PB, Benson DW, et al. Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circ Res. 2006;98:1431–1438. doi: 10.1161/01.RES.0000224114.65109.4e. doi: 10.1161/01.RES.0000224114.65109.4e. [DOI] [PubMed] [Google Scholar]
- 10.Mjaatvedt CH, Yamamura H, Capehart AA, Turner D, Markwald RR. The Cspg2 gene, disrupted in the hdf mutant, is required for right cardiac chamber and endocardial cushion formation. Dev Biol. 1998;202:56–66. doi: 10.1006/dbio.1998.9001. doi: 10.1006/dbio.1998.9001. [DOI] [PubMed] [Google Scholar]
- 11.Rodgers LS, Lalani S, Hardy KM, Xiang X, Broka D, Antin PB, et al. Depolymerized hyaluronan induces vascular endothelial growth factor, a negative regulator of developmental epithelial-to-mesenchymal transformation. Circ Res. 2006;99:583–589. doi: 10.1161/01.RES.0000242561.95978.43. doi: 10.1161/01.RES.0000242561.95978.43. [DOI] [PubMed] [Google Scholar]
- 12.Stern R. Devising a pathway for hyaluronan catabolism: are we there yet? Glycobiology. 2003;13:105R–115R. doi: 10.1093/glycob/cwg112. doi: 10.1093/glycob/cwg112. [DOI] [PubMed] [Google Scholar]
- 13.Andre B, Duterme C, Van Moer K, Mertens-Strijthagen J, Jadot M, Flamion B. Hyal2 is a glycosylphosphatidylinositol-anchored, lipid raft-associated hyaluronidase. Biochem Biophys Res Commun. 2011;411:175–179. doi: 10.1016/j.bbrc.2011.06.125. doi: 10.1016/j.bbrc.2011.06.125. [DOI] [PubMed] [Google Scholar]
- 14.Triggs-Raine B, Natowicz MR. Biology of hyaluronan: insights from genetic disorders of hyaluronan metabolism. World J Biol Chem. 2015;6:110–120. doi: 10.4331/wjbc.v6.i3.110. doi: 10.4331/wjbc.v6.i3.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jadin L, Wu X, Ding H, Frost GI, Onclinx C, Triggs-Raine B, et al. Skeletal and hematological anomalies in HYAL2-deficient mice: a second type of mucopolysaccharidosis IX? FASEB J. 2008;22:4316–4326. doi: 10.1096/fj.08-111997. doi: 10.1096/fj.08-111997. [DOI] [PubMed] [Google Scholar]
- 16.Chowdhury B, Hemming R, Faiyaz S, Triggs-Raine B. Hyaluronidase 2 (HYAL2) is expressed in endothelial cells, as well as some specialized epithelial cells, and is required for normal hyaluronan catabolism. Histochem Cell Biol. 2016;145:53–66. doi: 10.1007/s00418-015-1373-8. doi: 10.1007/s00418-015-1373-8. [DOI] [PubMed] [Google Scholar]
- 17.Corrigan N, Brazil DP, Auliffe FM. High-frequency ultrasound assessment of the murine heart from embryo through to juvenile. Reprod Sci. 2010;17:147–157. doi: 10.1177/1933719109348923. doi: 10.1177/1933719109348923. [DOI] [PubMed] [Google Scholar]
- 18.Metscher BD. MicroCT for comparative morphology: simple staining methods allow high-contrast 3D imaging of diverse non-mineralized animal tissues. BMC Physiol. 2009;9:11. doi: 10.1186/1472-6793-9-11. doi: 10.1186/1472-6793-9-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Armistead J, Patel N, Wu X, Hemming R, Chowdhury B, Basra GS, et al. Growth arrest in the ribosomopathy, Bowen-Conradi syndrome, is due to dramatically reduced cell proliferation and a defect in mitotic progression. Biochim Biophys Acta. 2015;1852:1029–1037. doi: 10.1016/j.bbadis.2015.02.007. doi: 10.1016/j.bbadis.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 20.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hemming R, Martin DC, Slominski E, Nagy JI, Halayko AJ, Pind S, et al. Mouse Hyal3 encodes a 45- to 56-kDa glycoprotein whose overexpression increases hyaluronidase 1 activity in cultured cells. Glycobiology. 2008;18:280–289. doi: 10.1093/glycob/cwn006. doi: 10.1093/glycob/cwn006. [DOI] [PubMed] [Google Scholar]
- 22.Fenderson BA, Stamenkovic I, Aruffo A. Localization of hyaluronan in mouse embryos during implantation, gastrulation and organogenesis. Differentiation. 1993;54:85–98. doi: 10.1111/j.1432-0436.1993.tb00711.x. [DOI] [PubMed] [Google Scholar]
- 23.Bernanke DH, Orkin RW. Hyaluronidase activity in embryonic chick heart muscle and cushion tissue and cells. Dev Biol. 1984;106:351–359. doi: 10.1016/0012-1606(84)90233-1. [DOI] [PubMed] [Google Scholar]
- 24.Zhu X, Deng X, Huang G, Wang J, Yang J, Chen S, et al. A novel mutation of Hyaluronan synthase 2 gene in Chinese children with ventricular septal defect. PLoS One. 2014;9:e87437. doi: 10.1371/journal.pone.0087437. doi: 10.1371/journal.pone.0087437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Araki T, Chan G, Newbigging S, Morikawa L, Bronson RT, Neel BG. Noonan syndrome cardiac defects are caused by PTPN11 acting in endocardium to enhance endocardial-mesenchymal transformation. Proc Natl Acad Sci USA. 2009;106:4736–4741. doi: 10.1073/pnas.0810053106. doi: 10.1073/pnas.0810053106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frieden LA, Townsend TA, Vaught DB, Delaughter DM, Hwang Y, Barnett JV, et al. Regulation of heart valve morphogenesis by Eph receptor ligand, ephrin-A1. Dev Dyn. 2010;239:3226–3234. doi: 10.1002/dvdy.22458. doi: 10.1002/dvdy.22458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leung DY, Boyd A, Ng AA, Chi C, Thomas L. Echocardiographic evaluation of left atrial size and function: current understanding, pathophysiologic correlates, and prognostic implications. Am Heart J. 2008;156:1056–1064. doi: 10.1016/j.ahj.2008.07.021. doi: 10.1016/j.ahj.2008.07.021. [DOI] [PubMed] [Google Scholar]
- 28.Tei C. New non-invasive index for combined systolic and diastolic ventricular function. J Cardiol. 1995;26:135–136. [PubMed] [Google Scholar]
- 29.Bruch C, Schmermund A, Marin D, Katz M, Bartel T, Schaar J, et al. Tei-index in patients with mild-to-moderate congestive heart failure. Eur Heart J. 2000;21:1888–1895. doi: 10.1053/euhj.2000.2246. doi: 10.1053/euhj.2000.2246. [DOI] [PubMed] [Google Scholar]
- 30.Poulsen SH, Nielsen JC, Andersen HR. The influence of heart rate on the Doppler-derived myocardial performance index. J Am Soc Echocardiogr. 2000;13:379–384. doi: 10.1016/s0894-7317(00)70007-1. [DOI] [PubMed] [Google Scholar]
- 31.Marian AJ. Modifier genes for hypertrophic cardiomyopathy. Curr Opin Cardiol. 2002;17:242–252. doi: 10.1097/01.HCO.0000013803.40803.6A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Störk T, Möckel M, Danne O, Völler H, Eichstädt H, Frei U. Left ventricular hypertrophy and diastolic dysfunction: their relation to coronary heart disease. Cardiovasc Drugs Ther. 1995;9(suppl 3):533–537. doi: 10.1007/BF00877866. [DOI] [PubMed] [Google Scholar]
- 33.Murdoch CE, Chaubey S, Zeng L, Yu B, Ivetic A, Walker SJ, et al. Endothelial NADPH oxidase-2 promotes interstitial cardiac fibrosis and diastolic dysfunction through proinflammatory effects and endothelial-mesenchymal transition. J Am Coll Cardiol. 2014;63:2734–2741. doi: 10.1016/j.jacc.2014.02.572. doi: 10.1016/j.jacc.2014.02.572. [DOI] [PubMed] [Google Scholar]
- 34.Zaid RR, Barker CM, Little SH, Nagueh SF. Pre- and post-operative diastolic dysfunction in patients with valvular heart disease: diagnosis and therapeutic implications. J Am Coll Cardiol. 2013;62:1922–1930. doi: 10.1016/j.jacc.2013.08.1619. doi: 10.1016/j.jacc.2013.08.1619. [DOI] [PubMed] [Google Scholar]
- 35.Dupuis LE, McCulloch DR, McGarity JD, Bahan A, Wessels A, Weber D, et al. Altered versican cleavage in ADAMTS5 deficient mice; a novel etiology of myxomatous valve disease. Dev Biol. 2011;357:152–164. doi: 10.1016/j.ydbio.2011.06.041. doi: 10.1016/j.ydbio.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kern CB, Wessels A, McGarity J, Dixon LJ, Alston E, Argraves WS, et al. Reduced versican cleavage due to Adamts9 haploinsufficiency is associated with cardiac and aortic anomalies. Matrix Biol. 2010;29:304–316. doi: 10.1016/j.matbio.2010.01.005. doi: 10.1016/j.matbio.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Braunlin EA, Harmatz PR, Scarpa M, Furlanetto B, Kampmann C, Loehr JP, et al. Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis and management. J Inherit Metab Dis. 2011;34:1183–1197. doi: 10.1007/s10545-011-9359-8. doi: 10.1007/s10545-011-9359-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fesslová V, Corti P, Sersale G, Rovelli A, Russo P, Mannarino S, et al. The natural course and the impact of therapies of cardiac involvement in the mucopolysaccharidoses. Cardiol Young. 2009;19:170–178. doi: 10.1017/S1047951109003576. doi: 10.1017/S1047951109003576. [DOI] [PubMed] [Google Scholar]