

Abstract
MUC1 is a glycoprotein expressed on the apical surface of ductal epithelial cells. Malignant transformation results in loss of polarization and overexpression of hypoglycosylated MUC1 carrying truncated carbohydrates known as T or Tn tumor antigens. Tumor MUC1 bearing Tn carbohydrates (Tn-MUC1) represent a potential target for immunotherapy. We evaluated the Tn-MUC1 glycopeptide in a human phase I/II clinical trial for safety that followed a preclinical study of different glycosylation forms of MUC1 in rhesus macaques, whose MUC1 is highly homologous to human MUC1. Either unglycosylated rhesus macaque MUC1 peptide (rmMUC1) or Tn-rmMUC1 glycopeptide were mixed with an adjuvant, or loaded on autologous dendritic cells (DCs), and responses compared. Unglycosylated rmMUC1 peptide induced negligible humoral or cellular responses compared to the Tn-rmMUC1 glycopeptide. Tn-rmMUC1 loaded on DCs induced the highest anti-rmMUC1 T-cell responses and no clinical toxicity. In the phase I/II clinical study, 17 patients with non-metastatic castrate-resistant prostate cancer (nmCRPC) were tested with aTn-MUC1 glycopeptide-DC vaccine. Patients were treated with multiple intradermal and intranodal doses of autologous DCs, which were loaded with the Tn-MUC1 glycopeptide (and KLH as a positive control for immune reactivity). PSA doubling time (PSADT) improved significantly in 11 of 16 evaluable patients (P = 0.037). Immune response analyses detected significant Tn-MUC1-specific CD4+ and/or CD8+ T-cell intracellular cytokine responses in 5 out of 7 patients evaluated. In conclusion, vaccination with Tn-MUC1-loaded DCs in nmCRPC patients appears to be safe, able to induce significant T-cell responses, and have biological activity as measured by the increase in PSADT following vaccination.
Keywords: Tn-MUC1, Glycopeptide, Dendritic Cell Vaccine, Prostate Cancer, Clinical Trial
INTRODUCTION
Adenocarcinoma of the prostate is the third leading cause of cancer-related deaths in men in North America (1). Primary treatment—including radical prostatectomy, radiotherapy, or brachytherapy—can be successful, especially for men less than 70 years of age and in good general health (2). Unfortunately, up to 40% of patients will develop local recurrence or distant metastases within 10 years of diagnosis. A rising prostate-specific antigen (PSA) is usually the earliest sign of prostate cancer recurrence in absence of radiologic evidence of metastasis. Patients with recurring elevated PSA levels after primary therapy are initially treated with androgen deprivation (3). Despite serum concentrations of testosterone similar to those after castration, treatment resistance often develops within 15 to 20 months. Invariably, patients with castrate-resistant prostate cancer (CRPC) progress to radiologic detectable metastatic disease (4) and begin treatment with second-generation anti-androgens or docetaxel (5). These therapies are not curative and may be associated with significant cumulative toxicity (3).
Cancer vaccines have emerged as a promising treatment for prostate cancer with excellent tolerability and safety (6). A cell-based therapy for prostate cancer was approved by the U.S. Food and Drug Administration (FDA) in 2012 (7) and other immune approaches are in active development (8–10). Currently, the optimal vaccine strategy or disease stage to target is unknown (8). Preclinical studies in a variety of cancer models have demonstrated superior results with immunotherapy when tumor burden is minimal (11). Thus, a specific immunotherapeutic intervention may be clinically effective in prostate cancer patients with a rising PSA, but no evidence of overt metastatic disease. An effective cancer vaccine could delay the time to overt metastatic disease, ideally with negligible toxicity. We therefore targeted this patient population, prostate cancer patients with rising PSA, for our clinical investigation of dendritic cell vaccination as described in this report.
Prostate cancer cells express molecules recognized by the host immune system (12). These tumor-associated antigens (TAAs) may be incorporated into cancer vaccines. Epithelial cell mucin-1 (MUC1) is overexpressed in an abnormal hypoglycosylated form on a variety of human adenocarcinomas including prostate cancer, as well as multiple myeloma (13). MUC1 currently belongs to a group of well-characterized high priority tumor antigens (14, 15). It is a large transmembrane glycoprotein containing long O-linked carbohydrates and some N-linked sugars. Much of the glycosylation is within the variable number of tandem repeat region (VNTR) in the extracellular domain (15).
When epithelial cells undergo malignant transformation, MUC1 continues to localize to the surface, but also accumulates in the cytoplasm and apical polarity is lost (16, 17). Cell transformation leads to MUC1 becoming hypersialylated and hypoglycosylated with truncated oligosaccharide chains such as GalNAc-Gal-α-O-Ser/Thr, (T antigen) or GalNAc-α-O-Ser/Thr (Tn antigen) (18). These carbohydrates along with T and Tn glycopeptides trigger immune recognition of the unglycosylated peptide backbone and the spontaneous development of low titer antibodies to MUC1 and low frequency MUC1-specific cytotoxic T lymphocytes (CTLs) (19–21). Active immunotherapy has the potential to enhance these immune responses or elicit de novo responses in patients with adenocarcinoma (20, 21).
To date, MUC1 peptide (unglycosylated) vaccines have had limited efficacy in patients with advanced stage cancer (22–26). The immunogenicity of MUC1 peptide was tested in patients with pre-malignant colonic polyps, thus without advanced cancer or having undergone immunosuppressive chemotherapy (27). The immune responses observed suggested that both stage and tumor burden influence the efficacy of a MUC1 peptide vaccine. MUC1 glycopeptide vaccines have not yet been tested in patients with cancer. In preclinical mouse models, MUC1 glycopeptide bearing Tn carbohydrates (Tn-MUC1) was efficiently taken up and processed into smaller peptides and glycopeptides by DCs (28). Dendritic cells loaded with Tn-MUC1 (Tn-MUC1-DC) elicited peptide- and glycopeptide-specific T-cell responses resulting in stronger immunity when compared to unglycosylated vaccines, and in tumor rejection (28–31). These findings suggest that tolerance towards MUC1 peptide epitopes may be overcome by employing Tn-MUC1 antigen.
We vaccinated rhesus macaque (Macaca mulatta) monkeys with either an unglycosylated 100-amino acid (aa)-long rhesus macaque-derived MUC1 peptide (rmMUC1) or a Tn-glycosylated rm-MUC1 glycopeptide (Tn-rmMUC1). The results of safety and immune analyses from this preclinical study are reported here. We also report on the results of a clinical trial evaluating vaccination with autologous DCs loaded with a synthetic 100-aa human Tn-MUC1 glycopeptide in 17 patients with nmCRPC and a rising PSA.
MATERIALS AND METHODS
Preparation of rhesus macaque MUC1 peptides
RmMUC1 and Tn-rmMUC1 peptides were synthesized by the University of Pittsburgh Genomics and Proteomics Core Laboratories (University Pittsburgh, Pennsylvania, USA). A 100-aa-long peptide was synthesized incorporating five identical repeats of the sequence GVVTSAPDTSAAPGSTGPPA from the rhesus macaque MUC1 VNTR region. Tn-rmMUC1 was generated by enzymatic addition of GalNAc to the synthetic peptide using recombinant human UDP-GalNAc:polypeptide N-acetyl-galactosaminyl rGalNAc-T1 as previously described (29, 30).
Generation of rhesus macaque DC vaccines
Peripheral blood mononuclear cells (PBMCs) were cultured with human GM-CSF (10 ng/ml, Sigma) and human IL15 (100 ng/ml, R&D Systems) in IMDM media with 10% FCS for 5 days to generate immature DCs (CD80, CD86, class II MHClow). To induce maturation, rmMUC1 or Tn-rmMUC1 peptides (50 μg/ml) were added to cells for 4 hours. Cells were then washed and incubated in media containing 10 ng/ml each of human IL6, IL1β, TNFα, (R&D Systems), and PGE2 (1 μg/ml, Sigma) for 2 days. Autologous DCs were cryopreserved and thawed immediately before immunization or for use in ELISPOT assays.
Administration of vaccines to rhesus macaques
Chinese origin rhesus macaques (n = 20) were immunized by intradermal vaccination (week 0) and boosted at weeks 3 and 8 using one of four vaccine preparations each comprised of : (i) rmMUC1 peptide (100 μg) emulsified with glucopyranosyl lipid adjuvant (GLA) stable emulsion (32), (ii) Tn-rmMUC1 (100 μg) emulsified with GLA, (iii) autologous DCs pulsed with 100 μg of rmMUC1 peptide, or (iv) autologous DCs pulsed with 100 μg of Tn-rmMUC1. Blood samples for immune monitoring were collected from each animal at biweekly intervals for 3 months and then monthly thereafter. Serum was stored at −20°C. PBMCs were isolated and stored in liquid nitrogen. Pre-immunization samples were used as controls in ELISA and ELISPOT assays.
Measurement of serum IgG in vaccinated rhesus macaques
ELISA plates were coated overnight with rmMUC1 or Tn-rmMUC1 peptide (5 μg/ml), washed with PBS, and blocked with BSA in PBS. Sera diluted in PBS were added and incubated overnight followed by PBS washes and addition of anti-human IgG conjugated to alkaline phosphatase (Sigma). Binding was detected using colorimetric detection with p-nitrophenyl phosphate substrate (Sigma).
ELISPOT assays
DCs pulsed with rmMUC1 or Tn-rmMUC1 peptides were thawed and incubated in 96-well multiscreen plates (Millipore) coated with capture antibody. Thawed pre- and post- immunization PBMCs were added in 10-fold excess to antigen-pulsed autologous DCs as stimulators in triplicate wells and cocultured for 7 days. IFNγ was detected using a monkey IFNγ ELISpot kit (MabTech).
Phase I/II clinical trial regulatory approval
A phase I/II trial was designed to evaluate toxicity, antitumor activity, and immune response to the Tn-MUC1-DC vaccine. The study protocol was reviewed and approved by Health Canada and the Hamilton Integrated Research Ethics Board and registered with ClinicalTrials.gov (Trial Registration ID: NCT00852007). The study was conducted at the Juravinski Cancer Centre in Hamilton, ON, Canada in accordance with the Declaration of Helsinki and Good Clinical Practice Guidelines. Written informed consent was obtained from each participant.
Patient eligibility criteria
Patients with nmCRPC with rising PSA were enrolled. Inclusion criteria required were: (i) histologically documented prostate cancer, (ii) surgical or pharmacological castration for a minimum of 3 months (pharmacological castration was maintained during study), (iii) a PSA value of ≥ 20 ng/mL (μg/L) obtained 12 months prior to study inclusion or a 50% rise in PSA values with a minimum rise of 1.0 ng/ml (μg/L) within 6 months prior to study inclusion or a rise in PSA defined by two sequential increases in PSA values, and (iv) absence of metastatic disease and withdrawal of first generation anti-androgens (e.g., bicalutamide, nilutamide, flutamide) for a minimum of 4 weeks. Participants with: (i) poor hematologic and liver function, (ii) an Eastern Cooperative Oncology Group (ECOG) status >1, (iii) other malignancies (with the exception of nonmelanoma skin cancers), or (iv), prescribed steroidal or immunosuppressive therapy were excluded.
MUC1 expression on primary tumors
When available, formalin-fixed and paraffin-embedded primary tumors (biopsy or radical prostatectomy specimens) were assayed for MUC1 expression by immunohistochemistry using mAbs against the Epithelial Membrane Antigen (EMA) (Clone E29, dilution 1:500, Dako) and the CD227 antigen (clone HMPV, dilution 1:500, BD Pharmingen). Antibodies bound to MUC1 were detected with a polymer-based HRP substrate system (ID-Labs).
Clinical protocol
The study objectives were to assess the safety and feasibility of the DC-Tn-MUC1 vaccine and establish the efficacy of vaccination as measured by cancer progression (time to radiographic or PSA progression) and immune response. A total of 17 eligible patients were entered into the study. Patients received up to a maximum of five vaccinations on an outpatient basis. Three Tn-MUC1-DC vaccinations were given at two week intervals. Two additional booster vaccinations were administered at 6 and 12 months in patients with stable disease. Patients were followed for an additional 12 months (2 years from trial entry). PSA levels were evaluated monthly for the first year and every 3 months during year 2. Radiographic examinations (CT chest, abdomen/pelvis, bone scan) were performed every 3 months.
Preparation of Tn-MUC1 glycopeptide
A certified clinical grade 100-aa-long synthetic MUC1 peptide containing five identical repeats of the sequence GVTSAPDTRPAPGSTAPPAH from the human MUC1 VNTR was manufactured at the University of Pittsburgh Genomics and Proteomics Core Laboratories. Following synthesis, the peptide was further purified by high-performance liquid chromatography and lyophilized. The peptide was glycosylated as described above for the rm-MUC1 peptide (29, 30).
Manufacture of the Tn-MUC1-DC vaccine
Eligible patients underwent 10–12 liter apheresis using a Cobe Spectra (TerumoBCT) via peripheral intravenous access. Leukapheresis products were processed to enrich monocytes by using an Elutra® Cell Separation System (TerumoBCT) as described elsewhere (33). The enriched monocyte product was cultured in CellGro® DC medium (CellGenix, GMP grade) supplemented with 1% human AB serum (SeraCare Life Sciences), 50 ng/mL GM-CSF and 50 ng/mL IL4 (CellGenix, CellGro® GMP grade). On day 4, immature DC were loaded with 40 μg/mL Tn-100mer-MUC1 or 30 μg/mL KLH (Vacmune Liquid, BioSyn, GMP grade). A cocktail consisting of IL1β (10 ng/mL), IL6 (5 ng/mL), TNFα (10 ng/mL) (CellGenix, CellGro® GMP grade) and prostaglandin E2 (PGE2, 1 μg/ml, Sigma-Aldrich, γ-irradiated) was added on day 5. The following day, cells were harvested and washed in DPBS (CTS™, Life Technologies,). Tn-MUC1 and KLH pulsed DCs were mixed 10:1 in a final formulation consisting of 44% RPMI (Invitrogen), 44% human AB serum (SeraCare Life Sciences), and 12% DMSO (Hybri-Max™, Sigma-Aldrich) at a concentration of 2×107 cells/ml and cryopreserved. Safety tests were performed on the final cryopreserved product to confirm the absence of bacteria, fungi, and mycoplasma in test articles, and determine that endotoxin levels were within specifications. Immunophenotyping of thawed cells confirmed the identity (mature DC markers), purity (presence of viable DCs and absence of T and B cell markers), and potency (presence of DC activation and DC presentation markers) of the DC vaccines. DCs were stained with anti-human CD80-PE (clone MEM-233, Life Technologies), CD83-PC5 (clone-HB15a), CD86-PE (clone HA5.2B7), CD54-FITC (clone 84H10), HLA-DR-FITC (clone Immu-357), CD3-PC-5, CD14-ECD, CD19-PE (Beckman Coulter) and 7AAD-PC-5. Cell staining was analyzed using an FC500 flow cytometer with CXP2.1 and 2.2 software (Beckman Coulter).
In vivo DC Tracking in Mice
Experiments were performed to assess the migratory capacity of DCs generated under conditions similar to those used for the clinical DCs using excess elutriated monocytes from study patients (n=3). The elutriated monocytes were further enriched by immunomagnetic negative selection of CD14+ and CD16+ cells (EasySep™, Stemcell Technologies). Monocytes were cultured in CellGro® DC medium (CellGenix) supplemented with human GM-CSF (50 ng/mL) and IL4 (50 ng/mL) (CellGenix). On day 4 cells were incubated at 37°C and 5% CO2 with 2.5 mg/mL Cell Sense, a 19Fluorine-perfluorcabon based cell labeling agent (Celsense, Inc.) for 24h (34). Unlabeled cells served as a control. On day 5 a cocktail consisting of IL6 (0.01 μg/mL), IL1β (10 ng/mL), TNFα (25 ng/mL) all from Life Technologies and PGE2 (1×106 units) from Sigma, was added to cultures. Cells were harvested on day 6. For the in vivo migration studies, 2×106 labeled DCs were injected into the footpads of nude Balb/c mice (n=2–4 per patient DC preparation). Migration of the labeled DCs to the draining popliteal lymph node (Figure S2) was assessed by acquiring a 1H MR image first and then 19F MR image second on a 9.4T Aligent MRI scanner with a dual-tuned 1H/19F mouse body coil. A 3D-balanced steady state free precession (bSSFP) sequence was employed with an image resolution of 1×1×1mm3 for 19F and 200×200×200μm3 for 1H, which equated to a scan time of less than 90 minutes. For in vivo quantification of 19F-labeled cells, nuclear magnetic resonance spectroscopy was performed on a DC pellet of known cell number to determine the number of fluorine atoms per cell as well as measuring and comparing the signal in a region of interest to the signal in a reference of known concentration of 19F atoms using Voxel TrackerTM software (Celsense Inc) (34). Images were constructed using Voxel Tracker™ software.
Administration of Tn-MUC1-DC vaccine
Thawed, antigen-loaded DC vaccines were administered intradermally (i.d.) into the area of a single lymph node basin. In addition to intradermal administration, the dose was also administered directly into a node with ultrasound guidance at week 0 for patients 2-01 to 2-08, and at week 0 and 6 months for patients 2-09 to 2-17. The intranodal dose was 0.2 × 107 DCs and the intradermal dose was either 1.0 × 107 or 1.2 × 107 DCs.
Monitoring of adverse events and toxicity
Patients were observed for one hour following vaccination and were instructed to observe and record any reactions at the injection site for 24 hours. Adverse events were graded according to the National Cancer Institute Common Toxicity Criteria (NCI CTCAE v 4.0).
Clinical response
Overall survival of patients at study entry was predicted by the prognostic model developed by Halabi et al. (35) using the web-based calculator (https://www.cancer.duke.edu/Nomogram/firstlinechemotherapy.html).
The PSA doubling times (PSADT) were calculated using the Memorial Sloan-Kettering Medical Center prostate cancer prediction tool (http://www.mskcc.org/mskcc/html/10088.cfm).
Time to radiographic progression (time from week 0 to the occurrence of any metastatic disease based on modified RECIST 1.0) and time to PSA progression (according to Prostate Cancer Working Group criteria) were monitored over 2 years.
Samples for immune monitoring
Peripheral blood (100 ml) was collected prior to each Tn-MUC1-DC administration and at two weeks after vaccination. PBMCs were isolated by ficoll gradient centrigugation (Ficoll-Paque™ PLUS, GE HealthCare Bioscience, AB) and cryopreserved for immune analysis at a later date. Vials of autologous DCs (Tn-MUC1-DC, KLH-DC, and non-pulsed (NP-DC)) were prepared during clinical manufacture of each Tn-MUC1-DC vaccine and stored for later use in immunoassays.
Analysis of immune responses to Tn-MUC1-DC vaccination
Sensitive multicolor flow-based methods were used to characterize cell-mediated immunity by intracellular cytokine staining of Tn-MUC1 and KLH antigens in the first seven subjects (2-01, 2-02, 2-03, 2-04, 2-05, 2-06, and 2-08). These analyses were performed by ImmuneCarta Services (www.immunecarta.com, Montreal, Canada), in compliance with applicable GLP and GCLP regulations. An intracellular cytokine staining (ICS) assay was used to quantitate cytokine secretion in total and memory CD4+ and CD8+ T cells. ICS was performed after stimulation with autologous Tn-MUC1-DC, KLH-DC as well as NP-DC and media, as negative controls. Stimulation with a CEF peptide pool consisting of CMV, EBV and influenza immunodominant 8–12-mer peptides covering diverse HLA alleles was used as an antigenic positive control. PMA/ionomycin polyclonal stimulation was used as a positive control to assess the overall functionality of the processed PBMC. Functional markers were used including IFNγ (PE-Cy7), TNFα (APC), IL2 (Alexa 700), lL4 (FITC) and IL17 (PerCP-Cy7) in combination with cell surface expression of CD107a (PE), as a marker of T cell degranulation. The main T cell populations were defined based on the following cell surface markers: CD3 (V450), CD4 (PE-Texas Red), CD8 (eFluro650), CD45RA (APC-Cy7), CD27 (Qdot605) and the LIVE/DEAD Fixable Aqua marker. The representative gating strategy is presented in the Supplementary Fig. S3. A Boolean gating strategy was applied using all functional markers and SPICE analyses (NIH, provided by M. Roederer) were performed to determine the frequency and combination of cytokine/CD107a responses to cognate stimuli (Tn-MUCl-DC, KLH-DC) after background subtraction from the NP-DC stimulated cultures. All functional signatures were summed to generate the memory CD4+/CD8+ T cells responses over time, except the CD107a mono-functional signature as it is not antigen-specific. Permutation tests and Wilcoxon signed rank tests were used to evaluate whether the measured functional signatures increased following treatment. To detect CD4+ Tregs, the flow-based panel included LIVE/DEAD Fixable Aqua marker (Invitrogen), CD3(V450), CD4 (PE-Texas red) and CD8 (eFluro650) for main T cell populations and CD45RA (APC-Cy7), CD27 (Qdot605), CCR7 (PE-Cy7) for memory subset characterization. FoxP3 (APC), CD25 (PE), Ki67 (FITC) and CD127 (PerCP-Cy5.5) were included in the panel for Treg characterization and activation. Subset populations were analyzed looking at the frequency of FoxP3+CD25+CD127lo Tregs among CD4+ T cells as well as by the combination of CD45RA, CD27 and CCR7. A Boolean gating strategy was used for analyzing the activation of Treg cells within the CD4+ memory compartment (Supplementary Fig. S6).
ELISA assays to detect serum IgG to Tn-MUC1 and KLH in the clinical samples were performed as described above for rhesus macaques.
RESULTS
Pre-clinical studies in rhesus macaques
Four groups of five rhesus macaques were immunized with either: (i) rmMUC1 plus GLA adjuvant, (ii) rmMUC1-loaded DCs, (iii) Tn-rmMUC1 plus GLA adjuvant, or (iv) Tn-rmMUC1-loaded DCs. Subsequent rmMUC1-specific antibody and T-cell responses were measured over time (Figs. 1 and 2). Four of 5 animals immunized with the rmMUC1 plus GLA adjuvant developed a weak transient anti-rmMUC1 IgG response (Fig. 1A). A weak anti-rmMUC1 IgG response was detected at week 6 in one animal and was no longer detectable at week 24. None of these animals developed measurable T-cell responses. Conversely, the group immunized with Tn-rmMUC1 and GLA (Fig. 1B) demonstrated significant and durable anti-rmMUC1 IgG responses in 3/5 animals. Responses peaked at weeks 10 and 12. All of these animals (5/5) developed strong and measurable INFγ+ T-cell responses at week 12 as determined by IFNγ ELISpot, including the two IgG negative animals.
Figure 1.
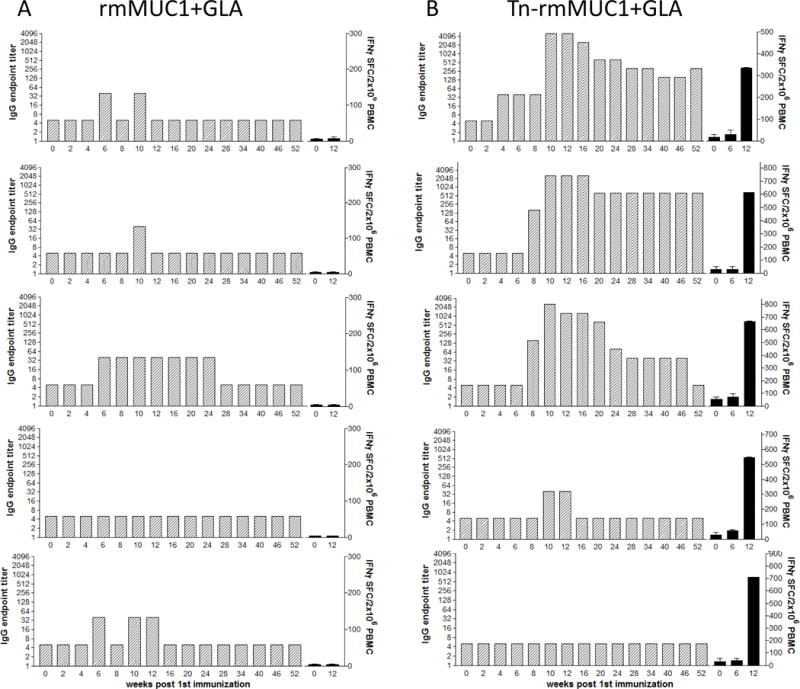
Groups of five rhesus macaques were immunized at baseline, week 3 and week 8 with either the unglycosylated rmMUC1 (A) or the Tn-rm-MUC1 (B) peptides emulsified with GLA adjuvant. Blood samples were taken from each animal at various time points for the analysis of humoral and cellular immune responses. IgG titers, as determined by ELISA (gray bars), and frequency of INFγ-positive PBMCs as determined by ELISPOT (black bars) are presented. Assays were performed in triplicate.
Figure 2.

Groups of five rhesus macaques were immunized at baseline, week 3 and week 8 with monocyte-derived dendritic cells loaded with either the unglycosylated rmMUC1 (A) or the Tn-rm-MUC1 (B) peptides. Blood samples were taken from each animal at various time points for the analysis of humoral and cellular immune responses. IgG titers, as determined by ELISA (gray bars), and frequency of INFγ+ PBMCs as determined by ELISPOT (black bars) are presented. Assays were performed in triplicate.
A similar analysis was performed using rmMUC1 and Tn-rmMUC1-loaded DCs. The non-glycosylated peptide did not induce rmMUC1-specific T cells nor a measurable antibody response (Fig. 2A). However, strong INFγ+ T-cell responses, but no antibody responses, were seen upon immunization with Tn-rmMUC1-DC vaccine (n = 5/5). Tn-rmMUC1-DC vaccines induced INFγ+ T-cell responses (Fig. 2B) that were approximately 60% stronger than seen with the soluble Tn-rmMUC1 glycopeptide plus GLA vaccine (Fig. 1B).
Characteristics of nmCRPC patients
Between January 2009 and January 2013, 17 patients were enrolled into two sequential cohorts (2-01 to 2-08 and 2-09 to 2-17). Patient characteristics are summarized in Table 1. Median age at enrollment was 72.8 years. The majority of patients had previous radiation treatment. The median PSA at enrollment was 11 μg/L (range 1.5 −71.1 μg/L). Although not an eligibility criteria, all patients that could be tested for antigen expression on their tumor had MUC1+ primary tumors. The average Halabi score (predicted median overall survival) was 31.6 months. According to the Halabi scores, all patients were in the low risk category except two, who were in the intermediate risk category (2-07 and 2-11).
Table 1.
Patient characteristics and clinical response.
| Patient ID | Age (y) | Previous treatments (a) | Gleason | PSA at entry (μg/L) | ECOG | Predicted Survival (months) (b) | MUC1 histology (c) | Nb of vaccine doses | PSADT pre-vacc (months) (d) | PSADT post-vacc (months) (d) | PSADT variation | Time to PSA prog. (days) (e) | Time to radiologic prog. (days) (f) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2-01 | 64 | RT, HT | 8 | 71.1 | 1 | 27.0 | n.a. | 3 | 5.91 | 7.52 | 1.3 | 66 | 169 |
| 2-02 | 72 | RT, HT | 7 | 2.7 | 0 | 33.7 | + | 3 | 6.17 | 4.13 | 0.7 | 27 | 139 |
| 2-03 | 49 | HT | 9 | 5.7 | 0 | 36.8 | + | 4 | 1.52 | 2.08 | 1.4 | 55 | no prog. (g) |
| 2-04 | 69 | RP, RT, HT | 8 | 18.0 | 0 | 35.3 | n.a. | 3 | 1.89 | 21.73 | 11.5 | no prog. | 174 |
| 2-05 | 72 | HT | 7 | 42.0 | 0 | 31.7 | + | 5 | 9.82 | 23.41 | 2.4 | 339 | no prog. (h) |
| 2-06 | 77 | RT, HT | 7 | 1.5 | 0 | 26.7 | + | 5 | 1.65 | 7.58 | 4.6 | 57 | no prog. (h) |
| 2-07 | 74 | RP, HT | n.a. | 38.0 | 0 | 23.8 | n.a. | 0 | − | − | − | − | − |
| 2-08 | 86 | RT, HT | 6 | 16.0 | 0 | 26.6 | + | 5 | 2.87 | 6.46 | 2.3 | 163 | no prog. (g) |
| 2-09 | 65 | RP, RT, HT | 7 | 42.0 | 0 | 35.3 | + | 3 | 2.36 | 3.94 | 1.7 | 55 | 159 |
| 2-10 | 87 | RT, HT | 6 | 6.9 | 1 | 28.3 | + | 5 | 2.72 | 26.28 | 9.7 | 193 | 487 |
| 2-11 | 76 | RRT, HT | 9 | 3.9 | 0 | 25.2 | + | 5 | 4.40 | 5.97 | 1.4 | 154 | 508 |
| 2-12 | 78 | RRT, HT | 7 | 1.8 | 0 | 42.4 | + | 5 | 3.07 | 5.14 | 1.7 | 63 | no prog. (g) |
| 2-13 | 75 | RRT, HT | 7 | 11.0 | 0 | 35.7 | + | 3 | 2.63 | 1.74 | 0.7 | 29 | 99 |
| 2-14 | 65 | RRT, HT | 7 | 29.0 | 1 | 27.9 | n.a. | 5 | 14.83 | 32.87 | 2.2 | no prog. | no prog. (h) |
| 2-15 | 65 | RT, HT | 7 | 11.0 | 0 | 35.3 | + | 4 | 5.34 | 3.08 | 0.6 | 87 | 344 |
| 2-16 | 75 | RRT, HT | 7 | 7.4 | 0 | 33.7 | n.a. | 5 | 6.48 | 5.06 | 0.8 | 87 | no prog. (g) |
| 2-17 | 54 | RP, RT, HT | 9 | 2.2 | 0 | 31.8 | n.a. | 3 | 1.17 | n.a. | − | 27 | 62 |
RP: Radical prostatectomy; RT: Radiotherapy; RRT: Radical Radiotherapy; HT; Hormonotherapy
Overall median survival was predicted using the Halabi 2014 nomogram (35)
MUC1 expression was assessed on FFPE primary tumor tissues using immunohistochemistry (see Material and Methods).
Pre-vaccination PSA doubling time (PSADT) was obtained using the web-based MSKCC PSADT calculator (see Material and Methods). Four PSA values obtained before the first vaccination, including the PSA at day 1 (just before vaccination) were used to calculate the pre-vaccination PSADT. All PSA values available during the first 200 days (about 6 months) after the first vaccination were used to calculate the post-vaccination PSADT.
See definition of PSA progression in the Results section.
See definition of Radiologic progression in the Material and Methods section.
No radiologic progression before the patient came off-study.
No radiologic progression at 2 years, i.e. at the end of the study.
Production and characterization of DC vaccines
Leukapheresis products contained an average of 19.8 ± 7.1×109 total white blood cells. Two patients required a second apheresis as the first elutriated product failed to meet the established limit for monocyte cell purity (≥60%). Elutriated products contained 21.0% to 90.3% CD14+ cells (mean, 63.2%). The average number of CD14+ cells seeded at day 0 was 1.4 ± 0.4×109. Vaccine DCs expressed high CD80, CD83, CD86, HLA-DR, and CD54 and little non-DC cell markers (CD14, CD3, and CD19) (Supplementary Fig. S1). Viability of thawed cells determined by trypan blue exclusion and 7-AAD was high (means 90.0 ± 3.3% and 97.7 ± 3.1%, respectively). All vaccines, with the exception of one vaccine with low CD80 expression, met release criteria for concentration, viability, lack of microbial contamination, surface expression of mature DC markers, and low expression of non-DC markers. The unreleased vaccine was for a patient removed from the study before treatment due to rapid disease progression. DCs prepared in the described manner from an aliquot of a patient’s leukophoresis product were demonstrated to be migration competent in vivo as the expected 5% of injected DC (36–38) migrated to a lymph node, detected by labeling with 19Fluorine-Cell Sense and injecting into nude BALB/c mice (39) as determined by 19F-cellular MRI (Supplementary Fig. S2).
Tn-MUC1-DC vaccination safety profile
Sixty-seven vaccines in total were safely given, including 23 intranodal injections administered under ultrasound guidance. The number of vaccinations received by each patient is indicated in Table 1. Adverse events were mostly grade 1 and 2 local reactions at injection sites (summarized in Table 2). No signs of acute toxicity or autoimmunity were observed throughout the study.
Table 2.
Summary of adverse events.
|
No. of events (No. of patients involved)
|
||
|---|---|---|
| Event | Grade 1 | Grade 2 |
| Erythema/induration at injection sites | 34 (16) | |
| Infection at injection site | 1 (1) | |
| Hematoma at injection site | 1 (1) | |
| Fatigue | 1 (1) | |
| Insomnia | 1 (1) | |
| Candidiasis (groin area) | 1 (1) | |
Adverse events, probably or possibly related to DC-Tn-MUC1 vaccination
Clinical Course of Disease
None of the 16 vaccinated patients presented a PSA response defined by a 50% decrease of their baseline PSA, but rather all experienced an increase of their PSA (Fig. 3). The mean PSADT for 11 of 16 patients was significantly increased compared to the pre-vaccination period (10.5 vs. 4.6 months, P = 0.037) during the on-study vaccination period (first 200 days after vaccination; Table 1). PSA progression was defined as a 25% increase over the baseline concentration and an absolute increase in PSA concentration of ≥ 5 ng/mL, and this was confirmed by a second value. Time to PSA progression ranged from 27 to 339 days. Two patients remained PSA progression-free at study end. Time to radiographic progression varied between 62 and 508 days as summarized in Table 1. Four patients had no radiologic progression when they came off study, and 3 patients were still free of radiologic progression at 24 months, i.e. at the end of the study. At 2 years, 2 of 16 patients were deceased (12.5% mortality). As of February 2016, 9 out of 16 patients were still alive.
Figure 3.
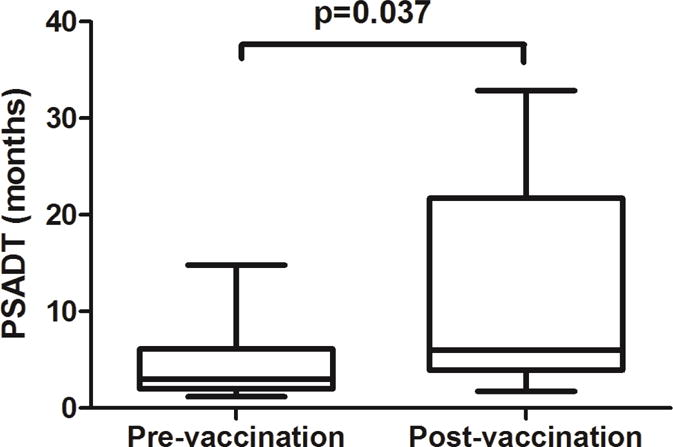
Box and whiskers plot of PSADT calculated for each patient at pre-vaccination and post-vaccination period comprising the first 200 days following the first immunization. Data represent the mean and range for the 16 patients. The mean PSADT during the on-study vaccination period is significantly increased compared to the pre-vaccination period (p=0.037, unpaired Student t-test).
Immunological response to Tn-MUC1-DC vaccination
Humoral and cellular immune responses were analyzed for the first seven vaccinated patients (patients 2-01 to 2-08). As in the macaque subjects, none of the patients developed an antibody response to Tn-MUC1, nor KLH (data not shown). Cellular immune responses were observed for all patients in ICS assays after stimulation of their PBMCs with Tn-MUC1 or KLH-loaded DCs (Fig. 4, Supplementary Figs. S3 to S6 and Supplementary Tables S1 and S2). Responses against Tn-MUC1 or KLH were detected for combinations of IFNγ, IL2, TNFα and/or CD107a in total and memory (CD45RA−) CD8+ and/or CD4+ T cells. Positive response was defined as a two-fold increase of the sum of cytokine/CD107a responses over baseline (i.e. pre-treatment response at week 0).
Figure 4.
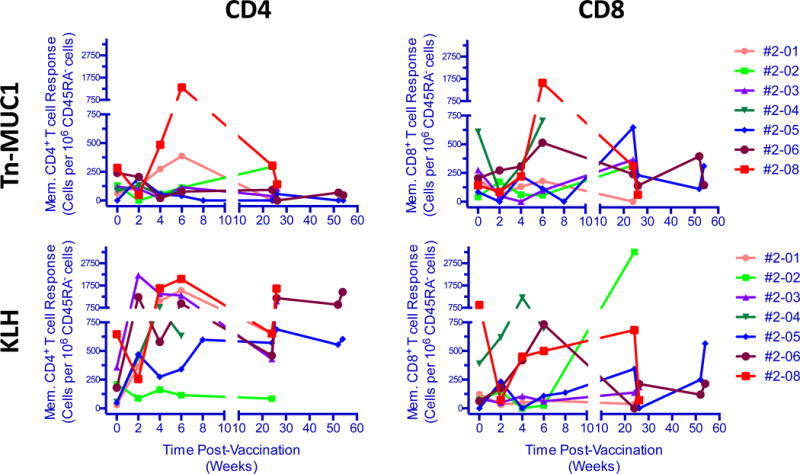
Total frequency of Tn-MUC1 or KLH-specific memory CD4+ and CD8+ T cells at different time points following vaccination. Patients’ PBMCs were stimulated in vitro with Tn-MUC1-DCs or KLH-DCs and the total frequency of functional subsets (either positive for IL2, TNFα, INFγ or CD107a) of CD4+ or CD8+ T cells was determined. Samples from each time point were assayed in monoplicate. Total functionality was calculated by summing all functional subsets and normalized to 106 memory CD45RA− CD4+ or CD45RA− CD8+ T cells.
CD4+ and CD8+ T-cell responses against the control KLH antigen were observed at various times post-vaccination for patients 2-03, 2-04, 2-05, and 2-06, whereas patients 2-01 and 2-08 exhibited KLH specific CD4+, but not CD8+ responses (Fig. 4, Supplementary Tables S1 and S2). A KLH-specific CD8+ T-cell response alone was observed for 2-02, and only at W24. The CD4+ cytokine profiles indicated that the dominant response to KLH involved IL2 production; cells were predominately IL2+ single-positive or TNFα+IL2+ double-positive and were negative for IFNγ and CD107a, concordant with a memory-type CD4+ response. These responses were significantly upregulated at < 2 months and/or >6 months compared to baseline (P < 0.05) (Supplementary Fig. S4). The overall CD4+ response was strong; KLH specific CD4+ cells represented ≥ 0.05% of memory T cells (Fig. 4, Supplementary Fig. S4, and Supplementary Table S1). KLH-specific CD4+ and CD8+ T-cell responses were observed within 2 weeks of vaccination, and were sustained for up to 6 months for some patients, as demonstrated by positive CD4+ responses for 2-05 and 2-06 at weeks 24 and 52, and CD8+ responses for 2-05 at week 52. Variability in response over time was low, patients positive for response tended to be positive at all time points post baseline.
Both CD4+ and CD8+ T-cell responses against Tn-MUC1 were observed for patients 2-02 and 2-08. A CD4+ response was only observed in 2-01, whereas 2-05 and 2-06 only had CD8+ responses. The dominant cytokine profiles in response to Tn-MUC1 were IL2+ single positive and TNFα+IL2+ double positive CD4+ T cells that were negative for IFNγ and CD107a. (Supplementary Fig. S5). Some triple positive TNFα+IL2+lFNγ+ CD4+ T-cell responses were observed. The magnitude of the response, summing up all possible combinations of cytokine and CD107a responses, was in the range of 0 to 1,315 responding cells per 106 memory CD4+ T cells and 0 to 1,567 responding cells per 106 memory CD8+ T cells (Fig. 4, Supplementary Table S1 and S2). Patient 2-08 showed the highest CD4+ response. Tn-MUC1 response was more variable than the response to KLH, responses in PBMC from multiple time points were seen only for 2-01 (CD4+) and 2-05 (CD8+).
The frequency of the Treg populations (CD4+CD25+Foxp3+) was highly variable, ranging between 1 to 6% of the CD4+ T-cell pool with a significant change observed post-vaccine compared to baseline. The frequency of Treg cells > 6 months after vaccination was significantly increased compared to baseline (P < 0.05; Fig. 5).
Figure 5.
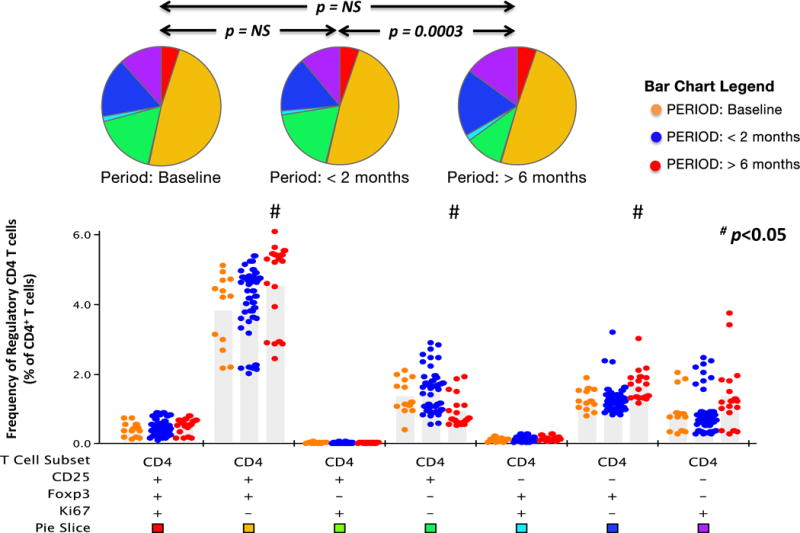
Frequency of regulatory T cells following vaccination. Patients’ PBMCs obtained at various time points were analyzed for the presence of CD4+ T cells expressing any combination of FoxP3, CD25 and Ki67. Data from weeks 2, 4, 6 and 8 were included in the <2 month group whereas data from weeks 24, 26, 52 and 54 were included in the >6 month group. Response profiles were generated using Boolean analysis of 3 functional response gates, resulting in 7 separate functional T cell subsets. The frequency of each CD4+ T cell subset at baseline, <2 months and > 6 months is presented in bar graphs. Each dot in the bar graphs represents data from one PBMC sample (see bar chart legend for color code). The bar graph provides fine detail on frequencies within each of the individual 7 categories; medians are shown. Assays were performed in triplicate. Pie charts represent the distribution of regulatory and non-regulatory subsets within the CD4+ T-cell pool at the three periods. Each color in the pie charts represents one of the CD4+ T-cell subsets (see pie slice color code presented below the bar graph). P values identify whether the distribution of subsets analyzed differed between periods. Subsets which were statistically different compared to the baseline time point are identified (#: p < 0.05, Wilcoxon test).
DISCUSSION
Several groups have reported on cell-based immunotherapy trials for prostate cancer (7, 40–44). These trials used antigen-presenting cells to elicit immune responses against well-characterized prostate-specific differentiation antigens such as PSA. These vaccine trials have proven to be safe with some evidence of antitumor immunity observed. Sipuleucel-T was the first FDA-approved cellular immunotherapy for prostate cancer (6, 8). Results of the phase III IMPACT trial demonstrated improved overall survival of 4.1 months over the control arm (7). Data from a subgroup analysis show the greatest benefit in patients with low burden of disease (45). This may correlate with the degree of immunocompetence and tolerance to putative TAAs and suggests that a proportion of older prostate cancer patients can respond to an immune intervention and mount a clinically relevant therapeutic response.
Based on studies in transgenic mice expressing human MUC1, we hypothesized that potential tolerance to MUC1 may be overcome using a DC-based Tn-MUC1 vaccination protocol. We confirmed here in macaques that, as in mice, the Tn-rmMUC1 glycopeptide antigen was immunogenic and superior to unglycosylated rmMUC1 peptide when administered with adjuvant or when loaded on autologous DCs ex vivo. Hence, Tn-MUC1 was selected as the best immunogen for a MUC1 vaccine trial in nmCRPC patients. The primary objective of this trial was to evaluate the feasibility and safety of vaccination with Tn-MUC1-DC and the secondary objective was to establish the efficacy and immunogenicity of the vaccine. We showed that the production and administration of the vaccine were feasible and safe and that it led, in some patients, to the induction of significant immune responses and biological effects as observed by a change in PSADT.
Our clinical vaccine was comprised of Tn-MUC1 glycopeptide and KLH (as a vaccination control antigen) loaded on autologous monocyte-derived DCs. The conditions used for generation of the DCs from elutriated monocytes efficiently produced cells with the desired phenotype, typical of mature DCs (46). Migration of DCs is highly dependent upon their state of maturation, with mature DCs showing superior migratory capacity in comparison to immature DCs (36). Migration studies of DCs produced under similar conditions demonstrated that the DCs generated were migration competent, although at a low frequency consistent with the findings of others (36–38).
Different routes of administration were investigated in various studies, yet no strong consensus exists as to which route is superior. Intranodal vaccination results in increased DC migration to efferent lymph nodes, yet despite this finding, the frequency of antigen-specific T cells after intranodal injection is no different than after intradermal injection (37). The presence of macrophages that infiltrate the lymph nodes and phagocytose DCs after intranodal, but not intradermal, injection of DCs may contribute to an unfavourable vaccination microenvironment (37). As our DC vaccine was administered by both routes, this study could not compare these routes in terms of ability to induce a response. We found intranodal injection of the DC vaccine to be feasible and safe. Intradermal DC administration resulted in local injection site reactions. These reactions were only seen after booster vaccination, not at week 0. We saw no evidence of delayed toxicity in any of the patients, unlike other standard treatments that may adversely affect patient’s life quality.
Consensus has not been reached on the optimal schedule for DC vaccination. Our immunization protocol was designed to have multiple vaccine administrations. However, the withdrawal of some patients due to disease progression at variable times makes it impossible to draw conclusions regarding response to the number of vaccine administrations. Since all 7 vaccinated patients in the first cohort received at least 3 vaccinations, a direct comparison of the immune responses induced after one, two, and three vaccinations can be made. We found that vaccination induced a CD4+ response to the control antigen KLH at weeks 2, 4, and 6 for five of the seven patients, and for weeks 4 and 6 for one patient (2-08). The CD4+ and CD8+ responses against Tn-MUC1 were more variable in comparison. Overall, these analyses revealed the following observations; (i) the immune responses were considered strong and (ii) they were persistently maintained over the year of observation in the two patients who could be evaluated. Taken together, these results provide evidence of the quality and immunological efficacy of our DC vaccines and immunization protocol.
However, none of the vaccinated patients showed an objective clinical response as measured by a PSA response. A significant increase in PSADT following vaccination was nevertheless observed in 11 out of 16 patients, suggesting the vaccine did have a biological impact. At the end of the study, 3 patients were well without any other treatment and were still free of radiologic progression. However, 9 of 16 patients (56%) had a radiologic progression within 17 months. The impact of vaccination on the time to radiologic progression is difficult to interpret without comparison to a control arm. Moreover, the natural history of the progression of patients with nmCRPC is not well documented. Our data, however, could be compared with data from placebo groups of trials performed in patient population having the same characteristics. Smith et al. reported that at 2 years, 33% and 46% of nmCRPC patients of the placebo arms of zoledronic acid and atrasentan studies developed bone metastases, and 21% and 20% died, respectively (47). This indicates that overall, there was no delay in the time to radiologic progression in our treated patients compared to reference placebo groups. The primary objective of the trial was to assess the safety and feasibility of DC vaccination, therefore conclusions about the clinical efficacy are made with caution. Nevertheless, the clinical response was seen to be comparable with responses observed in previous prostate cancer DC vaccine studies (48).
The oldest patient (2-08), who was 86 years-old at study entry, developed the highest cellular immune response against Tn-MUC1, despite his advanced age. This patient, with a Halabi nomogram-predicted survival of 26.6 months, completed the 2 year follow-up without radiographic progression.
A major advantage of the DC-Tn-MUC1 vaccine is that MUC1 presentation is HLA unrestricted, hence, endogenous processing of MUC1 glycopeptide results in MHC class I and II presentation of multiple peptides and induced immune responses are not restricted to a specific HLA type. However, predominately CD4+ T-cell responses were induced by vaccination, with variable CD8+ T-cell responses observed. This variability may be due to factors that determine the ability of ex vivo generated DCs to cross-present antigen and elicit a CD8+ T-cell response, such as interpatient variability in antigen uptake and processing, and subsequent cross presentation, and the influence of the tumor-induced immunosuppressive environment. The observed results may also reflect assay conditions that preferentially bias detection of a CD4+ T-cell response. Stimulation with 9-mer peptide pools rather than with 100-mer polypeptide pulsed DCs might have led to optimal detection of CD8+ T-cell responses. It is also possible that antigen-specific CD8+ T cells may preferentially localize to lymph nodes and the tumor, leaving very low numbers in the peripheral circulation. Vaccine strategies have focused on eliciting a CD8+ cytotoxic T cell–specific response. However, the importance of CD4+ T-cell support of CD8+ T-cell responses and direct antitumor effects are clear (49). CD8+ T-cell independent, antitumor activity mediated by CD4+ T cells through direct CD4+ T-cell cytotoxicity and activation of cytotoxic macrophages and NK cells, as well as cytokine-mediated mechanisms, has been reported (50). CD4+ T cells also enhance recruitment of CD8+ T cells to lymph nodes and tumors (51). Stimulation and maintenance of antigen specific CD4+ T cells promotes a sustained CD8+ T-cell response leading to an improved antitumor response and long-lasting memory against tumour antigen (51).
Analysis of immune response after vaccination is typically measured by the quantification of IFNγ producing cells after ex vivo antigen stimulation. However, IFNγ is a subdominant effector response (52). Our analysis enabled us to determine the capacity of antigen-specific T cells elicited by the vaccine to produce multiple cytokines. Stimulation with recall cytomegalovirus, Epstein-Barr virus, and influenza virus (CEF) peptides as controls, and PMA/ionomycin (P/I) to assess overall functionality, demonstrated interpatient variability, but consistency in responses over time for each patient (data not shown). The observed responses were not generally polyfunctional, with the exception of 2-06 (combinations of IL2+ TNFα+ IFNγ+) for P/I stimulation in CD4+ T cells, and 2-08 (combinations of IL2+ TNFα+ IFNγ+ CD107a+) in CD8+ T cells following stimulation with CEF peptides. It is not unexpected to find variability in the observed responses to KLH and Tn-MUC1 in vaccinated patients, and that the responses represent a skewed subset of the overall potential responses as shown by P/I stimulation (52). The predominant responses were IL2+ and/or TNFα+ and IFNγ−. The lack of polyfunctionality might be attributed to a lack of immungenicity of the vaccine, or a lack of immunocompetency in patients. However, IL2+IFNγ+ cells are short lived in vitro and in vivo, and IL2+IFNγ– cells efficiently develop into long-term memory cells (52). This could explain the predominance of IL2-producing cells found in the response to our vaccine.
No correlation was observed with regard to CD4+CD25+FoxP3+CD127lo Treg cells and the cell-mediated immunity results obtained. Moreover, the increased Treg frequency may be related to an overall restoration of functional immune CD4+ T-cell populations that includes Tregs.
In a previous clinical trial of MUC1 vaccine in patients with advanced colonic adenomas, which showed a high number of responders, high numbers of circulating myeloid-derived suppressor cells (MDSCs) prior to vaccination correlated with lack of response to the vaccine (27). However 6/6 of our patients had low MDSCs at baseline, and thus could not provide a possible explanation for our observed weak responses (data not shown).
The superior nature of Tn-MUC1 as a vaccine antigen has been validated in mice, primates, and now humans. It allows for the testing of other immune interventions such as immune checkpoint inhibitors, Treg suppressors, and novel adjuvants to improve antigen cross presentation in combination with Tn-MUC1 antigen, which may enhance the immune response and clinical activity against prostate cancer and other MUC1-expressing tumors (48).
In conclusion, the procedure and schedule used for generating and administering the Tn-MUC1-DC vaccine are feasible and safe. No patient demonstrated an objective PSA response but most patients displayed an increase in the PSADT, indicating biological activity. Based on the success of immune checkpoint inhibitors in clinical trials, their combination with Tn-MUC1-DC vaccine in nmCRPC patients could improve the biological activity and clinical efficacy of this vaccine.
Supplementary Material
Acknowledgments
The authors thank Mrs. Pamela O’Hoski for her technical assistance with monocyte elutriation, and Mr. Kazi Islam for the production of the rhesus and human MUC1 peptides and glycopeptides, Ms. Surita Singh for technical assistance with the rhesus experiments, Dr. John McKolanis for human antibody ELISAs and Dr. Steve Reed for the gift of GLA adjuvant.
Financial Support: The rhesus macaque studies were supported by NIH grant 5 R01 CA168392 to OJF and RDS. The Tn-MUC1 dendritic cell clinical study was supported by CANVAC with funds from the Networks of Centres of Excellence (NCE) and by the ORBiT Program with funds from the Ontario Institute for Cancer Research (OICR).
References
- 1.Center MM, Jemal A, Lortet-Tieulent J, Ward E, Ferlay J, Brawley O, et al. International variation in prostate cancer incidence and mortality rates. Eur Urol. 2012;61:1079–92. doi: 10.1016/j.eururo.2012.02.054. [DOI] [PubMed] [Google Scholar]
- 2.Heidenreich A, Bastian PJ, Bellmunt J, Bolla M, Joniau S, van der KT, et al. EAU guidelines on prostate cancer. part 1: screening, diagnosis, and local treatment with curative intent-update 2013. Eur Urol. 2014;65:124–37. doi: 10.1016/j.eururo.2013.09.046. [DOI] [PubMed] [Google Scholar]
- 3.Heidenreich A, Bastian PJ, Bellmunt J, Bolla M, Joniau S, van der KT, et al. EAU guidelines on prostate cancer. Part II: Treatment of advanced, relapsing, and castration-resistant prostate cancer. Eur Urol. 2014;65:467–79. doi: 10.1016/j.eururo.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 4.Cookson MS, Roth BJ, Dahm P, Engstrom C, Freedland SJ, Hussain M, et al. Castration-resistant prostate cancer: AUA Guideline. J Urol. 2013;190:429–38. doi: 10.1016/j.juro.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 5.Berthold DR, Pond GR, Soban F, de WR, Eisenberger M, Tannock IF. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer: updated survival in the TAX 327 study. J Clin Oncol. 2008;26:242–5. doi: 10.1200/JCO.2007.12.4008. [DOI] [PubMed] [Google Scholar]
- 6.Singh BH, Gulley JL. Therapeutic vaccines as a promising treatment modality against prostate cancer: rationale and recent advances. Ther Adv Vaccines. 2014;2:137–48. doi: 10.1177/2051013614539478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–22. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 8.Schweizer MT, Drake CG. Immunotherapy for prostate cancer: recent developments and future challenges. Cancer Metastasis Rev. 2014;33:641–55. doi: 10.1007/s10555-013-9479-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kwon ED, Drake CG, Scher HI, Fizazi K, Bossi A, van den Eertwegh AJ, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014;15:700–12. doi: 10.1016/S1470-2045(14)70189-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jochems C, Tucker JA, Tsang KY, Madan RA, Dahut WL, Liewehr DJ, et al. A combination trial of vaccine plus ipilimumab in metastatic castration-resistant prostate cancer patients: immune correlates. Cancer Immunol Immunother. 2014;63:407–18. doi: 10.1007/s00262-014-1524-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burotto M, Singh N, Heery CR, Gulley JL, Madan RA. Exploiting synergy: immune-based combinations in the treatment of prostate cancer. Front Oncol. 2014;4:351. doi: 10.3389/fonc.2014.00351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Di Lorenzo G, Buonerba C, Kantoff PW. Immunotherapy for the treatment of prostate cancer. Nat Rev Clin Oncol. 2011;8:551–61. doi: 10.1038/nrclinonc.2011.72. [DOI] [PubMed] [Google Scholar]
- 13.Choi C, Witzens M, Bucur M, Feuerer M, Sommerfeldt N, Trojan A, et al. Enrichment of functional CD8 memory T cells specific for MUC1 in bone marrow of patients with multiple myeloma. Blood. 2005;105:2132–4. doi: 10.1182/blood-2004-01-0366. [DOI] [PubMed] [Google Scholar]
- 14.Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15:5323–37. doi: 10.1158/1078-0432.CCR-09-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kimura T, Finn OJ. MUC1 immunotherapy is here to stay. Expert Opin Biol Ther. 2013;13:35–49. doi: 10.1517/14712598.2012.725719. [DOI] [PubMed] [Google Scholar]
- 16.Beatson RE, Taylor-Papadimitriou J, Burchell JM. MUC1 immunotherapy. Immunotherapy. 2010;2:305–27. doi: 10.2217/imt.10.17. [DOI] [PubMed] [Google Scholar]
- 17.Major P, Lavallee M, Minassian H, Kovac P, Wang NS. Ultrastructural localization of a breast tumor-associated antigen. J Histochem Cytochem. 1987;35:375–9. doi: 10.1177/35.3.3546485. [DOI] [PubMed] [Google Scholar]
- 18.Hanisch FG, Muller S. MUC1: the polymorphic appearance of a human mucin. Glycobiology. 2000;10:439–49. doi: 10.1093/glycob/10.5.439. [DOI] [PubMed] [Google Scholar]
- 19.Monzavi-Karbassi B, Pashov A, Kieber-Emmons T. Tumor-Associated Glycans and Immune Surveillance. Vaccines. 2013;1:174–203. doi: 10.3390/vaccines1020174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Madsen CB, Petersen C, Lavrsen K, Harndahl M, Buus S, Clausen H, et al. Cancer associated aberrant protein O-glycosylation can modify antigen processing and immune response. PLoS One. 2012;7:e50139. doi: 10.1371/journal.pone.0050139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Von Mensdorff-Pouilly S, Moreno M, Verheijen RH. Natural and induced humoral responses to MUC1. Cancers (Basel) 2011;3:3073–103. doi: 10.3390/cancers3033073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tempero RM, VanLith ML, Morikane K, Rowse GJ, Gendler SJ, Hollingsworth MA. CD4+ lymphocytes provide MUC1-specific tumor immunity in vivo that is undetectable in vitro and is absent in MUC1 transgenic mice. J Immunol. 1998;161:5500–6. [PubMed] [Google Scholar]
- 23.Hiltbold EM, Ciborowski P, Finn OJ. Naturally processed class II epitope from the tumor antigen MUC1 primes human CD4+ T cells. Cancer Res. 1998;58:5066–70. [PubMed] [Google Scholar]
- 24.Hiltbold EM, Alter MD, Ciborowski P, Finn OJ. Presentation of MUC1 tumor antigen by class I MHC and CTL function correlate with the glycosylation state of the protein taken Up by dendritic cells. Cell Immunol. 1999;194:143–9. doi: 10.1006/cimm.1999.1512. [DOI] [PubMed] [Google Scholar]
- 25.Chen D, Koido S, Li Y, Gendler S, Gong J. T cell suppression as a mechanism for tolerance to MUC1 antigen in MUC1 transgenic mice. Breast Cancer Res Treat. 2000;60:107–15. doi: 10.1023/a:1006332009414. [DOI] [PubMed] [Google Scholar]
- 26.Karanikas V, Hwang LA, Pearson J, Ong CS, Apostolopoulos V, Vaughan H, et al. Antibody and T cell responses of patients with adenocarcinoma immunized with mannan-MUC1 fusion protein. J Clin Invest. 1997;100:2783–92. doi: 10.1172/JCI119825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kimura T, McKolanis JR, Dzubinski LA, Islam K, Potter DM, Salazar AM, et al. MUC1 vaccine for individuals with advanced adenoma of the colon: a cancer immunoprevention feasibility study. Cancer Prev Res (Phila) 2013;6:18–26. doi: 10.1158/1940-6207.CAPR-12-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vlad AM, Muller S, Cudic M, Paulsen H, Otvos L, Jr, Hanisch FG, et al. Complex carbohydrates are not removed during processing of glycoproteins by dendritic cells: processing of tumor antigen MUC1 glycopeptides for presentation to major histocompatibility complex class II-restricted T cells. J Exp Med. 2002;196:1435–46. doi: 10.1084/jem.20020493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ryan SO, Vlad AM, Islam K, Gariepy J, Finn OJ. Tumor-associated MUC1 glycopeptide epitopes are not subject to self-tolerance and improve responses to MUC1 peptide epitopes in MUC1 transgenic mice. Biol Chem. 2009;390:611–8. doi: 10.1515/BC.2009.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ryan SO, Turner MS, Gariepy J, Finn OJ. Tumor antigen epitopes interpreted by the immune system as self or abnormal-self differentially affect cancer vaccine responses. Cancer Res. 2010;70:5788–96. doi: 10.1158/0008-5472.CAN-09-4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Finn OJ, Gantt KR, Lepisto AJ, Pejawar-Gaddy S, Xue J, Beatty PL. Importance of MUC1 and spontaneous mouse tumor models for understanding the immunobiology of human adenocarcinomas. Immunol Res. 2011;50:261–8. doi: 10.1007/s12026-011-8214-1. [DOI] [PubMed] [Google Scholar]
- 32.Coler RN, Bertholet S, Moutaftsi M, Guderian JA, Windish HP, Baldwin SL, et al. Development and characterization of synthetic glucopyranosyl lipid adjuvant system as a vaccine adjuvant. PLoS One. 2011;6:e16333. doi: 10.1371/journal.pone.0016333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berger TG, Strasser E, Smith R, Carste C, Schuler-Thurner B, Kaempgen E, et al. Efficient elutriation of monocytes within a closed system (Elutra) for clinical-scale generation of dendritic cells. J Immunol Methods. 2005;298:61–72. doi: 10.1016/j.jim.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 34.Helfer BM, Balducci A, Nelson AD, Janjic JM, Gil RR, Kalinski P, et al. Functional assessment of human dendritic cells labeled for in vivo (19)F magnetic resonance imaging cell tracking. Cytotherapy. 2010;12:238–50. doi: 10.3109/14653240903446902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Halabi S, Lin CY, Kelly WK, Fizazi KS, Moul JW, Kaplan EB, et al. Updated prognostic model for predicting overall survival in first-line chemotherapy for patients with metastatic castration-resistant prostate cancer. J Clin Oncol. 2014;32:671–7. doi: 10.1200/JCO.2013.52.3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Vries IJ, Krooshoop DJ, Scharenborg NM, Lesterhuis WJ, Diepstra JH, Van Muijen GN, et al. Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Res. 2003;63:12–7. [PubMed] [Google Scholar]
- 37.Verdijk P, Aarntzen EH, Lesterhuis WJ, Boullart AC, Kok E, van Rossum MM, et al. Limited amounts of dendritic cells migrate into the T-cell area of lymph nodes but have high immune activating potential in melanoma patients. Clin Cancer Res. 2009;15:2531–40. doi: 10.1158/1078-0432.CCR-08-2729. [DOI] [PubMed] [Google Scholar]
- 38.Baumjohann D, Hess A, Budinsky L, Brune K, Schuler G, Lutz MB. In vivo magnetic resonance imaging of dendritic cell migration into the draining lymph nodes of mice. Eur J Immunol. 2006;36:2544–55. doi: 10.1002/eji.200535742. [DOI] [PubMed] [Google Scholar]
- 39.Dekaban GA, Hamilton AM, Fink CA, Au B, de Chickera SN, Ribot EJ, et al. Tracking and evaluation of dendritic cell migration by cellular magnetic resonance imaging. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2013;5:469–83. doi: 10.1002/wnan.1227. [DOI] [PubMed] [Google Scholar]
- 40.Fong L, Brockstedt D, Benike C, Breen JK, Strang G, Ruegg CL, et al. Dendritic cell-based xenoantigen vaccination for prostate cancer immunotherapy. J Immunol. 2001;167:7150–6. doi: 10.4049/jimmunol.167.12.7150. [DOI] [PubMed] [Google Scholar]
- 41.Rini BI, Weinberg V, Fong L, Conry S, Hershberg RM, Small EJ. Combination immunotherapy with prostatic acid phosphatase pulsed antigen-presenting cells (provenge) plus bevacizumab in patients with serologic progression of prostate cancer after definitive local therapy. Cancer. 2006;107:67–74. doi: 10.1002/cncr.21956. [DOI] [PubMed] [Google Scholar]
- 42.Lodge PA, Jones LA, Bader RA, Murphy GP, Salgaller ML. Dendritic cell-based immunotherapy of prostate cancer: immune monitoring of a phase II clinical trial. Cancer Res. 2000;60:829–33. [PubMed] [Google Scholar]
- 43.Fuessel S, Meye A, Schmitz M, Zastrow S, Linne C, Richter K, et al. Vaccination of hormone-refractory prostate cancer patients with peptide cocktail-loaded dendritic cells: results of a phase I clinical trial. Prostate. 2006;66:811–21. doi: 10.1002/pros.20404. [DOI] [PubMed] [Google Scholar]
- 44.Jahnisch H, Fussel S, Kiessling A, Wehner R, Zastrow S, Bachmann M, et al. Dendritic cell-based immunotherapy for prostate cancer. Clin Dev Immunol. 2010;2010:517493. doi: 10.1155/2010/517493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schellhammer PF, Chodak G, Whitmore JB, Sims R, Frohlich MW, Kantoff PW. Lower baseline prostate-specific antigen is associated with a greater overall survival benefit from sipuleucel-T in the Immunotherapy for Prostate Adenocarcinoma Treatment (IMPACT) trial. Urology. 2013;81:1297–302. doi: 10.1016/j.urology.2013.01.061. [DOI] [PubMed] [Google Scholar]
- 46.Strasser EF, Eckstein R. Optimization of leukocyte collection and monocyte isolation for dendritic cell culture. Transfus Med Rev. 2010;24:130–9. doi: 10.1016/j.tmrv.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 47.Hong JH, Kim IY. Nonmetastatic castration-resistant prostate cancer. Korean J Urol. 2014;55:153–60. doi: 10.4111/kju.2014.55.3.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Westdorp H, Skold AE, Snijer BA, Franik S, Mulder SF, Major PP, et al. Immunotherapy for prostate cancer: lessons from responses to tumor-associated antigens. Front Immunol. 2014;5:191. doi: 10.3389/fimmu.2014.00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, et al. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med. 2008;358:2698–703. doi: 10.1056/NEJMoa0800251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim HJ, Cantor H. CD4 T-cell subsets and tumor immunity: the helpful and the not-so-helpful. Cancer Immunol Res. 2014;2:91–8. doi: 10.1158/2326-6066.CIR-13-0216. [DOI] [PubMed] [Google Scholar]
- 51.Aarntzen EH, Schreibelt G, Bol K, Lesterhuis WJ, Croockewit AJ, De Wilt JH, et al. Vaccination with mRNA-electroporated dendritic cells induces robust tumor antigen-specific CD4+ and CD8+ T cells responses in stage III and IV melanoma patients. Clin Cancer Res. 2012;18:5460–70. doi: 10.1158/1078-0432.CCR-11-3368. [DOI] [PubMed] [Google Scholar]
- 52.De Rosa SC, Lu FX, Yu J, Perfetto SP, Falloon J, Moser S, et al. Vaccination in humans generates broad T cell cytokine responses. J Immunol. 2004;173:5372–80. doi: 10.4049/jimmunol.173.9.5372. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.