

ABSTRACT
Inflammation is part of the complex biological response of body tissues to harmful stimuli, such as pathogens. It serves as a protective response that involves leukocytes, blood vessels and molecular mediators with the purpose to eliminate the initial cause of cell injury and to initiate tissue repair. Inflammation is tightly regulated by the body and is associated with transient crossing of leukocytes through the blood vessel wall, a process called transendothelial migration (TEM) or diapedesis. TEM is a close collaboration between leukocytes on one hand and the endothelium on the other. Limiting vascular leakage during TEM but also when the leukocyte has crossed the endothelium is essential for maintaining vascular homeostasis. Although many details have been uncovered during the recent years, the molecular mechanisms from the vascular part that drive TEM still shows significant gaps in our understanding. This review will focus on the local signals that are induced in the endothelium that regulate leukocyte TEM and simultaneous preservation of endothelial barrier function.
KEYWORDS: actin, endothelium, GEF, GTPase, transmigration, permeability
Introduction
The vascular system is a complex network formed by numerous connected blood vessels that are embedded in tissue throughout the human body. Removing all tissues leaving only the vascular system intact fully outlines the shape of the human body showing the high density of blood vessels in our tissues and organs. Proper functions of this high density network are essential for human health, since it provides our body with nutrients, oxygen and hormones and regulates body homeostasis such as temperature and pH. In addition the vascular system governs guidance to traveling immune cells and thereby supports protective immune functions that keep our body free of pathogens, cancer and foreign material.1,2 In case of inflammation or immune surveillance the cells lining the luminal site of blood vessels, known as endothelial cells (ECs), attract and direct traveling immune cells to suitable exit sites in the vasculature allowing cells to enter underlying tissue. ECs therefore fulfill an important supportive role in guidance and directional migration of trafficking immune cells. During inflammation ECs expose a variety of adhesion molecules at their surface that slow down and arrest traveling immune cells in the blood circulation. These adhesive molecules are thought to provide guidance cues to immune cells where to breech the blood vessel wall through a multi-step process known as transendothelial migration (TEM) or diapedesis.3 Although many adhesion molecules have been identified, the exact composition of adhesion molecules that determine a suitable exit site for immune cell diapedesis remains elusive. It is well appreciated that blood vessels in inflamed tissues are more permissive for macro molecules. This endothelial leakiness supports several inflammatory functions such as activation of the complement system and recruitment of innate immune cells. Paradoxically, recruitment of innate immune cells occurs through transient openings in the endothelium without plasma leakage.4-6 This indicates that vascular permeability for small macromolecules and immune cells are separately regulated. Which mechanisms protect the endothelial barrier during leukocyte diapedesis is currently poorly understood.
In several diseases, such as thrombocytopenia, ischemia and rheumatoid arthritis accumulation of immune cells evoke serious tissue damage. In case of thrombocytopenia it is known that the physical movement of immune cells through the ECs barrier elicits organ hemorrhages.7 This bleeding disorder is partly caused due to the incapability of ECs to maintain a tight barrier during the physical movement of immune cells through the EC layer. In the past years lots of effort has made into the development of blocking antibodies targeting leukocyte integrins or integrin ligands that are exposed at the endothelial surface. Blocking immune cell exit sites may prevent TEM and consequently reduce patients symptoms. However, 2 clinical trials that tried to interfere with adhesion molecules evoked serious side effects and aggravated the patients conditions, since the blocking antibodies used in the trial activated the immune cells in contrast to their predicted blocking effect.8 In order to improve treatments for diseases that involve immune cell traffic it is a necessity that we increase our understanding about what processes occur during leukocyte TEM, both at the cellular and molecular level. In this review, we focus on how immune cells travels through the endothelial barrier and discuss recent insights on how ECs protect their barrier function during immune cell trafficking.
Transendothelial migration ‘hotspots’
The regulation of immune cell trafficking is complex and goes far beyond our current understanding. Although much is yet to discover, intensive research over the past decade has revealed several fundamental principles that regulate cell migration in a variety of immune cell related responses such as hematopoiesis, immune surveillance and innate and adaptive immunity. The current paradigm of TEM is a refined version of the multi-step model that was first proposed by Butcher and Springer.9,10 The current order in the multistep paradigm are; leukocyte rolling, arrest, crawling, firm adhesion and transmigration. The latter occurs either through the endothelial junctions (paracellular route)11,12 or through the endothelial cell body (transcellular route).13-15 Interestingly, leukocytes diapedesis gives the impression to occur at predefined places in the endothelium lining. Some locations even favor the migration of multiple immune cells that breech the endothelial lining in rapid succession. In fact, when looking at a transmigrating leukocyte, just prior to exiting, the leukocyte changes its crawling morphology to a more round appearance. This raises some important questions, such as what factors determine these so called ‘hotspots for transmigration’, why do 2 routes exist and what defines the usage of one over the other. Judging on the recordings of transmigrating leukocytes, it appears that the leukocytes search the endothelial monolayer to find an exit point, indicating that they use the crawling step as a sort of searching period. However, strong evidence on this latter suggestion is missing and requires future investigations. So far, several key principles have been established, although it needs to be kept in mind that these principles are based on in vitro studies and therefore can only serve as a model that awaits confirmation in future in vivo studies. First of all, immune cells are attracted toward an optimal concentration of chemokines (chemotaxis), density of adhesion molecules (haptotaxis) or cellular stiffness (durotaxis). Secondly, migration into a tissue or organ is believed to follow the path of least resistance (tenertaxis). In addition, shear forces, vessel type and composition of the glycocalyx play an important regulatory role in dictating suitable exit sites. Each principle will be briefly delineated starting with chemotaxis.
Chemotaxis
Chemokines are of key importance for leukocyte TEM not only because of their involvement in chemotaxis but also because of their role in integrin activation inducing leukocyte arrest (Fig. 1a). Chemokines are immobilized by heparan sulfate (HS) proteoglycans that are part of a 50–100 nm negatively charged network on the apical surface of EC called the glycocalyx. Immobilized chemokines elicit integrin-mediated adhesion.16 Recently, it has been shown that perivascular macrophages located between the tissue and blood vessels, secrete chemokines that cause local “hotspots” for neutrophil diapedesis in vivo.17 These chemokines secreted in the extravascular space are bound to glycosaminoglycans (GAGs) and are subsequently transcytosed to the luminal side of the vasculature. There are some indications that oligomeric chemokine-forms activate leukocyte-integrins that direct leukocyte arrest and firm adhesion whereas monomeric-forms activate integrin subsets on the leukocyte that govern cell movement.18,19
Figure 1.

Model of factors that determine ´hotspots´ for leukocyte transendothelial migration. Several key principles are thought to govern leukocyte diapedesis at predefined places in the vasculature. In the first place leukocytes are attracted toward an optimal; (a) concentration of chemokines (chemotaxis), (b) density of adhesion molecules (haptotaxis) or (c) cellular stiffness (durotaxis). Oligomeric chemokine-forms bound to glycosaminoglycans (GAGs) are thought to direct leukocyte firm adhesion and arrest whereas monomeric chemokine-forms govern directional cell movement. (d) Secondly, migration into a tissue or organ is believed to follow the path of least resistance, i.e. tenertaxis. Tenertaxis may affect the decision making to go trans or paracellular. Additional factors such as (e) shear forces and (f) vessel type play an important regulatory role in dictating suitable exit sites. The major route of transmigration into lung, skin and cremaster is believed to be the paracellular route whereas the transcellular route is recognized as the dominant route to enter the lymph node, blood brain barrier (BBB) or the peritoneum.
Haptotaxis
Similar ideas have been suggested for integrin ligands presented at the apical surface of ECs where the amount of leukocyte-integrin ligands regulates leukocyte behavior. A good example of haptotaxis is the amount of ICAM-1 molecules present at the endothelial surface (Fig. 1b). Surface density and distribution of endothelial ICAM-1 induced a transition from paracellular to transcellular migration, while intermediate levels favored the paracellular route.20,21 Related to the amount of surface ligands, neutrophil-EC interactions during TEM does increase integrin expression at the surface of neutrophils thereby affecting their activity and behavior after transmigration.22
Durotaxis
Migrating cells sense environmental cues that give direction to their movement. Migrating cells are attracted to an optimal surface stiffness also called stiffness sensing or durotaxis (Fig. 1c). Leukocytes sense and respond to their physical surroundings, for example in vitro neutrophils migrate slower on soft (4 kPa) and very rigid (13 kPa) fibronectin coated surfaces whereas optimal crawling speeds were reached on 7 kPa. Interestingly fibronectin density also affected the outcome of migration speed. Using FN concentrations of 100 µg/ml the optimal stiffness for migration is 4 kPa while on 10 µg/ml the optimal rigidity for maximal migration is increased to 7 kPa.23 This suggests that leukocyte TEM in vivo depends on the combination between matrix rigidity (i.e. durotaxis) and the amount of available surface ligands (i.e., haptotaxis) for leukocytes to interact with.
Tenertaxis
Another phenomenon that is often observed in vitro when the endothelial barrier is very tight, is the predominant use of the transcellular route, whereas weak endothelial junctional integrity shows high association with paracellular diapedesis (Fig. 1d).24 To find these spots of low resistance, lymphocytes dynamically probe the underlying endothelium by extending invadosome-like protrusions into its surface that deform the plasma membrane, depolymerize F-actin filaments at the membrane cortex and ultimately breach the barrier.24,25 The authors suggest that leukocyte transmigration is guided by a common principle namely ‘the path of least resistance’.24 However, determining where the path of least resistance is present in vivo is very difficult, if not impossible. The observations of tenertaxis for TEM are so far only performed in in vitro studies. Another point is the seemingly contradictory effects of durotaxis and tenertaxis. According to tenertaxis, leukocytes would prefer a site of low endothelial cytoskeletal density, while according to durotaxis leukocytes need an optimum cytoskeletal stiffness to cross the vessel wall.24 This opposing arguments for determining the transendothelial migration ‘hotspot’ indicate that the mechanism that determines the TEM hotspot is likely an interdependent combination of the TEM factors chemo-, hapto-, duro- and tenertaxis, where, dependent on specific (patho)physiological conditions, one factor may play a more dominant role over the other in determining the site for leukocytes to cross.
Shear forces
The impact of shear forces on leukocyte behavior has been established by several research groups. Transmigration kinetics of neutrophils was significantly faster under shear stress than under static conditions (Fig. 1e).26 Leukocyte extravasation primarily takes place in the postcapillary venules of the inflamed tissue where the flow velocity is between 1–10 dyn/cm.2,27 Cinamon and co-workers showed that specifically for lymphocytes TEM was promoted by a continuous physiological flow between 0.75 and 5 dyn/cm.2,28 From these data it was concluded that flow-induced mechanical signals are coupled to Gi protein signaling at the luminal endothelial cell surface, resulting in further enhancing lymphocyte TEM.29 Additional work from the same group convincingly showed that shear stress promotes extensive filopodia formation by T-lymphocytes.19 Filopodia are small membrane “finger-like” protrusions that leukocytes use to probe the luminal endothelial surface before and during TEM. This process of lymphocyte probing the endothelial surface was underscored by a report by Carman and colleagues, who referred to these structures as invading podosomes.30
Although the majority of leukocyte extravasation occurs under low shear conditions in postcapillary venules, during some pathological conditions such as atherosclerosis, monocytes adhere and transmigrate through the endothelial lining of the artery wall where shear stress is much higher. It was been shown that leukocytes tethered to and rolled on platelet-decorated ultra-large Von Willebrand factor (ULVWF) string-like structures presented on the luminal side of the endothelium.31 Leukocytes scanned for activated platelets to interact via P-selectin glycoprotein ligand-1 (PSGL-1) resulting in clustering and activation of the β2 integrin Mac-1 that mediates neutrophil TEM.32,33 This simultaneous interaction with activated platelets and the endothelium results in rapid neutrophil exit and the onset of inflammation.34 Using platelets as intermediate substrates, monocytes are able to transmigrate under high shear stress varying between 20 and 40 dyn/cm.2,31 Thus, also matrices generated on the luminal surface of the endothelium can drive leukocyte TEM under high shear conditions.
Vascular beds
Leukocyte diapedesis through the blood brain barrier, into the peritoneum or lungs has been shown to be differentially regulated (Fig. 1f). For instance, neutrophil diapedesis in ICAM-1/P-selectin knock-out mice is normal in the lungs but totally abrogated in the peritoneum.35 Recently, it has been shown that locking the endothelial junctions prevented leukocyte diapedesis, but not in all tissues. Diapedesis into lung, skin and cremaster was severely reduced, establishing the paracellular route as the dominant route in these tissues. However, the migration of naïve lymphocytes into lymph nodes and transmigration of neutrophils into the peritoneum was not affected by junctional locking.36 Moreover, during inflammation in the respiratory tract of rats, plasma proteins leakage is predominantly observed in the post-capillary venules whereas capillaries and arterioles did not leak. Under these inflammatory conditions most leukocytes, in particular neutrophils, transmigrate in the collecting venules downstream of the leaky post-capillary venules.4 This landmark paper reveals that plasma protein leakage and leukocyte recruitment are 2 separable events that can occur side by side, but this leakage is not necessarily caused by the transmigration of immune cells through the ECs.
Leukocyte extravasation and vascular permeability
Inflammation is characterized by increased vasodilation, microvascular leakage and leukocyte recruitment. Whether the physical movement of leukocytes directly causes increased microvascular permeability has been debated for decades. Some studies propose leukocyte adhesion and transmigration to be acute events leading to tissue damage and organ failure during inflammation and ischemia-reperfusion.37,38 A strong argument that supports this hypothesis are the neutrophil depletion or CD11/CD18 blocking experiments that have been shown to attenuate vascular injury under inflammatory and ischemia-reperfusion conditions.38-41 However, when microvascular permeability was measured simultaneously with leukocyte-endothelial interactions, local plasma leakage sites were distinct from those of leukocyte adhesion or transmigration.4,5,42-45 Moreover, several studies have shown that the timing of leukocyte adhesion and transmigration are not well correlated with the evoked permeability change during acute inflammation.46-49 Recently, molecular evidence for the uncoupling between leukocyte TEM and vascular permeability has been presented by Wessel and colleagues. They mechanistically uncoupled leukocyte extravasation and vascular permeability by showing that opening of endothelial junctions in those distinct processes are controlled by different tyrosine residues of VE-cadherin in vivo.6,50 However, how the endothelium maintains a tight barrier during leukocyte TEM is still unknown.
Paracellular and transcellular migration
Paracellular migration is the main route taken by neutrophils to enter lung, skin or cremasteric tissue.12,36 Currently, 2 hypothesizes to open EC junctions dominate the field of leukocyte diapedesis. The first is based on research conducted on GPCR signaling in ECs, such as thrombin induced junctional opening51 and postulates that leukocytes induce actomyosin contraction in ECs triggering junctional opening.52-54 The second hypothesis anticipates that EC junctions are locally destabilized to allow migrating cells to squeeze through the transient opening in the junction. Recent evidence supporting the latter hypothesis shows that leukocytes trigger rapid dephosphorylation of Tyr731 on VE-cadherin via the tyrosine phosphatase SHP-2, which allows the adaptin AP-2 to bind and initiates endocytosis of VE-cadherin (Fig. 2a). This destabilizes VE-cadherin-based junctions, allowing junctional opening and consequent paracellular migration of leukocytes.6 Interestingly, the same group showed that VEGF and histamine, i.e. Gα signaling, increased the phosphorylation of Tyr685 on VE-cadherin, resulting in opening of cell-cell junctions. As described above for Tyr731 to be dephosphorylated, SHP2 is crucial and for Tyr685 to become phosphorylated, it is required that VE-PTP moves out of the way. However, both phosphorylation events have been shown to depend on Src-kinase.55,56 Also, both SHP2 and Gα signaling can result in RhoA activation.57,58 How then can these events occur specifically at one Tyr residue of the same protein with an almost opposite outcome? Although more detailed research should be performed to fully clarify this point, but we speculate that the signals require more proteins within one complex that can act in a very local and transient manner. So, for the above described events, RhoA may be activated in both signaling pathways, however, the presence of specific other triggering GTPase molecules like guanine-nucleotide exchange factors (GEFs) and/or GTPase activating proteins (GAPs) are likely to make the difference in the final outcome, resulting in local signals. We will discuss the contribution of these GEFs and GAPS in a separate sub-section below.
Figure 2.
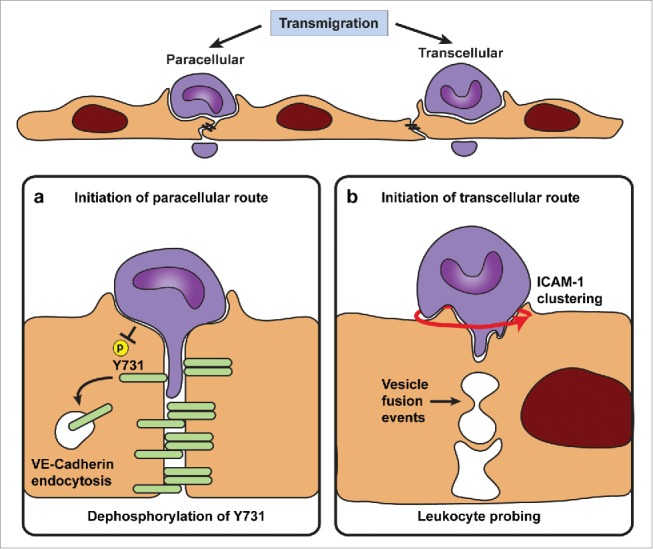
Leukocyte diapedesis through or between endothelial cells. (a) The initiation of paracellular and transcellular transmigration is believed to involve distinct molecular mechanisms that allow transient endothelial permeability to leukocytes. Destabilization of VE-cadherin based cell-cell contacts is recognized as the major mechanisms that initiates the opening of the paracellular pathway. It is thought that leukocytes trigger rapid dephosphorylation of Tyr731 via the tyrosine phosphatase SHP-2 allowing the adaptin AP-2 to bind and initiate endocytosis of VE-cadherin and thereby destabilize VE-cadherin based junctions. (b) The initiation of a transcellular passageway is thought to occur through fusion of ICAM-1 bearing endocytic vesicles forcing a transcellular pore allowing transcellular migration to occur. For both transmigration routes, endothelial pore opening is in part mediated by mechanical forces that are generated by migrating leukocytes. Polarized actin polymerization in the leukocyte elicits pulling and pushing forces that supports their movement through the confined endothelial pore.
Transcellular migration is the major transmigration route used by neutrophils to enter the peritoneum and for lymphocytes to enter lymph nodes.36 Several studies show that transcellular migration is ICAM-1-dependent.20,21,35,36,59 The initiation of a transcellular passageway is thought to occur through fusion of ICAM-1 containing endocytic vesicles forcing a transcellular pore.59 Lymphocytes induce transient ICAM-1 clustering and in areas with a high density of caveolin and actin stress fibers ICAM-1 could associate with, and induce fusion of, caveolae resulting in the formation of a transcellular pore.59 Moreover, local depolarization of F-actin at the endothelial cell cortex results in making the endothelial softer in a confined region underneath the adherent leukocyte.25 In combination with a local reduction in actomyosin-based contraction of the cell cortex, a transcellular pore can be formed.60 Chemotaxis during transcellular migration of lymphocytes is mediated by intra-endothelial vesicles containing chemokines rather than by extracellular released chemokines (Fig. 2b).29 Endocytic vesicle fusion thereby supports a simultaneous release of chemokines and initiation of a transcellular passageway.
For both transmigratory routes the trigger that initiates endothelial pore opening at the start of transmigration is heavily debated. The role of ICAM-1 as a potential trigger has been controversial for many years. An attractive hypothesis involves the combination of ICAM-1 clustering and mechanical forces of probing leukocytes.13 In case of transcellular migration ICAM-1 may soften the EC body by vesicle fusion events. But in paracellular migration ICAM-1 clustering may direct weakening of the junctions through recruitment of the tyrosine phosphatase SHP-2.6,61,62 For both transmigration routes, endothelial pore opening is in part mediated by mechanical forces that are generated by migrating leukocytes. Actin polymerization in the leukocyte elicits pulling and pushing forces that support the movement through the confined endothelial pore.63-65
The docking structure
A widely observed phenomenon associated with leukocyte TEM is the formation of endothelial membrane protrusions rich in Filamentous (F)-actin that surround transmigrating leukocytes. These endothelial structures were first described by Barreiro and colleagues who defined them as docking structures.66 Other researchers found similar endothelial structures but proposed different names, e.g. transmigratory cups, apical cups, dome structures, ICAM-1-enriched contact areas, or actin dynamic structures.14,67-71 The names were based on the hypothesized function or morphology of these structures. Work of Carman and coworkers showed that these structures, that formed both during para- and transcellular diapedesis (Fig. 2), were more frequently associated with leukocytes in the process of transmigration than with firm adherent leukocytes prior to diapedesis.14 Many of these F-actin structures comprise vertical microvilli-like protrusions. These protrusions have been suggested to anchor endothelial adhesion receptors, such as ICAM-1 and VCAM-1. As such they may serve as migration-supporting platforms or adhesion substrates to assist leukocyte transmigration.50,72-75 These platforms are controlled by tetraspannins, transmembrane proteins that keep the platforms together and organize it into small microdomains.75,76 It has been shown that assembly of F-actin, the major component and driving force to induce such apical protrusions, requires the activation small GTPases RhoG and Rac1.67,77 Currently, the major function of the docking structure is thought to provide guidance for transmigrating leukocytes.78
The regulation of the endothelial F-actin cytoskeleton
Regulation of leukocyte TEM and vascular integrity both depend on the ability to dynamically remodel the F-actin cytoskeleton in ECs. Failure to do so leads to disruption of junctional integrity and ineffective TEM.
Perhaps the best known example of F-actin remodeling is the formation of the lamellipodia structure at the leading edge of migrating cells. Lamellipodia contain an extensively branched network of polarized actin filaments with the plus ends directed toward the plasma membrane. The growing of actin monomers into a branched filamentous network produces mechanical force that drives the forward extension of the lamellipodia. Numerous actin-based responses are triggered by extracellular signals as gain or loss of cell-cell contact, contact inhibition, gain or loss of cell-matrix interaction, growth factor receptor mediated signaling, chemotaxis in response to attractive and repulsive guidance cues and so forth. These input signals are subsequently converted into an intracellular response. Three well known proteins that fulfill this task of signal conversion are the small Rho-GTPases Rac1, RhoA and Cdc42. These small Rho-GTPases act as molecular switches between receptor-mediated signaling and the actin polymerization machinery. Actin polymerization does not occur spontaneously, or if so at a very slow rate. To accelerate the polymerization, it requires a set of proteins forming the actin polymerization machinery, including nucleating promoting factors (NPFs) such as WAVE and N-WASP that activate actin nucleators like Arp2/3, mDia and Ena/VASP proteins. These processes as described above are reviewed extensively by Pollard and colleagues.79
GEFs and GAPs
Guanine-nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs) are the master regulators of Rho-family GTPases and therefore regulate numerous cellular responses. Endothelial cells express over 22 Rho-GTPases and more than 69 GEFs and an equal number of GAPs.80 GEF and GAP function is required to regulate the rate, location and timing of GTPase activity. This is probably why cells express a higher variety of GEFs and GAPs compared to the number of GTPases, to fine-tune complex cellular processes such as maintenance of stable endothelial cell-cell junctions or directional migration. Rho proteins cycle between GDP- and GTP-bound states. GEFs exchange the transition between the GDP (inactive) to the GTP (active) loaded state. Whereas GAPs enhance the relative slow intrinsic GTPase activity of Rho proteins. Another set of regulatory proteins are the GDI proteins, known as guanine nucleotide-dissociation inhibitors that keep GTPases in the inactive state in the cytosol. GEFs contain a DH domain, which catalyzes the exchange of GDP for GTP to activate Rho-GTPases. Another domain found in many GEFs is the PH domain which has been reported to target the GEF to the plasma membrane81 or to facilitate binding to the GTPase. For instance, the leukemia associated Rho-GEF (LARG) binds directly to RhoA through its PH domain.82 Interestingly, the PH domain does not only bind to phospholipids or GTPases but also binds to other proteins. For example, the first PH-DH domain of Trio directly interacts with the actin cross linker filamin. There is always an exception to the rule: some GEFs like Tiam1 and Vav1 use other protein domains to determine their subcellular distribution and do not require their PH domain for membrane binding.83 In addition to this exception, some GEFs (e.g., ARNO/DOCK family) completely lack a PH domain and use a BAR domain to interact with curved membrane.84 Nonetheless, PH domains in many GEFs are responsible for their subcellular localization and are required for proper GTPase activation. Binding of the PH domain to phospholipids orientates the associated DH domain correctly for proper GTPase activation. In addition to that, plasma membrane binding could lead to a conformational change between the PH and DH domain that enhances GEF activity. Some GEFs like Vav1 contain auto-inhibitory sequences at the N-terminus.85 In case of Vav1 phosphorylation terminates the auto-inhibition and as a result the DH domain becomes accessible and able to activate GTPases.86
Importantly, depending on their subcellular localization Rho-GEFs can globally and locally change the equilibrium of the Rho-GTP bound state. For instance, the GEF Net1 resides inactive in the nucleus but after translocation to the plasma membrane it activates RhoA.87 Similarly, Ect2 is normally localized in the nucleus during interphase but comes out of the nucleus during cell division to activate RhoA to regulate the cleavage furrow that separates the 2 cells during cell division.88 Another example is GEF-H1 that directly interacts with microtubules, inhibiting its exchange potential toward RhoA. Tubulin depolymerization disrupts this interaction resulting in local RhoA activation.89 Finally, GEFs can also function as scaffolding proteins supporting larger proteins complexes up- or downstream of Rho-GTPases. This scaffolding function does not require GEF activity. For instance α-Pix acts as a scaffold to integrate signals that arise from GPCRs with the activation of Cdc42 to drive chemotaxis.90 In a related example β-Pix tethers NADPH oxidase-1 to Rac1 for activation,91 a pathway that regulates production of reactive oxygen species that is important to kill pathogenic bacteria.
Dysregulation of Rho-GTPases can have numerous causes such as altered GTPase gene expression, deregulated function or gene expression of regulatory GAPs, GEFs or GDIs, including Rho-GTPase effectors such as WAVE. Cancer is often associated with altered GTPase regulation. For example, LARG has been identified in acute myelogenous leukemia. A missense mutation in Tiam1 has been recognized in inducing transforming activities of the cell. Developmental and neurological disorders such as ALS (amyotrophic lateral sclerosis) are caused by loss of function mutations in the GEF ALS2. And for viral and bacterial pathogenesis: some Rho-GEFs are hijacked to facilitate the pathogenic invasion of host cells.92 Current studies aim to unravel the spatiotemporal activation of GTPases using functional FRET-based biosensors to study local GEF and GAP activity. We recently reported that the GEFs LARG and Ect2 play an important role in maintaining vascular leakage during leukocyte diapedesis.93 The next section will elaborate on this.
Actin regulation during leukocyte TEM
Molecular evidence for the uncoupling between leukocyte TEM and vascular permeability has been presented6 and we recently reported that local RhoA-mediated F-actin rings contribute to endothelial pore confinement in order to maintain the endothelial barrier integrity during leukocyte diapedesis.93 Using a FRET-based DORA RhoA biosensor, we show that RhoA is transiently and locally activated during leukocyte diapedesis and not during the adhesion and crawling phase by inducing F-actin-rich rings around the spot where the leukocyte crosses. These rings show asymmetrical phosphorylation of myosin light chain, indicating that these contractile F-actin rings serve as elastic straps to limit leakage. A well-studied downstream signaling route of RhoA is through activation of Rho Kinase (ROCK).94 ROCK promotes the phosphorylation of the myosin-II regulatory light chains by Myosin Light Chain Kinase (MLCK). Myosin-II is a motor protein that moves along actin filaments toward the plus end. The movement of 2 opposing myosin-II complexes generates force on actin filaments causing antiparallel sliding of adjacent actin filaments relative to one another and contraction of the actin filaments.95 This may underlie the induction of the contractile ring and prevention of vascular leakage during TEM. Our recent data further support this signaling pathway being involved: a GEF screen indicates a role for the RhoA-GEFs LARG and Ect2 in this process, activating RhoA that in turn induces myosin-based contraction via ROCK2b and MLCK activation.93
However, how these F-actin-rich endothelial straps are initiated is not clear. We hypothesize that it all starts with a signaling receptor expressed on the surface of the endothelium that transmits the presence of a transmigrating leukocyte, finally resulting in local RhoA activation. Candidate receptors are the inflammatory adhesion molecule ICAM-1 and the junctional protein PECAM-1, both present at the site of pore formation during neutrophil transmigration, either paracellular or transcellular. Interestingly, upon mechanical tension both proteins have been reported to induce RhoA activity.96,97 In support of a prominent role for ICAM-1, our data show that depletion of ICAM-1 in TNF-α-stimulated endothelial cells resulted in an increase of neutrophil-induced dextran leakage compared to control endothelial cells, although the levels did not reach the levels when RhoA was silenced.93 PECAM-1 however appeared not to be involved in maintenance of barrier function, as depletion in ECs had no effect on neutrophil-induced dextran leakage and neutrophil transmigration numbers.93 Homophilic PECAM-1 interactions between ECs and leukocytes are described repeatedly to be important for leukocytes TEM guidance.98,99 But the fact that we observed no effect of PECAM-1 depletion on both neutrophil TEM efficiency and endothelial barrier function can be explained by the stimulus-dependent role of PECAM-1 in leukocyte diapedesis. Where PECAM-1 plays an important role in guiding leukocyte TEM elicited by IL-1β, diapedesis responses to TNF-α or fMLP were shown to be PECAM-1 independent.98 This elucidates the observed TNF-α-induced effects we found, and proposes PECAM-1 as in interesting target for endothelial barrier preservation during IL-1β-induced leukocytes diapedesis.
Junctional protein candidates
ICAM-1-deficient ECs compromised the barrier function leading to dextran leakage during neutrophil TEM, but to lesser extent when compared to RhoA or its downstream effector ROCK2 depletion in ECs.93 This discrepancy in the difference in leakage implicates that ICAM-1 is involved in activating RhoA but is not the only signal transducer necessary to induce formation of the actin ring and prevent leakage. The involvement of other proteins at cell-cell junctions such as junctional adhesion molecules (JAMs) or CD99 in RhoA activation and preservation of endothelial barrier function during leukocytes diapedesis are unknown and therefore of interest for future research. In vivo blockade of JAM-A activity or expression results in a decrease of both neutrophil and monocyte diapedesis98 caused by its involvement in directing leukocyte TEM via homophilic interactions between endothelial and leukocyte JAM-A molecules.100 Moreover, a recent report shows that tension imposed on JAM-A induces RhoA activation resulting in endothelial cell stiffening.101 Therefore, it is tempting to speculate that JAM-A may, in concert with ICAM-1, be involved as the initial signaling receptors that lead to local RhoA activation and ring formation. However, based on our work, the GEFs implicated in tension-induced RhoA activation through JAM-A, p115rhoGEF and GEF-H1, do not support F-actin-rich rings during leukocyte diapedesis.93 In addition, JAM-A is not essential as knockout mice are viable and show no major defects in vascular development or permeability indicating redundancy with the other family members JAM-B and JAM-C or other junctional proteins as PECAM-1.98 Indeed, JAM-C is found to interact with Mac-1 integrin on leukocytes.102 Therefore it might be necessary to deplete more than only one JAM family members, possibly in combination with PECAM-1 silencing to study the involvement of JAM proteins in the local induction of RhoA activation, and thereby formation of the F-actin-rich ring surrounding transmigrating leukocytes to retain vascular barrier function.
At endothelial cell-cell junctions is among several junctional proteins CD99 found, a heavily O-glycosylated 32-kD type I transmembrane protein that can interact in a homophilic manner with its neighbor on the adjacent endothelial cells. Interestingly, endothelial CD99 can also interact with the monocytic CD99 and thereby facilitates monocyte transmigration.103 Later in vitro and in vivo involvement of CD99 in directing diapedesis of monocytes, neutrophils and T-cells was established104,105 making this junctional protein of interest in the context of unraveling the initial receptor involved in formatting of a confined actin pore during leukocyte TEM. Intracellular CD99 signaling regulating leukocyte TEM has recently been described to involve soluble adenylyl cyclase (sAC) that activates protein kinase A (PKA). This activation occurs at the lysine-rich intracellular tail of CD99 using the A-kinase anchoring protein (AKAP) ezrin as a scaffold.104 Involvement of ezrin in this signaling complex could connect homophilic CD99 interaction to formation of the F-actin rich pore, because ezrin belongs to the ERM family (ezrin/radixin/moesin) of proteins that link actin filaments to the plasma membrane. Moreover, CD99 functions at a later stage during the diapedesis step, i.e., when the leukocyte has worked its way into the junction already.103 For our model, we noticed that the RhoA activity also occurs during these stages of diapedesis, the so-called mid-to-late diapedesis steps.93 Therefore it is interesting to test if CD99 depletion in ECs would impair the barrier function of the endothelium during leukocyte TEM.
De novo actin ring formation
Although RhoA GDP/GTP cycling is necessary to induce de novo actin polymerization and thereby contraction and confinement of the pore during neutrophil diapedesis, it is not proven that RhoA is also involved in de novo actin polymerization to form the diapedesis ring. As we showed that F-actin-positive rings surround the endothelial pore during all steps of diapedesis, including the very early initiation of pore formation,93 the question is if the initial ring formation is mediated by de novo actin polymerization, or that existing stress fibers are remodeled into a ring like structure. In the latter assumption, RhoA-induced actin fibers would be involved in contraction only. However, depletion of endothelial RhoA reduced the accumulation of lifeact-GFP around the pore, implicating that the F-actin-rich endothelial pores are formed on existing fibers that are supported and strengthened with de novo actin polymerization.93
Conclusion
Together, the phenomenon how the endothelium regulates its barrier function during leukocyte extravasation appears to be a local affair. Although the mechanism responsible for this action is not yet fully elucidated, we can conclude that it involves the local contractile machinery of the endothelium. In an attempt to summarize the current knowledge on how local vascular signals differentially control permeability and leukocyte TEM, we compiled a figure starting with local vascular injury in the capillaries (Fig. 3a). Inflammatory signals active the vasculature more downstream, i.e. in the post-capillaries, to mediate leukocyte TEM (Fig. 3b-c). In order to restore vascular homeostasis, transmigrated leukocytes crawl through the underlying tissue toward the injured site to remove damaged tissue (Fig. 3d). This then results in a full resolution of the inflammation and restored vascular homeostasis (Fig. 3e).
Figure 3.

Leukocyte diapedesis and vascular permeability are uncoupled events. Vascular injury in the skin is resolved by various sequential processes that initiate tissue repair and the clearance of pathogens and dirt to restore vascular homeostasis. (a) In the first step of the cascade, platelets adhere to exposed collagen forming a haemostatic plug of fibrin that arrests blood leakage (a-c). Activated platelets produce thrombin, a compound that activates the coagulation cascade to produce the haemostatic plug. Thrombin released by activated platelets and histamine released by tissue basophils and mast cells are thought to initiate endothelial activation, a transient increase in endothelial permeability provoked by RhoA-mediated actomyosin contractility, and local enhancement of blood flow. This results in chemokine and cytokine release by various cell types followed by inflammation close to the site of injury. (b) Downstream of vascular injury endothelial cells get activated and in turn expose a variety of adhesion molecules at their surface. Upstream the transient permeability increase is counterbalanced by Trio-Rac1-mediated JAILs that stabilize endothelial junctions. (c) Leukocyte recruitment downstream of vascular leakage and vascular damage is believed to follow the multistep paradigm of TEM. Vascular leakage during leukocyte diapedesis is prevented by RhoA-mediated endothelial pore confinement. (d) Infiltrating leukocytes scan and clear pathogens and dirt from the site of infection. Rac1-mediated wound healing and angiogenesis repair damaged tissue and vessels. (e) Finally, tissue macrophages secrete chemokines that resolve inflammation arresting leukocyte recruitment to restore vascular homeostasis.
Despite the current lack of the true initiation signals, we hypothesize that the leukocyte itself triggers the endothelium to start the local machinery that induce formation of the contractile rings to limit vascular leakage during TEM. However, the question remains if the mechanism to prevent leakage is the same during para- and transcellular migration modes. This is also true for the TEM events that occur in vivo in different tissues under specific conditions such as acute lung injury and ischemic reperfusion injury. Nevertheless, despite these remaining uncertainties it is clear that permeability and leukocyte TEM are regulated independently and can be seen as 2 separate events, in vivo occurring at different location, i.e., the capillaries and post-capillaries, but yet interconnected events that need each other to resolve the inflammation (Fig. 3).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Grivennikov SI, Greten FR, Karin M. Immunity, Inflammation, and Cancer. Cell 2010; 140:883-99; PMID:20303878; http://dx.doi.org/ 10.1016/j.cell.2010.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Nourshargh S, Hordijk PL, Sixt M. Breaching multiple barriers: leukocyte motility through venular walls and the interstitium. Nat Rev Mol Cell Biol 2010; 11:366-78; PMID:20414258; http://dx.doi.org/ 10.1038/nrm2889 [DOI] [PubMed] [Google Scholar]
- [3].Heemskerk N, Van Rijssel J, Van Buul JD. Rho-GTPase signaling in leukocyte extravasation: An endothelial point of view. Cell Adhes Migr 2014; 8:67-75; PMID:24621576; http://dx.doi.org/ 10.4161/cam.28244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Baluk P, Bolton P, Hirata A, Thurston G, McDonald DM. Endothelial gaps and adherent leukocytes in allergen-induced early- and late-phase plasma leakage in rat airways. Am J Pathol 1998; 152:1463-76; PMID:9626051 [PMC free article] [PubMed] [Google Scholar]
- [5].McDonald DM. Endothelial gaps and permeability of venules in rat tracheas exposed to inflammatory stimuli. Am J Physiol 1994; 66:L61-L83; PMID:7508201 [DOI] [PubMed] [Google Scholar]
- [6].Wessel F, Winderlich M, Holm M, Frye M, Rivera-Galdos R, Vockel M, Linnepe R, Ipe U, Stadtmann A, Zarbock A, et al.. Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-cadherin. Nat Immunol 2014; 15:223-30; PMID:24487320; http://dx.doi.org/ 10.1038/ni.2824 [DOI] [PubMed] [Google Scholar]
- [7].Hillgruber C, Pöppelmann B, Weishaupt C, Steingräber AK, Wessel F, Berdel WE, Gessner JE, Ho-Tin-Noé B, Vestweber D, Goerge T. Blocking neutrophil diapedesis prevents hemorrhage during thrombocytopenia. J Exp Med 2015; 212:1255-66; ; PMID:26169941; http://dx.doi.org/ 10.1084/jem.20142076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Becker KJ. Anti-leukocyte antibodies: LeukArrest (Hu23F2G) and Enlimomab (R6.5) in acute stroke. Curr Med Res Opin 2002; 18(Suppl 2):s18-s22; PMID:12365824; http://dx.doi.org/ 10.1185/030079902125000688 [DOI] [PubMed] [Google Scholar]
- [9].Butcher EC. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell 1991; 67:1033-6; PMID:1760836; http://dx.doi.org/ 10.1016/0092-8674(91)90279-8 [DOI] [PubMed] [Google Scholar]
- [10].Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell 1994; 76:301-14; PMID:7507411; http://dx.doi.org/ 10.1016/0092-8674(94)90337-9 [DOI] [PubMed] [Google Scholar]
- [11].Kroon J, Daniel AE, Hoogenboezem M, van Buul JD. Real-time imaging of endothelial cell-cell junctions during neutrophil transmigration under physiological flow. J Vis Exp 2014; e51766; PMID:25146919; http://dx.doi.org/ 10.3791/51766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Schulte D, Küppers V, Dartsch N, Broermann A, Li H, Zarbock A, Kamenyeva O, Kiefer F, Khandoga A, Massberg S, et al.. Stabilizing the VE-cadherin–catenin complex blocks leukocyte extravasation and vascular permeability. EMBO J. 2011; 30:4157-70; PMID:21857650; http://dx.doi.org/ 10.1038/emboj.2011.304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Carman CV. Mechanisms for transcellular diapedesis: probing and pathfinding by “invadosome-like protrusions”. J Cell Sci 2009; 122:3025-35; PMID:19692589; http://dx.doi.org/ 10.1242/jcs.047522 [DOI] [PubMed] [Google Scholar]
- [14].Carman CV, Springer TA. A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J Cell Biol 2004; 167:377-88; PMID:15504916; http://dx.doi.org/ 10.1083/jcb.200404129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Feng D, Nagy JA, Pyne K, Dvorak HF, Dvorak AM. Neutrophils emigrate from venules by a transendothelial cell pathway in response to FMLP. J Exp Med 1998; 187:903-15; PMID:9500793; http://dx.doi.org/ 10.1084/jem.187.6.903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bao X, Moseman EA, Saito H, Petryniak B, Thiriot A, Hatakeyama S, Ito Y, Kawashima H, Yamaguchi Y, Lowe JB, et al.. Endothelial heparan sulfate controls chemokine presentation in recruitment of lymphocytes and dendritic cells to lymph nodes. Immunity 2010; 33:817-29; PMID:21093315; http://dx.doi.org/ 10.1016/j.immuni.2010.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Abtin A, Jain R, Mitchell AJ, Roediger B, Brzoska AJ, Tikoo S, Cheng Q, Ng LG, Cavanagh LL, von Andrian UH, et al.. Perivascular macrophages mediate neutrophil recruitment during bacterial skin infection. Nat Immunol 2014; 15:45-53; PMID:24270515; http://dx.doi.org/ 10.1038/ni.2769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Salanga CL, Handel TM. Chemokine oligomerization and interactions with receptors and glycosaminoglycans: The role of structural dynamics in function. Exp Cell Res 2011; 317:590-601; PMID:21223963; http://dx.doi.org/ 10.1016/j.yexcr.2011.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shulman Z, Shinder V, Klein E, Grabovsky V, Yeger O, Geron E, Montresor A, Bolomini-Vittori M, Feigelson SW, Kirchhausen T, et al.. Lymphocyte crawling and transendothelial migration require chemokine triggering of high-affinity LFA-1 integrin. Immunity 2009; 30:384-96; PMID:19268609; http://dx.doi.org/ 10.1016/j.immuni.2008.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Abadier M, Haghayegh Jahromi N, Cardoso Alves L, Boscacci R, Vestweber D, Barnum S, Deutsch U, Engelhardt B, Lyck R. Cell surface levels of endothelial ICAM-1 influence the transcellular or paracellular T-cell diapedesis across the blood-brain barrier. Eur J Immunol 2015; 45:1043-58; PMID:25545837; http://dx.doi.org/ 10.1002/eji.201445125 [DOI] [PubMed] [Google Scholar]
- [21].Yang L, Froio RM, Sciuto TE, Dvorak AM, Alon R, Luscinskas FW. ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF- ⊔ – activated vascular endothelium under flow. Blood 2005; 106:584-93; PMID:15811956; http://dx.doi.org/ 10.1182/blood-2004-12-4942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gonzalez AL, El-Bjeirami W, West JL, McIntire LV, Smith CW. Transendothelial migration enhances integrin-dependent human neutrophil chemokinesis. J Leukoc Biol 2007; 81:686-95; PMID:17164427; http://dx.doi.org/ 10.1189/jlb.0906553 [DOI] [PubMed] [Google Scholar]
- [23].Stroka KM, Aranda-Espinoza H. Neutrophils display biphasic relationship between migration and substrate stiffness. Cell Motil Cytoskeleton 2009; 66:328-41; PMID:19373775; http://dx.doi.org/ 10.1002/cm.20363 [DOI] [PubMed] [Google Scholar]
- [24].Martinelli R, Zeiger AS, Whitfield M, Sciuto TE, Dvorak A, Van Vliet KJ, Greenwood J, Carman CV, et al.. Probing the biomechanical contribution of the endothelium to lymphocyte migration: diapedesis by the path of least resistance. J Cell Sci 2014; 127:3720-34; PMID:25002404; http://dx.doi.org/ 10.1242/jcs.148619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Isac L, Thoelking G, Schwab A, Oberleithner H, Riethmuller C. Endothelial f-actin depolymerization enables leukocyte transmigration. Anal Bioanal Chem 2011; 399:2351-8; PMID:20632161; http://dx.doi.org/ 10.1007/s00216-010-3978-z [DOI] [PubMed] [Google Scholar]
- [26].Kitayama J, Hidemura A, Saito H, Nagawa H. Shear stress affects migration behavior of polymorphonuclear cells arrested on endothelium. Cell Immunol 2000; 203:39-46; PMID:10915560; http://dx.doi.org/ 10.1006/cimm.2000.1671 [DOI] [PubMed] [Google Scholar]
- [27].Lawrence M, Springer T. Leukocytes roll on a selectin at physiologic flow rates: distinction from and prerequisite for adhesion through integrins. Cell 1991; 65:859-73; PMID:1710173; http://dx.doi.org/ 10.1016/0092-8674(91)90393-D [DOI] [PubMed] [Google Scholar]
- [28].Cinamon G, Shinder V, Alon R. Shear forces promote lymphocyte migration across vascular endothelium bearing apical chemokines. Nat.Immunol. 2001; 2:515-22; PMID:11376338; http://dx.doi.org/ 10.1038/88710 [DOI] [PubMed] [Google Scholar]
- [29].Shulman Z, Cohen SJ, Roediger B, Kalchenko V, Jain R, Grabovsky V, Klein E, Shinder V, Stoler-Barak L, Feigelson SW, et al.. Transendothelial migration of lymphocytes mediated by intraendothelial vesicle stores rather than by extracellular chemokine depots. Nat Immunol 2011; 13:67-76; PMID:22138716; http://dx.doi.org/ 10.1038/ni.2173 [DOI] [PubMed] [Google Scholar]
- [30].Carman CV, Sage PT, Sciuto TE, de la Fuente MA, Geha RS, Ochs HD, Dvorak HF, Dvorak AM, Springer TA. Transcellular Diapedesis Is Initiated by Invasive Podosomes. Immunity 2007; 26:784-97; PMID:17570692; http://dx.doi.org/ 10.1016/j.immuni.2007.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bernardo A, Ball C, Nolasco L, Choi H, Moake JL, Dong JF. Platelets adhered to endothelial cell-bound ultra-large von Willebrand factor strings support leukocyte tethering and rolling under high shear stress. J Thromb Haemost 2005; 3:562-70; PMID:15748247; http://dx.doi.org/ 10.1111/j.1538-7836.2005.01122.x [DOI] [PubMed] [Google Scholar]
- [32].Xu T, Zhang L, Geng ZH, Wang HB, Wang JT, Chen M, Geng JG. P-Selectin cross-links PSGL-1 and enhances neutrophil adhesion to fibrinogen and ICAM-1 in a Src kinase-dependent , but GPCR-independent mechanism. Cell Adh Migr 2007; 1:115-23; PMID:19262138; http://dx.doi.org/ 10.4161/cam.1.3.4984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wang HB, Wang JT, Zhang L, Geng ZH, Xu WL, Xu T, Huo Y, Zhu X, Plow EF, Chen M, et al.. P-selectin primes leukocyte integrin activation during inflammation. Nat Immunol 2007; 8:882-92; PMID:17632516; http://dx.doi.org/ 10.1038/ni1491 [DOI] [PubMed] [Google Scholar]
- [34].Sreeramkumar V, Adrover JM, Ballesteros I, Cuartero MI, Rossaint J, Bilbao I, Nácher M, Pitaval C, Radovanovic I, Fukui Y, et al.. Neutrophils scan for activated platelets to initiate inflammation. Science (80-. ) 2014; 346:1234-8; PMID:25477463; http://dx.doi.org/ 10.1126/science.1256478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bullard DC, Qin L, Lorenzo I, Quinlin WM, Doyle NA, Bosse R, Vestweber D, Doerschuk CM, Beaudet AL. P-selectin/ICAM-1 double mutant mice: Acute emigration of neutrophils into the peritoneum is completely absent but is normal into pulmonary alveoli. J Clin Invest 1995; 95:1782-8; PMID:7535798; http://dx.doi.org/ 10.1172/JCI117856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Küppers V, Vestweber D, Schulte D. Locking endothelial junctions blocks leukocyte extravasation, but not in all tissues. Tissue Barriers 2013; 1:e23805; PMID:24665379; http://dx.doi.org/ 10.4161/tisb.23805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Oliver MG, Specian RD, Perry MA, Granger DN. Morphologic assessment of leukocyte-endothelial cell interactions in mesenteric venules subjected to ischemia and reperfusion. Inflammation 1991; 15:331-46; PMID:1684573; http://dx.doi.org/ 10.1007/BF00917350 [DOI] [PubMed] [Google Scholar]
- [38].Hernandez LA, Grisham MB, Twohig B, Arfors KE, Harlan JM, Granger DN. Role of neutrophils in ischemia-reperfusion-induced microvascular injury. Am J Physiol 1987; 253:H699-703; PMID:3631303 [DOI] [PubMed] [Google Scholar]
- [39].Kubes P, Suzuki M, Granger DN. Modulation of PAF-induced leukocyte adherence and increased microvascular permeability. Am J Physiol 1990; 259:G859-64; PMID:2240225 [DOI] [PubMed] [Google Scholar]
- [40].Sumagin R, Lomakina E, Sarelius IH. Leukocyte-endothelial cell interactions are linked to vascular permeability via ICAM-1-mediated signaling. Am J Physiol Heart Circ Physiol 2008; 295:H969-77; PMID:18641276; http://dx.doi.org/ 10.1152/ajpheart.00400.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Carden DL, Smith JK, Korthuis RJ. Neutrophil-mediated microvascular dysfunction in postischemic canine skeletal muscle. Role of granulocyte adherence. Circ Res 1990; 66:1436-44; PMID:2159391; http://dx.doi.org/ 10.1161/01.RES.66.5.1436 [DOI] [PubMed] [Google Scholar]
- [42].McDonald DM, Thurston G, Baluk P. Endothelial gaps as sites for plasma leakage in inflammation. Microcirculation 1999; 6:7-22; PMID:10100186; http://dx.doi.org/ 10.1080/713773924 [DOI] [PubMed] [Google Scholar]
- [43].Baluk P, Bertrand C, Geppetti P, McDonald DM, Nadel JA. NK1 receptors mediate leukocyte adhesion in neurogenic inflammation in the rat trachea. Am J Physiol 1995; 268:L263-9; PMID:7864147 [DOI] [PubMed] [Google Scholar]
- [44].Gawlowski DM, Benoit JN, Granger HJ. Microvascular pressure and albumin extravasation after leukocyte activation in hamster cheek pouch. Am J Physiol 1993; 264:H541-6; PMID:7680539 [DOI] [PubMed] [Google Scholar]
- [45].Rosengren S, Ley K, Arfors KE. Dextran sulfate prevents LTB4-induced permeability increase, but not neutrophil emigration, in the hamster cheek pouch. Microvasc Res 1989; 38:243-54; PMID:2481803; http://dx.doi.org/ 10.1016/0026-2862(89)90003-4 [DOI] [PubMed] [Google Scholar]
- [46].Valeski JE, Baldwin AL. Effect of early transient adherent leukocytes on venular permeability and endothelial actin cytoskeleton. Am J Physiol 1999; 277:H569-75; PMID:10444481 [DOI] [PubMed] [Google Scholar]
- [47].Kim M-H, Curry F.-RE, Simon SI. Dynamics of neutrophil extravasation and vascular permeability are uncoupled during aseptic cutaneous wounding. Am J Physiol Cell Physiol 2009; 296:C848-56; PMID:19176758; http://dx.doi.org/ 10.1152/ajpcell.00520.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lewis RE, Granger HJ. Diapedesis and the permeability of venous microvessels to protein macromolecules: the impact of leukotriene B4 (LTB4). Microvasc Res 1988; 35:27-47; PMID:2830471; http://dx.doi.org/ 10.1016/0026-2862(88)90048-9 [DOI] [PubMed] [Google Scholar]
- [49].Lewis RE, Miller RA., Granger HJ. Acute microvascular effects of the chemotactic peptide N-formyl-methionyl-leucyl-phenylalanine: Comparisons with leukotriene B4. Microvasc Res 1989; 37:53-69; PMID:2537922; http://dx.doi.org/ 10.1016/0026-2862(89)90072-1 [DOI] [PubMed] [Google Scholar]
- [50].Vestweber D, Wessel F, Nottebaum AF. Similarities and differences in the regulation of leukocyte extravasation and vascular permeability. Semin Immunopathol 2014; 36:177-92; PMID:24638889; http://dx.doi.org/ 10.1007/s00281-014-0419-7 [DOI] [PubMed] [Google Scholar]
- [51].Amerongen GPVN, Delft SV, Vermeer MA, Collard JG, van Hinsbergh VWM. Activation of RhoA by Thrombin in Endothelial Hyperpermeability: Role of Rho Kinase and Protein Tyrosine Kinases. Circ Res 2000; 87:335-40; PMID:10948069; http://dx.doi.org/ 10.1161/01.RES.87.4.335 [DOI] [PubMed] [Google Scholar]
- [52].Hixenbaugh EA, Goeckeler ZM, Papaiya NN, Wysolmerski RB, Silverstein SC, Huang AJ. Stimulated neutrophils induce myosin light chain phosphorylation and isometric tension in endothelial cells. Am J Physiol Hear. Circ Physiol 1997; 273:H981-8; PMID:9277518 [DOI] [PubMed] [Google Scholar]
- [53].Huang AJ, Manning JE, Bandak TM, Ratau MC, Hanser KR, Silverstein SC. Endothelial cell cytosolic free calcium regulatesneutrophil migration across monolayers of endothelial cells. J Cell Biol 1993; 120:1371-80; PMID:8449983; http://dx.doi.org/ 10.1083/jcb.120.6.1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Saito H, Minamiya Y, Saito S, Ogawa J. Endothelial Rho and Rho kinase regulate neutrophil migration via endothelial myosin light chain phosphorylation. J Leukoc Biol 2002; 72:829-36; PMID:12377953 [PubMed] [Google Scholar]
- [55].Adam AP, Sharenko AL, Pumiglia K, Vincent PA. Src-induced tyrosine phosphorylation of VE-cadherin is not sufficient to decrease barrier function of endothelial monolayers. J Biol Chem 2010; 285:7045-55; PMID:20048167; http://dx.doi.org/ 10.1074/jbc.M109.079277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wallez Y, Cand F, Cruzalegui F, Wernstedt C, Souchelnytskyi S, Vilgrain I, Huber P. Src kinase phosphorylates vascular endothelial-cadherin in response to vascular endothelial growth factor: identification of tyrosine 685 as the unique target site. Oncogene 2007; 26:1067-77; PMID:16909109; http://dx.doi.org/ 10.1038/sj.onc.1209855 [DOI] [PubMed] [Google Scholar]
- [57].Schoenwaelder SM, Petch LA, Williamson D, Shen R, Feng GS, Burridge K. The protein tyrosine phosphatase Shp-2 regulates RhoA activity. Curr Biol 2000; 10:1523-6; PMID:11114521; http://dx.doi.org/ 10.1016/S0960-9822(00)00831-9 [DOI] [PubMed] [Google Scholar]
- [58].Mikelis CM, Simaan M, Ando K, Fukuhara S, Sakurai A, Amornphimoltham P, Masedunskas A, Weigert R, Chavakis T, Adams RH, et al.. RhoA and ROCK mediate histamine-induced vascular leakage and anaphylactic shock. Nat Commun 2015; 6:6725; PMID:25857352; http://dx.doi.org/ 10.1038/ncomms7725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Millán J, Hewlett L, Glyn M, Toomre D, Clark P, Ridley AJ. Lymphocyte transcellular migration occurs through recruitment of endothelial ICAM-1 to caveola- and F-actin-rich domains. Nat Cell Biol 2006; 8:113-23; PMID:16429128; http://dx.doi.org/ 10.1038/ncb1356 [DOI] [PubMed] [Google Scholar]
- [60].Lemichez E, Gonzalez-Rodriguez D, Bassereau P, Brochard-Wyart F. Transcellular tunnel dynamics: Control of cellular dewetting by actomyosin contractility and I-BAR proteins. Biol Cell 2013; 105:109-17; PMID:23189935; http://dx.doi.org/ 10.1111/boc.201200063 [DOI] [PubMed] [Google Scholar]
- [61].Thompson PW, Randi AM, Ridley AJ. Intercellular adhesion molecule (ICAM)-1, but not ICAM-2, activates RhoA and stimulates c-fos and rhoA transcription in endothelial cells. J Immunol 2002; 169:1007-13; PMID:12097408; http://dx.doi.org/ 10.4049/jimmunol.169.2.1007 [DOI] [PubMed] [Google Scholar]
- [62].Greenwood J, Amos CL, Walters CE, Couraud PO, Lyck R, Engelhardt B, Adamson P. Intracellular domain of brain endothelial intercellular adhesion molecule-1 is essential for T lymphocyte-mediated signaling and migration. J Immunol 2003; 171:2099-108; PMID:12902516; http://dx.doi.org/ 10.4049/jimmunol.171.4.2099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Toyjanova J, Flores-Cortez E, Reichner JS, Franck C. Matrix confinement plays a pivotal role in regulating neutrophil-generated tractions, speed, and integrin utilization. J Biol Chem 2015; 290:3752-63; PMID:25525264; http://dx.doi.org/ 10.1074/jbc.M114.619643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Rabodzey A, Alcaide P, Luscinskas FW, Ladoux B. Mechanical forces induced by the transendothelial migration of human neutrophils. Biophys J 2008; 95:1428-38; PMID:18390614; http://dx.doi.org/ 10.1529/biophysj.107.119156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Jannat RA, Dembo M, Hammer DA. Traction forces of neutrophils migrating on compliant substrates. Biophys J 2011; 101:575-84; PMID:21806925; http://dx.doi.org/ 10.1016/j.bpj.2011.05.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Barreiro O, Yanez-Mo M, Serrador JM, Montoya MC, Vicente-Manzanares M, Tejedor R, Furthmayr H, Sanchez-Madrid F. Dynamic interaction of VCAM-1 and ICAM-1 with moesin and ezrin in a novel endothelial docking structure for adherent leukocytes. J Cell Biol 2002; 157:1233-45; PMID:12082081; http://dx.doi.org/ 10.1083/jcb.200112126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Van Buul JD, Allingham MJ, Samson T, Meller J, Boulter E, García-Mata R, Burridge K. RhoG regulates endothelial apical cup assembly downstream from ICAM1 engagement and is involved in leukocyte trans-endothelial migration. J Cell Biol 2007; 178:1279-93; PMID:17875742; http://dx.doi.org/ 10.1083/jcb.200612053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Phillipson M, Kaur J, Colarusso P, Ballantyne CM, Kubes P. Endothelial domes encapsulate adherent neutrophils and minimize increases in vascular permeability in paracellular and transcellular emigration. PLoS One 2008; 3:e1649; PMID:18297135; http://dx.doi.org/ 10.1371/journal.pone.0001649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Petri B, Kaur J, Long EM, Li H, Parsons SA, Butz S, Phillipson M, Vestweber D, Patel KD, Robbins SM, Kubes P. Endothelial LSP1 is involved in endothelial dome formation, minimizing vascular permeability changes during neutrophil transmigration in vivo. Blood 2011; 117(3):942-52; http://dx.doi.org/ 10.1182/blood-2010-02-270561 [DOI] [PubMed] [Google Scholar]
- [70].Vestweber D, Zeuschner D, Rottner K, Schnoor M. Cortactin regulates the activity of small GTPases and ICAM-1 clustering in endothelium: Implications for the formation of docking structures. Tissue Barriers 2013; 1(1):e23862; http://dx.doi.org/10.4161/tisb.23862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Mooren OL, Li J, Nawas J, Cooper JA. Endothelial cells use dynamic actin to facilitate lymphocyte transendothelial migration and maintain the monolayer barrier. Mol Biol Cell 2014; 25:4115-29; PMID:25355948; http://dx.doi.org/ 10.1091/mbc.E14-05-0976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol 2007; 7:678-89; PMID:17717539; http://dx.doi.org/ 10.1038/nri2156 [DOI] [PubMed] [Google Scholar]
- [73].Luissint AC, Nusrat A, Parkos CA. JAM-related proteins in mucosal homeostasis and inflammation. Semin Immunopathol 2014; 36:211-26; PMID:24667924; http://dx.doi.org/ 10.1007/s00281-014-0421-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Sullivan DP, Muller WA. Neutrophil and monocyte recruitment by PECAM, CD99, and other molecules via the LBRC. Semin Immunopathol 2014; 36:193-209; PMID:24337626; http://dx.doi.org/ 10.1007/s00281-013-0412-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Barreiro O, Zamai M, Yáñez-Mó M, Tejera E, López-Romero P, Monk PN, Gratton E, Caiolfa VR, Sánchez-Madrid F. Endothelial adhesion receptors are recruited to adherent leukocytes by inclusion in preformed tetraspanin nanoplatforms. J Cell Biol 2008; 183:527-42; PMID:18955551; http://dx.doi.org/ 10.1083/jcb.200805076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Barreiro O, Yáñez-Mó M, Sala-Valdés M, Gutiérrez-López MD, Ovalle S, Higginbottom A, Monk PN, Cabañas C, Sánchez-Madrid F. Endothelial tetraspanin microdomains regulate leukocyte firm adhesion during extravasation. Blood 2005; 105:2852-61; PMID:15591117; http://dx.doi.org/ 10.1182/blood-2004-09-3606 [DOI] [PubMed] [Google Scholar]
- [77].Van Rijssel J, Kroon J, Hoogenboezem M, van Alphen FP, de Jong RJ, Kostadinova E, Geerts D, Hordijk PL, van Buul JD. The Rho-guanine nucleotide exchange factor Trio controls leukocyte transendothelial migration by promoting docking structure formation. Mol Biol Cell 2012; 23:2831-44; PMID:22696684; http://dx.doi.org/ 10.1091/mbc.E11-11-0907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Vestweber D. How leukocytes cross the vascular endothelium. Nat Rev Immunol 2015; 15:692-704; PMID:26471775; http://dx.doi.org/ 10.1038/nri3908 [DOI] [PubMed] [Google Scholar]
- [79].Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell 2003; 112:453-65; PMID:12600310; http://dx.doi.org/ 10.1016/S0092-8674(03)00120-X [DOI] [PubMed] [Google Scholar]
- [80].Van Buul JD, Geerts D, Huveneers S. Rho GAPs and GEFs: Controling switches in endothelial cell adhesion. Cell Adhes Migr 2014; 8:108-24; PMID:24622613; http://dx.doi.org/ 10.4161/cam.27599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Ferguson KM, Lemmon MA, Schlessinger J, Sigler PB. Structure of the high affinity complex of inositol trisphosphate with a phospholipase C pleckstrin homology domain. Cell 1995; 83:1037-46; PMID:8521504; http://dx.doi.org/ 10.1016/0092-8674(95)90219-8 [DOI] [PubMed] [Google Scholar]
- [82].Kristelly R, Gao G, Tesmer JJG. Structural determinants of RhoA binding and nucleotide exchange in leukemia-associated Rho guanine-nucleotide exchange factor. J Biol Chem 2004; 279:47352-62; PMID:15331592; http://dx.doi.org/ 10.1074/jbc.M406056200 [DOI] [PubMed] [Google Scholar]
- [83].Rossman KL, Cheng L, Mahon GM, Rojas RJ, Snyder JT, Whitehead IP, Sondek J. Multifunctional roles for the PH domain of Dbs in regulating Rho GTPase activation. J Biol Chem 2003; 278:18393-400; PMID:12637522; http://dx.doi.org/ 10.1074/jbc.M300127200 [DOI] [PubMed] [Google Scholar]
- [84].Premkumar L, Bobkov AA, Patel M, Jaroszewski L, Bankston LA, Stec B, Vuori K, Côté JF, Liddington RC. Structural basis of membrane targeting by the Dock180 family of Rho family guanine exchange factors (Rho-GEFs). J Biol Chem 2010; 285:13211-22; PMID:20167601; http://dx.doi.org/ 10.1074/jbc.M110.102517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Bustelo X. Regulation of Vav proteins by intramolecular events. Front Biosci 2002; 7:d24-30; PMID:11779690 [DOI] [PubMed] [Google Scholar]
- [86].Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol 2005; 6:167-80; PMID:15688002; http://dx.doi.org/ 10.1038/nrm1587 [DOI] [PubMed] [Google Scholar]
- [87].Schmidt S, Diriong S, Méry J, Fabbrizio E, Debant A. Identification of the first Rho-GEF inhibitor, TRIPα, which targets the RhoA-specific GEF domain of Trio. FEBS Lett 2002; 523:35-42; PMID:12123800; http://dx.doi.org/ 10.1016/S0014-5793(02)02928-9 [DOI] [PubMed] [Google Scholar]
- [88].Tatsumoto T, Xie X, Blumenthal R, Okamoto I, Miki T. G2 / M Phases , and Involved in Cytokinesis. Cell 1999; 147:921-7; http://dx.doi.org/ 10.1083/jcb.147.5.921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Krendel M, Zenke FT, Bokoch GM. Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat Cell Biol 2002; 4:294-301; PMID:11912491; http://dx.doi.org/ 10.1038/ncb773 [DOI] [PubMed] [Google Scholar]
- [90].Zhao ZS, Manser E, Loo TH, Lim L. Coupling of PAK-interacting exchange factor PIX to GIT1 promotes focal complex disassembly. Mol Cell Biol 2000; 20:6354-63; PMID:10938112; http://dx.doi.org/ 10.1128/MCB.20.17.6354-6363.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Park HS, Lee SH, Park D, Lee JS, Ryu SH, Lee WJ, Rhee SG, Bae YS. Sequential activation of phosphatidylinositol 3-kinase, beta Pix, Rac1, and Nox1 ingrowth factor-induced production of H2O2. Mol Cell Biol 2004; 24(10):4384–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Lemichez E, Lecuit M, Nassif X, Bourdoulous S. Breaking the wall: targeting of the endothelium by pathogenic bacteria. Nat Rev Microbiol 2010; 8:93-104; PMID:20040916 [DOI] [PubMed] [Google Scholar]
- [93].Heemskerk N, Schimmel L, Oort C, van Rijssel J, Yin T, Ma B, van Unen J, Pitter B, Huveneers S, Goedhart J, et al.. F-actin-rich contractile endothelial pores prevent vascular leakage during leukocyte diapedesis through local RhoA signalling. Nat Commun 2016; 7:10493; PMID:26814335; http://dx.doi.org/ 10.1038/ncomms10493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Huveneers S, Daemen MJ, Hordijk PL. Between Rho(k) and a hard place: the relation between vessel wall stiffness, endothelial contractility, and cardiovascular disease. Circ Res 2015; 116(5):895–908; http://dx.doi.org/10.1161/CIRCRESAHA.116.305720 [DOI] [PubMed] [Google Scholar]
- [95].Vicente-manzanares M, Choi CK, Horwitz AR. Integrins in cell migration- the actin connection Integrins in cell migration - the actin connection. Nat Rev Mol Cell Biol 2009; 1473:199-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Lessey-Morillon EC, Osborne LD, Monaghan-Benson E, Guilluy C, O'Brien ET, Superfine R, Burridge K. The RhoA guanine nucleotide exchange factor, LARG, mediates ICAM-1-dependent mechanotransduction in endothelial cells to stimulate transendothelial migration. J Immunol 2014; 192:3390-8; PMID:24585879; http://dx.doi.org/ 10.4049/jimmunol.1302525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Collins C, Guilluy C, Welch C, O'Brien ET, Hahn K, Superfine R, Burridge K, Tzima E. Localized tensional forces on PECAM-1 elicit a global mechanotransduction response via the integrin-RhoA pathway. Curr Biol 2012; 22:2087-94; PMID:23084990; http://dx.doi.org/ 10.1016/j.cub.2012.08.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Nourshargh S, Krombach F, Dejana E. The role of JAM-A and PECAM-1 in modulating leukocyte infiltration in inflamed and ischemic tissues. J Leukoc Biol 2006; 80:714-8; PMID:16857733; http://dx.doi.org/ 10.1189/jlb.1105645 [DOI] [PubMed] [Google Scholar]
- [99].Dangerfield J, Larbi KY, Huang MT, Dewar A, Nourshargh S. PECAM-1 (CD31) homophilic interaction up-regulates alpha6beta1 on transmigrated neutrophils in vivo and plays a functional role in the ability of α6 integrins to mediate leukocyte migration through the perivascular basement membrane. J Exp Med 2002; 196(9):1201–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Dejana E. Endothelial cell–cell junctions: happy together. Nat Rev Mol Cell Biol 2004; 5:261-70; PMID:15071551; http://dx.doi.org/ 10.1038/nrm1357 [DOI] [PubMed] [Google Scholar]
- [101].Scott DW, Tolbert CE, Burridge K. Tension on JAM-A activates RhoA via GEF-H1 and p115 RhoGEF. Mol Biol Cell 2016; 27(9):1420–30; http://dx.doi.org/10.1091/mbc.E15-12-0833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Santoso S, Sachs UJ, Kroll H, Linder M, Ruf A, Preissner KT, Chavakis T. The junctional adhesion molecule 3 (JAM-3) on human platelets is a counter receptor for the leukocyte integrin Mac-1. J Exp Med 2002; 196(5):679–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Schenkel AR, Mamdouh Z, Chen X, Liebman RM, Muller WA. CD99 plays a major role in the migration of monocytes through endothelial junctions. Nat Immunol 2002; 3:143-50; PMID:11812991; http://dx.doi.org/ 10.1038/ni749 [DOI] [PubMed] [Google Scholar]
- [104].Watson RL, Buck J, Levin LR, Winger RC, Wang J, Arase H, Muller WA. Endothelial CD99 signals through soluble adenylyl cyclase and PKA to regulate leukocyte transendothelial migration. J Exp Med 2015; 212:1021-41; PMID:26101266; http://dx.doi.org/ 10.1084/jem.20150354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Lou O, Alcaide P, Luscinskas FW, Muller WA. CD99 is a key mediator of the transendothelial migration of neutrophils. J Immunol 2007; 178:1136-43; PMID:17202377; http://dx.doi.org/ 10.4049/jimmunol.178.2.1136 [DOI] [PubMed] [Google Scholar]