

ABSTRACT
Lung cancer is the leading cause of cancer death in the US with ∼124,000 new cases annually, and a 5 y survival rate of ∼16%. Mutant KRAS-driven lung adenocarcinoma (KRAS LADC) is a particularly prevalent and deadly form of lung cancer. Protein kinase Cι (PKCι) is an oncogenic effector of KRAS that activates multiple signaling pathways that stimulate transformed growth and invasion, and maintain a KRAS LADC tumor-initiating cell (TIC) phenotype. PKCι inhibitors used alone and in strategic combination show promise as new therapeutic approaches to treatment of KRAS LADC. These novel drug combinations may improve clinical management of KRAS LADC.
KEYWORDS: lung adenocarcinoma, oncogenic Kras, Protein Kinase Ciota, therapeutic targeting
KRAS mutation is an oncogenic driver required for LADC tumor initiation and maintenance
Activating KRAS mutations are detected in approximately 33% of LADC. KRAS mutations are also present in preneoplastic lesions of the lung (atypical alveolar hyperplasias, AAH), suggesting early acquisition during lung neoplasia.1 Genomic sequencing of lung adenomas in situ revealed both regional histological and genomic heterogeneity; however, KRAS mutations were observed uniformly throughout individual lesions, indicating that mutant KRAS drives early tumor cell transformation, selection and evolution.2 Expression of mutant KrasG12D in the mouse lung is sufficient to drive LADC initiation, providing in vivo evidence that KRAS is a key oncogenic driver of LADC initiation.3,4 Interestingly, systemic delivery of KRAS siRNA significantly inhibited tumor growth and metastasis of mutant KRAS LADC tumors, demonstrating a continued dependency on mutant KRAS signaling for tumor maintenance.5
Therapeutic targeting of KRAS LADC
Small molecule inhibitors designed to directly target KRAS have focused on several approaches: agents designed to decrease exchange of GDP for GTP, increase the hydrolytic activity of mutant KRAS, or inhibit effector binding and/or activation (recently reviewed by Marcus and Mattos).6 Inhibitors of KRAS processing/membrane association, and disruption of interaction with chaperones that transfer KRAS to and from the cell membrane have also been investigated.7 However, despite these efforts, agents directly targeting KRAS have been of limited clinical utility. As a consequence, extensive efforts have more recently focused on defining key oncogenic KRAS signaling pathways, and targeting more druggable downstream signaling components of these KRAS-dependent pathways.
PKCι is required for KRAS LADC
Studies from our lab and others demonstrate that atypical PKCι promotes tumorigenesis in numerous human tumor types in vitro and in vivo.8,9 PKCι is frequently targeted for tumor-specific genetic alteration and/or overexpression in many human tumor types, including myelogenous leukemias,10 glioma,11 triple negative breast cancer12 and cancers of the lung,13 colon,14 pancreas15 and ovary.16-18 PKCι was the first PKC isozyme to be identified as a bonafide oncogene in human cancer; first demonstrated by our group in non-small cell lung cancer (NSCLC),13 and subsequently in ovarian and other cancers.18,19 Atypical PKCs directly interact with oncogenic RAS in vitro and in vivo, implicating them as mediators of oncogenic RAS signaling.20 PKCι is required for Kras LADC tumorigenesis in mice, and for the transformed growth of KRAS LADC cell lines, revealing PKCι as a critical effector in KRAS LADC.21,22
PKCι promotes the transformed growth and invasion of KRAS LADC cells by activating an oncogenic RAC1-MEK-ERK proliferative signaling pathway.22,23 In KRAS LADC cells, PKCι forms an oncogenic complex with the polarity protein PAR6 through PB1-PB1 domain interactions.23 The RHO family GTPase guanine nucleotide exchange factor ECT2 binds this complex and serves to activate the RHO family GTPase RAC1, which in turn drives a RAC1-PAK-MEK-ERK signaling cascade.23-25 PKCι directly phosphorylates ECT2 at T328, an event that promotes ECT2 binding to the PKCι-PAR6 complex, RAC1 activation, and transformed growth and invasion in NSCLC cells.23,24 PKCɩ-RAC1-MEK-ERK signaling drives proliferation and invasion, at least in part, by stimulating expression of matrix metalloproteinase 10 (MMP10).23,26,27 Taken together, these data demonstrate that PKCι drives a critical proliferative signaling pathway required for the transformed growth of KRAS LADC.
A critical factor regulating NSCLC susceptibility to chemotherapy-induced apoptosis is splice site selection within the BCL-X gene, yielding either pro-apopotic Bcl-x(s) or anti-apoptotic Bcl-x(L).28,29 Downregulation of PKCι in KRAS LADC A549 cells significantly decreased cell survival and reduced expression of SAP155, an RNA trans-acting factor that promotes Bcl-x pre-mRNA splice site selection to generate the pro-survival Bcl-x(L).30 Re-expression of Bcl-x(L) in PKCι-depleted cells rescued cell survival, demonstrating that PKCι promotes survival of KRAS NSCLC cells by regulating alternative splicing of the BCL-X pre-mRNA.30
PKCι drives a KRAS LADC tumor-initiating cell (TIC) phenotype
KRAS LADC tumors are comprised of a heterogeneous population of cells that exhibit a hierarchy of tumorigenic potential. Cells at the top of this hierarchy, tumor-initiating cells (TICs), exhibit a high level of tumor-initiating activity and an enhanced capacity to recapitulate KRAS LADC in vivo.31-33 TICs exhibit the unique capacity to both self-renew, and form a more differentiated, but highly proliferative tumor cell population, driving both tumor initiation and maintenance.34,35 Tumors harboring mutant KRAS exhibit enhanced chemoresistance,36 radiation resistance,37 and poor survival;38 all properties associated with the TIC phenotype, suggesting that KRAS is an important driver of the LADC TIC phenotype.39,40
We have identified PKCɩ as a key mediator of the KRAS LADC TIC phenotype.21,41 PKCɩ is highly expressed in mutant Kras transformed bronchioalveolar cells, a putative stem cell population and cell-of-origin of LADC in the mouse lung.21 PKCι deficiency dramatically reduces Kras-mediated transformation and expansion of the bronchioalveolar population, thereby inhibiting Kras-driven tumor formation.21 MMP10, a critical target of PKCɩ proliferative signaling, is also required for Kras-mediated bronchioalveolar stem cell expansion and LADC tumorigenesis.42 MMP10 is elevated in human KRAS LADC TICs, and both PKCɩ and MMP10 are required for LADC TIC behavior and tumor formation.26,41 These data demonstrate that PKCɩ is required for maintenance of the KRAS LADC TIC phenotype, and indicate that MMP10 is an important mediator of PKCɩ-driven growth of KRAS LADC TICs.
Notch3 is also required for KRAS LADC TICs self-renewal, tumor initiation and maintenance.32,33 We recently demonstrated that PKCɩ regulates Notch3 expression in KRAS LADC TICs by phosphorylating the ELF3 transcription factor, thereby promoting its occupancy of the NOTCH3 promoter. KRAS LADCs require expression of PKCɩ, ELF3 and Notch3 for TIC cell growth, clonal expansion and tumorigenesis.41 A critical feature of TICs is the ability to undergo symmetric cell division to self-renew, as well as asymmetric cell division to generate a more differentiated, highly proliferative population of tumor cells.34 PKCɩ-ELF3-Notch3 controls the TIC phenotype by regulating asymmetric cell division, a process required for tumor initiation and expansion.41 Interestingly, the PKCɩ-ELF3-Notch3 signaling axis is specific for mutant KRAS LADC TICs, since this cascade is not observed in LADC TICs harboring wild-type KRAS or in lung squamous cell carcinoma (LSCC). Our previous work demonstrated that PKCɩ drives a LSCC TIC phenotype by activating a distinct PKCι-SOX2-Hedgehog (Hh) signaling axis that drives Hh-dependent TIC growth.41,43
PKCι inhibitors as anti-tumor agents
Given the potential for PKCι as a therapeutic target, we and others have sought to identify PKC isotype-selective inhibitors of PKCι. Recently, an isotype selective, ATP-competitive inhibitor of PKCι activity (CRT0066854) was identified and characterized to suppress transformed growth of mutant Ras cancer cell lines.44 A separate screen for molecules that could bind a unique sequence in the PKCι catalytic domain and inhibit its kinase activity identified ICA-1, capable of blocking PKCι substrate phosphorylation and transformed growth in neuroblastoma cells.45 While both of these inhibitors hold promise as PKCι-targeted agents, evidence of their effectiveness as therapeutic agents awaits pre-clinical evaluation. To identify inhibitors of PKCι that could be rapidly translated to clinical application, we conducted a high-throughput screen of FDA-approved drugs for compounds that inhibit oncogenic PKCι signaling. We reasoned that compounds that can disrupt the PB1-PB1 domain interaction between PKCι and its oncogenic partner PAR6 would exhibit anti-tumor activity.46 Our screen identified the anti-rheumatoid agent aurothiomalate (ATM) as a selective inhibitor of the PKCι-PAR6 interaction.46,47 ATM, and the structurally related anti-rheumatoid drug, auranofin (ANF), selectively bind the PB1 domain of PKCι and block PAR6 binding, inhibit PKCι-mediated oncogenic signaling (i.e. the RAC1-MEK-ERK signaling axis) and block transformed growth and invasion of NSCLC cells in vitro and in vivo.27,46,47 The anti-tumor activity of these agents is dependent upon their ability to bind PKCι and inhibit PKCι-PAR6 signaling since a PB1 domain PKCι mutant that no longer binds ATM but retains binding to PAR6, can support transformed growth and confer resistance to the growth inhibitory effects of ATM.47 ANF is currently being evaluated clinically as an anti-tumor agent in lung and ovarian cancer patients.48,49 These compounds are well tolerated in the oncology setting and exhibit promising therapeutic potential.49
Though ATM and ANF exhibit efficacy as single agents in pre-clinical models, it is likely they will find optimal clinical use in strategic combination with other agents. In this regard, we have demonstrated synergistic pre-clinical efficacy of ANF in combination with a SMO inhibition in LSCC.43 In mutant KRAS LADC, combining ANF and a gamma-secretase inhibitor (GSI; inhibitor of Notch signaling) exhibited synergistic activity in KRAS LADC in vitro and in vivo, demonstrating effective “vertical blockade” of this critical signaling pathway (Fig. 1).41
Figure 1.
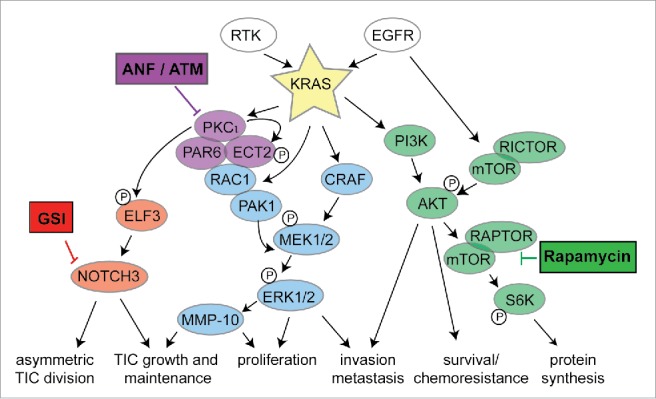
Clinically relevant PKCɩ-targeted combination therapies for treatment of KRAS LADC. “Vertical Blockade” of the PKCɩ-ELF3-Notch3 signaling axis and “Horizonal Blockade” of PKCɩ and mTOR signaling pathways are denoted.
We have also assessed ATM in combination with inhibitors of signaling pathways frequently activated in KRAS LADC, including rapamycin (mTOR), Erlotinib (EGFR) and the multikinase inhibitor, Sorafenib (Fig. 2A). Whereas each of these drug combinations showed synergistic growth inhibitory effects in one or more cell line, the combination of ATM and rapamycin showed highly significant synergistic activity in 3 out of 3 mutant KRAS LADC cell lines tested (Fig. 2A). Although mTOR inhibitors have shown clinical activity in lung cancer patients, a significant impediment to their use has been either intrinsic or acquired resistance. Combined ATM and rapamycin exhibits enhanced anti-tumor activity against KRAS LADC tumor growth in vivo (Fig. 2B). Taken together these data indicate that combined PKCι and mTOR inhibition may be an effective “horizontal blockade” strategy to treat mutant KRAS LADC, including those tumors that exhibit intrinsic or acquired resistance to mTOR inhibition (summarized in Fig. 1). A phase I trial is currently ongoing to assess this combination clinically.
Figure 2.
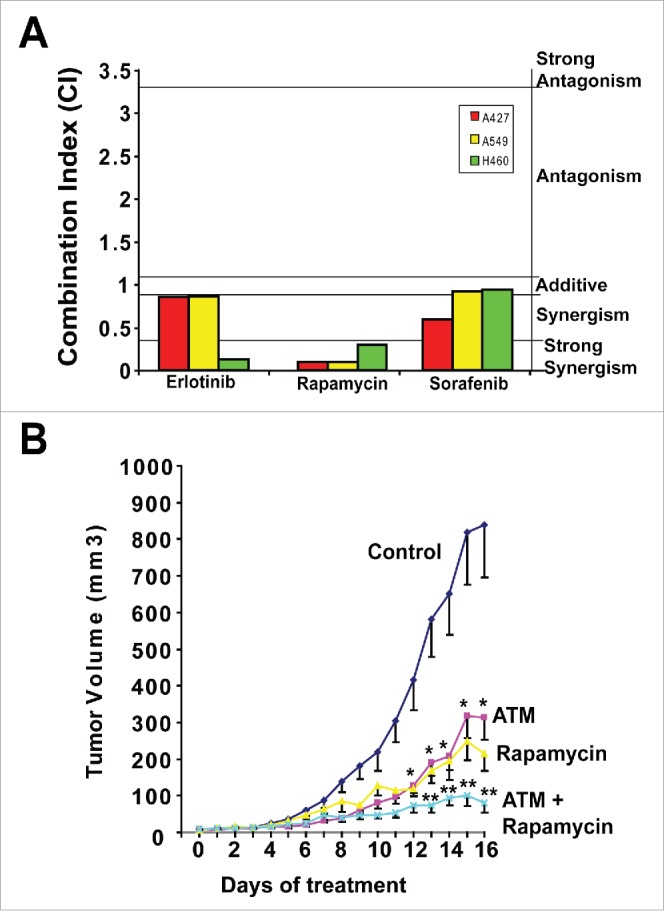
ATM and rapamycin exhibit synergistic growth inhibitory activity against KRAS LADC. (A) Combination Index (CI) analysis of ATM combined with Erlotinib, rapamycin and sorafenib for inhibition of anchorage independent growth of KRAS LADC cells. (B) Combined ATM and rapamycin significantly inhibits growth of H460 KRAS LADC xenograft tumors. Mice were treated with ATM (60 mg/kg/day) and/or rapamycin (5 mg/kg/day) or diluent alone (control). Mean +/− SE is plotted. n = 8–9/group. *p < 0.05 versus control; **p < 0.05 vs. rapamycin or ATM alone.
Summary
Preclinical studies demonstrate that PKCɩ inhibition represents a viable therapeutic approach to treatment of KRAS LADC, particularly when used in strategic combination with agents targeting other KRAS effectors. Beyond KRAS LADC, PKCɩ promotes the oncogenic phenotype of many other cancers; therefore clinical development of PKCɩ-targeted therapies may provide effective therapeutic options for many other oncology patients.
Materials and methods
Materials: KRAS mutant LADC cell lines, A549, A427 and H460, were purchased from American Type Culture Collection and maintained in culture conditions as recommended.
In vitro studies: Anchorage-independent growth in soft agar was assessed as previously described.27 Drug interactions were analyzed using the median effect combination index of Chou and Talalay, calculated using CalcuSyn software.50 n = 5/dose group and results are representative of 2 independent experiments.
In vivo studies. 5 × 106 H460 cells were injected into the flank of 4–8 week old immunocompromised nude mice (Jackson Labs). Tumor cells were allowed to engraft and tumor size was monitored 3×/week by caliper measurements. Tumor volume was calculated as length x width x height x 0.5236. Mice were randomly distributed into treatment group when tumor average size = 10 mm3. Mice were administered ATM (60 mg/kg) and/or rapamycin (1 mg/kg) daily by intraperitoneal injection and humanely harvested after 16 d treatment. Control mice were injected daily with equivalent volumes of diluent only.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors wish to acknowledge Dr. Lee Jamieson and Dr. Suzanne J. Randle for technical support.
Funding
The work described herein was supported by grants from the National Institutes of Health (R01 CA081436-17 and R21 CA151250-02 to APF; R01 CA14090-05 to NRM; and R21 CA204938-01 (VJ); the James and Esther King Biomedical Research Program (1KG-05-33971) to APF, the Mayo Clinic Center for Individualized Medicine (CIM) to APF; a National Institutes of Health Research Supplement to Promote Diversity in Health-related Research Award from the National Cancer Institute to VJ, and the George Haub Family Career Development Award (VJ). APF is the Monica Flynn Jacoby Professor of Cancer Research. SAA is the recipient of the Edward C. Kendall Fellowship in Biochemistry from the Mayo Clinic Graduate School.
References
- [1].Westra WH, Baas IO, Hruban RH, Askin FB, Wilson K, Offerhaus GJ, Slebos RJ. K-ras oncogene activation in atypical alveolar hyperplasias of the human lung. Cancer Res 1996; 56:2224-8; PMID:8616876 [PubMed] [Google Scholar]
- [2].Izumchenko E, Chang X, Brait M, Fertig E, Kagohara LT, Bedi A, Marchionni L, Agrawal N, Ravi R, Jones S, et al.. Targeted sequencing reveals clonal genetic changes in the progression of early lung neoplasms and paired circulating DNA. Nat Commun 2015; 6:8258; PMID:26374070; http://dx.doi.org/ 10.1038/ncomms9258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 2001; 15:3243-8; PMID:11751630; http://dx.doi.org/ 10.1101/gad.943001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, Jacks T. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 2001; 410:1111-6; PMID:11323676; http://dx.doi.org/ 10.1038/35074129 [DOI] [PubMed] [Google Scholar]
- [5].Pecot CV, Wu SY, Bellister S, Filant J, Rupaimoole R, Hisamatsu T, Bhattacharya R, Maharaj A, Azam S, Rodriguez-Aguayo C, et al.. Therapeutic silencing of KRAS using systemically delivered siRNAs. Mol Cancer Ther 2014; 13:2876-85; PMID:25281617; http://dx.doi.org/ 10.1158/1535-7163.MCT-14-0074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Marcus K, Mattos C. Direct Attack on RAS: Intramolecular Communication and Mutation-Specific Effects. Clin Cancer Res 2015; 21:1810-8; PMID:25878362; http://dx.doi.org/ 10.1158/1078-0432.CCR-14-2148 [DOI] [PubMed] [Google Scholar]
- [7].Cox AD, Der CJ, Philips MR. Targeting RAS Membrane Association: Back to the Future for Anti-RAS Drug Discovery? Clin Cancer Res 2015; 21:1819-27; PMID:25878363; http://dx.doi.org/ 10.1158/1078-0432.CCR-14-3214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Fields AP, Frederick LA, Regala RP. Targeting the oncogenic protein kinase Ciota signalling pathway for the treatment of cancer. Biochem Soc Trans 2007; 35:996-1000; PMID:17956262; http://dx.doi.org/ 10.1042/BST0350996 [DOI] [PubMed] [Google Scholar]
- [9].Fields AP, Regala RP. Protein kinase C iota: human oncogene, prognostic marker and therapeutic target. Pharmacol Res 2007; 55:487-97; PMID:17570678; http://dx.doi.org/ 10.1016/j.phrs.2007.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gustafson WC, Ray S, Jamieson L, Thompson EA, Brasier AR, Fields AP. Bcr-Abl regulates protein kinase Ciota (PKCiota) transcription via an Elk1 site in the PKCiota promoter. J Biol Chem 2004; 279:9400-8; PMID:14670960; http://dx.doi.org/ 10.1074/jbc.M312840200. [DOI] [PubMed] [Google Scholar]
- [11].Patel R, Win H, Desai S, Patel K, Matthews JA, Acevedo-Duncan M. Involvement of PKC-iota in glioma proliferation. Cell Prolif 2008; 41:122-35; PMID:18211289; http://dx.doi.org/ 10.1111/j.1365-2184.2007.00506.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Paul A, Gunewardena S, Stecklein SR, Saha B, Parelkar N, Danley M, Rajendran G, Home P, Ray S, Jokar I, et al.. PKClambda/iota signaling promotes triple-negative breast cancer growth and metastasis. Cell Death Differ 2014; 21:1469-81; PMID:24786829; http://dx.doi.org/ 10.1038/cdd.2014.62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Regala RP, Weems C, Jamieson L, Khoor A, Edell ES, Lohse CM, Fields AP. Atypical protein kinase C iota is an oncogene in human non-small cell lung cancer. Cancer Res 2005; 65:8905-11; PMID:16204062; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-2372 [DOI] [PubMed] [Google Scholar]
- [14].Murray NR, Jamieson L, Yu W, Zhang J, Gokmen-Polar Y, Sier D, Anastasiadis P, Gatalica Z, Thompson EA, Fields AP. Protein kinase Ciota is required for Ras transformation and colon carcinogenesis in vivo. J Cell Biol 2004; 164:797-802; PMID:15024028; http://dx.doi.org/ 10.1083/jcb.200311011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Scotti ML, Bamlet WR, Smyrk TC, Fields AP, Murray NR. Protein kinase Cι is required for pancreatic cancer cell transformed growth and tumorigenesis. Cancer Research 2010; 70:2064-74; PMID:20179210; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-2684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Eder AM, Sui X, Rosen DG, Nolden LK, Cheng KW, Lahad JP, Kango-Singh M, Lu KH, Warneke CL, Atkinson EN, et al.. Atypical PKCiota contributes to poor prognosis through loss of apical-basal polarity and cyclin E overexpression in ovarian cancer. Proc Natl Acad Sci U S A 2005; 102:12519-24; PMID:16116079; http://dx.doi.org/ 10.1073/pnas.0505641102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Weichert W, Gekeler V, Denkert C, Dietel M, Hauptmann S. Protein kinase C isoform expression in ovarian carcinoma correlates with indicators of poor prognosis. Int J Oncol 2003; 23:633-9; PMID:12888898 [PubMed] [Google Scholar]
- [18].Zhang L, Huang J, Yang N, Liang S, Barchetti A, Giannakakis A, Cadungog MG, O'Brien-Jenkins A, Massobrio M, Roby KF, et al.. Integrative genomic analysis of protein kinase C (PKC) family identifies PKCiota as a biomarker and potential oncogene in ovarian carcinoma. Cancer Res 2006; 66:4627-35; PMID:16651413; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-4527 [DOI] [PubMed] [Google Scholar]
- [19].Wang Y, Hill KS, Fields AP. Protein Kinase Ciota maintains a tumor-initiating cell phenotype that is required for ovarian tumorigenesis. Mol Cancer Res 2013; 11(12):1624-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Diaz-Meco MT, Lozano J, Municio MM, Berra E, Frutos S, Sanz L, Moscat J. Evidence for the in vitro and in vivo interaction of Ras with protein kinase C zeta. J Biol Chem 1994; 269:31706-10; PMID:7989344 [PubMed] [Google Scholar]
- [21].Regala RP, Davis RK, Kunz A, Khoor A, Leitges M, Fields AP. Atypical protein kinase C{iota} is required for bronchioalveolar stem cell expansion and lung tumorigenesis. Cancer Res 2009; 69:7603-11; PMID:19738040; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-2066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Regala RP, Weems C, Jamieson L, Copland JA, Thompson EA, Fields AP. Atypical protein kinase Ciota plays a critical role in human lung cancer cell growth and tumorigenicity. J Biol Chem 2005; 280:31109-15; PMID:15994303; http://dx.doi.org/ 10.1074/jbc.M505402200 [DOI] [PubMed] [Google Scholar]
- [23].Frederick LA, Matthews JA, Jamieson L, Justilien V, Thompson EA, Radisky DC, Fields AP. Matrix metalloproteinase-10 is a critical effector of protein kinase Ciota-Par6alpha-mediated lung cancer. Oncogene 2008; 27:4841-53; PMID:18427549; http://dx.doi.org/ 10.1038/onc.2008.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Justilien V, Fields AP. Ect2 links the PKCiota-Par6alpha complex to Rac1 activation and cellular transformation. Oncogene 2009; 28:3597-607; PMID:19617897; http://dx.doi.org/ 10.1038/onc.2009.217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Justilien V, Jameison L, Der CJ, Rossman KL, Fields AP. Oncogenic activity of Ect2 is regulated through protein kinase C iota-mediated phosphorylation. J Biol Chem 2011; 286:8149-57; PMID:21189248; http://dx.doi.org/ 10.1074/jbc.M110.196113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Justilien V, Regala RP, Tseng IC, Walsh MP, Batra J, Radisky ES, Murray NR, Fields AP. Matrix metalloproteinase-10 is required for lung cancer stem cell maintenance, tumor initiation and metastatic potential. PLoS ONE 2012; 7:e35040; PMID:22545096; http://dx.doi.org/ 10.1371/journal.pone.0035040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Regala RP, Thompson EA, Fields AP. Atypical protein kinase Cι expression and aurothiomalate sensitivity in human lung cancer cells. Cancer Research 2008; 68:5888-95; PMID:18632643; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-0438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Taylor JK, Zhang QQ, Wyatt JR, Dean NM. Induction of endogenous Bcl-xS through the control of Bcl-x pre-mRNA splicing by antisense oligonucleotides. Nat Biotechnol 1999; 17:1097-100; PMID:10545916; http://dx.doi.org/ 10.1038/15079 [DOI] [PubMed] [Google Scholar]
- [29].Mercatante DR, Mohler JL, Kole R. Cellular response to an antisense-mediated shift of Bcl-x pre-mRNA splicing and antineoplastic agents. J Biol Chem 2002; 277:49374-82; PMID:12381725; http://dx.doi.org/ 10.1074/jbc.M209236200 [DOI] [PubMed] [Google Scholar]
- [30].Shultz JC, Vu N, Shultz MD, Mba MU, Shapiro BA, Chalfant CE. The Proto-oncogene PKCiota regulates the alternative splicing of Bcl-x pre-mRNA. Mol Cancer Res 2012; 10:660-9; PMID:22522453; http://dx.doi.org/ 10.1158/1541-7786.MCR-11-0363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hassan KA, Wang L, Korkaya H, Chen G, Maillard I, Beer DG, Kalemkerian GP, Wicha MS. Notch pathway activity identifies cells with cancer stem cell-like properties and correlates with worse survival in lung adenocarcinoma. Clin Cancer Res 2013; 19:1972-80; PMID:23444212; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-0370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sullivan JP, Spinola M, Dodge M, Raso MG, Behrens C, Gao B, Schuster K, Shao C, Larsen JE, Sullivan LA, et al.. Aldehyde dehydrogenase activity selects for lung adenocarcinoma stem cells dependent on notch signaling. Cancer Res 2010; 70:9937-48; PMID:21118965; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-0881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zheng Y, de la Cruz CC, Sayles LC, Alleyne-Chin C, Vaka D, Knaak TD, Bigos M, Xu Y, Hoang CD, Shrager JB, et al.. A rare population of CD24(+)ITGB4(+)Notch(hi) cells drives tumor propagation in NSCLC and requires Notch3 for self-renewal. Cancer Cell 2013; 24:59-74; PMID:23845442; http://dx.doi.org/ 10.1016/j.ccr.2013.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lathia JD, Hitomi M, Gallagher J, Gadani SP, Adkins J, Vasanji A, Liu L, Eyler CE, Heddleston JM, Wu Q, et al.. Distribution of CD133 reveals glioma stem cells self-renew through symmetric and asymmetric cell divisions. Cell Death Dis 2011; 2:e200; PMID:21881602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Pine SR, Ryan BM, Varticovski L, Robles AI, Harris CC. Microenvironmental modulation of asymmetric cell division in human lung cancer cells. Proceedings of the National Academy of Sciences of the United States of America 2010; 107:2195-200; PMID:20080668; http://dx.doi.org/ 10.1073/pnas.0909390107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Campos-Parra AD, Zuloaga C, Manriquez ME, Aviles A, Borbolla-Escoboza J, Cardona A, Meneses A, Arrieta O. KRAS mutation as the biomarker of response to chemotherapy and EGFR-TKIs in patients with advanced non-small cell lung cancer: clues for its potential use in second-line therapy decision making. Am J Clin Oncol 2015; 38:33-40; PMID:23538866; http://dx.doi.org/ 10.1097/COC.0b013e318287bb23 [DOI] [PubMed] [Google Scholar]
- [37].Hallqvist A, Enlund F, Andersson C, Sjogren H, Hussein A, Holmberg E, Nyman J. Mutated KRAS Is an Independent Negative Prognostic Factor for Survival in NSCLC Stage III Disease Treated with High-Dose Radiotherapy. Lung Cancer Int 2012; 2012:587424; PMID:26316935; http://dx.doi.org/ 10.1155/2012/587424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Johnson ML, Sima CS, Chaft J, Paik PK, Pao W, Kris MG, Ladanyi M, Riely GJ. Association of KRAS and EGFR mutations with survival in patients with advanced lung adenocarcinomas. Cancer 2013; 119:356-62; PMID:22810899; http://dx.doi.org/ 10.1002/cncr.27730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, Parada LF. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012; 488:522-6; PMID:22854781; http://dx.doi.org/ 10.1038/nature11287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Phillips TM, McBride WH, Pajonk F. The response of CD24(−/low)/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst 2006; 98:1777-85; PMID:17179479; http://dx.doi.org/ 10.1093/jnci/djj495 [DOI] [PubMed] [Google Scholar]
- [41].Ali SA, Justilien V, Jamieson L, Murray NR, Fields AP. Protein Kinase Ciota Drives a NOTCH3-dependent Stem-like Phenotype in Mutant KRAS Lung Adenocarcinoma. Cancer Cell 2016; 29:367-78; PMID:26977885; http://dx.doi.org/ 10.1016/j.ccell.2016.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Regala RP, Justilien V, Walsh MP, Weems C, Khoor A, Murray NR, Fields AP. Matrix metalloproteinase-10 promotes Kras-mediated bronchio-alveolar stem cell expansion and lung cancer formation. PLoS ONE 2011; 6:e26439; PMID:22022614; http://dx.doi.org/ 10.1371/journal.pone.0026439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Justilien V, Walsh MP, Ali SA, Thompson EA, Murray NR, Fields AP. The PRKCI and SOX2 oncogenes are coamplified and cooperate to activate Hedgehog signaling in lung squamous cell carcinoma. Cancer Cell 2014; 25:139-51; PMID:24525231; http://dx.doi.org/ 10.1016/j.ccr.2014.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kjaer S, Linch M, Purkiss A, Kostelecky B, Knowles PP, Rosse C, Riou P, Soudy C, Kaye S, Patel B, et al.. Adenosine-binding motif mimicry and cellular effects of a thieno[2,3-d]pyrimidine-based chemical inhibitor of atypical protein kinase C isoenzymes. Biochem J 2013; 451:329-42; PMID:23418854; http://dx.doi.org/ 10.1042/BJ20121871 [DOI] [PubMed] [Google Scholar]
- [45].Pillai P, Desai S, Patel R, Sajan M, Farese R, Ostrov D, Acevedo-Duncan M. A novel PKC-iota inhibitor abrogates cell proliferation and induces apoptosis in neuroblastoma. Int J Biochem Cell Biol 2011; 43:784-94; PMID:21315177; http://dx.doi.org/ 10.1016/j.biocel.2011.02.002 [DOI] [PubMed] [Google Scholar]
- [46].Stallings-Mann M, Jamieson L, Regala RP, Weems C, Murray NR, Fields AP. A novel small-molecule inhibitor of protein kinase Ciota blocks transformed growth of non-small-cell lung cancer cells. Cancer Res 2006; 66:1767-74; PMID:16452237; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-3405 [DOI] [PubMed] [Google Scholar]
- [47].Erdogan E, Lamark T, Stallings-Mann M, Lee J, Pellecchia M, Thompson EA, Johansen T, Fields AP. Aurothiomalate inhibits transformed growth by targeting the PB1 domain of protein kinase Ciota. J Biol Chem 2006; 281:28450-9; PMID:16861740; http://dx.doi.org/ 10.1074/jbc.M606054200 [DOI] [PubMed] [Google Scholar]
- [48].Jatoi A, Radecki Breitkopf C, Foster NR, Block MS, Grudem M, Wahner Hendrickson A, Carlson RE, Barrette B, Karlin N, Fields AP. A mixed-methods feasibility trial of protein kinase C iota inhibition with auranofin in asymptomatic ovarian cancer patients. Oncology 2015; 88:208-13; PMID:25502607; http://dx.doi.org/ 10.1159/000369257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Mansfield AS, Fields AP, Jatoi A, Qi Y, Adjei AA, Erlichman C, Molina JR. Phase I dose escalation study of the PKCiota inhibitor aurothiomalate for advanced non-small-cell lung cancer, ovarian cancer, and pancreatic cancer. Anticancer Drugs 2013; 24(10):1079-83; PMID:23962904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 2010; 70:440-6; PMID:20068163; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-1947 [DOI] [PubMed] [Google Scholar]