

ABSTRACT
T cell acute lymphoblastic leukemia/lymphoma (T-ALL) is an aggressive bone marrow cancer in children and adults, and chemotherapy often fails for relapsing patients. Molecularly targeted therapy is hindered by heterogeneity in T-ALL and mechanistic details of the affected pathways in T-ALL are needed. Deregulation of Ras signals is common in T-ALL. Ras is genetically mutated to a constitutively active form in about 15% of all haematopoietic malignancies, but there is a range of other ways to augment signaling through the Ras pathway. Several groups including our own uncovered that RasGRP1 overexpression leads to T-ALL in mouse models and in pediatric T-ALL patients, and we reported that this Ras guanine nucleotide exchange factor, RasGRP1, cooperates with cytokines to drive leukemogenesis. In our recent study by Ksionda et al. we analyzed the molecular details of cytokine receptor-RasGRP1-Ras signals in T-ALL and compared these to signals from mutated Ras alleles, which yielded several surprising results. Examples are the striking differences in flux through the RasGDP/RasGTP cycle in distinct T-ALL or unexpected differences in wiring of the Ras signaling pathway between T-ALL and normal developing T cells, which we will discuss here.
KEYWORDS: cancer, heterogeneity, leukemia, mouse models, PI3K, patients, PI3K, Ras, RasGAP, RasGRP1, therapy
T cell acute lymphoblastic leukemia (T-ALL) or T cell lymphoblastic lymphoma (T-LL) are currently treated on separate clinical trials, although it is controversial whether T-ALL and T-LL are different clinical presentations (progeny) of the same disease or separate diseases. ETP T-ALL (Early T cell precursor acute lymphoblastic leukemia) is a third type characterized by expression of early developmental markers and usually relatively poor clinical outcome.1 Here we collectively refer to T-ALL, T-LL, and ETP T-ALL as “T-ALL.” T-ALL is an aggressive bone marrow cancer that affects children and adults,2 and despite improvements with modern chemotherapy treatment still fails for a substantial number of patients who relapse.2,3 Current stratification efforts with cell surface markers or gene expression have not yielded clear insights in terms of subgrouping T-ALL patients for therapy.4-7 We favor the idea that truly understanding the different types of T-ALL (and perhaps knowing what precision therapy to use for a specific T-ALL patient) will require full biochemical stratification, which is our motivation for studying perturbed pathways operating in T-ALL. One pathway involved is the Notch pathway,8 which is often activated by retroviral integrations and point mutations in mouse T-ALL9 and is a common target of somatic mutations in human T-ALL.10 A second commonly affected pathway is the Ras pathway with ∼50% of all T-ALL cases demonstrating hyperactive Ras signals.11
Various cell surface receptors activate the small GTPase Ras through RasGEFs (guanine nucleotide exchange factors). Once GTP-loaded, H-, N-, and K-Ras function as powerful signaling switches and connect to different effector pathways.12-15 RasGTP (active Ras) signals to RAF-MEK-ERK and PI3 kinase (PI3K)-Akt kinase effector pathways that in turn stimulate cell cycle, survival, and metabolic changes.16,17 Three RasGEF families can activate Ras: SOS (Son of Sevenless), RasGRP (Ras guanine nucleotide releasing protein), and RasGRF (Ras guanine nucleotide releasing factor). SOS1 and SOS2 are ubiquitously expressed. RasGRP1, -3, and -4 are activators of Ras, whereas RasGRP2 activates Rap.18 RasGRP1 and -3 are highly homologous, play unique roles in immune cells, and appear to be more frequently dysregulated than other RasGEFs in cancers cells.19,20 Rasgrp1-deficient mice have a severe T cell development defect in the thymus21 whereas Rasgrp3-deficient mice show subtle defects in B cell development.22 RasGRP4 has been show to play an important role in mast cells and neutrophils functions.20 The default path to inactivate Ras prevails in normal cells via the intrinsic, self-inactivating GTPase function of Ras. This function is enhanced by the action of RasGAPs (Ras GTPase activating protein). Ten different RasGAPs have been identified in mammals with distinct regulatory domains. Most cell types express different combinations of various RasGAPs, arguing that the cellular function to enhance Ras inactivation needs to be covered by multiple RasGAPs.23 Ras is genetically mutated to a constitutively active form with impaired GTPase function in about 15% of all haematopoietic malignancies,24 we will focus on T-ALL here.
Deregulation of the Ras signaling pathway is a common event in T-ALL11 but somatic Ras mutations (termed RasMUT here) such as KRasG12D that lock Ras in the GTP-loaded state are relatively rare in most T-ALL.25-27 In addition to lesions in Ras itself, mutations can occur in genes encoding upstream receptors or in other pathway constituents, chromosomal translocations can have deregulating activities, and alterations in expression levels of key components can have major impacts on the activity through the Ras pathway.20,28 Recent studies have started to map out the pathways that can lead to aberrant Ras signals in T-ALL. Whole genome sequencing revealed activating mutations in the cytokine receptor IL7R (Interleukin 7 receptor) and in JAK1 and JAK3 (Janus kinase 1 and 3),1 and the IL7R mutations found in T-ALL constitutively activate downstream JAK1 kinase.29 Studies with different mouse models and with pediatric T-ALL patient samples pointed to a role for RasGRP1 in T-ALL leukemogenesis.20,30-33 Likewise, the inactivating function of RasGAPs on the Ras pathway also appears critical; combined T cell-specific deletion of NF1 and p120 RasGAPs in mice leads to development of T-ALL34 and NF1 is mutated in both pediatric and adult T-ALL patients.35-37 In our recent study by Ksionda et al.38 we describe the details of cytokine receptor-Ras pathways in T-ALL and how Ras signals in T-ALL with RasGRP1 overexpression not only differ from those in developing normal T cells (thymocytes) but also from Ras signals in RasMUT T-ALL. Here we will highlight four different vignettes from our study.38
The first topic highlights the fact that the wiring for cytokine-induced Ras activation is unique in RasGRP1-overexpressing T-ALL and not seen in normal thymoctes. This is somewhat surprising since T-ALL are, in general, arrested at an early developmental stage that have the appearance (and gene expression features) of distinct thymocyte subsets. Most of both T-ALL and developing thymocytes express the T cell receptor (TCR) and cytokine receptors (such as IL7R). While both T-ALL and thymocytes load Ras with GTP in response to TCR stimulation, only T-ALL cells activate Ras following cytokine stimulation. Overexpression of RasGRP1 is critical for this cytokine receptor-Ras pathway33 and thus RasGRP1 created a rewiring that is not seen in normal thymocytes. Both TCR stimulation and cytokine-induced Ras signaling via RasGRP1 yield strong and sustained PI3K-Akt signals in T-ALL.38 However, the pathways involved are fundamentally different. The first clue for this was provided by the fact that TCR stimulation but not cytokine stimulation leads to strong calcium influx in T-ALL cells. PLCγ1 generates diacylglycerol (DAG) and Inositol trisphosphate (IP3) and IP3 triggers a pathway that generates the calcium flux. We noted that, just like in normal thymocytes, PLCγ1 signaling is essential for a fully functional TCR signaling cascade to Ras via RasGRP1 in T-ALL. In the canonical RasGRP1 pathway, PLCγ1-generated DAG recruits RasGRP1 to the membrane where RasGRP1 is phosphorylated and activates Ras.20 Indeed, both TCR-induced RasGRP1 phosphorylation and increases in RasGTP were blocked when inhibiting PLCγ1 in T-ALL. By stark contrast, PLCγ1 is dispensable for effective cytokine-induced Ras activation via RasGRP1. Instead, baseline levels of DAG already seem to be sufficient to recruit (some of the) overexpressed RasGRP1 to the membrane at steady state in T-ALL. In agreement, these T-ALL reveal low and constitutive levels of RasGRP1 phosphorylation that are not increased upon cytokine stimulation. These results implied that cytokine-induced Ras activation in RasGRP1-overexpressing T-ALL does not follow a canonical PLCγ1-RasGRP1-Ras pathway, and we will come back to this point. Our results also suggest that PLCγ1 inhibition as possible therapy for T-ALL would likely be unsuccessful.
PI3K-Akt signaling has a critical function in cell cycle progression, cell proliferation, and regulation of apoptosis. Accordingly, this pathway is frequently involved in oncogenic signaling, and PIK3CA and PTEN (phosphatase and tensin homolog, PI3K's negative regulator) have been identified as second and third most frequently mutated genes in human cancer, respectively.39 During normal T cell development, the PI3K-Akt pathway controls the differentiation of thymocytes and their transition from the double-negative to the double-positive stage, as well as the survival of double-positive thymocytes after initiation of pre-TCR signals.40 Genetic mutations in RAS,1 PI3K, and PTEN41 have provided concrete proof for the frequent involvement of the Ras-PI3K pathway in T-ALL. Several groups including our own observed that Ras-PI3K signaling is universally utilized in T-ALL, but with heterogeneous activation patterns.41-47 By contrast, the high usage of PI3K-Akt signals is not seen in other hematologic malignancies.48 Given that PI3K inhibitors like GDC0941 are very potent agents,49 such inhibition may be useful as therapy for T-ALL, although the heterogeneous activation patterns observed in different T-ALL may complicate a general application. We found that cytokine stimulation of T-ALL preferentially triggers PI3K-Akt signaling over RAF-MEK-ERK, and that Akt activation is highly dependent on RasGRP1 expression.38 The exact molecular mechanism for such differential PI3K-Akt signaling is not known to date, but perhaps reflects the fact that T-ALL blasts require PI3K but not MEK for viability and proliferation.50 The notion that RasGRP1 preferentially signals to PI3K highlights the importance of the PI3K-Akt pathway in T-ALL and makes it an attractive target for anti-cancer therapy in leukemia.
The third vignette covers surprising insights that we obtained regarding the RasGDP/RasGTP cycle in T-ALL. The presence of high levels of RasGTP at basal state has been widely reported in RasMUT leukemic cell lines and patient samples (or other cancers), particularly when harboring the GTPase-crippling KRasG12D mutation (see Fig. 1, left). In contrast, cells featuring wild type Ras but elevated expression of RasGRP1 do not display high basal levels of RasGTP when measured by traditional RasGTP pulldown assays.46 However, RasGTP pulldowns only measure the net amount of RasGTP at a certain time point by fishing in the cell lysate with the portion of RAF that binds RasGTP and miss the turnover rate of nucleotide exchange on Ras. This RasGDP/GTP cycling can be directly assessed employing a digitonin nucleotide exchange assay that our collaborators in Dr. Ignacio Rubio's group optimized. This assay measures a radiolabeled GTP that is loaded onto Ras. The radiolabeled phosphate is at the α position so that Ras remains radiolabeled when Ras' GTPase function hydrolyzes the RasGTP to RasGDP (both are radiolabeled). This assay effectively measures (cumulative) flux through the RasGDP/GTP cycle (See Fig. 6a in ref. 38). Using this assay, we find that non-immune cells usually have a very low rate of nucleotide exchange. Normal immune cells expressing (some of the) RasGRP family members show a much higher basal rate of GTP loading onto Ras. Strikingly, T-ALL with high RasGRP1 expression display exceptionally high exchange rates, which is in stark contrast to RasMUT T-ALL that show almost no nucleotide exchange (see Fig. 1). Altogether this leads to a novel model that RasMUT cells - which usually feature low expression of RasGRP1 - display high levels of basal RasGTP but low levels of RasGDP/GTP turnover, while Ras wild type cells with high expression of RasGRP1 only have low (net) levels of basal RasGTP yet a very high RasGDP/GTP turnover. Per definition these result show that hydrolysis of RasGTP to RasGDP is also extremely high in RasGRP1-overexpressing T-ALL, pointing to the RasGAPs that are counterbalancing and reducing the net RasGTP. We noted a broad coverage of expression of different RasGAP family members in 100 pediatric T-ALL samples,38 arguing that joint activity of several RasGAP family members regulate the RasGTP levels in T-ALL and not the activity of one particular RasGAP.
Figure 1.
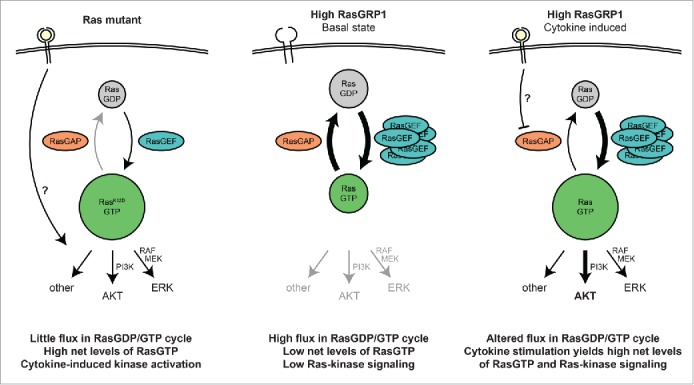
Schematic comparison of different oncogenic Ras signaling in RasMUT and RasGRP1-overexpressing T-ALL. Left: Leukemic cells with constitutively active Ras display high net levels of active RasGTP and very low nucleotide exchange rates due to the defective GTPase function of Ras. Middle: RasGRP1-overexpressing T-ALL show high levels of flux through the RasGDP/GTP cycle, yet low net levels of RasGTP, presumably due to the high compensatory activity of RasGAP family members. Right: After cytokine activation, RasGRP1-overexpressing T-ALL reveal elevated RasGTP levels and active downstream signaling, primarily through the PI3K-Akt pathway.
Fourth, our results allow us to speculate on the physiological or cell biological differences between RasMUT and RasGRP1-overexpressing T-ALL. Strong and constitutive Ras signaling in T-ALL - as driven by classic oncogenic mutations locking Ras into it´s active RasGTP state - can also trigger cell cycle arrest programs. We noted in the past that not all RasMUT T-ALL can uncouple the arrest programs to allow growth of leukemic blasts.46 Perhaps this helps explain why RasMUT is relatively rare in T-ALL? In contrast, overexpression of RasGRP1 in T-ALL does not lead to constitutive Ras signaling but only to a high RasGDP/GTP turnover, and should not induce cell cycle arrest. Indeed, this is what we observed.46 Instead, RasGRP1-overexpressing T-ALL can rapidly accumulate RasGTP following receptor signal input (IL7R), resulting in strong Ras downstream signaling and sustained proliferative effects. To date it is still not fully understood how such a cytokine receptor signal is integrated into the proliferative signaling network in T-ALL cells. We could not find evidence for cytokine-induced RasGRP1 activation. RasGRP1 does not appear to couple to cytokine receptors in the canonical, PLCγ1-dependent manner as seen following TCR stimulation. A plausible model involves decreased activity of RasGAPs upon cytokine stimulation, which would automatically ramp up RasGTP due to the high levels of constitutive GTP loading onto Ras by RasGRP1 (see Fig. 1, right).
Furthermore, our findings illustrate a novel type of oncogenic Ras signaling in cancer, driven by cytokine receptor input together with high flux through the RasGDP/RasGTP cycle caused by RasGRP1 overexpression. Clearly, this mode is very distinct from signals sent by mutant forms of Ras but this does not mean that the RasGDP/RasGTP cycle should be ignored in cancers with RasMUT. Our work in T-ALL suggests that we should pay more attention to the flux through this cycle in all cancer types and the roles of the RasGEFs and RasGAPs herein. Work from others point in the same direction; the importance of a balanced activity of RasGEF and RasGAP effectors on the Ras signaling network was highlighted by a recent study from the Lim lab, which analyzed in greater detail how changes in those players can effectively alter the amplitude and duration of the RasGTP levels. In addition, they could also show that oncogenic RasMUT still depends on the RasGEF/RasGAP circuit further shaping the Ras signaling outcome.51 Development of small molecules that specifically bind to the RasMUT KRASG12C and decrease the viability of cancer cells with this mutation led to the realization that there is a pocket in KRASG12C that may be a good target for cancer therapy.52 Two subsequent studies reveal that KRASG12C is still subjective to nucleotide exchange and does not fully get “locked in” it´s constitutively active form. This provided the opportunity to target KRASG12C with a chemical inhibitor and successfully arrest it in the inactive GDP-bound state, which resulted in robust growth retardation of KRASG12C mutant cell lines.53,54 These studies provide a true shift in paradigm and we should consider the flux through the RasGDP/RasGTP cycle in future studies of oncogenic Ras signals in different cancer types.
Lastly, it is of interest to return to the topic of heterogeneity observed between T-ALL patients. It is tempting to speculate that different oncogenic Ras signals underlie the heterogeneity that is observed and future in-depth analysis of the differential signaling patterns involved is essential to understand the underlying causes and to tailor effective molecular therapies.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
We are supported by grants from the Leukemia & Lymphoma Society (to MM) and the Alex Lemonade Stand Foundation as well as the NIH - R01 CA187318 from the NCI and R01 HL120724 from the NHLBI (all three to JPR).
References
- [1].Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, et al.. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012; 481:157-63; PMID:22237106; http://dx.doi.org/ 10.1038/nature10725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Pui CH, Relling MV, Downing JR. Acute lymphoblastic leukemia. N Engl J Med 2004; 350:1535-48; PMID:15071128; http://dx.doi.org/ 10.1056/NEJMra023001 [DOI] [PubMed] [Google Scholar]
- [3].Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med 2006; 354:166-78; PMID:16407512; http://dx.doi.org/ 10.1056/NEJMra052603 [DOI] [PubMed] [Google Scholar]
- [4].Thiel E, Kranz BR, Raghavachar A, Bartram CR, Löffler H, Messerer D, Ganser A, Ludwig WD, Büchner T, Hoelzer D. Prethymic phenotype and genotype of pre-T (CD7+/ER-)-cell leukemia and its clinical significance within adult acute lymphoblastic leukemia. Blood 1989; 73:1247-58; PMID:2467704 [PubMed] [Google Scholar]
- [5].Pullen J, Shuster JJ, Link M, Borowitz M, Amylon M, Carroll AJ, Land V, Look AT, McIntyre B, Camitta B. Significance of commonly used prognostic factors differs for children with T cell acute lymphocytic leukemia (ALL), as compared to those with B-precursor ALL. A Pediatric Oncology Group (POG) study. Leukemia 1999; 13:1696-707; PMID:10557041; http://dx.doi.org/ 10.1038/sj.leu.2401555 [DOI] [PubMed] [Google Scholar]
- [6].Pui CH, Hancock ML, Head DR, Rivera GK, Look AT, Sandlund JT, Behm FG. Clinical significance of CD34 expression in childhood acute lymphoblastic leukemia. Blood 1993; 82:889-94; PMID:7687897 [PubMed] [Google Scholar]
- [7].Graux C, Cools J, Michaux L, Vandenberghe P, Hagemeijer A. Cytogenetics and molecular genetics of T-cell acute lymphoblastic leukemia: from thymocyte to lymphoblast. Leukemia 2006; 20:1496-510; PMID:16826225; http://dx.doi.org/ 10.1038/sj.leu.2404302 [DOI] [PubMed] [Google Scholar]
- [8].Grabher C, von Boehmer H, Look AT. Notch 1 activation in the molecular pathogenesis of T-cell acute lymphoblastic leukaemia. Nat Rev Cancer 2006; 6:347-59; PMID:16612405; http://dx.doi.org/ 10.1038/nrc1880 [DOI] [PubMed] [Google Scholar]
- [9].Lin YW, Nichols RA, Letterio JJ, Aplan PD. Notch1 mutations are important for leukemic transformation in murine models of precursor-T leukemia/lymphoma. Blood 2006; 107:2540-3; PMID:16282337; http://dx.doi.org/ 10.1182/blood-2005-07-3013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Weng AP, Ferrando AA, Lee W, Morris JP 4th, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004; 306:269-71; PMID:15472075; http://dx.doi.org/ 10.1126/science.1102160 [DOI] [PubMed] [Google Scholar]
- [11].von Lintig FC, Huvar I, Law P, Diccianni MB, Yu AL, Boss GR. Ras activation in normal white blood cells and childhood acute lymphoblastic leukemia. Clin Cancer Res 2000; 6:1804-10; PMID:10815901 [PubMed] [Google Scholar]
- [12].Mitin N, Rossman KL, Der CJ. Signaling interplay in Ras superfamily function. Curr Biol 2005; 15:R563-574; PMID:16051167; http://dx.doi.org/ 10.1016/j.cub.2005.07.010 [DOI] [PubMed] [Google Scholar]
- [13].Mor A, Philips MR. Compartmentalized Ras/MAPK signaling. Annu Rev Immunol 2006; 24:771-800; PMID:16551266; http://dx.doi.org/ 10.1146/annurev.immunol.24.021605.090723 [DOI] [PubMed] [Google Scholar]
- [14].Vetter IR, Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science 2001; 294:1299-304; PMID:11701921; http://dx.doi.org/ 10.1126/science.1062023 [DOI] [PubMed] [Google Scholar]
- [15].Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell 2007; 129:865-77; PMID:17540168; http://dx.doi.org/ 10.1016/j.cell.2007.05.018 [DOI] [PubMed] [Google Scholar]
- [16].Downward J. Cancer biology: signatures guide drug choice. Nature 2006; 439:274-5; PMID:16421553; http://dx.doi.org/ 10.1038/439274a [DOI] [PubMed] [Google Scholar]
- [17].Schubbert S, Bollag G, Shannon K. Deregulated Ras signaling in developmental disorders: new tricks for an old dog. Curr Opin Genet Dev 2007; 17:15-22; PMID:17208427; http://dx.doi.org/ 10.1016/j.gde.2006.12.004 [DOI] [PubMed] [Google Scholar]
- [18].Stone JC. Regulation and Function of the RasGRP family of ras activators in blood cells. Genes Cancer 2011; 2:320-34; PMID:21779502; http://dx.doi.org/ 10.1177/1947601911408082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jun JE, Rubio I, Roose JP. Regulation of Ras exchange factors and cellular localization of ras activation by lipid messengers in T Cells. Frontiers Immunol 2013; 4:239; http://dx.doi.org/ 10.3389/fimmu.2013.00239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ksionda O, Limnander A, Roose JP. RasGRP Ras guanine nucleotide exchange factors in cancer. Front Biol (Beijing) 2013; 8:508-32; PMID:24744772; http://dx.doi.org/ 10.1007/s11515-013-1276-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dower NA, Stang SL, Bottorff DA, Ebinu JO, Dickie P, Ostergaard HL, Stone JC. RasGRP is essential for mouse thymocyte differentiation and TCR signaling. Nat Immunol 2000; 1:317-21; PMID:11017103; http://dx.doi.org/ 10.1038/80799 [DOI] [PubMed] [Google Scholar]
- [22].Coughlin JJ, Stang SL, Dower NA, Stone JC. RasGRP1 and RasGRP3 regulate B cell proliferation by facilitating B cell receptor-Ras signaling. J Immunol 2005; 175:7179-84; PMID:16301621; http://dx.doi.org/ 10.4049/jimmunol.175.11.7179 [DOI] [PubMed] [Google Scholar]
- [23].King PD, Lubeck BA, Lapinski PE. Nonredundant functions for Ras GTPase-activating proteins in tissue homeostasis. Sci Signal 2013; 6:re1; PMID:23443682; http://dx.doi.org/ 10.1126/scisignal.2003669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ward AF, Braun BS, Shannon KM. Targeting oncogenic Ras signaling in hematologic malignancies. Blood 2012; 120:3397-406; PMID:22898602; http://dx.doi.org/ 10.1182/blood-2012-05-378596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ahuja HG, Foti A, Bar-Eli M, Cline MJ. The pattern of mutational involvement of RAS genes in human hematologic malignancies determined by DNA amplification and direct sequencing. Blood 1990; 75:1684-90; PMID:2183888 [PubMed] [Google Scholar]
- [26].Yeoh EJ, Ross ME, Shurtleff SA, Williams WK, Patel D, Mahfouz R, Behm FG, Raimondi SC, Relling MV, Patel A, et al.. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell 2002; 1:133-43; PMID:12086872; http://dx.doi.org/ 10.1016/S1535-6108(02)00032-6 [DOI] [PubMed] [Google Scholar]
- [27].Wiemels JL, Zhang Y, Chang J, Zheng S, Metayer C, Zhang L, Smith MT, Ma X, Selvin S, Buffler PA, et al.. RAS mutation is associated with hyperdiploidy and parental characteristics in pediatric acute lymphoblastic leukemia. Leukemia 2005; 19:415-9; PMID:15674422; http://dx.doi.org/ 10.1038/sj.leu.2403641 [DOI] [PubMed] [Google Scholar]
- [28].Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev 2014; 13:140-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zenatti PP, Ribeiro D, Li W, Zuurbier L, Silva MC, Paganin M, Tritapoe J, Hixon JA, Silveira AB, Cardoso BA, et al.. Oncogenic IL7R gain-of-function mutations in childhood T-cell acute lymphoblastic leukemia. Nat Genet 2011; 43:932-9; PMID:21892159; http://dx.doi.org/ 10.1038/ng.924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kim R, Trubetskoy A, Suzuki T, Jenkins NA, Copeland NG, Lenz J. Genome-based identification of cancer genes by proviral tagging in mouse retrovirus-induced T-cell lymphomas. J Virol 2003; 77:2056-62; PMID:12525640; http://dx.doi.org/ 10.1128/JVI.77.3.2056-2062.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Klinger MB, Guilbault B, Goulding RE, Kay RJ. Deregulated expression of RasGRP1 initiates thymic lymphomagenesis independently of T-cell receptors. Oncogene 2004; 24:2695-704; http://dx.doi.org/ 10.1038/sj.onc.1208334 [DOI] [PubMed] [Google Scholar]
- [32].Oki T, Kitaura J, Watanabe-Okochi N, Nishimura K, Maehara A, Uchida T, Komeno Y, Nakahara F, Harada Y, Sonoki T, et al.. Aberrant expression of RasGRP1 cooperates with gain-of-function NOTCH1 mutations in T-cell leukemogenesis. Leukemia 2011; 26(5):1038-45. [DOI] [PubMed] [Google Scholar]
- [33].Hartzell C, Ksionda O, Lemmens E, Coakley K, Yang M, Dail M, Harvey RC, Govern C, Bakker J, Lenstra TL, et al.. Dysregulated RasGRP1 Responds to Cytokine Receptor Input in T Cell Leukemogenesis. Sci Signal 2013; 6:ra21; PMID:23532335; http://dx.doi.org/ 10.1126/scisignal.2003848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lubeck BA, Lapinski PE, Oliver JA, Ksionda O, Parada LF, Zhu Y, Maillard I, Chiang M, Roose J, King PD. Cutting Edge: Codeletion of the Ras GTPase-Activating Proteins (RasGAPs) Neurofibromin 1 and p120 RasGAP in T Cells Results in the Development of T Cell Acute Lymphoblastic Leukemia. J Immunol 2015; 195:31-5; PMID:26002977; http://dx.doi.org/ 10.4049/jimmunol.1402639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Balgobind BV, Van Vlierberghe P, van den Ouweland AM, Beverloo HB, Terlouw-Kromosoeto JN, van Wering ER, Reinhardt D, Horstmann M, Kaspers GJ, Pieters R, et al.. Leukemia-associated NF1 inactivation in patients with pediatric T-ALL and AML lacking evidence for neurofibromatosis. Blood 2008; 111:4322-8; PMID:18172006; http://dx.doi.org/ 10.1182/blood-2007-06-095075 [DOI] [PubMed] [Google Scholar]
- [36].Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, et al.. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012; 481:157-63; PMID:22237106; http://dx.doi.org/ 10.1038/nature10725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Neumann M, Heesch S, Schlee C, Schwartz S, Gökbuget N, Hoelzer D, Konstandin NP, Ksienzyk B, Vosberg S, Graf A, et al.. Whole-exome sequencing in adult ETP-ALL reveals a high rate of DNMT3A mutations. Blood 2013; 121:4749-52; PMID:23603912; http://dx.doi.org/ 10.1182/blood-2012-11-465138 [DOI] [PubMed] [Google Scholar]
- [38].Ksionda O, Melton AA, Bache J, Tenhagen M, Bakker J, Harvey R, Winter SS, Rubio I, Roose JP. RasGRP1 overexpression in T-ALL increases basal nucleotide exchange on Ras rendering the Ras/PI3K/Akt pathway responsive to protumorigenic cytokines. Oncogene 2015; PMID:26549032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al.. Mutational landscape and significance across 12 major cancer types. Nature 2013; 502:333-9; PMID:24132290; http://dx.doi.org/ 10.1038/nature12634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Juntilla MM, Koretzky GA. Critical roles of the PI3K/Akt signaling pathway in T cell development. Immunol Lett 2008; 116:104-10; PMID:18243340; http://dx.doi.org/ 10.1016/j.imlet.2007.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gutierrez A, Sanda T, Grebliunaite R, Carracedo A, Salmena L, Ahn Y, Dahlberg S, Neuberg D, Moreau LA, Winter SS, et al.. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood 2009; 114:647-50; PMID:19458356; http://dx.doi.org/ 10.1182/blood-2009-02-206722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Maser RS, Choudhury B, Campbell PJ, Feng B, Wong KK, Protopopov A, O'Neil J, Gutierrez A, Ivanova E, Perna I, et al.. Chromosomally unstable mouse tumours have genomic alterations similar to diverse human cancers. Nature 2007; 447:966-71; PMID:17515920; http://dx.doi.org/ 10.1038/nature05886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Palomero T. et al.. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med 2007; 13:1203-10; PMID:17873882; http://dx.doi.org/ 10.1038/nm1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lonetti A, Antunes IL, Chiarini F, Orsini E, Buontempo F, Ricci F, Tazzari PL, Pagliaro P, Melchionda F, Pession A, et al.. Activity of the pan-class I phosphoinositide 3-kinase inhibitor NVP-BKM120 in T-cell acute lymphoblastic leukemia. Leukemia 2013; 28(6):1196-206. [DOI] [PubMed] [Google Scholar]
- [45].Piovan E, Yu J, Tosello V, Herranz D, Ambesi-Impiombato A, Da Silva AC, Sanchez-Martin M, Perez-Garcia A, Rigo I, Castillo M, et al.. Direct reversal of glucocorticoid resistance by AKT inhibition in acute lymphoblastic leukemia. Cancer Cell 2013; 24:766-76; PMID:24291004; http://dx.doi.org/ 10.1016/j.ccr.2013.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Hartzell C, Ksionda O, Lemmens E, Coakley K, Yang M, Dail M, Harvey RC, Govern C, Bakker J, Lenstra TL, et al.. Dysregulated RasGRP1 Responds to Cytokine Receptor Input in T Cell Leukemogenesis. Sci Signal 2013; 6:ra21; PMID:23532335; http://dx.doi.org/ 10.1126/scisignal.2003848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ikezoe T, Nishioka C, Bandobashi K, Yang Y, Kuwayama Y, Adachi Y, Takeuchi T, Koeffler HP, Taguchi H. Longitudinal inhibition of PI3K/Akt/mTOR signaling by LY294002 and rapamycin induces growth arrest of adult T-cell leukemia cells. Leuk Res 2007; 31:673-82; http://dx.doi.org/ 10.1016/j.leukres.2006.08.001 [DOI] [PubMed] [Google Scholar]
- [48].Silva A, Yunes JA, Cardoso BA, Martins LR, Jotta PY, Abecasis M, Nowill AE, Leslie NR, Cardoso AA, Barata JT. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J Clin Invest 2008; 118:3762-74; PMID:18830414; http://dx.doi.org/ 10.1172/JCI34616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Knight ZA, Lin H, Shokat KM. Targeting the cancer kinome through polypharmacology. Nat Rev Cancer 2010; 10:130-37; PMID:20094047; http://dx.doi.org/ 10.1038/nrc2787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Barata JT, Silva A, Brandao JG, Nadler LM, Cardoso AA, Boussiotis VA. Activation of PI3K is indispensable for interleukin 7-mediated viability, proliferation, glucose use, and growth of T cell acute lymphoblastic leukemia cells. J Exp Med 2004; 200:659-69; PMID:15353558; http://dx.doi.org/ 10.1084/jem.20040789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Coyle SM, Lim WA. Mapping the functional versatility and fragility of Ras GTPase signaling circuits through in vitro network reconstitution. Elife 2016; 5:e12435; PMID:26765565; http://dx.doi.org/ 10.7554/eLife.12435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013; 503:548-51; PMID:24256730; http://dx.doi.org/ 10.1038/nature12796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Patricelli MP, Janes MR, Li LS, Hansen R, Peters U, Kessler LV, Chen Y, Kucharski JM, Feng J, Ely T, et al.. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov 2016; 6:316-29; PMID:26739882; http://dx.doi.org/ 10.1158/2159-8290.CD-15-1105 [DOI] [PubMed] [Google Scholar]
- [54].Lito P, Solomon M, Li LS, Hansen R, Rosen N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 2016; 351:604-8; PMID:26841430; http://dx.doi.org/ 10.1126/science.aad6204 [DOI] [PMC free article] [PubMed] [Google Scholar]