

Abstract
The ribosome has been described as a ribozyme in which ribosomal RNA is responsible for peptidyl-transferase reaction catalysis. The W255C mutation of the universally conserved ribosomal protein uL3 has diverse effects on ribosome function (e.g., increased affinities for transfer RNAs, decreased rates of peptidyl-transfer), and cells harboring this mutation are resistant to peptidyl-transferase inhibitors (e.g., anisomycin). These observations beg the question of how a single amino acid mutation may have such wide ranging consequences. Here, we report the structure of the vacant yeast uL3 W255C mutant ribosome by X-ray crystallography, showing a disruption of the A-site side of the peptidyl-transferase center (PTC). An additional X-ray crystallographic structure of the anisomycin-containing mutant ribosome shows that high concentrations of this inhibitor restore a “WT-like” configuration to this region of the PTC, providing insight into the resistance mechanism of the mutant. Globally, our data demonstrate that ribosomal protein uL3 is structurally essential to ensure an optimal and catalytically efficient organization of the PTC, highlighting the importance of proteins in the RNA-centered ribosome.
Keywords: structure, crystallography, uL3, mutant, anisomycin
Introduction
The first high-resolution structures of the ribosome obtained by X-ray crystallography demonstrated that catalysis of the peptidyl-transferase reaction is achieved exclusively by ribosomal RNA (rRNA), thus identifying the ribosome as a ribozyme [1]. Structural comparison of ribosomes from various origins highlights a common organization around a conserved rRNA core, with the gradual addition of layers of rRNA and proteins functioning to increase complexity throughout evolution [2]. Although ribosomal proteins do not directly participate in the catalytic activity of the ribosome, their contribution is essential for efficient translation. In addition to their protective and structural role for rRNA, many ribosomal proteins are essential for ribosome assembly, for the formation of the peptidyl-transferase and the decoding centers, for binding of the different ribosomal ligands (mRNA, tRNAs, translation factors), and for signal transduction [3,4].
Among all the ribosomal proteins, the universally conserved ribosomal protein L3 (uL3) is the largest and is situated closest to the peptidyl-transferase center (PTC; see Fig. 1a). In the context of the ribosome, uL3 folds as a globular domain which interfaces with the solvent side of the large subunit (LSU) with three finger-like projections that extend into the central core of the LSU: the N-terminal extension, the “tryptophan finger” (W finger) and the “basic thumb” (the latter two both being part of the central loop) (Fig. 1b). These three “fingers” are situated close to the PTC, and several studies have shown that mutations in these regions are important for optimal ribosome functionality. Among these, the most well-characterized is the tryptophan 255 to cysteine (W255C) mutant, located in the so-called “W finger” (unless otherwise specified, Saccharomyces cerevisiae numbering is used throughout the text and corresponding bases and residues from various organisms are listed in Supplementary Table 1). This mutant was first identified as responsible for resistance to the peptidyl-transferase inhibitors trichodermin and anisomycin in yeast [5–7] as well as for promoting the inability of cells to maintain the M1 killer virus [8]. More recently, this mutant was shown to promote decreased rates of cell growth and protein synthesis, increased affinities for aminoacyl- and peptidyl-tRNAs (aa-tRNA and pep-tRNA), decreased affinity for eukaryotic elongation factor 2, decreased peptidyl-transferase activity [9], increased efficiency of programmed −1 ribosomal frameshifting [10], and an inability to recruit the pokeweed antiviral protein [11].
Fig. 1.

Global view of protein uL3 in the context of the ribosome. Localization and folding of the protein uL3 in the ribosome. Figures were generated by using the structure of the S. cerevisiae vacant wild-type ribosome (PDB 4V88) [12] with A- and P-site aa-tRNAs extracted from the superimposed Thermus thermophilus structure (PDB 4V5D) [19]. (a) Schematic top and A site views of the translating ribosome. The large ribosomal subunit is represented in gray, the small ribosomal subunit in blue, the A-site tRNA in yellow, the P-site tRNA in orange, the E-site tRNA in red, the PTC in green, and ribosomal protein uL3 in purple. (b) Structure of the protein uL3 in the context of the ribosome. uL3 folds as a 3-fingered platform formed by its globular domain (in deep purple), and the N-terminal extension (in light blue), the “tryptophan finger” (W finger; in wheat), and the “basic thumb” (in pale green). Those 3 “fingers” extend into the central core of the LSU, their tips reaching less than 12 Å away from the PTC (in green).
In order to better understand how a single mutation could have such severe consequences on the translation mechanisms, structural studies of the uL3 W255C ribosome were performed using X-ray crystallography. The structures reported here represent the first high-resolution structures of a mutant and drug-resistant eukaryotic ribosome. Structures of the S. cerevisiae uL3 W255C mutant ribosome were determined in its vacant form and in complex with the peptidyl-transferase inhibitor anisomycin at 3.4 and 3.1 Å respectively (I/σI = 1; Table 1). The data reveal that the structural changes caused by the uL3 W255C mutation are not limited to the proximal mutation environment, but rather extend through a long-range network of structural rearrangements leading to the disruption of the PTC, most likely accounting for the altered A-site tRNA binding and introducing a new drug resistance mechanism.
Table 1.
Data collection and refinement statistics
| 80S L3W255C Vacant | 80S L3W255C ANI | |
|---|---|---|
| Data collection | ||
| No. of crystals | 1 | 2 |
| Space group | P21 | P21 |
| Cell parameters | ||
| a, b, c (Å) | 435.45, 287.66, 303.76 | 436.11, 287.31, 303.99 |
| α, β, γ (°) | 90.000, 98.915, 90.000 | 90.000, 98.860, 90.000 |
| Resolution | 49.993–3.400 (3.500–3.400) | 49.958–3.100 (3.200–3.100) |
| Rmeas (%) | 38.2 (166.9) | 40.7 (229.0) |
| I/σI | 5.32 (1.02) | 6.77 (0.93) |
| CC1/2 (%) | 97.5 (36.3) | 98.6 (31.0) |
| Completeness (%) | 99.9 (100.0) | 100.0 (100.0) |
| Redundancy | 4.8 | 9.0 |
| Refinement | ||
| Resolution | 49.993–3.400 | 49.958–3.100 |
| No. of unique reflections | 1,009,263 | 1,333,169 |
| Rwork/Rfree (%) | 23.43/28.45 | 23.44/29.06 |
| No. of atoms | ||
| Protein | 180,135 | 179,857 |
| RNA | 222,826 | 222,826 |
| Ions and ligands | 8690 | 8236 |
| B factors | ||
| Protein | 80.74 | 72.65 |
| RNA | 75.46 | 67.70 |
| Ions and ligands | 107.46 | 86.92 |
| RMS deviations | ||
| Bond lengths (Å) | 0.010 | 0.009 |
| Bond angles (°) | 1.319 | 1.221 |
| PDB IDs | 5FCI | 5FCJ |
Values in parenthesis are for highest-resolution shell statistics. uL3 W255C mutant 80S ribosomes from S. cerevisiae were purified, crystallized, and treated essentially as previously described [12]. The post-crystallization treatments were modified to increase glycerol concentration to 20% in all intermediate solutions. Ribosome complexes containing anisomycin were formed by soaking 80S ribosome crystals with 500 μM of inhibitor for ~30 min at 4 °C in a buffer containing 80 mM Tris–acetate (pH 7.0), 70 mM KSCN, 40 mM potassium acetate, 7.5 mM ammonium acetate, 10 mM magnesium acetate, 6.5 mM spermidine, 5% PEG 20,000, 20% vol/vol glycerol, 2 mM DTT, 1.26 mM Deoxy Big CHAP, and 15% wt/vol PEG 6000 before the transfer to a cryo-protecting buffer containing 80 mM Tris–acetate (pH 7.0), 70 mM KSCN, 40 mM potassium acetate, 7.5 mM ammonium acetate, 10 mM magnesium acetate, 6.5 mM spermidine, 5% PEG 20,000, 20% vol/vol glycerol, 2 mM DTT, 1.26 mM Deoxy Big CHAP, 20% wt/vol PEG 6000, and 3 mM osmium hexamine supplemented with 100 μM anisomycin for ~2 h. Data collection was performed at synchrotron SOLEIL on PROXIMA 1 beamline. We applied the previously described data collection strategy, optimized for single-photon counting detectors [12,20], attenuating the beam to 20% of the incoming photon flux and collecting highly redundant data when possible. Diffraction data were processed and reduced using the XDS suite [21]. The structures were determined by molecular replacement using the S. cerevisiae 80S ribosome structure (PDB 4V88) [12] as a search model and then subjected to refinement using phenix.refine [22]. Restraints for anisomycin were generated with JLigand [23] and ReadySet from the Phenix suite [22]. Ligands building, fitting, remodeling of ribosomal binding sites, and analysis of Ramachandran plots were performed using Coot [24]. Final refinement was performed with phenix.refine [22]. Ligands geometry was validated with the software Mogul from the CCDC package [25]. Compared to the original model of S. cerevisiae ribosome (PDB 4V88), conformation of several rRNA nucleotides was corrected in both monomers, including residues A2397, A2872, U2873, G2874, and U2975 in 25S rRNA, and metal ions were modeled de novo. Figures were prepared using PyMOL (Schrödinger, LLC). To simplify comparisons of the yeast 80S ribosome with ribosomes from other species, ribosomal proteins were named throughout the manuscript according to the recently established nomenclature [26]. Coordinates and structure factors for vacant and anisomycin-containing 80S yeast ribosomes were deposited at the Worldwide Protein Data Bank with PDB IDs 5FCI and 5FCJ, respectively.
Structure of the vacant uL3 W255C mutant 80S yeast ribosome
The structure of the uL3 W255C mutant 80S ribosome from S. cerevisiae was determined by X-ray crystallography to better understand the mechanisms of action of this mutant. The same ribosome purification and crystallization protocols as for the S. cerevisiae WT 80S ribosome [12] enabled the structure of the uL3 W255C mutant ribosome to be solved to 3.4 Å (I/σI = 1; Table 1). The electron density map unambiguously revealed the W255C mutation itself, as well as reorganization of specific 25S residues (Fig. 2). In the structure of the WT ribosome from S. cerevisiae [12], a nearby 25S residue—A2397—adopts a syn conformation (Fig. 2a). In the structure of the uL3 W255C mutant ribosome, as cysteine is less bulky than tryptophan, A2397 is flipped toward uL3 “W finger” and adopts an anti conformation (Fig. 2b). The superposition of WT and mutant structures shows that hydrogens protruding from uL3 W255 and 25S A2397 in an anti conformation would be too close, suggesting that repulsive electrostatic interactions force 25S A2397 to flip into the syn conformation (Fig. 2c). Comparison of WT and mutant structures shows that movement of this single nucleotide induces destabilization of the 25S region corresponding to residues A2872 to U2875 (Fig. 2a and b), which form the A-site tRNA amino acid binding pocket of the PTC. Indeed, in the WT structure, A2397 is stacked with U2873, thus bracing residues A2872 to U2875 in—presumably—an optimal conformation for stabilization of the A-site tRNA amino acid in the correct position for the peptidyl-transfer reaction to occur. In the case of the uL3 W255C mutant structure, as A2397 adopts a syn conformation, U2873 is shifted to avoid a clash with A2397 (Fig. 2d) and residues A2872 to U2875 are no longer stabilized. This may result in the widening of the aa-tRNA binding pocket, explaining the observed increase in affinity, as it was previously suggested [13].
Fig. 2.
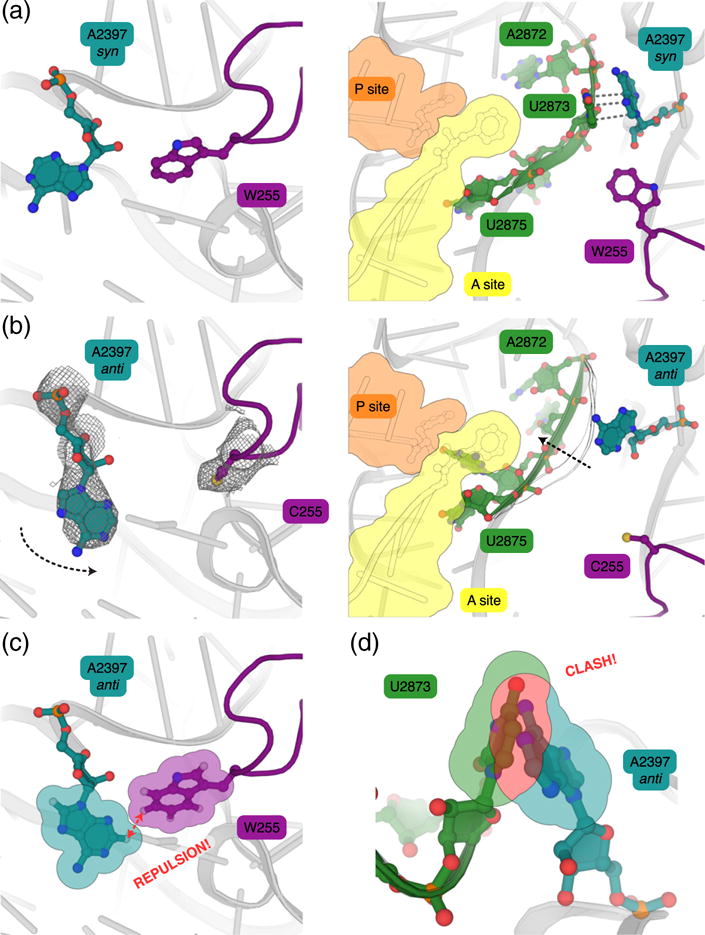
Structure of the uL3 W255C mutant ribosome shows that uL3 helps the 25S rRNA adopt an optimal conformation for peptidyl-transfer catalysis. Views comparing the organization of the A-site side of the PTC in the vacant wild-type (WT; PDB 4V88) [12] and uL3 W255C mutant ribosomes (this study). A- and P-site aa-tRNAs extracted from the superimposed T. thermophilus structure (PDB 4V5D) [19] were placed in an informative way. (a) Structure of the vacant WT ribosome. In the close environment of the WT uL3 residue W255 (purple), the 25S residue A2397 (blue-gray) adopts a syn conformation. Residues A2872–U2875 (in green) are stabilized due to the stacking interaction between U2873 and A2397 in this syn conformation. (b) Structure of the vacant uL3 W255C mutant ribosome. In the presence of the uL3 W255C mutation (purple), the 25S residue A2397 (blue-gray) is flipped to adopt an anti conformation. The 2Fo–Fc electron density map, contoured at 1σ, is indicated for C255 as a gray mesh. The stacking interaction between A2397 and U2873 is broken by the reorientation of A2397, destabilizing residues A2872 to U2875. (c) Superposition of A2397 anti conformation to the structure of the WT ribosome shows that this conformation is not possible in the presence of W255 because of repulsive electrostatic interactions between hydrogens of W255 and A2397 aromatic rings. Only the static hydrogens of carbon atoms of the aromatic rings are represented here. (d) Superposition of A2397 anti conformation to the structure of the WT ribosome shows that the destabilization of residues A2872 to U2875 is due to a clash between the anti conformation of A2397 and U2873.
It was also suggested that W255 may normally interact with A2941, which is positioned next to one of the 25S “tRNA gates”—C2942—and might help to open it, thus facilitating aa-tRNA passage to the A site [14,15]. The structure of the WT ribosome from S. cerevisiae [12] shows that A2941 is flanked by W255 on one side and the 25S residue C2374 on the other side (Supplementary Fig. 1). A2941 is thus very stable, with the neighboring tRNA-gate C2842 in an “open” conformation. It was proposed that the higher affinity of aa-tRNAs in uL3 W255C mutants could be explained by a more open conformation of the A-site vicinity of the PTC and of the aa-tRNA accommodation corridor, facilitating aa-tRNA binding [14]. Our structure of the uL3 W255C mutant ribosome shows that A2397 in anti conformation takes over the flanking of A2941. A slight reorientation of both A2941 and C2374 is visible, to adapt to the orientation of the anti conformation of A2397, but no rearrangements of tRNA gates are observed in this structure.
Structure of the anisomycin-containing uL3 W255C mutant 80S yeast ribosome
Given that the destabilized 25S region also corresponds to the anisomycin binding site [16], the structure of the uL3 W255C mutant 80S ribosome from S. cerevisiae in complex with anisomycin was also determined by X-ray crystallography. In this study, we modified the post-crystallization treatments and supplemented the soaking solutions with anisomycin. The use of high concentrations (500 μM) of anisomycin enabled the determination of the structure of anisomycin-containing S. cerevisiae 80S ribosome to 3.1-Å resolution (I/σI = 1; Table 1). In the structure of the WT ribosome from S. cerevisiae in complex with anisomycin [16], the inhibitor binds in a pocket formed by PTC residues, including A2872 to U2875, and its pyrrolidine and acetyl moieties are particularly well stabilized by direct or magnesium-mediated polar contacts (Fig. 3a and b). The 25S residues A2872 to U2875 maintain the same organization as in the structure of the vacant ribosome, with U2873 stabilized by stacking with A2397, with the exception of U2875 which adopts an “up” conformation (Fig. 3c) in comparison to the “down” conformation in the structure of the vacant ribosome (Fig. 2a). The structure of the anisomycin-containing uL3 W255C mutant ribosome shows that the inhibitor generally binds similarly to the mutant ribosome: the binding pocket is formed by the same residues, but the position of the methyl-tyrosine moiety of anisomycin is shifted by ~1 Å toward the A2872–U2875 region, and its pyrrolidine and acetyl moieties are not as well stabilized as in the WT ribosome (Fig. 3a and b). Nevertheless, binding of anisomycin forces the mutant ribosome to adopt a WT-like conformation: U2873 is pushed toward A2397, compelling it to adopt a syn conformation to avoid a clash with U2873, hence re-creating the stacking interaction observed in the WT ribosome (Fig. 3c). This may explain why uL3 W255C mutant cells grow better in the presence of anisomycin [9]. However, even though the phosphate backbone of residues A2872 to U2875 follows the same path as in the WT ribosome, A2872 and U2875 adopt different conformations: A2872 is in a syn conformation (versus anti conformation in both vacant and anisomycin-containing WT ribosomes), and U2875 adopts a “down” conformation similar to that found in the vacant WT ribosome (versus “up” conformation in the anisomycin-containing WT ribosome) (Fig. 3c). While it is not possible to deduce how the different A2872 conformations may mechanistically contribute to anisomycin resistance, it appears that U2875 may act as a bolt, closing access to the A-site side of the PTC. Indeed, superposition of A-site aa-tRNA shows a clash between the “up” conformation of A2872 and the amino acid of the A-site tRNA (Fig. 3d), whereas the “down” conformation seems compatible with the simultaneous presence of an aa-tRNA (Fig. 3d). Early studies showed that the W255C mutation does not affect anisomycin binding [17], suggesting that structural rearrangements in mutant ribosomes might reduce the ability of anisomycin to compete with aa-tRNA. In the uL3 W255C mutant ribosome, the “down” conformation of A2872 may allow the aa-tRNA to enter the A-site side of the PTC and compete with anisomycin. Two outcomes are then possible: (i) the aa-tRNA drives the inhibitor out of the PTC, or (ii) it pushes the inhibitor further into its binding pocket, in a conformation that will allow the binding of both substrates, as previously suggested [13]. This drug resistance mechanism is different from the ones previously described, where either the ribosome or the drug is modified, preventing binding of the drug due to novel steric clashes.
Fig. 3.

The uL3 W255C mutation is responsible for rearrangements in the anisomycin binding pocket. The 2Fo–Fc electron density map, contoured at 1σ, is indicated for anisomycin as a gray mesh in the right side of panels b and c. The structure of the anisomycin-containing mutant ribosome was obtained using “artificial” conditions, by forcing the inhibitor inside the “resistant” ribosomes, thanks to high concentrations of inhibitor (500 μM). As a consequence, occupancy is not a 100%, but the drug could nevertheless be placed unambiguously into the electron density map. (a) Chemical structure of anisomycin. For simplicity of description, the inhibitor has been divided into three moieties: the methyl-tyrosine moiety (in red), the pyrrolidine ring (in green), and the acetyl moiety (in blue). (b) Polar contacts implicated in anisomycin. The structure of anisomycin bound to the WT yeast 80S ribosome (on the left; PDB 4U3M) [16] shows that the 25S region destabilized by the uL3 W255C mutation (residues A2872 to U2875) flanks the anisomycin binding pocket. The pyrrolidine and acetyl moieties of the inhibitor are particularly well stabilized by direct or magnesium-mediated polar contacts with A2872, G2403, G2816, C2821, U2869, and C2870. The structure of anisomycin bound to the uL3 W255C mutant yeast 80S ribosome (on the right; this study) shows that the inhibitor binds in the same binding pocket as for the WT ribosome. Even though the methyl-tyrosine moiety binds in a very similar manner, the pyrrolidine and acetyl moieties are not as well stabilized (less polar contacts) and are more flexible. (c) Effects of anisomycin binding on A-site side of PTC reorganization. In the case of the WT ribosome (on the left; PDB 4U3M) [16], the presence of anisomycin does not cause particular rearrangements of the residues A2872 to U2875: this region is still stabilized because of the stacking between U2873 and A2397. The only difference concerns U2875 which is lifted up. In the case of the uL3 W255C mutant ribosome (on the right; this study), anisomycin binding pushes residues A2872 to U2875 to adopt a conformation “similar” to the WT ribosome one: because of anisomycin, an “artificial” stacking between U2873 and A2397 is created. However, not all residues match the WT ribosome: A2872 is in syn conformation, and U2875 remains pointed down. (d) Superposition of A-site aa-tRNA from T. thermophilus structure (PDB 4V5D) [19] and the anisomycin-containing ribosome structures. In the case of the WT ribosome (on the left), an important clash between the U2875 “up” conformation and the amino acid of the tRNA can be observed. In contrast, in the case of the uL3 W255C mutant ribosome (on the right), the U2875 “down” conformation does not clash with a aa-tRNA bound to the A site.
Discussion
In previous studies, it was shown that cysteine is the only viable mutation of uL3 W255 besides aromatic or basic amino acids [14], and genetic evidence suggested that C255 forms a disulfide bridge with another cysteine of the W finger—C251—causing displacement of the loop so that the role of W255 was played by another aromatic amino acid of the loop (either H256 or H259) [14]. Our structures do not highlight any disulfide bond between C255 and C251, or any other reorganization of the W finger in general (Supplementary Fig. 2). As dithiothreitol (DTT) was used during purification and crystallization, we thought that its reducing activity might be responsible for C255–C251 disulfide bridge cleavage. Thus, attempts were made to reproduce purification and crystallization of uL3 W255C mutant ribosome without DTT. The same experiments were performed for WT ribosomes as a control. Consistent with the requirement for uL3 in ribosome subunit assembly, purification profiles show that mutant ribosomes are characterized by higher subunit dissociation propensity (Supplementary Fig. 3a). However, we observed that both WT and mutant ribosomes purified in DTT-deprived buffers are less stable and are prone to aggregation. Unsurprisingly—considering the decrease in the macromolecule stability—crystallization assays were unsuccessful for WT and mutant ribosomes (Supplementary Fig. 3b). Given that the same effects were observed for both WT and mutant ribosomes, this instability cannot be specifically due to the uL3 W255C mutation, but are more likely caused by various effects on the whole ribosome. Thus, no conclusions could be drawn concerning the presence or absence of a C255-C251 disulfide bridge under “natural” conditions.
Overall, we show that the uL3 single mutation W255C disrupts the organization of the PTC through a cascade of structural rearrangements, and more specifically the 25S region forming the A-site tRNA amino acid binding pocket (Fig. 2). The increased flexibility of this region may facilitate entrance of aa-tRNA into the PTC, but this may come at the cost of precision of amino acid positioning in the A site, and consequently of peptidyl-transfer efficiency [9,18]. Distortion of this region also affects the binding and mechanisms of action of PTC-occupying inhibitors such as anisomycin (Fig. 3), introducing a novel drug resistance mechanism. Indeed, the structural analysis is consistent with a previously proposed model of anisomycin resistance in which destabilization of the A-site proximal region of the PTC may allow room for binding of both anisomycin and the aa-tRNA acceptor stem (see Fig. 5 in [13]). Taken together, our data suggest that ribosomal protein uL3 is structurally essential to fine-tune the organization of the PTC in order to ensure optimal catalytic efficiency, highlighting the importance of proteins in the RNA-directed function of the ribosome.
Supplementary Material
Acknowledgments
We are grateful to the staff of PROXIMA 1 beamline at the synchrotron SOLEIL (France), and in particular to Andrew Thompson and Pierre Legrand for providing rapid access and assisting with data collection. This work was supported by the French National Research Agency (ANR-11-BSV8-006 01; to G.Y.), the European Research Council (advanced grant 294312), and the Human Frontier Science Program (grant RGP0062/2012; both to M.Y.), the Russian Government Program of Competitive Growth of Kazan Federal University (both to M.Y. and G.Y.), and in part by the National Institutes of Health (grants R01 GM117177 and R01 HL119439; both to J.D.D.).
Abbreviations used
- rRNA
ribosomal RNA
- tRNA
transfer RNA
- PTC
peptidyl-transferase center
- LSU
large subunit
- W finger
tryptophan finger
- aa-tRNA
aminoacyl-tRNA
- pep-tRNA
peptidyl-tRNA
- DTT
dithiothreitol
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jmb.2016.02.013.
Footnotes
Accession numbers
The structures were deposited to the protein data bank with accession codes 5FCI (vacant) and 5FCJ (with anisomycin).
References
- 1.Simonović M, Steitz TA. A structural view on the mechanism of the ribosome-catalyzed peptide bond formation. Biochim Biophys Acta. 2009;(1789):612–623. doi: 10.1016/j.bbagrm.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steitz TA, Moore PB. RNA, the first macromolecular catalyst: the ribosome is a ribozyme. Trends Biochem Sci. 2003;28:411–418. doi: 10.1016/S0968-0004(03)00169-5. [DOI] [PubMed] [Google Scholar]
- 3.Wilson DN, Nierhaus KH. Ribosomal proteins in the spotlight. Crit Rev Biochem Mol Biol. 2005;40:243–267. doi: 10.1080/10409230500256523. [DOI] [PubMed] [Google Scholar]
- 4.Voorhees RM, Ramakrishnan V. Structural basis of the translational elongation cycle. Annu Rev Biochem. 2013;82:203–236. doi: 10.1146/annurev-biochem-113009-092313. [DOI] [PubMed] [Google Scholar]
- 5.Ben-Shem A, GarreaudeLoubresse N, Melnikov S, Jenner L, Yusupova G, Yusupov M. The structure of the eukaryotic ribosome at 3.0 Å resolution. Science. 2011;334:1524–1529. doi: 10.1126/science.1212642. [DOI] [PubMed] [Google Scholar]
- 6.Voorhees RM, Weixlbaumer A, Loakes D, Kelley AC, Ramakrishnan V. Insights into substrate stabilization from snapshots of the peptidyl transferase center of the intact 70S ribosome. Nat Struct Mol Biol. 2009;16:528–533. doi: 10.1038/nsmb.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schindler D, Grant P, Davies J. Trichodermin resistance–mutation affecting eukaryotic ribosomes. Nature. 1974;248:535–536. doi: 10.1038/248535a0. [DOI] [PubMed] [Google Scholar]
- 8.Jimenez A, Sanchez L, Vazquez D. Simultaneous ribosomal resistance to trichodermin and anisomycin in Saccharomyces cerevisiae mutants. Biochim Biophys Acta. 1975;383:427–434. doi: 10.1016/0005-2787(75)90312-3. [DOI] [PubMed] [Google Scholar]
- 9.Grant PG, Schindler D, Davies JE. Mapping of trichodermin resistance in Saccharomyces cerevisiae: a genetic locus for a component of the 60S ribsomal subunit. Genetics. 1976;83:667–673. doi: 10.1093/genetics/83.4.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wickner RB, Leibowitz MJ. Chromosomal genes essential for replication of a double-stranded RNA plasmid of Saccharomyces cerevisiae: the killer character of yeast. J Mol Biol. 1976;105:427–443. doi: 10.1016/0022-2836(76)90102-9. [DOI] [PubMed] [Google Scholar]
- 11.Petrov A, Meskauskas A, Dinman JD. Ribosomal protein L3: influence on ribosome structure and function. RNA Biol. 2004;1:59–65. doi: 10.4161/rna.1.1.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meskauskas A, Harger JW, Jacobs KL, Dinman JD. Decreased peptidyltransferase activity correlates with increased programmed −1 ribosomal frameshifting and viral maintenance defects in the yeast Saccharomyces cerevisiae. RNA. 2003;9:982–992. doi: 10.1261/rna.2165803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hudak KA, Dinman JD, Tumer NE. Pokeweed antiviral protein accesses ribosomes by binding to L3. J Biol Chem. 1999;274:3859–3864. doi: 10.1074/jbc.274.6.3859. [DOI] [PubMed] [Google Scholar]
- 14.Meskauskas A, Petrov AN, Dinman JD. Identification of functionally important amino acids of ribosomal protein L3 by saturation mutagenesis. Mol Cell Biol. 2005;25:10863–10874. doi: 10.1128/MCB.25.24.10863-10874.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meskauskas A, Dinman JD. Ribosomal protein L3: gatekeeper to the A site. Mol Cell. 2007;25:877–888. doi: 10.1016/j.molcel.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rakauskaite R, Dinman JD. Mutations of highly conserved bases in the peptidyltransferase center induce compensatory rearrangements in yeast ribosomes. RNA. 2011;17:855–864. doi: 10.1261/rna.2593211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garreau de Loubresse N, Prokhorova I, Holtkamp W, Rodnina MV, Yusupova G, Yusupov M. Structural basis for the inhibition of the eukaryotic ribosome. Nature. 2014;513:517–522. doi: 10.1038/nature13737. [DOI] [PubMed] [Google Scholar]
- 18.Jimenez A, Vazquez D. Quantitative binding of antibiotics to ribosomes from a yeast mutant altered on the peptidyl-transferase center. Eur J Biochem. 1975;54:483–492. doi: 10.1111/j.1432-1033.1975.tb04160.x. [DOI] [PubMed] [Google Scholar]
- 19.Leung EK, Suslov N, Tuttle N, Sengupta R, Piccirilli JA. The mechanism of peptidyl transfer catalysis by the ribosome. Annu Rev Biochem. 2011;80:527–555. doi: 10.1146/annurev-biochem-082108-165150. [DOI] [PubMed] [Google Scholar]
- 20.Mueller M, Wang M, Schulze-Briese C. Optimal fine ϕ-slicing for single-photon-counting pixel detectors. Acta Crystallogr D Biol Crystallogr. 2012;68:42–56. doi: 10.1107/S0907444911049833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lebedev AA, Young P, Isupov MN, Moroz OV, Vagin AA, Murshudov GN. JLigand: a graphical tool for the CCP4 template-restraint library. Acta Crystallogr D Biol Crystallogr. 2012;68:431–440. doi: 10.1107/S090744491200251X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bruno IJ, Cole JC, Kessler M, Luo J, Motherwell WD, Purkis LH, et al. Retrieval of crystallographically-derived molecular geometry information. J Chem Inf Comput Sci. 2004;44:2133–2144. doi: 10.1021/ci049780b. [DOI] [PubMed] [Google Scholar]
- 26.Ban N, Beckmann R, Cate JH, Dinman JD, Dragon F, Ellis SR, et al. A new system for naming ribosomal proteins. Curr Opin Struct Biol. 2014;24:165–169. doi: 10.1016/j.sbi.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.