

Abstract
Aim:
We compared predictive modeling approaches to estimate placental methylation using cord blood methylation.
Materials & methods:
We performed locus-specific methylation prediction using both linear regression and support vector machine models with 174 matched pairs of 450k arrays.
Results:
At most CpG sites, both approaches gave poor predictions in spite of a misleading improvement in array-wide correlation. CpG islands and gene promoters, but not enhancers, were the genomic contexts where the correlation between measured and predicted placental methylation levels achieved higher values. We provide a list of 714 sites where both models achieved an R2 ≥0.75.
Conclusion:
The present study indicates the need for caution in interpreting cross-tissue predictions. Few methylation sites can be predicted between cord blood and placenta.
Keywords: : 450k arrays, cord blood, DNA methylation, epigenetics, methylation prediction, placenta, support vector machine
Background
Epigenetic mechanisms contribute to differences in the biologic functions of different tissues [1–3]. Understanding these differences and how we can borrow information or contrast tissue types can enhance epigenomic studies characterizing human populations. Previous research has suggested that DNA methylation levels in one tissue may serve as surrogate markers of DNA methylation levels in another tissue [4,5]. Since cord blood is a more available and frequently collected tissue than placenta, a predictive model between these two tissues may be useful to researchers in reproductive health. The general methylomic patterns across tissues would also be informative regardless of which sites are correlated, as we need to understand these patterns to better understand the role of methylation in tissue differentiation. Moreover, some studies suggest that DNA methylation patterns within some genomic regions are largely conserved across tissues, although intraindividual variation exceeds interindividual variation [6–8]. A recent study [9] proposed to use support vector machine (SVM) to predict locus-specific methylation in a target tissue based on methylation in a surrogate tissue (e.g., predicting methylation in atrial tissue using peripheral blood). SVM is a supervised learning method used to analyze data and recognize patterns [10]. SVM represents a powerful technique for general classification, regression and outlier detection and has been widely used in many bioinformatics applications. DNA methylation prediction based on a surrogate tissue could be especially useful when the target tissue of interest is difficult to collect, such as brain or heart tissue. Furthermore, these methods could be advantageous even for tissues that are more readily available. The placenta, for example, is an accessible tissue that can be noninvasively collected at delivery and has been proposed to harbor molecular signals that may reflect fetal programming from in utero exposures and risk factors. Yet, while collection of placenta is noninvasive, protocols for sampling of placental tissue and storage are less standardized and more time consuming than those for cord blood, a source of fetal DNA that is also available at birth. As a result, fewer cohort studies have banked placenta samples as more have focused on collecting cord blood. Further, the placenta is a unique organ that – different from other human tissues – grows rapidly during pregnancy, is perfused by both maternal and fetal blood, and ends its functions at delivery. Therefore, we need to determine whether DNA methylation prediction methods developed for other target tissues can be used for placenta.
In this study, we examined genome-wide methylation in 174 pairs of cord blood and placental samples. We present first a descriptive comparison of methylation levels in cord blood and placenta throughout the genome. We then performed locus-specific prediction of methylation in placenta based on methylation in cord blood using both linear regression and SVM prediction models. This comparison was conducted to determine whether methylation in cord blood can predict methylation in placenta, and to identify subsets of CpG sites and genomic contexts in which the prediction model performed better or worse than average.
Materials & methods
Subject & tissue collection
Cord blood and placental tissues were collected from 174 participants from the dual-site PRogramming of Intergenerational Stress Mechanisms study, a prospective pregnancy cohort of mother–child pairs originally designed to examine how perinatal stress influences respiratory health in children. Women were recruited from prenatal clinics during pregnancy (26.9 ± 8.1 weeks gestation) from Beth Israel Deaconess Medical Center in Boston (MA, USA) and the Icahn School of Medicine at Mount Sinai in New York (NY, USA) from March 2011 to August 2014. Eligibility criteria included: English- or Spanish-speaking; age ≥18 years at enrollment; and singleton pregnancy. Cord blood was collected before delivery of the placenta. Cord blood was separated into plasma and buffy coat by centrifugation and buffy coat was stored at -20°C. Placentas were sampled per a published protocol [11], immediately after birth. Each of four samples (∼1 cm3) was taken on the fetal side approximately 4 cm from the cord insertion site and approximately 1–1.5 cm below the fetal membrane to avoid membrane contamination. The decidua and fetal membranes were removed, the sample was rinsed in a cold phosphate-buffered saline bath, cut into smaller pieces (∼0.1 cm3) and placed into 1 ml of RNAlater™ RNA Stabilization Reagent (Qiagen) to allow for isolation of RNA in addition to DNA from the same samples. Samples in RNAlater were placed at -4°C for ≤24 h; excess RNAlater was then removed and samples were placed at -80°C until DNA extraction. DNA was isolated using the Gentra Puregene kit from Qiagen (MD, USA) and quantified using an Implen Nanophotometer Pearl (CA, USA). The origin of placental tissue from the fetal side of the organ was confirmed by the near-perfect agreement of placenta and cord blood samples in 64 genotyping probes used for identity verification – indicating no meaningful contamination with maternal DNA. Across these 64 genotyping probes the range of the Pearson correlation between cord blood and placenta for each of the 174 sample pairs was (0.99, 1). Procedures for PRogramming of Intergenerational Stress Mechanisms were approved by the Institutional Review Boards at the Brigham and Women’s Hospital and the Icahn School of Medicine at Mount Sinai. Beth Israel Deaconess Medical Center relied on Brigham and Women’s Hospital for review and oversight of the protocol. Written informed consent was obtained from all participants.
DNA methylation profiling
HumanMethylation450 BeadChips (Illumina, Inc., CA, USA) were used to interrogate 485,577 DNA methylation sites and to generate a measure of the methylation proportion at each site. Specifically, single-CpG-site methylation values were quantified after bisulfite conversion using fluorescence measures at site-specific probes, which was computed as the methylated intensity divided by the sum of both the methylated and unmethylated intensities. Methylation values ranged from zero (for a fully unmethylated CpG site) to one (for a fully methylated CpG site). As others have found that the differences between plates or chips are often the largest source of technical variation [12,13], all pairs of placenta and cord blood (from the same individual) were arrayed on the same chip with a randomized position (row and column).
Quality control & preprocessing
The presence of failed arrays or outliers was checked with detection p-values (all samples passed with detection p-values <0.05 in >99% of probes) and through visualization of principal components. Principal components plots and the analysis of five pairs of technical replicates that were arranged across chips and plates were further used to assess potential batch effects. Sample identity was checked via imputed sex and agreement of genotype with paired tissues. Data were preprocessed using background correction [14], dye bias and probe type adjustment [15]. BMIQ (Beta Mixture Quantile dilation) normalization strategy was applied to all probes to adjust the methylation values of Infinium II probes into a statistical distribution characteristic of Infinium I probes. The BMIQ function in the R package wateRmelon [16] was used.
Probe filtering
Linear and SVM prediction models were not performed on all 485,577 methylation probes. Filtered-out probes included 1217 probes with a detection p-value > 0.05 in >1% of the samples, 64 genotyping SNPs, 3089 CpH sites, 29,127 cross-reactive probes [17], 74,645 CpG sites with variants within ten base pairs that are common to Asian, American, African and European populations with a frequency of >1%, and 9371 probes on chromosome X or Y [18]. We performed three further steps to prevent unknown SNPs from driving DNA methylation prediction as these would be shared across tissues. First, we conducted an empirical check for additional probes under the influence of a SNP and excluded from both tissues 3992 CpG sites with a multimodal distribution of methylation (Dip test p-value < 0.05) in either cord blood or placental samples. Second, we dropped 5132 probes with >10% of extreme outliers (<25th percentile – 3IQR or >75th percentile + 3IQR) either in cord blood or placental samples and, third, we trimmed extreme outliers in both cord blood and placental samples. The resulting dataset for analysis included 358,940 probes. In order to assess the contribution of subject-level characteristics to the concordance of DNA methylation in these probes between cord blood and placenta, we used linear regression models to test whether within-person R2 values for different sample pairs were associated with gestational age, sex, race/ethnicity, education or collection site.
Statistical models for prediction
Linear prediction model
Let y
ij and x
ij be, respectively, the placental and cord blood methylation values referring to the j-th CpG site in the i-th participant. Let z
ij be the mean of the cord blood methylation values across all the nearby DNA methylation sites that are correlated to the j-th CpG site in the i-th subject (not including the j-th site), as defined below. The linear regression model for prediction of the j-th CpG site is yij = β0j + β1j xij + β2j zij + εij. In order to estimate the coefficients of this model and to assess its prediction accuracy, data were randomly divided into ten sets for cross-validation (cv) using a training and a testing set. The samples of the training set were used to fit the linear regression model and to compute the predictor coefficients’ estimates 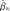 ,
,  and
and 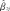 . For the i-th sample in the testing dataset, the predicted placental methylation value in the j-th CpG site is
. For the i-th sample in the testing dataset, the predicted placental methylation value in the j-th CpG site is 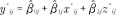 , where x*ij and z*ij are, respectively, the cord blood methylation value and the mean of the correlated nearby cord blood methylation sites from the sample being predicted. The assign.to.clusters function in the R package Aclust [19] was used to define the sets of neighboring CpG sites that are correlated with each other, using default parameters. This function implements a clustering method to discover methylation regions by clustering together adjacent probes within a genomic distance constraint according to their Spearman correlation between samples (within a single tissue). This covariate was added as an additional predictor, where available (33% of the analyzed probes), as a more stable measure of the regional methylation pattern in the surrogate tissue.
, where x*ij and z*ij are, respectively, the cord blood methylation value and the mean of the correlated nearby cord blood methylation sites from the sample being predicted. The assign.to.clusters function in the R package Aclust [19] was used to define the sets of neighboring CpG sites that are correlated with each other, using default parameters. This function implements a clustering method to discover methylation regions by clustering together adjacent probes within a genomic distance constraint according to their Spearman correlation between samples (within a single tissue). This covariate was added as an additional predictor, where available (33% of the analyzed probes), as a more stable measure of the regional methylation pattern in the surrogate tissue.
SVM prediction model
SVM is a supervised learning method used to analyze data and recognize patterns. SVM represents a powerful technique for both classification and regression purposes and has been widely used in many bioinformatic applications. In this analysis we used SVM for regression, introduced by Vapnik [10] in 1963. This statistical model is based on the computation of a linear regression function in a high dimensional feature space where the input data are mapped via a nonlinear function called the kernel function (for an introduction to SVMs see Chapter 12 of [20]). The main advantages of SVM over linear regression are that it can account for nonlinear relationships, it avoids overfitting, and it is robust to noise. The SVM model for prediction is constructed in a similar manner as the linear regression model. Let f(x,z) denote the SVM model, then, for the i-th sample in the testing data set, the predicted placental methylation value in the j-th CpG site is 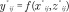 , where x*ij and z*ij are, respectively, the cord blood methylation value and the mean of the cord blood methylation values across the correlated sites from the sample being predicted. The train function in caret R package [21] was used, with parameter ‘method’ equal to svmRadial (SVMs with Radial Basis Function Kernel), σ = 1 and C = 1.
, where x*ij and z*ij are, respectively, the cord blood methylation value and the mean of the cord blood methylation values across the correlated sites from the sample being predicted. The train function in caret R package [21] was used, with parameter ‘method’ equal to svmRadial (SVMs with Radial Basis Function Kernel), σ = 1 and C = 1.
In order to better understand the relationship between these two tissues, we also ran our models in the reverse order – using placenta as a surrogate tissue to predict methylation in cord blood. For these models we used the same prediction strategies (linear model [LM] and SVM), and model formulation, including the mean of the correlated nearby sites where applicable (after running Aclust in the placenta) in the same set of probes as specified above.
Assessment of prediction accuracy
In order to compare the two regression procedures and assess their prediction accuracy we used k-fold cv, which is commonly employed in predictive modeling to use all of the data without overfitting. Specifically, we first randomly divided the samples into ten subsets or ‘folds’. At each of ten iterations, all the samples in a specific fold were removed (left out) and the remaining samples constituted a training dataset which was used to estimate the prediction models (linear and SVM). The left-out samples were then used as a testing data set. We applied the prediction model (linear and SVM) to obtain predicted values for the left-out samples. We used the square of the Pearson’s correlation coefficient (R2) and the root mean square error to estimate the accuracy of the predicted relative to the actual values. The steps above were repeated ten times, that is, once for each of the folds. We used the trainControl function in the caret R package [21] by setting the ‘method’ and ‘number’ parameters to cv and 10, respectively. The choice of the number of folds is arbitrary: a larger number is more computationally expensive and less biased, but can suffer from large variability, while a lower value is usually less computationally expensive and has less variance, but may be more biased. It is often reported that the optimal number of folds is between five and ten, because the statistical performance does not increase with larger values of folds, and averaging over fewer than ten splits remains computationally feasible (Chapter 7 of [20]). Although we are primarily interested in prediction, with a sample size of 174 we would expect a power of 0.8 to detect linear associations with an R2 as low as 0.04.
To understand whether prediction performance related to the genomic context of each probe, we stratified results by the Illumina annotation categories (version 1.2) [22] for promoters, CpG islands and enhancers, and compared the probes in each of these categories with the remainder of the analytic dataset. Because imprinted genes have a unique pattern of epigenetic control that is established in the germline, we also investigated prediction performance among a set of 620 probes in the analytic dataset that fell within imprinted regions previously described in leukocytes [23]. All data processing and analyses were conducted using R version 3.3.1 [24] and additional R packages as cited throughout.
Results
In this study we examined cord blood and placental samples from 174 participants. The average gestational age was 39.2 (range 34.6–41.6) weeks. Most of the participants self-reported white (38%) or black (including Haitian) (38%) race, and 13% reported Hispanic ethnicity.
Methylation patterns across cord blood & placenta
DNA methylation profiles were measured at single-CpG-site resolution for 485,577 DNA methylation sites on 174 pairs of cord blood and placental samples using Illumina HumanMethylation450 BeadChips. The preprocessing and filtering steps described in Methods reduced the number of sites for comparison to 358,940 CpGs across the 22 autosomal chromosomes. We first examined the distribution of cord blood and placental DNA methylation values across all 174 samples. For descriptive purposes, we labeled methylation levels >0.7 as hypermethylated and methylation levels <0.3 as hypomethylated. These thresholds are arbitrary but approximately capture the two large modes seen when plotting the density of the 450k array, as most probes are largely methylated or unmethylated. In cord blood, the majority of CpG sites were either hyper- or hypomethylated: methylation levels were >0.7 and <0.3 across all the samples in 41.54 and 40.59% of the CpG sites, respectively. Moreover, in almost half of the cord blood CpG sites (48.04%) methylation levels were ≥0.5 in all individuals. In placenta, methylation levels showed different distributions: 23.84% of the CpG sites had methylation >0.7, whereas 34.98% had methylation <0.3 in all individuals. Also, the percentage of CpG sites with methylation levels ≥0.5 across all subjects was lower in placenta (34.25%) than in cord blood. This greater proportion of intermediate methylation in human placenta has been previously reported [23]. Table 1 shows cord blood and placental DNA methylation distributions according to the functional categories of the Illumina annotation. CpG islands and promoters were the functional regions with the most consistently similar methylation levels between cord blood and placenta. 19.6% of the CpG sites had a difference in methylation >0.1 for all 174 samples. Cross-tissue differences in methylation were largely conserved across individuals: a CpG site with a methylation level higher in placenta than in cord blood in one of the participants generally had higher methylation levels in placenta than in cord blood in most other participants (Supplementary Figure 1). In addition, the magnitude of difference was similar across individuals. For each subject, we computed the square of the Pearson correlation coefficient (R2) between the two tissues, using all probes, and this ranged from 0.52 to 0.72 with a mean of 0.65 (Table 2). In order to assess the contribution of individual characteristics to the concordance of DNA methylation between cord blood and placenta, we used linear regression models to test whether within-person R2 values for different sample pairs were associated with gestational age, sex, race/ethnicity, education, or collection site, and we did not find any statistically significant associations.
Table 1. . Cord blood and placental DNA methylation distributions according to the Illumina annotation functional categories.
| Annotation category | n |
Hypomethylated status (β < 0.3) (%) |
Hypermethylated status (β > 0.7) (%) |
Methylated status (β ≥ 0.5) (%) |
|||
|---|---|---|---|---|---|---|---|
| Cord blood | Placenta | Cord blood | Placenta | Cord blood | Placenta | ||
| All probes |
358,940 |
40.6 |
35.0 |
41.5 |
23.8 |
48.0 |
34.3 |
| Island |
118,945 |
79.3 |
68.2 |
11.8 |
6.6 |
13.8 |
10.1 |
| Shelf |
32,537 |
7.0 |
5.8 |
72.4 |
46.5 |
82.2 |
62.3 |
| Shore |
86,626 |
42.0 |
35.2 |
33.8 |
19.8 |
41.4 |
30.2 |
| Other |
120,832 |
10.5 |
10.0 |
68.0 |
37.6 |
77.3 |
53.4 |
| Enhancer |
79,889 |
24.6 |
18.8 |
51.8 |
29.2 |
60.4 |
43.7 |
| Not enhancer |
279,051 |
45.2 |
39.6 |
38.6 |
22.3 |
44.5 |
31.5 |
| Promoter |
25,921 |
88.3 |
83.5 |
6.6 |
4.7 |
7.6 |
7.0 |
| Not promoter | 333,019 | 36.9 | 31.2 | 44.3 | 25.3 | 51.2 | 36.4 |
Table 2. . Mean and range of the overall correlation R2 from the same individual between cord blood and placental methylation values (first line) and between placental and predicted placental methylation values obtained with both support vector machine model (second line) and linear regression model (third line).
| Tissue pair |
All probes |
|
|---|---|---|
| Mean R2 | Range | |
| Cord blood – placenta |
0.65 |
0.52–0.72 |
| Placenta – SVM predicted placenta |
0.98 |
0.96–0.99 |
| Placenta – LM predicted placenta | 0.98 | 0.96–0.99 |
R2 is the square of the Pearson correlation coefficient.
LM: Linear regression model; SVM: Support vector machine.
Cross-tissue prediction of methylation levels
We explored methylation prediction in placenta based on methylation in cord blood by using linear regression and SVM prediction models. We applied tenfold cv to assess prediction accuracy and avoid overfitting. At most CpG sites, both models gave inaccurate predictions: 75% of CpG sites had an R2 between measured and predicted placental methylation values lower than 0.25 (Figure 1). Also, we found no substantial differences between the performance of predictions generated by the linear model and by the SVM model (R2 and root mean square error distributions were very similar between the two models). The averages of the R2 among all 358,940 linear and SVM prediction models were equal to 0.17 and 0.15, respectively.
Figure 1. . Prediction accuracy of both linear models and support vector machine models was evaluated by using R2 and root mean square error between measured and predicted methylation values.

For both models, the distribution of R2 (A) and root mean square error (B) in all CpG sites are shown. The filled square and triangle represent the mean. LM: Linear model; RMSE: Root mean square error; SVM: Support vector machine.
We also calculated from each pair of samples the R2 between placental and SVM/LM-predicted placental methylation values among all the CpG sites. The average R2 from the same individual increased from 0.65 (between cord blood and placental methylation values) to 0.98 (between placental and SVM/LM-predicted placental methylation values) (Table 2).
In order to compare different genomic contexts, we stratified the distribution of the site-specific correlations by Illumina annotation category (Figure 2). We found that CpG islands and promoters, but not enhancers, were the annotation subsets where the R2 between measured and predicted placental methylation values achieved higher values in both the linear and SVM models. Among the 620 included probes that fell in imprinted regions [23], predictions were slightly worse than the remainder of probes for both LM and SVM models (mean R2 of 0.159 vs 0.174 for linear models, t-test p-value = 0.0004).
Figure 2. . R2 and root mean square error distributions stratified by annotation category in both linear models and support vector machine models.

The categories were designated using Illumina annotation. The filled square and triangle represent the mean. LM: Linear regression model; RMSE: Root mean square error; SVM: Support vector machine.
We next explored those CpG sites where the SVM prediction model achieved high or low R2 values in the tenfold cv. The highest values of R2 (≈0.94) were obtained in CpG sites where cord blood and placental methylation distributions were very similar to each other (Figure 3). In contrast, in the CpG sites that showed different methylation levels between the two tissues, the correlation between measured and SVM-predicted methylation levels computed by tenfold cv were low. For example, Figure 3B shows three CpG sites where R2 is almost equal to zero: cg14182041 and cg12255501 are hypermethylated while cg18824446 is hypomethylated in cord blood but not in placenta. Overall, we found more CpG sites hyper- or hypomethylated in cord blood but not in placenta than the opposite situation (Supplementary Figure 2). The lighter diagonals in Figure 4A & B show that the SVM prediction model performed better when the means and the standard deviations of cord blood and placental methylation levels across all samples were similar to each other. In particular, the correlation between measured and SVM-predicted methylation values achieved the highest values in the CpG sites where there was a higher standard deviation in methylation across all samples (Supplementary Figure 3). We found 714 CpG sites with R2 ≥0.75 in both linear and SVM models. The proportion of these probes across autosomal chromosomes ranged from 0.1% (in chromosome 18) to 0.3% (in chromosome 19), but did not differ significantly (χ2 test, p = 0.74). We performed a χ2 test for the equality of proportions of the Illumina functional categories in the subset of probes where both models achieved good predictions versus the remainder. We found that good prediction CpG sites were enriched in shelves (14 vs 9%, p-value = 2.10e-07) and promoters (13 vs 7%, p = 5.48e-12), but depleted among CpG sites in shores (17 vs 24%, p = 3.02e-05) and enhancers (11 vs 22%, p = 8.21e-13). However, these 714 good prediction probes did not share any pathways associated with biologic processes in a Gene Ontology enrichment analysis (all False Discovery Rate corrected p-values near 1) using the gometh function of the missMethyl package [25]. We also performed a sensitivity analysis using the methylation data before any preprocessing: 407 (57%) of these 714 CpG sites still presented an R2 ≥0.75 in both linear and SVM models. The list of the 714 CpG sites with R2 ≥0.75 in both models is provided in the Supplementary Material along with their relevant annotation and descriptive statistics from our results.
Figure 3. . Scatter plots between measured placental methylation values (y-axis) and both support vector machine-predicted placenta (blue) and cord blood (red) methylation values (x-axis) in three CpG sites where the R2 achieves, respectively, high (A) and low (B) values.

Above each plot is the corresponding CpG site’s Illumina ID and the value of the R2 between measured and predicted placental methylation values obtained by tenfold cross-validation.
SVM: Support vector machine.
Figure 4. . R2 between measured and support vector machine-predicted placental methylation levels in terms of the average (A) and the standard deviation (B) of both cord blood and placental methylation values across all subjects.

Each hexagon bin’s color corresponds to the mean of the R2 of all the CpG sites contained in that bin and the overlaid red contour lines show the density of the set of analyzed sites. SVM: Support vector machine.
As a sensitivity analysis, we used the function estimateCellCounts in the R package minfi [26,27] to estimate the cell type proportions in the cord blood samples and reran both LM and SVM approaches using five out of six of the estimated proportions (which sum to one) as covariates. Adjusting for cell type proportions did not improve the LM predictions (the average [range] of the R2 across all 358,940 models was 0.17 [0.00–0.94]) and substantially worsened the SVM predictions (the average [range] of the R2 across all 358,940 models was 0.08 [0.00–0.70]).
As an additional exploration of our data, we also reversed our question of interest and performed prediction of cord blood methylation using the placenta with the same model formulation in the same set of probes. We found overall consistency in the performance of the reversed models with only 736 probes meeting the criteria for good prediction, of which 603 (82%) were among the 714 good prediction CpGs for estimating placental methylation.
Discussion
In this study we used statistical models to predict locus-specific DNA methylation levels in placenta using cord blood methylation. In particular, we built a linear prediction model for each of the 358,940 CpG sites we considered. In each site, prediction accuracy was assessed with tenfold cv by computing the R2 between measured and predicted methylation values across 174 subjects. The average R2 among all 358,940 linear prediction models, which can be considered an overall estimate of linear prediction accuracy, was equal to 0.17. We repeated the same steps using a SVM prediction model: the average R2 among all 358,940 SVM models was equal to 0.15. These results suggest that, overall, both modeling approaches perform poorly in predicting locus-specific methylation levels between the two tissues.
Our analysis attempted to replicate a previous study that contrasted linear and SVM models to predict locus-specific methylation in a target tissue based on methylation in a surrogate tissue [9]. In each pair of tissues, the overall (array-wise) R2 from the same individual between surrogate and target methylation values was always lower than the R2 from the same individual between measured and predicted methylation values (R2 increases from 0.38 between tissues to 0.89 for peripheral blood leukocytes [PBL]-to-artery prediction; from 0.39 to 0.95 for PBL-to-atrium; and from 0.81 to 0.98 for lymphoblastoid cell line-to-PBL). Consistent with their results, in our study, the overall array-wise correlation R2 from the same individual increased from 0.65 (between cord blood and placental methylation values) to 0.98 (between placental and SVM/LM-predicted placental methylation values) (Table 2). However, the summary of the R2 of the entire array measured on the same individual (e.g., measured vs predicted methylation) is misleading and does not indicate reasonable overall predictions for two reasons. The first reason the array-wide correlation is misleading is because most probes take values close to 0 or 1 and this bimodal array-wide distribution is highly influential on the correlation as a summary measure (whereas individual probes are more normally distributed across individuals). More importantly, as shown in Figure 3B, at many CpG sites both models simply shifted cord blood methylation values toward the mean of placental methylation values. This change in the intercept improves the within-person comparison while the correlation between measured and predicted placental methylation values remained very low at the probe level. Therefore, also considering that the standard deviation of the placental methylation was very small in the majority of sites (Figure 4B), it is not surprising that the array-wide correlation from the same individual between measured and predicted placental methylation values is much higher than the one between cord blood and placental methylation values. For example, at the CpG sites reported in Panel B of Figure 3 (Illumina IDs: cg14182041, cg18824446 and cg12255501), although the overall correlation R2 from the same individual increased from 0.24 between cord blood and placental methylation values to 0.79 between placental and LM-predicted placental methylation values and to 0.78 between placental and SVM-predicted placental methylation values, the average correlation R2 among the three CpG sites between measured and predicted placental methylation values was very low in both linear (0.005) and SVM (0.004) models.
We did find that probe-level methylation prediction performed better in CpG islands and promoters than in enhancers. A previous study showed that most CpG sites in CpG islands are hypomethylated across the genome [28], perhaps suggesting that our better performance in these regions is related to their biologic role and stable methylation, while higher variability of methylation across tissues in enhancers has also been reported [29]. By contrast, we report that the prediction was slightly worse in probes within imprinted regions, which we had anticipated might have better agreement across tissues due to the shared origins of epigenetic control in these regions originating in the germline.
Even though we did not expect all probes to be good surrogates for predicting between tissues (e.g. those with low overall variation), we were surprised by the low total number of probes that we found that achieved R2 ≥0.75 in both models (714 probes, listed in supplemental material). In these CpG sites, mean and standard deviation of methylation levels across all participants were very similar between cord blood and placenta. This result is consistent with Figure 4 and suggests that greater similarity between cord blood and placental methylation level distributions leads to better locus-specific methylation prediction. Lastly, we found that this group of probes was depleted of CpG sites in shores and enhancers, but enriched in CpG sites in shelves and promoters. These results are consistent with the overall distribution of the R2 from predictions across the Illumina annotation functional categories (Figure 2). Although we did not have methylation data from the mothers, we might anticipate that the inclusion methylation from a relevant surrogate tissue in the mother (e.g., peripheral blood) would also improve prediction – particularly among probes under genetic control. Because the methylome of the human placenta is known to have unique characteristics [23], it is unclear whether the results we report here on probe-specific prediction modeling would be improved if comparing cord blood with a more similar tissue. Nonetheless, we anticipate that our caution to avoid overinterpretation of the within-subject correlation as an indicator of prediction at the probe level remains generally applicable.
Conclusion
In this study we used both linear and SVM models to predict locus-specific DNA methylation levels in placenta using cord blood methylation in 174 pairs of an epigenome-wide array. Overall, both linear and SVM models gave poor predictions across these tissues: 75% of CpG sites had an R2 between measured and predicted placental methylation values lower than 0.25. CpG islands and gene promoters, but not enhancers, were enriched genomic contexts in the small subset of probes where the R2 between measured and predicted placental methylation levels achieved higher values. We found that the use of the array-wide correlation of predictions versus measured values was misleading because both models simply shifted the cord blood methylation values toward the mean of placental methylation while the probe-level predictions remained poor. While there remains a great interest in using surrogate tissues in epigenetics, care is needed in summarizing the performance of prediction methods in this challenging domain.
Executive summary.
In the present study we compared predictive modeling approaches to estimate methylation in placenta based on methylation in cord blood.
We performed locus-specific methylation prediction using both linear regression and support vector machine models with 174 matched pairs of 450k arrays.
At most CpG sites, both approaches gave poor predictions: 75% of CpG sites had an R2 between measured and predicted placental methylation values lower than 0.25.
We found that the use of the array-wide correlation of predictions versus measured values was misleading and did not indicate reasonable overall predictions.
CpG islands and gene promoters, but not enhancers, were the enriched genomic contexts where the correlation between measured and predicted placental methylation levels achieved higher values.
We provide a list of 714 CpG sites where both models achieved an R2 ≥ 0.75 and thus cord blood predicts placental methylation.
The present study indicates the need for caution in interpreting cross-tissue predictions. Few methylation sites can be predicted between cord blood and placenta.
Supplementary Material
Acknowledgements
The authors thank Alison Brown at the Partners HealthCare Center for Personalized Genetic Medicine Genotyping Facility for running the Illumina Arrays.
Footnotes
Financial & competing interests disclosure
This research was supported by NIH grants R01 HL095606, R01 HL114396, P30 ES023515, R01 ES021357 and R01 NR013945. AC Just was supported by grant R00 ES023450. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
References
- 1.Barrero MJ, Boue S, Izpisua Belmonte JC. Epigenetic mechanisms that regulate cell identity. Cell Stem Cell. 2010;7(5):565–570. doi: 10.1016/j.stem.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 2.Cedar H, Bergman Y. Programming of DNA methylation patterns. Annu. Rev. Biochem. 2012;81:97–117. doi: 10.1146/annurev-biochem-052610-091920. [DOI] [PubMed] [Google Scholar]
- 3.Kiefer JC. Epigenetics in development. Dev. Dyn. 2007;236(4):1144–1156. doi: 10.1002/dvdy.21094. [DOI] [PubMed] [Google Scholar]
- 4.Barault L, Ellsworth RE, Harris HR, Valente AL, Shriver CD, Michels KB. Leukocyte DNA as surrogate for the evaluation of imprinted Loci methylation in mammary tissue DNA. PLoS ONE. 2013;8(2):e55896. doi: 10.1371/journal.pone.0055896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ursini G, Bollati V, Fazio L, et al. Stress-related methylation of the catechol-O-methyltransferase Val 158 allele predicts human prefrontal cognition and activity. J. Neurosci. 2011;31(18):6692–6698. doi: 10.1523/JNEUROSCI.6631-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Byun HM, Siegmund KD, Pan F, et al. Epigenetic profiling of somatic tissues from human autopsy specimens identifies tissue- and individual-specific DNA methylation patterns. Hum. Mol. Genet. 2009;18(24):4808–4817. doi: 10.1093/hmg/ddp445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caliskan M, Cusanovich DA, Ober C, Gilad Y. The effects of EBV transformation on gene expression levels and methylation profiles. Hum. Mol. Genet. 2011;20(8):1643–1652. doi: 10.1093/hmg/ddr041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fan S, Zhang X. CpG island methylation pattern in different human tissues and its correlation with gene expression. Biochem. Biophys. Res. Commun. 2009;383(4):421–425. doi: 10.1016/j.bbrc.2009.04.023. [DOI] [PubMed] [Google Scholar]
- 9.Ma B, Wilker EH, Willis-Owen SA, et al. Predicting DNA methylation level across human tissues. Nucleic Acids Res. 2014;42(6):3515–3528. doi: 10.1093/nar/gkt1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vapnik VN. Statistical Learning Theory. Wiley; New York: 1998. [Google Scholar]
- 11.Janssen BG, Byun HM, Gyselaers W, Lefebvre W, Baccarelli AA, Nawrot TS. Placental mitochondrial methylation and exposure to airborne particulate matter in the early life environment: an ENVIRONAGE birth cohort study. Epigenetics. 2015;10(6):536–544. doi: 10.1080/15592294.2015.1048412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fortin JP, Labbe A, Lemire M, et al. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014;15(12):503. doi: 10.1186/s13059-014-0503-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buhule OD, Minster RL, Hawley NL, et al. Stratified randomization controls better for batch effects in 450K methylation analysis: a cautionary tale. Front. Genet. 2014;5:354. doi: 10.3389/fgene.2014.00354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Triche TJ, Jr, Weisenberger DJ, Van Den Berg D, Laird PW, Siegmund KD. Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res. 2013;41(7):e90. doi: 10.1093/nar/gkt090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Teschendorff AE, Marabita F, Lechner M, et al. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 2013;29(2):189–196. doi: 10.1093/bioinformatics/bts680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pidsley R, Cc YW, Volta M, Lunnon K, Mill J, Schalkwyk LC. A data-driven approach to preprocessing Illumina 450k methylation array data. BMC Genomics. 2013;14:293. doi: 10.1186/1471-2164-14-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen YA, Lemire M, Choufani S, et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013;8(2):203–209. doi: 10.4161/epi.23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Butcher L. Illumina450ProbeVariants.db: Annotation Package combining variant data from 1000 Genomes Project for Illumina HumanMethylation450 Bead Chip probes. R package version 1.6.0. 2013 https://bioc.ism.ac.jp/packages/3.2/data/experiment/html/Illumina450ProbeVariants.db.html [Google Scholar]
- 19.Sofer T, Schifano ED, Hoppin JA, Hou L, Baccarelli AA. A-clustering: a novel method for the detection of co-regulated methylation regions, and regions associated with exposure. Bioinformatics. 2013;29(22):2884–2891. doi: 10.1093/bioinformatics/btt498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hastie TT, Friedman J. The Elements of Statistical Learning. Data Mining, Inference, and Prediction (2nd Edition). Springer-Verlag; NY, USA: 2009. [Google Scholar]
- 21.Kuhn M. caret: Classification and Regression Training. R package version 6.0–71. https://CRAN.R-project.org/package=caret [Google Scholar]
- 22.Illumina. http://support.illumina.com/array/array_kits/infinium_humanmethylation450_beadchip_kit/downloads.html
- 23.Court F, Tayama C, Romanelli V, et al. Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of human imprinting and suggests a germline methylation-independent mechanism of establishment. Genome Res. 2014;24(4):554–569. doi: 10.1101/gr.164913.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.R Core Team. R: a language and environment for statistical computing. www.R-project.org/
- 25.Phipson B, Maksimovic J, Oshlack A. missMethyl: an R package for analyzing data from Illumina’s HumanMethylation450 platform. Bioinformatics. 2016;32(2):286–288. doi: 10.1093/bioinformatics/btv560. [DOI] [PubMed] [Google Scholar]
- 26.Aryee MJ, Jaffe AE, Corrada-Bravo H, et al. Minfi: a flexible and comprehensive bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30(10):1363–1369. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reinius LE, Acevedo N, Joerink M, et al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS ONE. 2012;7(7):e41361. doi: 10.1371/journal.pone.0041361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012;13(7):484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 29.Hwang W, Oliver VF, Merbs SL, Zhu H, Qian J. Prediction of promoters and enhancers using multiple DNA methylation-associated features. BMC Genomics. 2015;16(Suppl. 7):S11. doi: 10.1186/1471-2164-16-S7-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.