

Abstract
Inducible dimers are powerful tools for controlling biological processes through co-localizing signaling molecules. To be effective, an inducible system should have dissociation constants in the “off” and “on” state that bracket the concentrations of the molecules that are being controlled. Here, we reengineer the light inducible dimer, iLID, to better control proteins present at high effective concentrations (5–100 μM). The new variant of the switch displays a 42-fold change in binding affinity when activated with blue light (from 3 ± 2 μM to 125 ± 40 μM), and allows for light activated co-localization of transmembrane proteins in neurons, where a higher affinity switch (0.8 μM to 47 ;M) was less effective because more co-localization was seen in the dark. Additionally, we lengthened the reversion half-life of the photoswitch. This expanded suite of light induced dimers increases the variety of cellular pathways that can be targeted with light.
Keywords: iLID, Neurochemistry, Optogenetics, Protein Engineering, Lov2
Graphical Abstract
Optogenetics provides temporal and subcellular spatial control over biochemical pathways in living systems. In particular, genetically encoded light inducible dimers have emerged as a tool for controlling signaling. In the most common approach, one component of the dimer is fused to a protein that directs localization and the other component to a protein of interest, imparting control of localization and activity of the target protein with light. This strategy has been used to control transcription, GTPase protein signaling, epigenetic modifications, and nuclear localization 1–6. Ideally, inducible dimers would have no binding affinity in the dark, and very strong binding affinity in the light. However, we recently characterized the binding affinities of an established set of light inducible dimers and determined that binding is not typically all or none7. For instance, one version of the improved Light Inducible Dimer (iLID)1 system engineered by our lab, iLID/SspB_nano, has a dissociation constant for dimerization of 132 nM in the light and 4700 nM in the dark. This indicates that this switch will be most effective where the component proteins are functional at high nanomolar concentrations. However, proteins with functional ranges in mid-micromolar or low nanomolar are incompatible with this switch. At high concentrations it will dimerize in the absence of light and at low concentrations it will remain monomeric, even when activated. In many cases, protein concentrations in a cell or organism are often not known, highlighting the need for a suite of light inducible tools. In the case of proteins found in constrained locales, like the plasma or mitochondrial membranes, avidity effects and high effective concentrations require low affinity switches to properly modulate dimerization. To address these issues, we have engineered a panel of light-inducible switches that function over a range of affinities that can be empirically tested in localization experiments. Here, we expand the panel by creating a new variant of the iLID system that functions at higher protein concentrations, and show that this re-engineered system is more effective at regulating the localization of trans-membrane proteins in neuronal cells.
iLID is derived from the LOV2 domain of Avena sativa Phototropin (AsLOV2). When excited with blue light, the flavin mononucleotide (FMN) cofactor in iLID forms a metastable covalent bond with Cys450, leading to a conformational change exposing a peptide sequence (ssrA) that binds to the bacterial SspB protein8,9. SspB_nano and SspB_micro are engineered variants of the SspB protein. Through direct comparison to other light inducible dimers we have found that the iLID switches have many advantages including a large dynamic range of binding, tunable affinities, strictly monomeric components, easy to use in many organisms, and have no restrictions on fusion location7. To further expand iLID system’s capabilities we focused on two aspects: (1) utility when effective proteins concentrations are on the order of 100 micromolar and (2) less frequent requirement of light stimulation. The rate at which AsLOV2, and hence iLID, reverts to its dark state following photoactivation is relatively fast (T½ = ~20 s) which means light must be applied frequently in order to sustain the induced interaction. It has been shown that excess light can have undesired experimental effects on the cell or animal being studied10. Slowing the reversion rate will allow iLID to be kept in the lit state longer, reducing necessary light, and improving experimental conditions.
Experimental Procedures
Molecular cloning
pQE80L-sLID, pLL7.0-Venus-sLID-CAAX, and pLL7.0-Venus-sLID-Mito were generated by quick change site directed mutagenesis of the corresponding iLID constructs using the following primers: CTGGAACGGATCGAGAAACTGTTCGTGATTACTGATCCG; CGGATCAGTAATCACGAACAGTTTCTCGATCCGTTCCAG. pQE80L-SspB_milli and pLL7.0-tgRFPt-SspB_milli were generated by quick change site directed mutagenesis of the corresponding SspB_micro constructs using the following primers: CTGTCTGCAAGTGTGACCGGCAACCT; CAGGTTGCCGGTCACACTTGCAGACAG. GluA1-mCh-SspBnano, GluA1-mCh-SspBmicro, and GluA1-mCh-SspBmilli were generated by PCR amplifying SspBnano (addgene #60415), SspBmicro (addgene #60416), and SspBmilli, respectively, and cloning into a custom GluA1-mCh mammalian expression vector with using the chick beta-actin promoter to drive expression. Homer1c-YFP-iLID and Homer1c-YFP-sLID were generated by PCR amplifying iLID and sLID, respectively, and cloning into Homer1c-YFP. The cDNA for Homer1c-YFP was a gift from Shigeo Okabe (University of Tokyo). GluA1 cDNA was a gift from Michael Ehlers (Pfizer, Inc). Constructs were generated using standard cloning techniques in either a custom chick beta-actin promoter (pCAG; for GluA1-mCh-SspB constructs) or cytomegalovirus promoter (pCMV; for iLID constructs).
Protein expression and purification
Proteins were expressed as described in Hallett et. al. (2015). Briefly, BL21(DE3) cells were transformed by heat shock with each expression vector and used to inoculate 1.5 L of LB media. Cells were grown at 37°C to OD 0.6 and induced with 333mM IPTG at which point proteins were expressed for 16 hours at 18°C. Cells were then lysed and pelleted and the proteins of interest were purified from the supernatant by sequential HisTrap affinity purification and size exclusion chromatography on a Superdex 75 column (GE). All proteins were stored, quantified, and characterized in PBS buffer (10 mM dibasic sodium phosphate, 1.8 mM monobasic potassium phosphate, 137 mM NaCl, 2.7 mM KCl) + 5mM BME.
Fluorescent polarization Binding Assay
Fluorescent polarization binding assays were performed as described in Hallett et. al. (2015). Briefly, Fluorescence polarization measurements were recorded using a Jobin Yvon Horiba FluoroMax3 fluorescence spectrometer. All binding assays were performed in PBS buffer in either a 1 cm or 3 mm quartz cuvette at 25 °C. Polarization of a TAMRA labeled peptide was measured with an excitation of 555 nm and 584 nm emission. Initial affinities of the labelled peptides were measured through direct binding titrations to each SspB. For the competition assay, at each titration point, the sample chamber was illuminated with 6.0 mW cm−2 blue light using a collimated blue LED (ThorLabs). A lit state time point was taken immediately after removal of the blue light. For iLID a dark state measurement was made 5 min later while for sLID a dark state measurement was made 15 min later. Starting peptide concentrations were 25 nM.
Absorption Recovery after Activation
Absorption recovery after activation was performed as in Hallett et. al. (2015) Briefly, excited state recovery times were measured using a Cary 50 UV-Visible Spectrophotometer. Using a blue light collimated LED (ThorLabs), samples were irradiated (6.0 mW cm−2) for 30 seconds and absorbance at 450 nm was recorded until recovery.
Mammalian Cell Culture and Transfection
Mouse IA32 fibroblasts were cultured in DMEM supplemented with 10% (vol/vol) FBS (HyClone), 100 U/ml penicillin, 100 μg/mL streptomycin, and 292 μg/mL L-glutamine. Cells were cultured at a constant 37 °C and 5% (vol/vol) CO2. Transfections were performed in 6 well or 35 mm cell culture dishes using 1 μg total DNA at 1:1 ratio. NanoJuice (EMD Millipore) transfection reagent was used as recommended by the manufacturer.
Mammalian cell localization microscopy
Experiments were performed according to the methods found in Guntas et al. Briefly, cells were co-transfected with vectors encoding the sequence of each switch piece at a 1:1 stochiometry. 24 hr later tranfected cells were trypsonized and transferred to 10 ug/ml fibronectin coated 3.5 cm MatTek glass bottom dishes. 24 – 48 hr later cells were imaged and photo-activated with an Olympus FV1000 confocal microscope equipped with a 1.30 N.A. 40× oil immersion objective. The Fluoview software Time Controller was used to produce a timeline of image acquisition and photo-activation using the same standard parameters found in Guntas et al. Standard settings were maintained for each image to ensure similar protein expression levels which fell within the dynamic range of the instrument under these settings. Activation parameters were also kept constant between samples. In short laser power was set at 1% for the 488nm line. For whole cell activation the entire field of view was activated in a 512×512 pixel grid with a 2us/pixel dwell time and repeated 5x. before the next image was acquired. For ROI activation a 60×60 pixel grid was activated with a 8us/pixel dwell time and repeated 10× before the next image was taken.
Image analysis and quantification
All images were analyzed using FIJI software. Spot localization was quantified according to Guntas et al. Briefly the tgRFPt fluorescence intensity was measured within the activated ROI and an initial intensity and size matched area outside the activated ROI. A ratio of fluorescence intensity inside : outside the ROI was analyzed throughout time. The values that correspond to the period of activation were fit to the equation Y= 1 + Ymax*(1-exp(-K*X)). The values that correspond to the period of reversion were normalized to the maximum values and fit to the equation Y=(Y0 - Plateau)*exp(-K*X) + Plateau. Whole cell activation was quantified according to Hallett et. al. (2015) In short, computationally defined mitochondrial and cytoplasmic ROIs were determined based on the images captured. The ROIs were used to measure the tRFPt fluorescence intensity in each area over time. The ration of mitochondrial to cytoplasmic intensity was plotted though time and curves were fit to the values during the activation and reversion periods using the equations Y=S+Ymax*(1-exp(-K*X)) & Y=(Y0 - Plateau)*exp(-K*X) + Plateau respectively. The fold change was determined by (S+Ymax)/S. All curve fittings were performed using Prism (GraphPad) software.
Cultured Neuron Preparation and Transfection
Primary hippocampal neurons were prepared from neonatal Sprague Dawley rats. Hippocampi were dissected from the brains of postnatal day 0–2 rats and dissociated by papain digestion. Neurons were plated at 100,000 cells/ml in MEM, 10% FBS (Hyclone) containing penicillin/streptomycin on 18 mm poly-D lysine coated glass coverslips. After 1 d the media was replaced with Neurobasal-A supplemented with B27 (Invitrogen). The neurons were then fed with Neurobasal-A, B27, and mitotic inhibitors (uridine + fluorodeoxyuridine [Ur+FUdR]) by replacing half the media on day 4 or 5 and then twice weekly. Neurons were transfected between 16 and 18 days in vitro (DIV) with 1.0 μg of plasmid/1.5 μl of Lipofectamine according to the manufacturer’s protocol and imaged on DIV 18–21. All animal procedures were performed in accordance with the University of Colorado School of Medicine guidelines.
Neuronal live-cell Imaging
Live cell imaging of dissociated hippocampal neurons was carried out at 32°C on an Olympus IX71 equipped with a spinning disc scan head (Yokogawa) with a 60× NA1.4 objective. Excitation illumination was delivered from an AOTF controlled laser launch (Andor) and images were collected on a 1024×1024 pixel Andor iXon EM-CCD camera. Data acquisition and analysis were performed with Metamorph (Molecular Devices) and ImageJ (National Institutes of Health). Some images were smoothed (averaging over 3 × 3 pixels) using ImageJ for display, but never before quantification. We simultaneously activated iLID and imaged GFP with a fiber coupled 488 nm laser through the microscope objective (typical photoexcitation conditions were 25% laser power from a 50 mW 488 nm laser, 25–50 ms exposure time).
Results and Discussion
To create SspB_milli, a switch that functions at higher protein concentrations, we introduced a mutation, A58V, in the ssrA binding site of SspB_micro, which was predicted to form a steric clash. Using a competitive fluorescence polarization assay, the dissociation constant for lit state binding was measured to be 56 ± 6 μM. In the dark, non-specific interactions interfered with our measurements when concentrations were raised above 1 mM, but we were able to estimate that dark state affinities were at least 25 fold weaker than lit state affinities (Figure 1A, C).
Figure 1.

In vitro characterization of iLID SspB_milli and sLID. A) Left panel: Binding of iLID to SspB SspB_milli under blue light (blue) and in dark (black) measured by fluorescence polarization. Right panel: Binding of sLID to SspB_nano (circles) or SspB_milli (squares) under blue light (blue) and in dark (black). B) Reversion of FMN-LOV2 covalent adduct is measured by recovery of absorbance at 450nm after 30 sec of blue light activation for WT iLID (blue) and sLID (green). C) Schematic displaying dynamic range of each characterized photoswitch. Blue circles denote the lit state affinity, dark circles mark the dark state affinity, and the connecting line represents change due to blue light.
To generate a slower reversion time variant of iLID (sLID (slow Light Inducible Dimer)), we used a previously discovered mutation in the AsLOV2 domain, N414L, which increases the time of the metastable bond formed between Cys450 and the FMN cofactor in the lit state11. To determine the kinetics of sLID, we monitored recovery of FMN absorbance at 450 nm after blue light photoactivation and measured the lit state half-life of sLID to be 214 ± 4 s, 12-fold slower than the dark-state recovery time for iLID (Figure 1B).
To determine if SspB affinity for sLID is altered due to the N414L mutation, we measured the affinity of sLID for SspB_nano, SspB_micro, and SspB_milli. Surprisingly, in all cases the mutation increased binding affinity in both the light and the dark; SspB_nano lit: 7 ± 2 nM, dark: 310 ±90 nM, SspB_micro lit: 110 ± 10 nM, dark: 2.5 ± 0.4 μM and SspB_milli lit: 3 ± 2 μM, dark 125 ± 40 μM (Figure 1A and C, Figure S1). Strikingly, the 7 nM affinity for SspB_nano with sLID in the lit state is tighter than the affinity between the free ssrA peptide and SspB_nano, suggesting that N414L may result in additional favorable interactions between the LOV2 domain and SspB_nano. This result was unanticipated, but potentially useful, as it expands the range of affinities over which the iLID variants function.
Next, we tested the performance of sLID and SspB_milli in protein localization assays in mouse fibroblasts. By expressing SspB_milli fused to tagRFPt (tRFPt-SspB_milli) and a Venus labeled iLID fused to either a plasma membrane or a mitochondrial targeting sequence (Venus-iLID-CAAX, Venus-iLID-Mito) we controlled and monitored the localization of SspB_milli before and after irradiation with blue light. With the Venus-iLID-CAAX localization, we activated a specific region of interest (ROI) and over time compared the average tRFPt fluorescence intensity in that ROI to similar ROI elsewhere in the cell. When using iLID/SspB_milli, we observed weaker recruitment of tRFPt-SspB_milli to the activated ROI when compared to SspB_nano or SspB_micro (SspB_nano ~6.4x, SspB_micro ~4.9x, SspB_milli ~1.5x) (Figure 2A and B). This result suggests the lit state dissociation constant between SspB_milli and iLID (56 μM) is greater than the effective concentration of the molecules. However, when using SspB_milli with sLID we observed effective recruitment, consistent with the stronger affinity between sLID and SspB_milli in the lit state (3 μM). Interestingly, sLID combined with SspB_micro produces only ~1.8× change in localization (Figure 2A and B). In this case, dynamic range was limited because much of the protein was co-localized in the dark, consistent with the relatively high affinity between sLID and SspB_micro in the dark (2.5 μM).
Figure 2.

Membrane localization A) Representative images of membrane recruitment and reversion analyzed in B and C. Cells transfected with each membrane bound switch pair were imaged and activated by confocal microscopy. Blue square marks the ROI activated with blue light. B) Ratio of RFP fluorescence intensity inside the activated ROI to outside the ROI during activation. C) Ratio of RFP fluorescence intensity inside the activated ROI to outside the ROI after activation. D and E) Venus-iLID-CAAX/RFP-SspB_micro or Venus-sLID-CAAX/SspB_milli were activated with 15, 30, or 60 s between activations. Plots represent the normalized ratio of RFP fluorescence intensity inside the activated ROI to outside the ROI during activation. F) The maximum normalized fluorescence intensity ratio from D and E determined by fitting the curves. (Data for Nano and Micro has been previously published 7.)
In the mitochondrial localization assays, the entire field of view was irradiated with blue light and the average mitochondrial tRFPt fluorescence intensity was compared to the average cytoplasmic fluorescence intensity. As with the membrane localization assay we found that iLID/SspB_milli showed a smaller dynamic range (~1.9x) than SspB_nano (5.4x) and SspB_micro (5.2x), as only a small amount of the protein was recruited to the mitochondria (Figure 3A and B). Similar to the membrane localization experiments, the largest dynamic range observed for sLID was with SspB_milli (2.2x). sLID/SspB_micro experiments showed that high dark state affinity localized SspB_micro to the mitochondria pre-activation (Figure 3A and B).
Figure 3.

Mitochondrial localization A) Representative images of mitochondrial recruitment and reversion analyzed in B. Cells transfected with each mitochondrial bound switch pair were imaged and activated by confocal microscopy. B) Ratio of mitochondrial to cytoplasmic RFP fluorescence intensity for each switch pair. Fold change in localization is represented by the bars to the right. (Data for Nano and Micro has been previously published 7.)
In the mitochondrial and plasma membrane localization experiments we also measured the kinetics of reversion to equilibrium after the light was turned off. As expected, dark state recovery times (t1/2 ) were slower with sLID than with iLID in both experiments; plasma membrane experiments were ~60 and ~100 s respectively for iLID and sLID (Figure 2C), and mitochondrial experiments were ~30 and ~430 s (Figure 3B; S. Table 1). Since the in vitro and in vivo reversion half-lives of sLID were longer than iLID we hypothesized that sLID would maintain maximum recruitment of SspB_milli to the membrane with less frequent light stimulation. To test this, we compared Venus-iLID-CAAX/tRFPt-SspB_micro with Venus-sLID-CAAX/tRFPt-SspB_milli as these two combinations showed large dynamic ranges simplifying our ability to observe a change due to activation frequency. For each pair, we repeated the spot localization experiment while lengthening the times (15, 30, and 60 s intervals) between activations. We found that for iLID/SspB_micro the 30 and 60 s intervals recruited ~60% and ~15% of the SspB_micro that was observed for the 15 second interval (Figure 2D and F). However, with sLID/SspB_milli, the amount of SspB_milli recruitment for the 30 s interval was indistinguishable from the 15 s and the 60 s interval recruited ~30% of the SspB_milli that was observed with the 15 s interval (Figure 2E and F). This suggests that sLID requires less frequent light stimulation to maintain maximum activation.
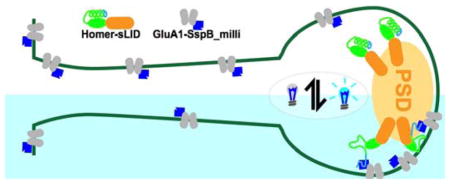
Next, we were curious if there are scenarios in which it would be advantageous to use SspB_milli instead of the tighter binding SspB variants. In particular, we hypothesized that weaker binding affinities may be needed to conditionally control interactions between two membrane-associated proteins, (i.e. their effective concentrations are increased by being restricted to the membrane). To test this hypothesis, we examined the ability of sLID and SspB variants to conditionally recruit neurotransmitter receptor proteins to the postsynaptic density (PSD) of neuronal excitatory synapses. The PSD is a protein dense region associated with the plasma membrane of dendritic spines, the major sites of excitatory synaptic contact in the central nervous system. The PSD anchors neurotransmitter receptors and signaling proteins apposed to presynaptic terminals. sLID was fused to YFP and the PSD scaffold Homer1c (h1c), which strongly localizes to the PSD12. This construct was co-expressed with fusion proteins containing the trans-membrane AMPA receptor subunit GluA1, mCherry, and variants of SspB. AMPA receptors are iontotropic glutamate receptors that mediate most fast neurotransmission in the central nervous system and their dynamic regulation at the PSD is critical for synaptic function and plasticity13. As expected, h1c-YFP-sLID was primarily localized to the PSD in neurons. Before activation, GluA1-mCh-SspB_nano was almost exclusively co-localized with the YFP signal and no appreciable difference in localization upon light stimulation (Figure 4A). The majority of GluA1-mCh-SspB_micro was also localized to the PSD in the dark. However, upon activation, there was a 1.2× increase in maximal signal (Figure 4A and C). In contrast, GluA1-mCh-SspB_milli was evenly distributed throughout the dendrite before activation, signifying minimal dark state binding between SspB_milli and sLID. With light activation, a 1.4× increase in signal at the PSD was measured. To further quantitate dark and lit state binding, we measured the GluA1-mCh-SspB intensity along the spine (which houses the PSD), and dendritic shaft in lit and dark conditions. Low dark-state binding and maximal dynamic range of sLID/SspB_milli is exemplified by the robust increase in the spine/shaft ratio of GluA1-mCh-SspB fluorescence, which increased from 1.5 ± 0.09 in the dark to 2.4 ± 0.18 in the light compared to sLID/SspB_micro (1.5 ± 0.05, dark vs 1.8 ± 0.06 light) and sLID/SspB_nano (2.8 ± 0.20, dark vs 2.9 ± 0.19, light) (Figure 4B and D). In addition, the kinetics of localization and reversion for both SspB_micro and SspB_milli with sLID were slowed in comparison to what we measured in fibroblasts (Activation ~3 min; Reversion ~6.5 min). The discrepancy between accumulation/dissociation rates using PSD localized sLID compared to our fibroblast experiments likely reflects slower diffusion of GluA1 within the plasma membrane and geometric constraints on diffusion imposed by dendritic spines.
Figure 4.

Induced localization of membrane protein, GluA1, is enhanced by use of sLID/SspB_milli. A) Representative images of GluA1-mCh-SspB localization using SspB nano (bottom), micro (middle) and milli (top) as analyzed in B, C and D. Arrowheads mark PSD. B) Representative change in GluA1-mCh-SspB fluorescence intensity over the spine and shaft before and after activation for SspB nano (bottom), micro (middle) and milli (top). Linescan profiles were generated using the grey dotted lines in A. C) PSD-localized mCherry fluorescence intensity is plotted over time normalized to the initial intensity for GluA1-mCh fused to SspB nano (n=89 spines from 5 neurons), micro (n=50 spines from 5 neurons) and milli (n=53 spines from 7 neurons). D) Ratio of spine to shaft mCherry fluorescence intensity before and after activation for GluA1-mCh fused to SspB nano (n=30 spines from 3 neurons), micro (n=30 spines from 2 neurons) and milli (n=30 spines from 5 neurons). ***p<0.001, paired student’s t-test.
In summary, the iLID suite of optogenetic dimers has been expanded to include SspB_milli and sLID. SspB_milli has been developed to shift the dynamic range of binding to higher protein concentrations with minor effects on the breadth of the dynamic range. sLID lengthened the half-life of reversion, allowing for a reduction in the frequency of light exposure while maintaining the same activity. In vitro measurements of affinity and kinetics correlated with the in vivo localization experiments, and can be used as a guide when designing future experiments with the switches. In a case study, we showed that when applying these switches to proteins with high effective concentrations, such as GluA1 concentrated in the plasma membrane, the weaker affinity SspB_milli reduced unwanted dark state binding. In addition, sLID allowed for less frequent light exposures, potentially maintaining the structural integrity of the postsynaptic density. Because of these advantages this expanded tool set should permit the use of iLID and its variants in a wider set of applications.
Supplementary Material
Acknowledgments
We would like to thank the following funding sources: NIH DA036877(B.K.); R01GM111557 (J.E.B.); NINDS RO1 NS082271, McKnight Foundation and Pew Charitable Trusts (M.J.K); NSF HRD-1301885 (A.M.B.)
References
- 1.Guntas G, Hallett Ra, Zimmerman SP, Williams T, Yumerefendi H, Bear JE, Kuhlman B. Engineering an improved light-induced dimer (iLID) for controlling the localization and activity of signaling proteins. Proc Natl Acad Sci U S A. 2014 doi: 10.1073/pnas.1417910112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kennedy MJ, Hughes RM, Peteya La, Schwartz JW, Ehlers MD, Tucker CL. Rapid blue-light-mediated induction of protein interactions in living cells. Nat Methods. 2010;7:973–5. doi: 10.1038/nmeth.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strickland D, Lin Y, Wagner E, Hope CM, Zayner J, Antoniou C, Sosnick TR, Weiss EL, Glotzer M. TULIPs: tunable, light-controlled interacting protein tags for cell biology. Nat Methods. 2012;9:379–84. doi: 10.1038/nmeth.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levskaya A, Weiner OD, Lim Wa, Voigt Ca. Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature. 2009;461:997–1001. doi: 10.1038/nature08446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Konermann S, Brigham MD, Trevino AE, Hsu PD, Heidenreich M, Cong L, Platt RJ, Scott Da, Church GM, Zhang F. Optical control of mammalian endogenous transcription and epigenetic states. Nature. 2013;500:472–6. doi: 10.1038/nature12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang X, Jost APT, Weiner OD, Tang C. A light-inducible organelle-targeting system for dynamically activating and inactivating signaling in budding yeast. Mol Biol Cell. 2013;24:2419–30. doi: 10.1091/mbc.E13-03-0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hallett RA, Zimmerman SP, Yumerefendi H, Bear JE, Kuhlman B. Correlating in Vitro and in Vivo Activities of Light-Inducible Dimers: A Cellular Optogenetics Guide. ACS Synth Biol. 2016;5:53–64. doi: 10.1021/acssynbio.5b00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harper SM, Neil LC, Gardner KH. Structural basis of a phototropin light switch. Science. 2003;301:1541–4. doi: 10.1126/science.1086810. [DOI] [PubMed] [Google Scholar]
- 9.Salomon M, Christie JM, Knieb E, Lempert U, Briggs WR. Photochemical and mutational analysis of the FMN-binding domains of the plant blue light receptor, phototropin. Biochemistry. 2000;39:9401–10. doi: 10.1021/bi000585+. [DOI] [PubMed] [Google Scholar]
- 10.Cheng KP, Kiernan EA, Eliceiri KW, Williams JC, Watters JJ. Blue Light Modulates Murine Microglial Gene Expression in the Absence of Optogenetic Protein Expression. Sci Rep. 2016;6:21172. doi: 10.1038/srep21172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zayner JP, Sosnick TR. Factors that control the chemistry of the LOV domain photocycle. PLoS One. 2014:9. doi: 10.1371/journal.pone.0087074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Okabe S, Urushido T, Konno D, Okado H, Sobue K. Rapid redistribution of the postsynaptic density protein PSD-Zip45 (Homer 1c) and its differential regulation by NMDA receptors and calcium channels. J Neurosci. 2001;21:9561–9571. doi: 10.1523/JNEUROSCI.21-24-09561.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kennedy MJ, Ehlers MD. Organelles and Trafficking Machinery for Postsynaptic Plasticity. Annu Rev Neurosci. 2006;29:325–362. doi: 10.1146/annurev.neuro.29.051605.112808. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.