

Abstract
The FMS-like tyrosine kinase-3 (FLT3) gene is the most commonly mutated gene in acute myeloid leukemia (AML), and patients carrying internal tandem duplication (ITD) mutations have a poor prognosis. Long-term inhibition of FLT3 activity in these patients has been elusive. To provide a more complete understanding of FLT3 biology, a mass spectroscopy-based screen was performed to search for FLT3-interacting proteins. The screen identified dedicator of cytokinesis 2 (DOCK2), which is a guanine nucleotide exchange factor for Rho GTPases, and its expression is limited to hematolymphoid cells. We show that DOCK2 is expressed in leukemia cell lines and primary AML samples, and DOCK2 co-immunoprecipitates with wild-type FLT3 and FLT3/ITD. Knock-down (KD) of DOCK2 by shRNA selectively reduced cell proliferation and colony formation in leukemia cell lines with increased FLT3 activity, and greatly sensitized these cells to cytarabine treatment, alone and in combination with FLT3 tyrosine kinase inhibitors. DOCK2 KD in a FLT3/ITD-positive leukemia cell line also significantly prolonged survival in a mouse xenograft model. These findings suggest that DOCK2 is a potential therapeutic target for novel AML treatments, as this protein regulates the survival of leukemia cells with elevated FLT3 activity and sensitizes FLT3/ITD leukemic cells to conventional anti-leukemic agents.
INTRODUCTION
Acute myeloid leukemia (AML) is a hematologic malignancy characterized by clonal expansion of myeloid blasts in the bone marrow and other tissues.1 The FMS-like tyrosine kinase-3 (FLT3) receptor gene is the most commonly mutated gene in AML2, and the most frequent of these mutations is an internal tandem duplication (ITD) in the juxtamembrane domain.3,4 FLT3/ITD mutations result in constitutive activation of the kinase, and patients with FLT3/ITD AML have a particularly poor prognosis,5,6 making inhibition of this tyrosine kinase an attractive therapeutic target.7 However, despite continuing progress in the development of FLT3 inhibitors, long-term inhibition of FLT3 activity in AML patients remains elusive.8,9
In order to achieve a better understanding of FLT3 biology and to develop more effective strategies for the inhibition of FLT3 activity and treatment of acute leukemia with activating mutations of FLT3, we performed a screen that utilized immunoprecipitation coupled with mass spectroscopy to identify proteins that interact with FLT3 and FLT3/ITD in human leukemia cell lines. Numerous candidate interactors were identified, including proteins involved in cell motility and proliferation, the regulation of reactive oxygen species, signal transduction in hematopoietic malignancies, and intracellular trafficking. One of the proteins identified in this screen was dedicator of cytokinesis 2 (DOCK2).
The DOCK family of proteins act as guanine nucleotide exchange factors (GEFs) for Rho GTPases, including Rac1.10 Rac1 is widely expressed in both neoplastic and normal epithelial and hematolymphoid cells, and is important for cell motility and growth.11,12 We have previously shown that FLT3/ITD activation results in increased reactive oxygen species (ROS) production partly through Rac1 activation.13 DOCK2 activates Rac1 but, unlike Rac1, DOCK2 expression is limited to hematopoietic tissues.14 DOCK2 is known to regulate several crucial processes including lymphocyte migration,14 activation and differentiation of T cells,15 cell-cell adhesion,16 and bone marrow homing of various immune cells.17,18 Since DOCK2 expression is limited to hematopoietic tissues, it is a particularly attractive drug target for the treatment of AML, since it would theoretically limit side effects by avoiding Rac1 inhibition in non-hematolymphoid tissues.
Here we confirm that DOCK2 interacts with FLT3 in both cell lines and primary leukemic cells. In cells with elevated FLT3 activity, knockdown (KD) of DOCK2 results in decreased cell proliferation and increased susceptibility to cytarabine (ARA-C), both in the presence and absence of FLT3 inhibitors. Additionally, mice transplanted with human leukemia cell lines that express mutated FLT3 show significantly increased survival when DOCK2 expression is suppressed. These findings suggest that targeting the Rac1 pathway via DOCK2 inhibition may be a feasible and novel therapeutic strategy for the treatment of FLT3/ITD acute leukemias.
MATERIALS AND METHODS
Cell lines and primary cells
Cells were cultured at 37 °C with 5% CO2 in DMEM (293T and HS5), or RPMI medium 1640 (all other cell lines), containing 10% fetal bovine serum, 100 units/ml penicillin and 100 units/ml streptomycin. Culture media for TF-1 cells that are FLT3/ITD-negative were supplemented with GM-CSF (2 ng/ml, Peprotech, Rocky Hill, NJ, USA). The Ba/F3:FLT3/D835Y cell line was previously described.13 Molm 14 and SEM K2 cells were obtained from the DSMZ (Deutsche Sammlung von Mikroorganismen und Zelkulturen, Braunschweig, Germany). The HB11;19 cell line was obtained from the laboratory of Dr. Michael Cleary (Stanford University, CA, USA). All other cells were obtained from American Type Culture Collection (Manassas, VA, USA). All cells were freshly thawed from stocks that were confirmed to be free of mycoplasma and frozen in 2010.
Peripheral blood (PB) and bone marrow (BM) samples from human AML patients were collected under a protocol approved by the Johns Hopkins Medicine Institutional Review Board. Proper consent was obtained for all subjects in accordance with the Declaration of Helsinki. Viable mononuclear cells were isolated from freshly thawed samples by Ficoll centrifugation. Human normal CD34+ cells were isolated using magnetically labeled microbeads (Miltenyi Biotec, San Diego, CA, USA) bound to MS columns (Miltenyi Biotec).
Reagents
Cytarabine (ARA-C) was obtained from Sigma-Aldrich (St. Louis, MO, USA). Sorafenib, quizartinib (AC220) and lestaurtinib (CEP701) were obtained from LC Laboratories (Woburn, MA, USA). NSC23766 was obtained from Tocris Bioscience (Ellisville, MO, USA).
Immunoprecipitation and Western blotting
Immunoprecipitation experiments were performed as previously described.19 Each immunoprecipitation experiment has been replicated at least 3 times. Protein samples were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis, transferred to a nitrocellulose membrane (BIO-RAD, Hercules, CA, USA), and detected by indicated antibodies followed by horseradish peroxidase-conjugated secondary antibodies and chemiluminescence. Anti-FLT3 (480), anti-DOCK2 (365242), anti-β-Actin (8432) and anti-β-Tubulin (5274) antibodies were obtained from Santa Cruz Biotechnology, Inc (Dallas, TX, USA). Antibodies against phospho-FLT3 (Y589/591; 3474), phospho-STAT5 (Y694; 9351), and STAT5 (9363) were obtained from Cell Signaling Technology (Danvers, MA, USA). Antibodies against phospho-tyrosine (05-321) were obtained from Millipore (Billerica, MA, USA). Rac-1 activation (Rac1-GTP) was assessed using the Rac1 activation assay (Millipore), and the G-LISA activation assay (Cytoskeleton, Inc., Denver, CO, USA). Quantifications of band densities were performed with a GS-900 Calibrated Densitometer (BIO-RAD) according to manufacturer’s instruction. For MG132 treatment, cells were incubated with 5 μM MG132 (Sigma-Aldrich) at 37°C for 1 hr prior to cell lysis. For DSP treatment, cells were incubated with 1 mM dithiobis(sccinimidyl propionate) (DSP; ThermoFisher Scientific, Waltham, MA, USA) at 4°C for 1.5 hr prior to cell lysis.
Mass spectroscopy
Cells were solubilized using NP-40 (1%), and the post-nuclear supernatant was immunoprecipitated with anti-FLT3 antibody. For each sample, the immunoprecipitated proteins were separated by SDS-polyacrylamide gel electrophoresis on 4–12% gradient gels, and the lanes were excised and sliced into bands. The bands were then proteolytically digested and the peptides subjected to mass spectroscopic analysis (Tackett laboratory; University of Arkansas). The results were uploaded into Scaffold 4 for viewing protein and peptide information.
Cell transduction
Stable cell lines with knockdown of DOCK2 were created using shRNA directed against DOCK2 from The RNAi Consortium (antisense: TTCTCAACTGTCACTTCCTTG ; Sigma-Aldrich). Cells were transduced with lentiviral vectors (pLKO1) with shRNA directed against DOCK2 and GFP (CCGGTACAACAGCCACAACGTCTATCTC-GAGATAGACGTTGTGGCTGTTGTATTTTT) under puromycin selection (2 μg/ml; Sigma-Aldrich).
Inducible cell lines were prepared using lentiviral pTRIPZ vectors expressing shRNAs against human DOCK2 (V3THS_358138: TTCTCAACTGTCACTTCCT and V3THS_358139: TGATATTCACACAGCATCA) and scrambled control shRNA (RHS4743:TGCTGTTGACAGTGAGCGATCTCGCTTGGGCGAGAGTAAGTAGTGAAGCCACAGATGTACTTACTCTCGCCCAAGCGAGAGTGCCTACTGCCTCGGA; Thermo Scientific). Cells were transduced according to manufacturer’s instructions, cultured in the presence of 5 μg/ml puromycin, and sorted for high RFP expression 48 hours after doxycycline (1 μg/ml) induction using FACSAria (BD Biosciences, San Jose, CA, USA).
Cell culture assays
Apoptosis assays were performed according to the manufacturer’s instructions (BD Biosciences) using Annexin V-APC and 7-Amino-actinomycin D (7-AAD). Late apoptosis was defined as cells positive for both 7-AAD and Annexin V. The IC50 values of the drugs for each cell line were determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-dimethyltetrazolium bromide (MTT) assays according to the manufacturer’s instruction (Roche Diagnostics, Indianapolis, IN, USA) (Supplemental Figure S3). For cells expressing inducible shRNA, the expression of shRNA was induced with doxycycline (1 ug/ml, Sigma-Aldrich) 24 hours prior to the start of the assay.
To measure cell proliferation, cells in logarithmic growth phase were diluted, placed in 24-well plate in triplicate, and cell densities were measured.
The colony formation assay was performed using MethoCult H4230 (STEMCELL Technologies, Vancouver, BC, Canada) according to manufacturer’s instruction. Each culture dish started with 200 cells.
The levels of reactive oxygen species (ROS) in cells were measured using ROS detection reagent CM-H2DCFDA (ThermoFisher Scientific) according to manufacturer’s instruction.
Each data point represents the average of three biological replicates.
Mouse transplantation experiments
NSG (NOD/Shi-scid/IL-2Rγnull) mice (in-house breeding; female, 4–6 weeks) were sublethally irradiated (250 rads), and each mouse was injected with 0.6 × 106 cells via lateral tail vein. Engraftment was assessed by flow cytometric measurement of human and mouse CD45 expression on the cell surface (APC mouse anti-human CD45 and FITC rat anti-mouse CD45, BD Biosciences). 15 mice were transplanted with MV4;11-C cells, 7 of which were assessed for engraftment. 10 mice were transplanted with MV4;11-KD cells, 5 of which were assessed for engraftment. For REH-C and REH-KD cells, 5 mice were transplanted with each cell line, and all were assessed for engraftment. The sample sizes were chosen based on efficiency of resource use, past experience with similar experiments, and pilot experiments to determine the effect size. No animal was excluded from the analysis, randomization was not used, and the investigator was not blinded. All animal procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (National Institute of Health, Bethesda, MD, USA) and were approved by the Institutional Animal Care and Use Committee at Johns Hopkins University.
Statistics
Statistical analysis was performed with Student t test (two-tailed), repeated measure ANOVA, and log-rank test using GraphPad software (GraphPad Software, Inc., La Jolla, CA, USA). All data are presented as the mean ± standard error of the mean. The variance was similar between all groups being compared. P values <.05 were considered to be statistically significant.
RESULTS
DOCK2 interacts with FLT3 in hematopoietic cells
An antibody that interacts with both wild-type and ITD-mutant FLT3 was used to perform immunoprecipitations (IP) from two different human leukemia cell lines: Molm14 and TF-1. The Molm14 cell line was derived from a patient with acute monocytic leukemia,20 and contains both wild type FLT3 and FLT3/ITD alleles. IP with rabbit serum was used as a negative control for this experiment. We also performed IP from a TF-1 cell line that was engineered to express a FLT3/ITD construct that had been initially identified in a patient with AML.21 The TF-1 parental cell line, which was derived from a patient with erythroid leukemia22 and lacks expression of FLT3, was used as a control. Following IP, a mass-spectroscopy based screen was performed to identify proteins that potentially interact with FLT3/ITD (Supplemental Figure S1A–C).
Through this screen, we identified a panel of proteins that co-IP with the antibody directed against FLT3, but were absent from the control lanes. Dedicator of cytokinesis 2 (DOCK2) was one of the potential interactors that were identified in IP from both the Molm14 and TF-1/ITD cell lines (Supplemental Figure S1C). In the Molm14 screen, 16 DOCK2 peptides were identified in immunoprecipitation from Molm14 cells, and 1 DOCK2 peptide was identified in the control lane. In the TF-1 screen, 6 DOCK2 peptides were identified in immunoprecipitation from the TF1:FLT3/ITD cells, and no DOCK2 peptides were identified in the immunoprecipitation from the parental TF-1 cells. We verified that DOCK2 was expressed in various established hematopoietic cell lines (Figure 1A) including: K562 cells (derived from an erythroleukemia arising from chronic myeloid leukemia that lacks expression of FLT3);23 Molm14 cells; Ba/F3 cells (derived from a murine pro-B-cell line) that were engineered to express the most frequent activating point mutation in the kinase domain (D835Y);24 HB11;19 cells (derived from a patient with acute leukemia carrying a D835H point mutation)25 (Supplemental Figure S2); SEM K2 cells (derived from a human leukemia overexpressing wild-type FLT3 that is constitutively activated by FL autocrine signaling);26 and MV4;11 cells (derived from a biphenotypic B/myeloid leukemia that expresses homozygous FLT3/ITD).27 The bone marrow stroma cell line HS5 was used as a negative control. DOCK2 was expressed in all primary AML samples tested (Figure 1B), including AMLs that lack FLT3 mutations and those with FLT3/ITD mutations. DOCK2 was also expressed in CD34+ cells from normal bone marrow, albeit at a lower level than in the primary AML samples.
Figure 1. DOCK2 is expressed in hematopoietic cell lines and leukemic blasts from patient samples, and interacts with FLT3, FLT3/D835 and FLT3/ITD.
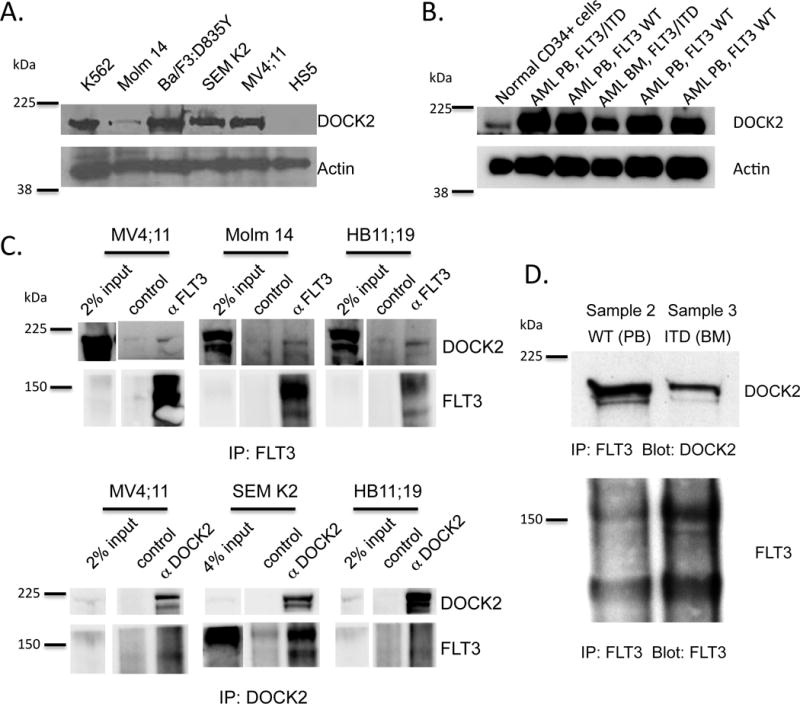
(A) Expression of DOCK2 in established hematopoietic cell lines that lack FLT3 (K562) or express wild-type FLT3 (SEM K2), FLT3/D835 (Ba/F3:FLT3/D835Y) or FLT3/ITD (MV4;11 and Molm 14) by Western blotting. DOCK2 is absent in the HS5 bone marrow stroma cell line. (B) Expression of DOCK2 in patient samples of AML from the peripheral blood (PB) and bone marrow (BM) that express either wild-type FLT3 (WT) or FLT3/ITD (ITD). (C) Co-immunoprecipitation of FLT3, FLT3/D835 and FLT3/ITD with DOCK2 in cell lines and (D) AML patient samples.
Co-immunoprecipitation experiments using antibodies against DOCK2 and FLT3 revealed that the two proteins co-IP in cell lines expressing FLT3/ITD mutations (Molm 14 and MV4;11), FLT3 D835 mutation (HB11;19) and activated wild-type FLT3 (SEM K2) (Figure 1C and Supplemental Figure S2), as well as AML patient samples expressing wild-type FLT3 and FLT3/ITD (Figure 1D). Consistent with the relatively low number of DOCK2 peptides detected in the initial screen (Supplemental Figure S1C), in many of the cell lines, the immunoprecipitation was relatively weak, likely reflecting the low level of expression of these proteins in many of these cell lines and the fact that this interaction is most likely to be transient and dynamic. Accordingly, the level of DOCK2 co-immunoprecipitating with FLT3 significantly increased, when Molm 14 cells were treated with the proteasome inhibitor MG132 or the protein cross-linker DSP prior to cell lysis (Supplemental Figure S2B). Interestingly, in Western blot studies DOCK2 appears to predominantly interact with the unphosphorylated form of wild type FLT3, FLT3/D835 and FLT3/ITD, regardless of whether FLT3 was stimulated with FLT3 ligand (FL; 10 ng/ml) or inhibited by sorafenib (25 nM) (Supplemental Figure S2).
Knock-down of DOCK2 leads to reduced cell proliferation and colony formation in cell lines with elevated FLT3 activity
We next examined the effects of reduced expression of DOCK2 on cell viability. Efficient knock-down (KD) of DOCK2 expression was achieved by either stable expression of an shRNA against DOCK2 (Figure 2A), or induced expression of shRNAs against DOCK2 in the presence of doxycycline (Figure 2B). Cell lines investigated include K562, REH cells (derived from a patient with acute leukemia that expresses wild-type FLT3 which is not constitutively active),28 HB11;19, Molm14, MV4;11 and SEM K2 cells. As predicted, shRNA KD of DOCK2 in Molm14, HB11;19 and MV4;11 cells led to decreased Rac1 activity, demonstrating that DOCK2 is functionally active in AML cells with constitutively active FLT3 (Figure 2C). Additionally, decreased expression of DOCK2 led to a statistically significant decrease in STAT5 activation in MV4;11 cells but not K562 cells (Figure 2D), suggesting that DOCK2 is involved in STAT5 activation resulted from activated FLT3 signaling.
Figure 2. DOCK2 expression is decreased by stable and inducible expression of shRNA directed against DOCK2, and suppression of DOCK2 results in decreased Rac1 and STAT5 activity.
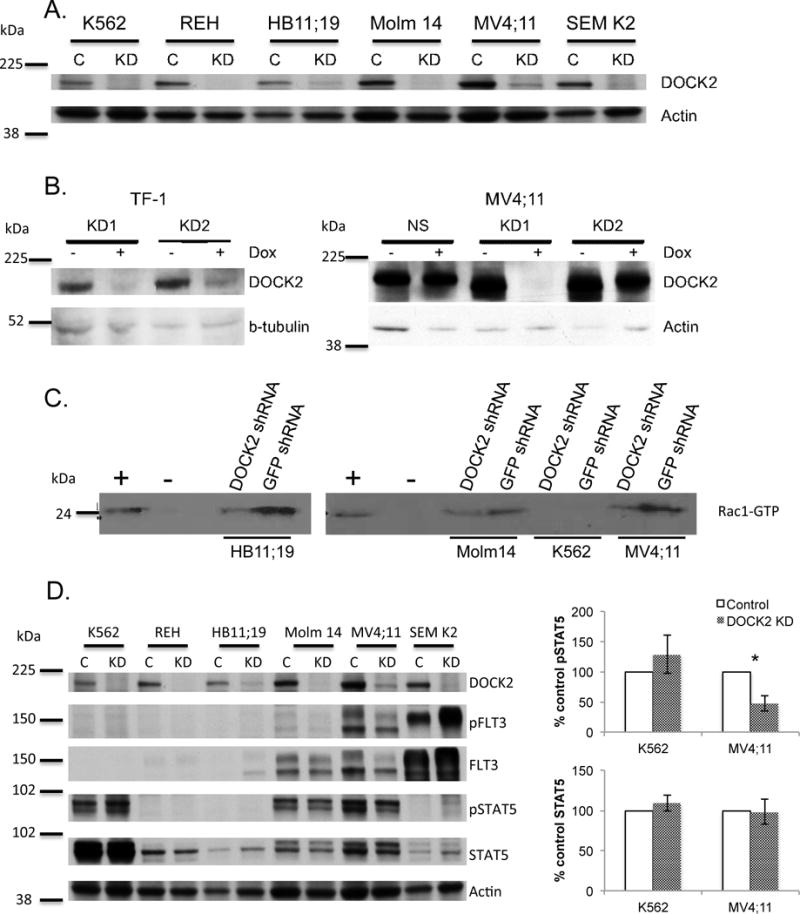
(A) Stable expression of shRNA directed against DOCK2 results in decreased DOCK2 expression (C: anti-GFP shRNA; KD: anti-DOCK2 shRNA). (B) Inducible expression of shRNAs directed against DOCK2 results in decreased expression of DOCK2 in TF-1 and MV4;11 cell lines (NS: non-specific control shRNA; KD1 and KD2: two different shRNAs directed against DOCK2). (C) Stable knockdown of DOCK2 results in decreased Rac1 activity (as measured by Rac1-GTP binding) in HB11;19, Molm14 and MV4;11 cell lines. Positive and negative controls were provided in the kit. (D) The effect of DOCK2 KD on STAT5 phosphorylation. The assay was performed in triplicate, and a statistically significant decrease in STAT5 phosphorylation was seen in MV4;11 cells (P ≤ 0.05, *) but not K562 cells.
The proliferation rate of cells with elevated FLT3/ITD activity (MV4;11) was markedly reduced upon stable knockdown of DOCK2 expression, and cells with relatively lower FLT3 activity (Molm 14, HB11;19) and cells expressing wild-type FLT3 (REH and SEM K2) also showed significant reduction, while cells with no FLT3 activity (K562) exhibited no difference in cell proliferation rate (Figure 3A). Similarly, the proliferation rate of MV4;11 cells was significantly reduced using inducible anti-DOCK2 shRNAs, while no difference was observed in TF-1 cells, which do not express FLT3 (Figure 3B).
Figure 3. Knockdown of DOCK2 results in decreased cell proliferation and decreased colony formation in cell lines that express FLT3.
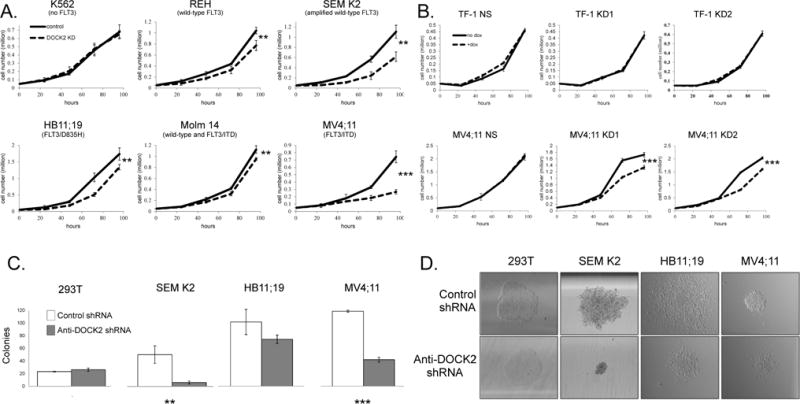
(A) Cells with stable expression of anti-DOCK2 shRNA were seeded at less than 0.1 million cells/ml to maximize the observable difference in proliferation, and counted every 24 hours. Cell lines expressing FLT3 showed decreased cell proliferation when expression of DOCK2 was reduced (REH, HB11;19, Molm 14, and SEM K2, P ≤ 0.01, **), and pronounced changes were observed in MV4;11 cells (P=0.0002, ***). Assays were performed in triplicate. (B) Induced expression of two shRNAs directed against DOCK2 in MV4;11 cells also resulted in decreased cell proliferation in assays that were performed in triplicate. TF-1 cells that lack FLT3 expression served as negative control. (C) Colony formation assays performed in triplicate showed that stable knock-down of DOCK2 led to significantly decreased colony formation in SEM K2 (P ≤ 0.01, **) and MV4;11 cells (P ≤ 0.001, ***). HB11;19 cells showed a trend in keeping with the SEM K2 and MV4;11 cells, but the difference was not statistically significant. HEK 293T cells that lack both DOCK2 and FLT3 were used as a negative control. Colony numbers were counted two weeks after plating. (D) Qualitative changes in colony morphology were seen in cell lines that express FLT3. Images were acquired at 40X using a Nikon Eclipse Ti microscope at room temperature two weeks after plating. All images are shown at the same scale.
To explore potential effects on clonability, KD of DOCK2 was performed in 293T, SEM K2, HB11;19 and MV4;11 cell lines followed by plating in methylcellulose. Cell lines with elevated FLT3 activity (MV4;11 and SEM K2) exhibited a significant reduction in colony-forming capacity in the presence of stably expressed anti-DOCK2 shRNA, while there was modest reduction in HB11;19 cells. No difference in colony formation was observed in 293T cells, which express neither FLT3 nor DOCK2 (Figure 3C). DOCK2 knock-down also resulted in smaller, more compact colonies in FLT3 expressing cells, while no such morphologic variability was observed in the 293T cells (Figure 3D). These results indicate that DOCK2 plays a role in cell proliferation and colony formation in cells with elevated FLT3 activity.
Decreased DOCK2 expression in cells with elevated FLT3 activity increases sensitivity to cytarabine treatment
Cytarabine (ARA-C) is a chemotherapeutic agent that is widely used during both induction and consolidation regimens in the treatment of AML.29 We investigated the effect of DOCK2 knock-down on leukemic cells in the presence of ARA-C. Cells with modest FLT3 activity (Molm14, HB11;19, REH) or no FLT3 activity (K562) showed no significant change or only modest response to ARA-C treatment upon KD of DOCK2; however, cells with elevated FLT3 activity (MV4;11 and SEM K2) exhibited significantly increased apoptosis in the presence of ARA-C when the expression of DOCK2 was stably reduced (Figure 4A). Similar results were found upon doxycycline treatment in cell lines expressing inducible DOCK2 shRNAs. The percent of late apoptosis was increased in MV4;11 cells treated with ARA-C when knock-down of DOCK2 was induced, while no difference was observed in TF-1 cells expressing the same shRNAs (Figure 4B). MV4;11 cells expressing an anti-DOCK2 shRNA exhibited reduced viable cell counts upon ARA-C treatment, while cells stably expressing a control shRNA continued to proliferate, albeit slowly (Figure 4C). In contrast, suppression of DOCK2 expression resulted in no observable difference in the response of REH cells to ARA-C treatment.
Figure 4. Knockdown of DOCK2 results in increased sensitivity to ARA-C in cell lines that express FLT3.
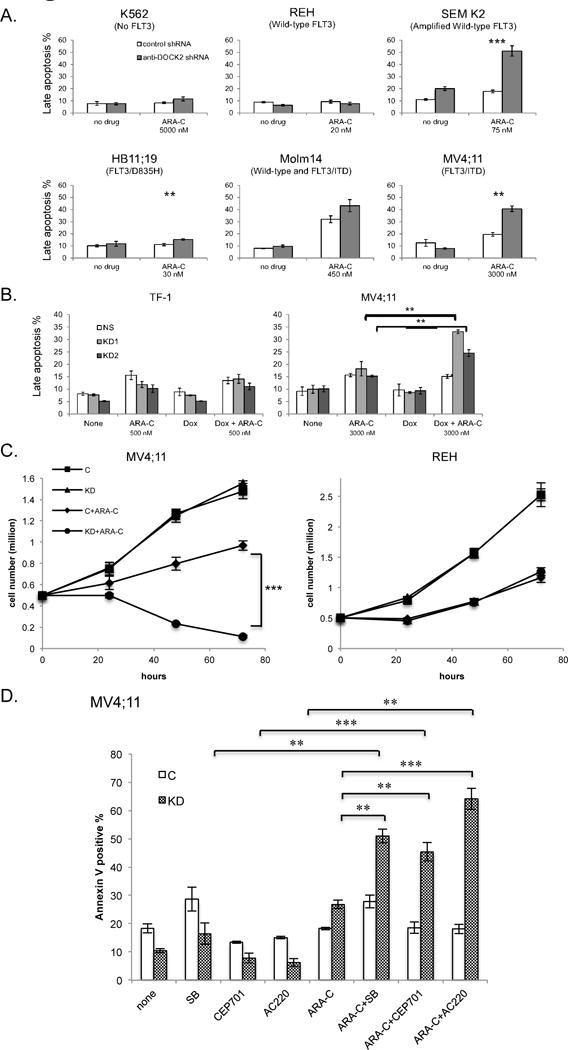
(A) Stable knockdown of DOCK2 expression increases the fraction of cells in late apoptosis upon treatment with ARA-C (48 hr) in cell lines with elevated FLT3 activity (SEM K2: P ≤ 0.001, ***; HB11;19: P ≤ 0.01, **; MV4;11: P ≤ 0.01, **). The ARA-C concentration used for each cell line was the IC50 as determined by MTT assay. (B) Induction of anti-DOCK2 shRNA by doxycycline treatment increases the fraction of cells in late apoptosis upon treatment with ARA-C (48 hr) in MV4;11 cells (P ≤ 0.01, **) but not TF-1 cells. (C) Proliferation assays demonstrate that stable knockdown of DOCK2 results in increased cell death upon treatment with ARA-C in MV4;11 cells (P<0.0001, ***) but not REH cells. Cells were seeded at a relatively high density (0.5 million cells/ml) to visualize differences in the proliferation of ARA-C treated cells. (D) Combination treatment of DOCK2-deficient MV4;11 cells with ARA-C (2500 nM) and FLT3 inhibitors increases the percentage of apoptotic cells over that seen with ARA-C or FLT-3 inhibitor treatment alone. Cells were treated for 48 hr. FLT3 inhibitors include sorafenib (SB; 25 nM), CEP-701 (5 nM), and AC220 (4 nM). Statistically significant differences were seen between ARA-C alone and ARA-C+SB (P ≤ 0.01, **), ARA-C+CEP-701(P ≤ 0.01, **) and ARA-C+AC220 (P ≤ 0.001, ***), as well as SB alone and ARA-C+SB (P ≤ 0.01, **), CEP-701 and ARa-C+CEP-701(P ≤ 0.001, ***), and AC220 and ARA-C+AC220(P ≤ 0.001, ***). All assays were performed in triplicate.
As the effect of DOCK2 knock-down on the sensitivity of cells to ARA-C treatment was most pronounced in MV4;11 and SEM K2 cells, both of which have elevated FLT3 activity, we next investigated the effect of FLT3 inhibition on the sensitivity of cells to ARA-C treatment. To better observe the combinatorial effect of FLT3 tyrosine kinase inhibitors (TKI) and ARA-C, cells were treated with lower doses of ARA-C and TKI, either of which exhibited modest effects when administered alone. MV4;11 cells expressing anti-DOCK2 shRNA showed a slightly lower late apoptotic rate in the presence of FLT3 TKI sorafenib (SB), quizartinib (AC220) and lestaurtinib (CEP701) as compared to control cells; however, upon the addition of ARA-C the cells expressing anti-DOCK2 shRNA showed a significant increase in apoptosis, which was further significantly increased by treatment with FLT3 TKI (Figure 4D). These results suggest that decreased DOCK2 expression in cells with elevated FLT3 activity renders cells more sensitive to ARA-C.
Mice transplanted with MV4;11 cells have prolonged survival when DOCK2 is knocked-down
To assess the effect of DOCK2 inhibition in vivo, we monitored disease progression in NSG mice transplanted with leukemia cell lines. Following injection of 0.6 × 106 leukemic cells via lateral tail vein, engraftment of the cells was observed by day 7 post-transplantation, as indicated by the presence of leukemic blasts in the peripheral blood. Mice transplanted with MV4;11 cells stably expressing control shRNA succumbed to disease within approximately 2 weeks (mean=15 days), whereas mice transplanted with MV4;11 cells stably expressing the anti-DOCK2 shRNA succumbed to disease by 6 to 8 weeks (mean = 55.5 days; Figure 5A). Peripheral blood samples from morbid mice transplanted with control cells revealed high proportions of human blasts among mononuclear cells (mean: 39%), while time-matched mice transplanted with MV4;11 DOCK2 KD cells had much lower percentage of human blasts in the peripheral blood (mean 8%; Figure 5B). Mice transplanted with the DOCK2 KD cells eventually achieved a similarly high percentage of human blasts in PB when they succumbed to disease 6–8 weeks post transplantation. Transplantation experiments carried out with REH cells +/− DOCK2 KD demonstrated no significant difference in survival (Figure 5C) or percentage of blasts in peripheral blood (Figure 5D) in control versus DOCK2 KD cells.
Figure 5. Knock-down of DOCK2 in transplanted cells with elevated FLT3 activity prolonged disease progression in NSG mice.
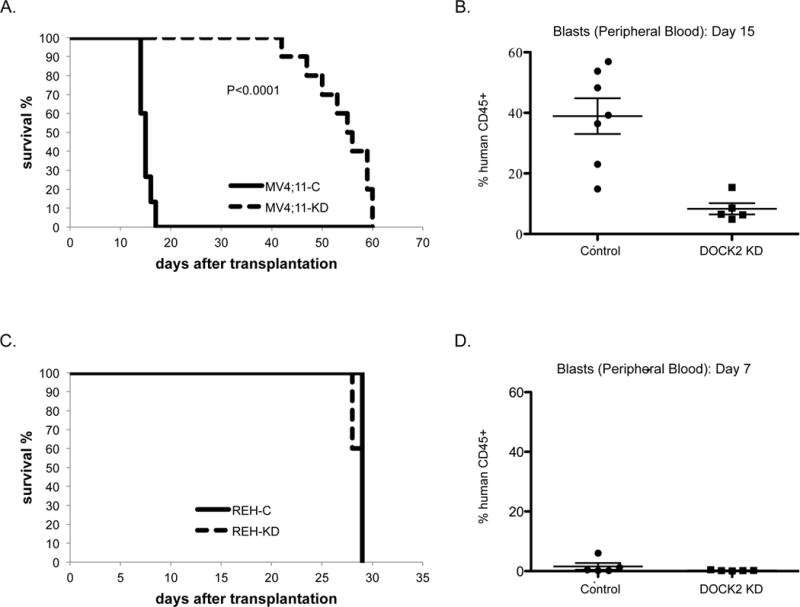
(A) Survival of sublethally irradiated immunodeficient mice (NSG mice; KD: n=10, Control: n=15) transplanted with MV4;11 cells (0.6 × 106 cells) stably expressing anti-DOCK2 shRNA or control shRNA. Median time to death for mice transplanted with cells expressing anti-DOCK2 shRNA and control shRNA was 55.5 and 15 days, respectively (P<0.0001; log-rank test). (B) The percentage of peripheral blood blasts on day 15 was significantly higher (P=0.002; Student t test) in the mice transplanted with control blasts (n=7, mean: 38.9 %) than the mice transplanted with leukemic blasts expressing anti-DOCK2 shRNA (n=5, mean: 8.3 %). (C) Survival of sublethally irradiated immunodeficient mice (NSG mice; KD and Control: n=5) transplanted with REH cells (0.6 × 106 cells) stably expressing anti-DOCK2 shRNA or control shRNA. Median time to death was 29 days (P=0.13) for both the KD and control mice. (D) The percentage of peripheral blood blasts on day 7 was similar in the mice with control (n=5, mean: 1.6 %) and anti-DOCK2 leukemic blasts (n=5, mean: 0.23 %; P=0.26).
DISCUSSION
We have shown that the Rho GEF DOCK2 interacts with FLT3, and that a decrease in DOCK2 expression inhibits the survival of leukemic cells that express activated FLT3 in both in vitro and in vivo models. These findings implicate DOCK2 as a potential druggable target for acute leukemias with elevated FLT3 activity.
DOCK2 is a tissue-specific GEF that mediates GTP-GDP exchange for Rac1 and Rac2. The Rho GTPases have been considered as potential druggable targets for the treatment of various tumors since misregulation of these proteins, particularly Rac1, has been described in both hematopoietic neoplasms and epithelial neoplasms including breast, lung, oral squamous cell and gastric carcinomas.30–34 While the function of Rac1 in neoplastic processes has been investigated, Rac1 signaling is also important for non-oncologic processes including endothelial function.
Small molecular Rac1 inhibitors are available, including NSC23766, which inhibits the Rac-specific GEFs Trio or Tiam1;35 however, due to the widespread expression and protean cellular functions of Rac1, the potential side effect profile is worrisome. Targeting tissue-specific Rho effectors, such as DOCK2, may help to improve the selectivity of Rac1 inhibition, making it a feasible drug target. Interestingly, DOCK2 knock-down and NSC23766 showed similar effects on Rac1 activity, reactive oxygen species (ROS) levels and sensitivity to ARA-C treatment in MV4;11 cells (Supplemental Figure S4), confirming that the anti-leukemic effects of DOCK2 inhibition in FLT3/ITD-positive leukemic cells were exerted through the inhibition of Rac1 pathway. DOCK2 inhibitors are not currently widely available, although small molecular inhibitors of DOCK2 have been reported in the literature.36,37 Additionally, the screening of preexisting drug libraries for potential DOCK2 inhibitors may be warranted.
While Rac1 has been extensively studied, DOCK2 is not particularly well-characterized. Most of the research on DOCK2 has focused on immunologic research, as DOCK2 deficiency has been described in patients with inborn errors of T-cell immunity,38 DOCK2 deficient mice show abnormalities of the immune system,39,40 and abrogation of DOCK2 expression is associated with decreased formation and proliferation of mature B-cell lymphomas.41 Recent reports have described mutations in DOCK2 that are associated with colorectal and esophageal carcinoma;42,43 however, these studies are descriptive and the functional consequences of the DOCK2 mutations found in solid tumors is unclear.
One clue as to the mechanism of action of downregulation of DOCK2 in FLT3/ITD cells is the concomitant decrease in STAT5 activity. Of note, STAT5 activity is associated with decreased survival in patients with FLT3/ITD AML.44 FLT3 has been shown to directly phosphorylate STAT5;45 however, STAT5 activation can also be mediated by alternative pathways, including FLT3/ITD-mediated activation of FAK with resultant activation of Tiam1, Rac1, PAK1 and finally phosphorylation of STAT5.46 Other mechanistic possibilities for DOCK2 inhibition include the changes in the generation of reactive oxygen species,47 changes in DNA damage repair pathways,48 and alterations in the interaction between the leukemic cells and the bone marrow microenvironment.49
Taken together, our findings suggest that DOCK2 inhibition may provide a feasible strategy for Rac1 inhibition in hematopoietic neoplasms, and could improve the efficacy of current therapeutic regimens for FLT3/ITD acute leukemia.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Cancer Institute (R21CA175667 to A.D. and R01CA90668 to D.S.), and a Clinician Scientist Career Award from Johns Hopkins University (A.D.). D.S. is also supported by the Kyle Haydock Professorship. We thank Dr. Alan Tackett for expert assistance with mass spectrometry experiments and data analysis.
Footnotes
CONFLICT OF INTEREST
The authors declare no competing financial interests.
AUTHORSHIP CONTRIBUTIONS
M. W. performed the experiments, analyzed the results and wrote the manuscript; M. H. performed the experiments, analyzed the results and revised the manuscript; L. L. performed the experiments and revised the manuscript; D. S. analyzed the results and revised the manuscript; A. D. designed the study, performed the experiments, analyzed the results and wrote the manuscript.
Supplementary information is available at Leukemia’s website.
References
- 1.McKenzie SB. Advances in understanding the biology and genetics of acute myelocytic leukemia. Clin Lab Sci. 2005;18(1):28–37. [PubMed] [Google Scholar]
- 2.Gilliland DG, Griffin JD. The role of FLT3 in hematopoiesis and leukemia. Blood. 2002;100(5):1532–1542. doi: 10.1182/blood-2002-02-0492. [DOI] [PubMed] [Google Scholar]
- 3.Nakao M, Yokota S, Iwai T, Kaneko H, Horiike S, Kashima K, et al. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia. 1996;10:1911–1918. [PubMed] [Google Scholar]
- 4.Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R, Kodera Y, Miyawaki S, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001;97:2434–2439. doi: 10.1182/blood.v97.8.2434. [DOI] [PubMed] [Google Scholar]
- 5.Moreno I, Martin G, Bolufer P, Barragán E, Rueda E, Román J, et al. Incidence and prognostic value of FLT3 internal tandem duplication and D835 mutations in acute myeloid leukemia. Haematologica. 2003;88(1):19–24. [PubMed] [Google Scholar]
- 6.Levis M, Small D. ITDoes matter in leukemia. Leukemia. 2003;17(9):1738–1752. doi: 10.1038/sj.leu.2403099. [DOI] [PubMed] [Google Scholar]
- 7.Smith CC, Wang Q, Chin CS, Salerno S, Damon LE, Levis MJ, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485(7397):260–263. doi: 10.1038/nature11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith BD, Levis M, Beran M, Giles F, Kantarjian H, Berg K, et al. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood. 2004;103:3669–3676. doi: 10.1182/blood-2003-11-3775. [DOI] [PubMed] [Google Scholar]
- 9.Levis M, Small D. FLT3 tyrosine kinase inhibitors. Int J Hematol. 2005;82(2):100–107. doi: 10.1532/IJH97.05079. [DOI] [PubMed] [Google Scholar]
- 10.Gadea G, Blangy A. Dock-family exchange factors in cell migration and disease. Eur J Cell Biol. 2014;93(10–12):466–477. doi: 10.1016/j.ejcb.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 11.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 12.Cancelas JA, Jansen M, Williams DA. The role of chemokine activation of Rac GTPases in hematopoietic stem cell marrow homing, retention, and peripheral mobilization. 2006;34(8):976–985. doi: 10.1016/j.exphem.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 13.Sallmyr A, Fan J, Datta K, Kim KT, Grosu D, Shapiro P, et al. Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: implications for poor prognosis in AML. Blood. 2008;111(6):3173–3182. doi: 10.1182/blood-2007-05-092510. [DOI] [PubMed] [Google Scholar]
- 14.Fukui Y, Hashimoto O, Sanui T, Oono T, Koga H, Abe M, et al. Haematopoietic cell-specific CDM family protein DOCK2 is essential for lymphocyte migration. Nature. 2001;412(6849):826–831. doi: 10.1038/35090591. [DOI] [PubMed] [Google Scholar]
- 15.Nishikimi A, Kukimoto-Niino M, Yokoyama S, Fukui Y. Immune regulatory functions of DOCK family proteins in health and disease. Exp Cell Res. 2013;319(15):2343–2349. doi: 10.1016/j.yexcr.2013.07.024. [DOI] [PubMed] [Google Scholar]
- 16.Garcia-Bernal D, Sotillo-Mallo E, Nombela-Arrieta C, Samaniego R, Fukui Y, Stein JV, et al. DOCK2 is required for chemokine-promoted human T lymphocyte adhesion under shear stress mediated by the integrin alpha4beta1. J Immunol. 2006;177(8):5215–5225. doi: 10.4049/jimmunol.177.8.5215. [DOI] [PubMed] [Google Scholar]
- 17.Kikuchi T, Kubonishi S, Shibakura M, Namba N, Matsui T, Fukui Y, et al. Dock2 participates in bone marrow lympho-hematopoiesis. Biochem Biophys Res Commun. 2008;367(1):90–96. doi: 10.1016/j.bbrc.2007.12.093. [DOI] [PubMed] [Google Scholar]
- 18.Wen Y, Elliott MJ, Huang Y, Miller TO, Corbin DR, Hussain LR, et al. DOCK2 is critical for CD8(+) TCR(−) graft facilitating cells to enhance engraftment of hematopoietic stem and progenitor cells. Stem Cells. 2014;32(10):2732–2743. doi: 10.1002/stem.1780. [DOI] [PubMed] [Google Scholar]
- 19.Tse KF, Mukherjee G, Small D. Constitutive activation of FLT3 stimulates multiple intracellular signal transducers and results in transformation. Leukemia. 2000;14(10):1766–1776. doi: 10.1038/sj.leu.2401905. [DOI] [PubMed] [Google Scholar]
- 20.Matsuo Y, MacLeod RA, Uphoff CC, Drexler HG, Nishizaki C, Katayama Y, et al. Two acute monocytic leukemia (AML-M5a) cell lines (MOLM-13 and MOLM-14) with interclonal phenotypic heterogeneity showing MLL-AF9 fusion resulting from an occult chromosome insertion, ins(11;9)(q23;p22p23) Leukemia. 1997;11(9):1469–1477. doi: 10.1038/sj.leu.2400768. [DOI] [PubMed] [Google Scholar]
- 21.Zheng R, Bailey E, Nguyen B, Yang X, Piloto O, Levis M, et al. Further activation of FLT3 mutants by FLT3 ligand. Oncogene. 2011;30(38):4004–4014. doi: 10.1038/onc.2011.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kitamura T, Tange T, Terasawa T, Chiba S, Kuwaki T, Miyagawa K, et al. Establishment and characterization of a unique human cell line that proliferates dependently on GM-CSF, IL-3, or erythropoietin. J Cell Physiol. 1989;140(2):323–334. doi: 10.1002/jcp.1041400219. [DOI] [PubMed] [Google Scholar]
- 23.Lozzio CB, Lozzio BB. Human chronic myelogenous leukemia cell-line with positive Philadelphia chromosome. Blood. 1975;45(3):321–334. [PubMed] [Google Scholar]
- 24.Zhang W, Konopleva M, Shi YX, McQueen T, Harris D, Ling X, et al. Mutant FLT3: a direct target of sorafenib in acute myelogenous leukemia. J Natl Cancer Inst. 2008;100(3):184–198. doi: 10.1093/jnci/djm328. [DOI] [PubMed] [Google Scholar]
- 25.Tkachuk DC, Kohler S, Cleary ML. Involvement of a homolog of Drosophila trithorax by 11q23 chromosomal translocation in acute leukemias. Cell. 1992;71(4):691–700. doi: 10.1016/0092-8674(92)90602-9. [DOI] [PubMed] [Google Scholar]
- 26.Greil J, Gramatzki M, Burger R, Marschalek R, Peltner M, Trautmann U, et al. The acute lymphoblastic leukaemia cell line SEM with t(4;11) chromosomal rearrangement is biphenotypic and responsive to interleukin-7. Br J Haematol. 1994;86(2):275–283. doi: 10.1111/j.1365-2141.1994.tb04726.x. [DOI] [PubMed] [Google Scholar]
- 27.Lange B, Valtieri M, Santoli D, Caracciolo D, Mavilio F, Gemperlein I, et al. Growth factor requirements of childhood acute leukemia: establishment of GM-CSF-dependent cell lines. Blood. 1987;70(1):192–199. [PubMed] [Google Scholar]
- 28.Rosenfeld C, Goutner A, Venuat AM, Choquet C, Pico JL, Dore JF, et al. An effect human leukaemic cell line: Reh. Eur J Cancer. 1977;13(4–5):377–379. doi: 10.1016/0014-2964(77)90085-8. [DOI] [PubMed] [Google Scholar]
- 29.Dohner H, Estey EH, Amadori S, Appelbaum FR, Büchner T, Burnett AK, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115(3):453–474. doi: 10.1182/blood-2009-07-235358. [DOI] [PubMed] [Google Scholar]
- 30.Alan JK, Lundquist EA. Mutationally activated Rho GTPases in cancer. Small GTPases. 2013;4(3):159–163. doi: 10.4161/sgtp.26530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ridley AJ. Rho proteins and cancer. Breast Cancer Res Treat. 2004;84:13–19. doi: 10.1023/B:BREA.0000018423.47497.c6. [DOI] [PubMed] [Google Scholar]
- 32.Fritz G, Just I, Kaina B. Rho GTPases are over-expressed in human tumors. Int J Cancer. 1999;81:682–687. doi: 10.1002/(sici)1097-0215(19990531)81:5<682::aid-ijc2>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 33.Lin Y, Zheng Y. Approaches of targeting Rho GTPases in cancer drug discovery. Expert Opin Drug Discov. 2015;10(9):991–1010. doi: 10.1517/17460441.2015.1058775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bid HK, Roberts RD, Manchanda PK, Houghton PJ. RAC1: an emerging therapeutic option for targeting cancer angiogenesis and metastasis. Mol Cancer Ther. 2013;12(10):1925–1934. doi: 10.1158/1535-7163.MCT-13-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci USA. 2004;101(20):7618–7623. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nishikimi A, Uruno T, Duan X, Cao Q, Okamura Y, Saitoh T, et al. Blockade of inflammatory responses by a small-molecule inhibitor of the Rac activator DOCK2. Chem Biol. 2012;19(4):488–497. doi: 10.1016/j.chembiol.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 37.Watanabe M, Terasawa M, Miyano K, Yanagihara T, Uruno T, Sanematsu F, et al. DOCK2 and DOCK5 act additively in neutrophils to regulate chemotaxis, superoxide production, and extracellular trap formation. J Immunol. 2014;193(11):5660–5667. doi: 10.4049/jimmunol.1400885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dobbs K, Domínguez Conde C, Zhang SY, Parolini S, Audry M, Chou J, et al. Inherited DOCK2 Deficiency in Patients with Early-Onset Invasive Infections. N Engl J Med. 2015;372(25):2409–2422. doi: 10.1056/NEJMoa1413462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang H, Pan F, Erickson LM, Jang MS, Sanui T, Kunisaki Y, et al. Deletion of DOCK2, a regulator of the actin cytoskeleton in lymphocytes, suppress cardiac allograft rejection. J Exp Med. 2005;202(8):1121–1130. doi: 10.1084/jem.20050911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yasuda K, Nundel K, Watkins AA, Dhawan T, Bonegio RG, Ubellacker JM, et al. Phenotype and function of B cells and dendritic cells from interferon regulatory factor 5-deficient mice with and without a mutation in DOCK2. Int Immunol. 2013;25(5):295–306. doi: 10.1093/intimm/dxs114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang L, Nishihara H, Kimura T, Kato Y, Tanino M, Nishio M, et al. DOCK2 regulates cell proliferation through Rac and ERK activation in B cell lymphoma. Biochem Biophys Res Commun. 2010;395(1):111–115. doi: 10.1016/j.bbrc.2010.03.148. [DOI] [PubMed] [Google Scholar]
- 42.Yu J, Wu WK, Li X, He J, Li XX, Ng SS, et al. Novel recurrently mutated genes and a prognostic mutation signature in colorectal cancer. Gut. 2015;64(4):636–645. doi: 10.1136/gutjnl-2013-306620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dulak AM, Stojanov P, Peng S, Lawrence MS, Fox C, Stewart C, et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat Genet. 2013;45(5):478–486. doi: 10.1038/ng.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lim Y, Gondek L, Li L, Wang Q, Ma H, Chang E, et al. Integration of Hedgehog and mutant FLT3 signaling in myeloid leukemia. Sci Transl Med. 2015;7(291):291ra96. doi: 10.1126/scitranslmed.aaa5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choudhary C, Brandts C, Schwable J, Tickenbrock L, Sargin B, Ueker A, et al. Activation mechanisms of STAT5 by oncogenic Flt3-ITD. Blood. 2007;110(1):370–374. doi: 10.1182/blood-2006-05-024018. [DOI] [PubMed] [Google Scholar]
- 46.Chatterjee A, Ghosh J, Ramdas B, Mali RS, Martin H, Kobayashi M, et al. Regulation of Stat5 by FAK and PAK1 in oncogenic FLT3- and KIT-driven leukemogenesis. Cell Rep. 2014;9(4):1333–1348. doi: 10.1016/j.celrep.2014.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Selvakumar B, Hess DT, Goldschmidt-Clermont PJ, Stamler JS. Co-regulation of constitutive nitric oxide synthases and NADPH oxidase by the small GTPase Rac. FEBS Lett. 2008;582:2195–2202. doi: 10.1016/j.febslet.2008.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Espinha G, Osaki JH, Magalhaes YT, Forti FL. Rac1 GTPase-deficient HeLa cells present reduced DNA repair, proliferation, and survival under UV or gamma irradiation. Mol Cell Biochem. 2015;404(1–2):281–297. doi: 10.1007/s11010-015-2388-0. [DOI] [PubMed] [Google Scholar]
- 49.Canclas JA, Lee AW, Prabhakar R, Stringer KF, Zheng Y, Williams DA. Rac GTPases differentially integrate signals regulating hematopoietic stem cell localization. Nat Med. 2005;11(8):886–891. doi: 10.1038/nm1274. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.