

Abstract
Background
Chronic pain and stress-related psychopathologies, such as depression and anxiety-associated abnormalities, are mutually reinforcing; however, the neuronal circuits and mechanisms that underlie this reinforcement are still not well understood. Pituitary adenylate cyclase activating polypeptide (PACAP; Adcyap1) and its cognate PAC1 receptor (Adcyap1r1) are expressed in peripheral nociceptive pathways, participate in anxiety-related responses and have been have been linked to posttraumatic stress disorder (PTSD) and other mental health afflictions.
Methods
Using immunocytochemistry, pharmacological treatments and behavioral testing techniques, we have used a rodent partial sciatic nerve chronic constriction model (CCI; n = 5-8 per group per experiment) to evaluate PACAP plasticity and signaling in nociceptive and stress-related behaviors.
Results
We show that chronic neuropathic pain increases PACAP expression at multiple tiers along the spino-parabrachioamygdaloid tract. Furthermore, CCI bilaterally augments nociceptive amygdala (CeA) PACAP immunoreactivity, ERK phosphorylation and c-Fos activation, in parallel with heightened anxiety-like behavior and nociceptive hypersensitivity. Acute CeA infusions with the PACAP receptor antagonist PACAP(6-38) blocked CCI-induced behavioral responses. Additionally, pretreatments with inhibitors of MEK or endocytosis to block endosomal PACAP receptor ERK signaling attenuated PACAP-induced CeA neuronal activation and nociceptive responses.
Conclusions
Our data suggest that chronic pain-induced PACAP neuroplasticity and signaling in spino-parabrachioamygdaloid projections impact CeA stress- and nociception-associated maladaptive responses which can be ameliorated upon receptor antagonism even during injury progression. Thus the PACAP pathway provides for an important mechanism underlying the intersection of stress and chronic pain pathways via the amygdala.
Keywords: PACAP, PAC1 receptor, parabrachial nucleus, amygdala, nociception, stress-related behavior
Introduction
Pain carries an aversive emotional component that can severely impact physiological and behavioral responses. Accordingly, chronic pain has been well associated with a number of stress-related psychopathologies, including depression, sleep dysregulation, panic disorders, obsessive compulsive behavior, anxiety abnormalities and post-traumatic stress disorder (PTSD) (1). Although the mechanisms underlying this association are not well understood, the high comorbidity between pain and stress-related behavioral disorders suggests that the two may be interrelated maladaptive processes (2). Among brain regions, the amygdala is centrally situated to integrate the many descending and ascending signals to modulate the sensory and emotional components of pain. Highly processed descending polymodal anti-nociceptive information is conveyed from the somatosensory cortex and thalamus to the central nucleus of the amygdala (CeA). The resulting CeA efferents signals are relayed to other central nuclei, including those traveling with hypothalamic - periaqueductal grey projections for autonomic control and anti-nociception to dampen pain stimuli (3). Among several direct ascending pathways carrying nociceptive transmission to the CeA, the most prominent is the spino-parabrachioamygdaloid tract (3-6). Peripheral nociceptive signals carried via primary sensory A - and C-fibers terminate on spinal projection neurons in lamina I/II and IV of the dorsal horn where the second order neurons send projections via the spino-parabrachial pathway to pontine lateral parabrachial nuclei (LPBn) (7). In turn, the third-order LPBn neurons relay sensory information to the lateral (CeL) and lateral capsular (CeLC) subdivisions of the CeA. Hence the PBn collects cutaneous (mechanical and thermal), deep (muscular and articular) and visceral nociceptive signals and relays the information in a highly organized topographical manner principally to the nociceptive amygdala.
Although the integration of these inputs with amygdala circuits is a key mechanism underlying the emotional aspects of stress and pain, the neurochemistry, neuroplasticity and regulatory events that drive the maladaptive responses are still not completely understood. In the CeA, chronic pain can alter glutamate receptor, extracellular-regulated kinase (ERK) and c-Fos expression or function, facilitate synaptic transmission to the CeLC and dysregulate anti-nociceptive signaling (2, 8-12). But in addition to diminished inhibitory neurocircuit function, persistent pain may also augment stimulatory CeA nociceptive neuropeptide levels including corticotrophin releasing hormone (CRH) and calcitonin gene-related peptide (CGRP) as complementary means to facilitate the stress- and pain-induced changes in neural function (6, 11, 13).
Among brain peptides, there is accumulating evidence implicating pituitary adenylate cyclase activating polypeptide (PACAP) and its cognate PAC1 receptor in mediating the behavioral and physiological responses to homeostatic challenges (14). Altered PACAP levels and PAC1 receptor polymorphism have been associated with PTSD (15-19). Mice lacking PACAP or the PAC1 receptor exhibit blunted anxiety-like behavior, show hypothalamic-pituitary-adrenal (HPA) axis and autonomic system dysregulation, and fail to develop hypersensitivity to nociceptive stimuli in inflammatory pain paradigms (20-27). Furthermore, chronic but not acute stress leads to an upregulation of PACAP and PAC1 receptor transcript expression in the bed nucleus of the stria terminalis (BNST) (28-30). BNST PACAP signaling increases anxiety-like behaviors and HPA axis activation, and mediates many of the behavioral consequences of chronic stress. The BNST and the amygdala share similar circuit connectivity; as in the BNST, dense PACAP immunoreactivity has been identified in the CeLC/CeL which represented LPBn PACAP projections from the spino-parabrachioamygdaloid tract (31). Importantly, infusions of PACAP or a specific PAC1 receptor agonist directly into the CeA of naive rats produced both anxiety-like behaviors and nociceptive hypersensitivity, suggesting that LPBn PACAP activity via the spino-parabrachioamygdaloid circuit carries signals that may in part alter the emotional responses to pain. Using a partial sciatic nerve ligation chronic constriction injury (CCI) model, we have now examined whether persistent neuropathic pain alters PACAP expression in the spino-parabrachioamygdaloid tract and whether PAC1 receptor antagonism can mitigate CCI-induced nociceptive hypersensitivity and anxiety-like behaviors. As PACAP signaling potently and efficaciously activates MAPK/ERK, a central mechanism in synaptic plasticity and CeA-dependent behaviors and pain hypersensitivity, we also assessed CeA PAC1 receptor mechanisms in vivo. The studies in aggregate suggest that endogenous PACAP signaling in the spino-parabrachioamygdaloid pathway and the resulting endosomal PAC1 receptor-stimulated activation of ERK in the CeA mediate the adverse emotional consequences of chronic pain. These results implicate PACAP mechanisms in the comorbidities between chronic pain and other stress-related pathologies.
Materials and Methods
Detailed Materials and Methods and extended data are available in Supplementary Information.
Animals
Adult male Sprague-Dawley rats were from Charles River Laboratories, Wilmington, MA. PACAP-EGFP BAC transgenic mice were generated by the GENSAT (Gene Expression Nervous System Atlas). All procedures were approved by the Institutional Animal Care and Use Committee at the University of Vermont.
Surgical Procedures
For chronic constriction injury (CCI), rats and PACAP-EGFP mice were anesthesized and loose ties (4-0 chromic gut, Ethicon) were placed proximal to the trifurcation of the sciatic nerve. Following recovery, only animals that developed thermal hypersensitivity were used for testing. For intra-amygdalar (CeA) infusions, 2 stainless steel cannulae (22GA, PlasticsOne, Roanoke, VA) were placed bilaterally using coordinates (from bregma in mm) AP: -2.6, ML: 4.5, DV: -7.2. Drug infusions at a rate of 0.5 ∼l/min (Harvard Apparatus, Holliston, MA) included PACAP38, PACAP(6-38), Pitstop 2 and PD98059.
Histochemistry and imaging
Anesthetized PACAP-EGFP mice were perfused transcardially with paraformaldehyde and tissue cryosections (30 ∼m) were mounted on subbed slides for endogenous EGFP fluorescence imaging. Tissues were processed similarly for immunocytochemistry using procedures detailed in supplementary information. PACAP antibody was from Jens Hannibal (Bisperg Hospital, Copenhagen, Denmark); other antisera included those for pERK and ˆ- arrestin 1/2 (Cell Signaling Technology, Danvers, MA), c-Fos (Santa Cruz Biotechnology, Dallas, TX), and vGlut1, vGlut2 and GAD (all from Millipore Billerica, MA). Images were acquired under identical settings and parameters using a Nikon E800 scanning confocal and Olympus fluorescence microscopes for analyses using ImageJ.
Open field and thermal sensitivity tests
In open field tests, rats were individually placed into the corner of a 75 cm × 75 cm open arena and center entries and total distance traveled (5 min) were digitally captured for analyses using EthoVision XT (Noldus Information Technology, The Netherlands). A Hargreave's apparatus (IITC Life Science, Inc., Woodland Hills, CA) was used to assess thermal stimuli responses. After baseline testing, a beam of focused radiant light (25% active intensity) was targeted onto the plantar surface of each hindpaw. Animal reaction time was automatically recorded. The hindpaws were randomly selected at trial initiation and 3 trials were performed on each hindpaw.
Statistics
All statistical tests were performed in SPSS and GraphPad PRISM. Two-way analysis of variance (ANOVA) was performed to examine main effects and interactions, and Bonferrroni's multiple comparisons tests were used to compare different groups.
Results
Neuropathic pain augments PACAP expression in the spino-parabrachioamygdaloid pathway
Our previous studies identified LPBn PACAP neuronal projections to CeLC and demonstrated that CeA PACAP infusions resulted in heightened nociceptive sensitivity and anxiety-like behaviors (31). As previous studies implicated PACAP plasticity in sensory systems (32, 33) we examined whether chronic neuropathic pain in a unilateral sciatic nerve CCI model regulated endogenous PACAP expression along the spino-parabrachioamygdaloid pathway. The CCI procedure reliably heightened nociceptive sensitivity as reflected by decreased thermal latency responses and increased anxiety-like behavior compared to sham controls (see below). Quantitative PCR analyses of micropunched PBn tissues demonstrated that CCI specifically elevated PBn PACAP transcript levels approximately 1.5 - fold compared to tissues from sham animals (t(12) = 2.36, p = 0.036) without changing PAC1 receptor transcript expression (Figure S1). CCI did not augment PACAP transcript levels in other regions tested (Figure S1). As an alternative to PACAP immunocytochemistry, which preferentially identified fiber peptide immunoreactivity rather than soma, we evaluated CCI-induced LPBn PACAP expression using PACAP-EGFP mice. LPBn PACAP-EGFP+ cells were identified under basal conditions and unilateral CCI increased the number of PACAP neurons nearly 2-fold compared to sham operated animals (F(1,22) = 7.99, p = 0.01, Figure 1A - 1C). The CCI-induced PACAP-EGFP+ neurons appeared throughout the LPBn; notably, the LPBn PACAP induction was observed both ipsilateral and contralateral to the injury which reflected bilateral dorsal horn neuronal projections to LPBn (Figure 1C).
Figure 1.
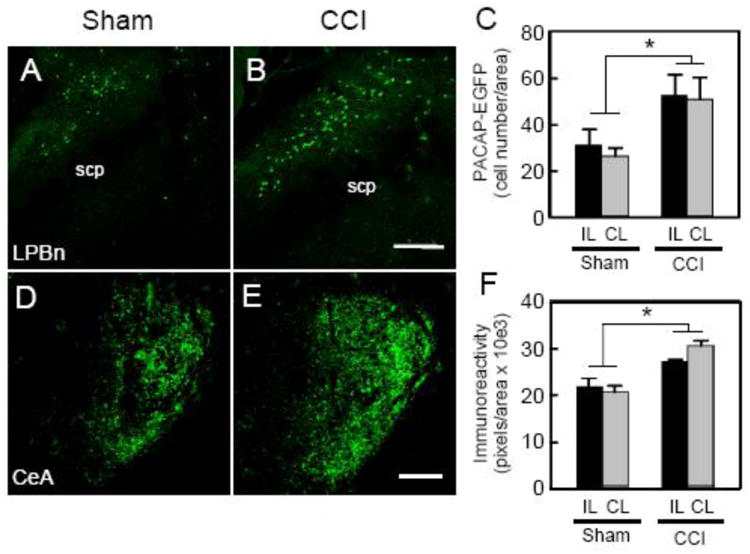
CCI increases LPBn and CeA PACAP levels. Control sham surgery (A) or CCI (B) were performed on transgenic PACAP-EGFP mice and native EGFP fluorescence was examined in LPBn tissues 2 weeks following surgery. The number of LPBn PACAP-EGFP cells was increased bilaterally in CCI compared to sham with a main effect of condition (C; sham ipsilatera/contralateral = 31.0 7.1 cells/26.3 3.5 cells vs CCI ipsilateral/contralateral = 52.5 9.2 cells/50.8 9.7 cells; F(1,22) = 7.99, *p = 0.01, n = 6 - 7 per group, 3 sections enumerated per side per animal). CeA PACAP immunoreactivity was also increased after CCI (E) compared to sham controls (D). From image analyses with thresholded area, there was a main effect of CCI (F; sham ipsilatera/contralateral = 21.7 2.0 units/20.6 1.3 units vs CCI ipsilateral/contralateral = 27.3 0.4 units/30.6 1.1 units; F(1,28) = 14.74, *p = 0.0006; n = 8 per group) but no main effect (F(1,28) = 0.32, p = 0.6) or interaction (F(1,28) = 1.17, p = 0.3) with respect to side. Data represent mean cells/unit area or fluorescence units/unit area SEM; scp, superior cerebellar peduncle; IL, ipsilateral; CL, contralateral; Scale bar = 200 ∼m.
In good correspondence to the increases in LPBn PACAP transcripts and neurons, CCI also augmented CeLC fiber PACAP staining from parabrachioamygdaloid projections (Figure 1D - 1F). Image analyses after thresholding fluorescence intensity revealed a 1.4-fold increase in PACAP staining density in the CeLC fiber terminals and varicosities of CCI animals compared to that in sham animals (F(1,28) = 14.74, p = 0.0006, Figure 1F). As anticipated from bilateral dorsal horn neuronal projections to the LPBn, the increase in CeLC PACAP immunoreactivity was also bilateral after unilateral CCI; however, there was a notable bias toward greater PACAP immunoreactivity in the right CeLC irrespective of the side of the CCI, in agreement with CeA lateralization described previously (34, 35) (Figure S2).
Given PACAP roles in neuroplasticity, we also evaluated whether neuropathic pain from CCI similarly affected other PACAPergic neurons within the spino-parabrachioamygdaloid tract in the PACAP-EGFP mice. While the sciatic nerve in sham operated and the contralateral leg of CCI animals demonstrated minimal PACAP-EGFP fluorescence, the sciatic nerve segment proximal to CCI ligation demonstrated marked sensory fiber PACAP expression (condition × side, F(1,10) = 57.22, p < 0.0001, Figure 2D - 2F). In correspondence, CCI increased the number of PACAP-EGFP+ L3 - L5 DRG neurons (supplying the sciatic nerve) ipsilateral to the injury (L4 DRG condition × side, F(1,8) = 93.12, p < 0.0001, Figure 2A - 2C, Figure S3) and dramatically augmented DRG central axon EGFP fluorescence in laminae III-V of the ipsilateral dorsal horn and in the gracile fasciculus (Figure 2G) projecting to higher order central nuclei. Interestingly, as in previous work, CCI also induced PACAP-EGFP in some ipsilateral ventral horn motor neurons (33). Accordingly, these studies demonstrate that chronic neuropathic pain elevates PACAP expression along multiple neuronal elements in the spino- parabrachioamygdaloid pathway.
Figure 2.
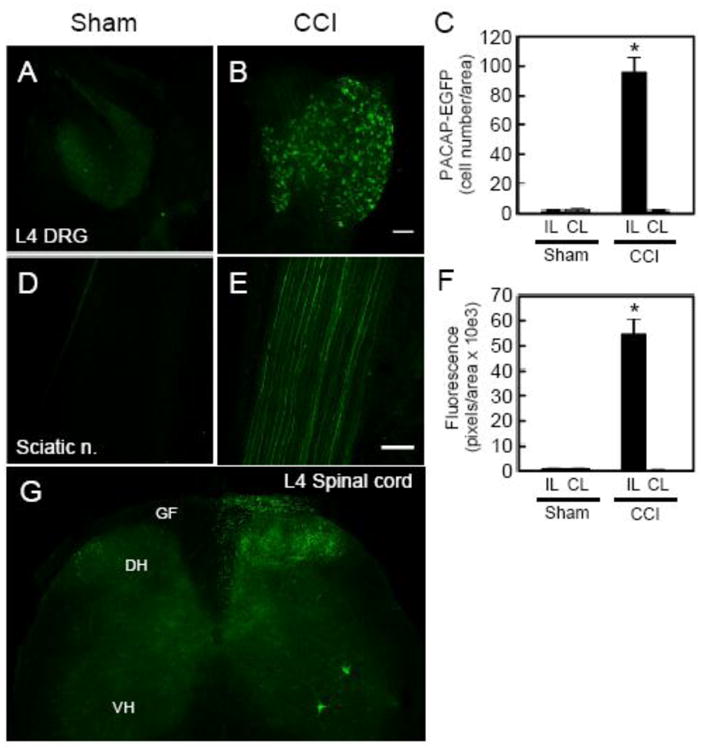
Sensory pathway PACAP expression is enhanced by CCI. Compared to sham surgery controls (A), unilateral partial sciatic nerve CCI (B) induced PACAP-EGFP expression in the ipsilateral L4 dorsal root ganglion (DRG) sensory neurons (C; sham ipsilateral/contralateral = 1.3 0.9 cells/2.0 1.0 cells vs CCI ipsilateral/contralateral = 96.0 9.6 cells/1.3 0.3 cells, F(1,8) = 95.78, *p < 0.0001 Bonferroni's multiple comparison, n = 3 per group). L4 DRG represents the major contributor to mouse sciatic nerve; similar PACAP-EGFP inductions were observed in L3 and L5 DRGs (Figure S3). The increase in CCI induced DRG PACAP expression was also reflected in peripheral and central DRG axons. The ipsilateral sciatic nerve fibers proximal to the ligation demonstrated pronounced PACAP-EGFP fluorescence (E) compared to sham (D) or contralateral control tissues (F; sham ipsilateral/contralateral = 0.8 0.5 units/0.9 0.4 units vs CCI ipsilateral/contralateral = 54.7 6.1 units/0.2 0.1 units; interaction side × condition F(1,10) = 57.22, *p < 0.0001, Bonferroni's multiple comparison, n = 3 - 4 per group). The CCI-induced PACAP-EGFP fluorescence in the central DRG axons were observed in the dorsal horn of L4 spinal cord with prominent projections in the white matter tracts (G). Few PACAP-EGFP neurons were also observed in laminae I of the dorsal horn; there were no apparent differences in the number of second order PACAP neurons between the ipsilateral and contralateral dorsal horn after sciatic nerve injury. CCI also induced PACAP-EGFP expression in the ipsilateral ventral horn motor neurons. Data represent mean cells/unit area or fluorescence units/unit area SEM; DH, dorsal horn; VH, ventral horn; GF, gracile fasciculus. Scale bars = 200 ∼m
CeA PACAP signaling facilitates neuropathic pain-related anxiety-like behaviors and thermal hypersensitivity
We next examined if elevated CeA PACAP signaling in CCI contributes to heightened anxiety-like behaviors and nociceptive sensitivity. Our previous work demonstrated that CeA administration of PACAP or the PAC1 receptor specific agonist maxadilan was capable of producing both anxiety-like behaviors and thermal hypersensitivity (31). But to evaluate the contribution of sustained endogenous CeA PACAP signaling in chronic neuropathic pain, the PAC1 receptor antagonist PACAP(6-38) was infused bilaterally into the CeA of CCI rats before behavioral and hypersensitivity testing (Figure 3A). Similar to chronic stress models, CCI attenuated weight gain; daily weight gain in the CCI group was smaller than that compared to sham (t(28) = 2.92, p = 0.007, Figure 3B). In open field tests 14 days post-surgery, CCI decreased the number of open field center entries more than 50% compared to sham vehicle controls (Figure 3C, Bonferroni's multiple comparison, t(22) = 2.246, p = 0.035). While CeA PACAP(6-38) infusions into the sham control group had no effects on center entries (Bonferroni's multiple comparison t(22) = 0.47, p = 0.9), demonstrating that the antagonist alone had no apparent behavior effects, the pain-associated stress responses in the CCI animals were completely blocked upon CeA PACAP(6-38) administration (Bonferroni's multiple comparison, t(22) = 3.12, p = 0.03, Figure 3C - 3D). These responses were paralleled by a trend for PACAP(6-38) to increase center duration times in CCI (Bonferroni's multiple comparison, t(22) = 2.22, p = 0.07; data not shown). The CCI procedure did not impair locomotion or affect the total distance traveled (F(1,20) = 0.46, p = 0.6); similarly, treatment with PACAP(6-38) did not alter total distance traveled (F(1,20) = 1.22, p = 0.3).
Figure 3.
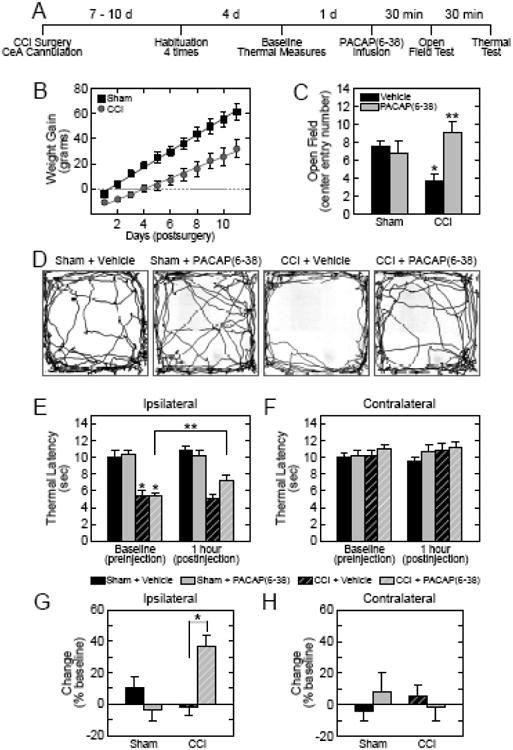
Blocking CeA PACAP signaling attenuates CCI-mediated anxiety-like behavior and thermal nociceptive hypersensitivity. CCI and CeA cannulations were performed concurrently in rats for behavior and nociception studies in the experimental timeline shown (A). The CCI-mediated pain- and stress-related responses was associated with attenuated weight gain compared to sham control animals during the post-surgical recovery period (B). Sham operated animals gained on average 5.7 0.6 gm/day (1.9 0.1%) whereas weight gain in CCI animals was 3.0 0.7 gm/day (1.2 0.4%), (t(28) = 2.92, p = 0.007, n = 8 per group). The pain- and stress-related behavior in CCI was also reflected in decreased center entries in open field tests compared to sham controls (C; sham-vehicle = 7.5 0.7 vs CCI-vehicle = 3.71 0.8, t(22) = 2.246, *p = 0.035, Bonferroni's multiple comparison). CeA infusions in sham operated animals with the PACAP receptor antagonist PACAP(6-38) had no effects on center field entries over the 5 min test period (sham-vehicle = 7.5 0.7 vs sham-PACAP(6-38) = 6.75 1.4, t(22) = 0.47, p = 0.9, Bonferroni's multiple comparison) but blocked the stress- and anxiety-like open field responses in CCI (CCI-vehicle = 3.7 0.8 vs CCI-PACAP(6-38) = 9.0 1.3, t(22) = 3.12, **p = 0.03, Bonferroni's multiple comparison; condition × treatment F(1,22) = 6.78, p = 0.02; n = 5 - 8 per group). Representative movement tracks in open field area for the 4 groups are shown in panel D. There were no significant differences in total distance traveled for either condition or treatment. In Hargreave's thermal nociception assays, CCI increased thermal sensitivity as reflected by decreased baseline latency times in the ipsilateral hindpaw compared to the contralateral leg or in sham animals (E, F(1,21) = 14.13, *p = 0.001). PACAP(6-38) infusions into the CeA attenuated the CCI-induced thermal hypersensitivity compared to baseline (E); simple effect of day in CCI-PACAP6-38 on ipsilateral side (baseline 5.3 0.6 secs vs 7.2 0.7 sec at 1 hour, F(1,21) = 12.21, **p = 0.002) and interaction of condition × side × treatment × day (F(1,21) = 6.29, p = 0.02, n = 5 - 8 animals per group) and within group PACAP(6-38) ameliorated the nociceptive sensitivity. The effects were amplified when the responses of each animal were normalized to their own baseline measures prior to antagonist treatment (G; CCI-Vehicle = -3.4 7.2% vs CCI-PACAP(6-38) = 36.2 6.6%, simple effect of treatment F(1,21) = 16.40, *p = 0.001; interaction of condition × treatment F(1,21) = 15.49, p = 0.001). There were no effects of PACAP(6-38) on thermal latency in the contralateral leg (F and H).
Concurrent with anxiety-related behaviors, CeA PACAP infusions also reliably facilitated nociceptive hypersensitivity in Hargreave's thermal assays that persisted for several hours (31). Sciatic nerve CCI has been used extensively to produce thermal nociceptive responses and comparable to previous work, CCI heightened nociceptive sensitivity as reflected by a 40 - 50% decrease in thermal latency in the ipsilateral hindpaw compared to sham control groups or to the contralateral hindpaw (F(1,21) = 55.02, p < 0.001, Figure 3E). The same animal groups were treated with either vehicle or PAC1 receptor antagonist; 1 h following bilateral CeA PACAP(6-38) administration, the PAC1 receptor antagonist attenuated the heightened thermal nociceptive responses compared to baseline measures (F(1,21) = 12.21, p = 0.009, Figure 3E). The effects were more marked when the responses in each animal were normalized to their own latency baseline immediately prior to the injections (F(1,21) = 16.40, p = 0.001, Figure 3G). There were no significant effects of CeA PACAP(6-38) on the uninjured contralateral hindpaw or in the sham operated condition (Figure 3F and 3H). Accordingly, these results mirrored previous PAC1 receptor antagonist studies, demonstrating that PACAP has no apparent behavioral effects under control sham handling conditions (29), but contributes to heightened anxiety-like behavior and nociceptive sensitivity in chronic neuropathic pain.
CeLC PACAP-mediated ERK signaling in chronic neuropathic pain
One of the most consistent amygdala responses to persistent pain is increased ERK activation in a subset of CeLC neurons. Enhanced amygdala ERK signaling can increase behavioral sensitivity in normal conditions and ERK signaling contributes to PBn - CeLC neurotransmission in persistent pain (10, 36, 37). Conversely, MEK/ERK inhibition in inflammatory pain can decrease behavioral hypersensitivity (10). Comparable to previous work and PACAP data above, unilateral CCI on either the right or left hind limb increased bilaterally the number of CeLC pERK+ cells compared to sham (F(1,26) = 7.62, p = 0.01 Figure 4A - 4C). There was no apparent difference in response relative to the side of injury (ipsilateral vs contralateral, F(1,26) = 0.01, p = 0.9); however, for all CCI, there was an apparent trend towards a greater pERK+ cell number in the right CeLC (left vs right, F(1,26) = 3.16, p = 0.09, Figure S2). Hence similar to other pain models, chronic neuropathic pain enhances ERK signaling in the CeLC.
Figure 4.
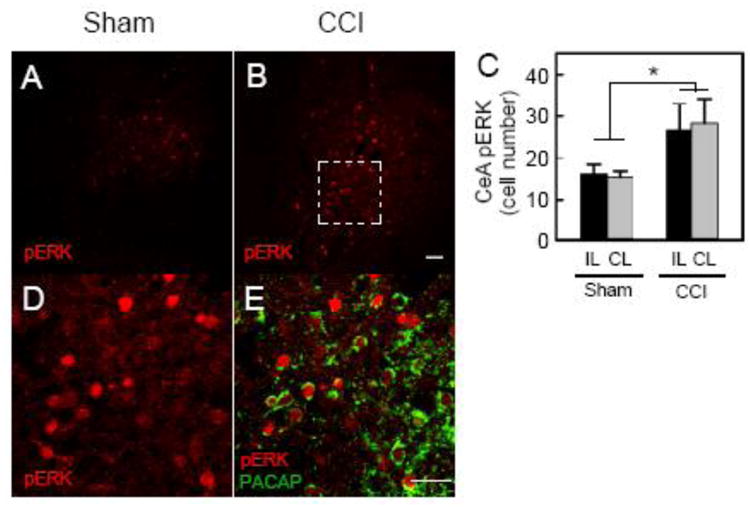
PACAPergic fibers contact CeA activated ERK cells in CCI. CCI produced a bilateral increase in the number CeLC activated pERK+ neurons (Cy3, red) compared to that in the sham condition (A - C; sham ipsilateral/contralateral = 32.1 4.5 cells/30.6 2.8 cells vs CCI ipsilateral/contralateral = 53.4 12.6 cells/56.6 11.9 cells, F(1,26) = 7.62, *p = 0.01, n = 7 - 8 animals per group). When the same sections were dually processed for PACAP immunoreactivity (AlexaFluor 488, green), a majority of the CeLC pERK+ neurons were found in apposition to PACAP-immunoreactive fibers and varicosities (D - E). Data represent mean cells/unit area SEM; IL, ipsilateral; CL, contralateral. Scale bars = 50 ∼m
To evaluate whether CeA PACAP fibers can affect amygdala ERK activation in CCI, pERK and PACAP colocalization was performed (Figure 4D - 4E). Notably, the majority of the CCI pERK+ cells (84.5 5.0%) were in close apposition (< 2 ∼m) with CeLC PACAPergic fibers suggestive of perisomatic signaling. PACAP(6-38) can blocked PAC1 receptor-mediated ERK signaling as in a PAC1-EGFP receptor cell line, and in accord with the biochemical and behavioral studies (Figures 3 and S4A), CeA PACAP(6-38) infusions attenuated pERK+ cell number in CCI (Figure S4B). In good correspondence with previous neuronal PACAP characterizations, the CeLC PACAP fibers were mainly glutamatergic from PACAP colocalization with vGlut2 immunoreactivity (Figure S5); there was little overlap with vGlut1 or glutamic acid decarboxylase (GAD) staining (Figure S6).
Since the BNST displays structural and functional homology with the CeA and also receives LPBn PACAP projections (31), the effects of CCI on BNST pERK were also examined. As in the CeLC, CCI induced a robust increase in anterolateral BNST pERK+ cell number (F(1,8) = 15.04, p = 0.005, Figure S7) which were in close contact with glutamatergic PACAP fibers (83.1 0.5%, Figures S5 and S6). Unlike the CeLC, lateralization of CCI-induced pERK+ cells was not apparent in the BNST. These results implied that BNST PACAP signaling may also have roles in the behavioral consequences of persistent pain which complements previous work (38).
To establish whether CeA PACAP signaling via ERK can evoke thermal hypersensitivity, the MEK inhibitor PD98059 (20 ∼M) was infused into the CeA prior to PACAP38 injection. While infusions of PACAP38 alone resulted in marked increases in CeA c-Fos and pERK immunoreactivity in the same neurons (Figure 5D, 5G and 5J; co-incidence = 87%), MEK inhibitor pretreatments abolished the PACAP-stimulated responses, demonstrating that ERK activation is an essential component of PACAP signaling to instigate CeA neuronal activity (Figure 5E, 5H, 6A and 6b). CeA PACAP infusion and signaling within the same study heightened nociception sensitivity as shown by decreased thermal latency times (Figure 6C and 6D); the PACAP responses were completely abolished by MEK inhibition, corroborating that PACAP-mediated ERK signaling is central to CeA nociception processes (Bonferroni's multiple comparison, t(41) = 3.59, p = 0.002, Figure 6C and 6D).
Figure 5.
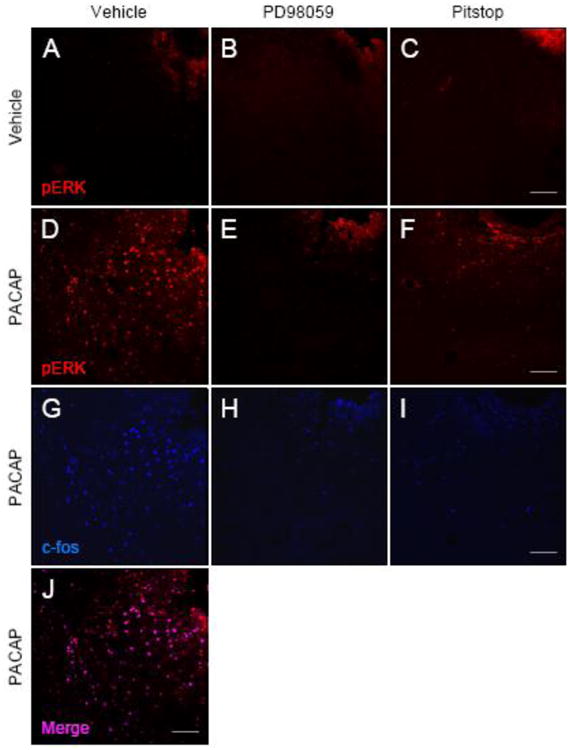
PACAP-mediated CeA ERK and c-Fos activation are blocked by MEK and endocytosis inhibitors. Compared to vehicle (A), CeA PACAP infusion increased the number of activated phosphorylated ERK neurons (D, Cy3 red) which coincided with the increase in neuronal activity marker c- Fos (G, blue; J, pERK/c-Fos merge). Pretreatments with MEK inhibitor PD98059 (B, E, H) or clathrin-mediated endocytosis inhibitor Pitstop 2 (C, F, I) blocked the ability of PACAP to induce ERK phosphorylation or c-Fos in CeA neurons. Representative data from n = 7 – 8 per group. Scale bar: 100 ∼m.
Figure 6.
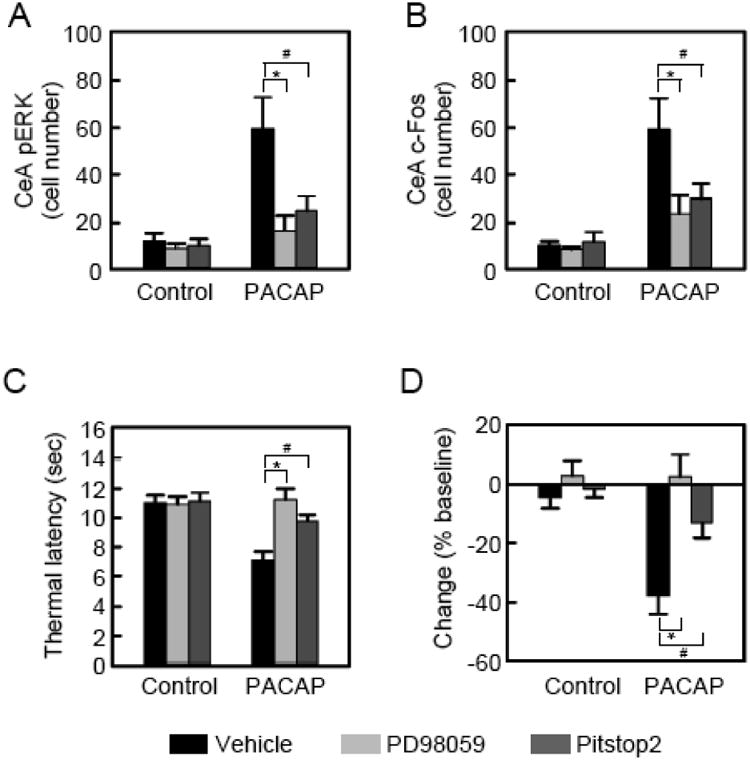
PACAP receptor internalization and ERK activation participate in CeA-mediate nociceptive hypersensitivity. A, Quantitative data from Figure 5. PACAP infusions increased the number of pERK+ neurons in the CeA compared to vehicle controls (F(1,40) = 17.09, p = 0.0002). The increase in PACAP-stimulated ERK activation was attenuated approximately 60 -70% by PD98059 (vehicle + PACAP = 149.0 33.1 cells vs PD98059 + PACAP = 41.9 15.9 cells, t(40) = 4.49, *p = 0.0001, Bonferroni's multiple comparison) and Pitstop 2 (veh + PACAP = 149.0 33.1 cells vs Pitstop2 + PACAP = 62.7 14.2 cells, t(40) = 3.50, #p = 0.002, Bonferroni's multiple comparison; pretreatment × treatment F(2,40) = 4.67, p = 0.02). B, Quantitative data from Figure 5. PACAP injections increased the number of c-Fos+ neurons in the CeA compared to vehicle controls (F(1,40) = 22.45, p < 0.0001). The increase in PACAP-stimulated c-Fos levels activation was attenuated approximately 50 - 60% by PD98059 (cells/unit area, vehicle + PACAP = 148.5 32.3 cells vs. PD98059 + PACAP = 59.8 18.7 cells, t(40) = 3.62, *p=0.002, Bonferroni's multiple comparison) and Pitstop 2 (vehicle + PACAP = 148.5 32.3 cells vs. Pitstop2 + PACAP = 74.4 15.8 cells, t(40) = 2.92, #p=0.02, Bonferroni's multiple comparison; pretreatment × treatment F(2,40) = 3.46, p = 0.04). Data represent mean cell number SEM; n = 7 - 8 per group. C, Commensurate with ERK activation, CeA PACAP injection induced nociceptive hypersensitivity in decreasing thermal latency (F(1,41 = 11.53, p = 0.002). Inhibition of either MEK or endocytosis blocked the PACAP-induced thermal sensitivity (M; latency in sec, Veh + PACAP = 7.1 0.6 sec vs PD98059 + PACAP = 11.2 0.8, t(42) = 5.05, *p < 0.0001, Bonferroni's multiple comparison; Veh + PACAP = 7.1 0.6 vs Pitstop2 + PACAP = 9.8 0.4, t(41) = 3.31, #p = 0.004, Bonferroni's multiple comparison, pretreatment × treatment F(2,41) = 6.64, p = 0.003). D, Expressed as percent change from baseline measures of each animal before drug administration, MEK inhibition (% latency change from vehicle control; PACAP = -37.8 5.9% vs. PD98059 + PACAP = 2.6 4.8%, t(41) = 5.58, *p=0.0001 Bonferroni's multiple comparison) and endocytosis inhibitor Pitstop 2 (PACAP = -37.8 5.9% vs Pitstop + PACAP = -13.5 4.9%, t(40) = 3.36, #p=0.003, Bonferroni's multiple comparison) attenuated nociceptive hypersensitivity. Data represent mean SEM, n = 7 - 8 per group.
There are several potential mechanisms for PAC1 receptors to engage MEK/ERK pathways including PKA and/or PKC activation (39-42). However, PAC1 receptor internalization into signaling endosomes has also been shown to be an alternative and efficacious means of ERK phosphorylation to sustain cell stimulation (41, 42). Blocking PAC1 receptor internalization at ambient temperature conditions or with endocytosis inhibitors substantially attenuated ERK phosphorylation. Even brief PACAP treatments of PAC1-EGFP receptor cells in vitro resulted in PAC1 receptor internalization into vesicles that colocalized with endosomal markers (data not shown) and ˆ-arrestin, a GPCR-associated scaffolding protein important for endosomal MEK/ERK signaling (Figure S8) (43, 44). Further, pre-treatments of the PAC1 receptor cell line with PACAP(6-38) blocked PACAP-stimulated PAC1-EGFP receptor internalization, in parallel with diminished ERK activation (Figure S9). These observations implicated endogenous CeA PAC1 receptor endosomal ERK signaling in nociception, and hence, contiguous with the previous MEK inhibitor experiment, a separate experimental group was pretreated with Pitstop 2 (30 ∼M), an inhibitor of clathrin-mediated endocytosis, prior to PACAP infusion. Consistent with cell culture data (42), Pitstop 2 pretreatments markedly block PACAP-mediated ERK phosphoryation and c-Fos expression in the CeA (Figure 5F and 5I). Importantly, inhibition of clathrin-mediated endocytosis reduced PACAP-induced hypersensitivity (Bonferroni's multiple comparison, t(41) = 2.57, p = 0.03, Figure 6C and 6D). Neither PD98059 nor Pitstop 2 produced CeA damage or cellular apoptosis (Figure S10). In aggregate, these studies provide in vivo evidence that GPCR PAC1 receptor internalization and downstream ERK signaling can modulate CeA nociception responses.
Discussion
The current studies establish roles for CeA PACAP signaling as an effector conveying the behavioral and sensory consequences of chronic neuropathic pain. Among several lines of evidence, CCI increased PACAP transcripts and neurons in the LPBn which correlated with enhanced LPBn PACAP projection fiber immunoreactivity in the CeLC, and increased PACAP expression in the spino-parabrachioamygdaloid tract. In good agreement with previous studies demonstrating the anxiety-related and nociceptive hypersensitivity responses following CeA PACAP administration (31), blockade of endogenous PACAP signaling in CCI with PAC1 receptor antagonist attenuated the CCI neuropathic pain-induced heightened anxiety-like behavior in the open field tests and nociceptive hypersensitivity in thermal assays. Importantly, both CCI and PACAP stimulated CeA ERK activation and c-Fos expression, which were diminished upon pretreatments with MEK or clathrin-mediated endocytosis inhibitors in parallel with diminished PACAP-induced nociceptive hypersensitivity. These results further our understandings of CNS PACAP mechanisms and functions, and how maladaption in PACAP signaling in intersecting stress-related and pain circuits may negatively impact the course of psychopathologies.
Previous studies have shown PACAP neurophenotypic plasticity and demonstrated that central and peripheral neuronal PACAP expression can be upregulated in response to diverse homeostatic challenges. In a chronic stress paradigm, heightened PACAP and PAC1 receptor transcript expression was observed in the BNST and paraventricular nucleus of the hypothalamus (28). In several nerve injury models, PACAP was elevated in sensory, autonomic and motor neurons (32, 33, 45). The recent availability of the PACAP-EGFP mice has illustrated the importance of that plasticity. Whereas basal endogenous PACAP levels appeared low in many neuronal systems, physiological challenges especially nerve injury significantly induced PACAP expression. Consistent with previous results, CCI increased DRG PACAP expression which augmented dramatically PACAP levels in both peripheral sciatic sensory nerve fibers and central DRG axons in the dorsal horn and spinal pathways. Potential second order PACAP producing neurons were found in lamina I/II of the dorsal horn but notably CCI increased PACAP expression centrally in the LPBn and CeA as a consequence of enhanced nociceptive signaling in the spino-parabrachioamygdaloid pathway. The second order dorsal horn neurons project to the brain bilaterally, yet upon completion of all analyses, PACAP and pERK immunoreactivity was preferentially heightened in the right CeA, irrespective of the side of injury. These studies agreed with those suggesting CeA lateralization, with the right CeA displaying greater increases in pERK and synaptic potentiation in response to pain (34, 35). Interestingly, despite evidence for bilateral LPBn to BNST projections, BNST pERK lateralization was not apparent in these studies. The injury mechanisms underlying the induction of phenotypically plastic peptides are not well understood but uniquely, these studies demonstrate PACAP expression at multiple levels along the spino-parabrachio-amygdaloid pathway suggesting that PACAP is a prominent physiological neuroregulator in this circuit.
Following CCI, a two week postsurgical recovery period was established to allow locomotor return from transient deficits, injury-induced PACAP expression, and the development of chronic pain hypersensitivity and stress-related behaviors for multiple nociceptive and behavioral assessments. As many weeks of CCI have been shown to facilitate anxiodepressive-like disorders (46) and PACAP has been implicated in anxiety- and depression-related behaviors (15, 29, 47, 48), the increase in PACAP expression and signaling may be a mechanism underlying the development of psychopathologies.
To evaluate whether continued CeA PACAP signaling participates in these heightened pain and behavioral responses, the PAC1/VPAC2 receptor antagonist PACAP(6-38) was infused into the CeA before testing. The infusion of PACAP(6-38) alone into sham control animals had no effects on either pain or stress-related behaviors, suggesting that PACAP signaling under basal conditions may be low and not to significantly impact the normal course of CeA functions. The ability for acute PACAP(6-38) treatments to mitigate anxiety-like behavior and thermal hypersensitivity responses during chronic injury suggested that the increase in CeA PACAP levels and signaling was sustained during the course of CCI to facilitate the pain-related behavioral responses. The involvement of CeA PACAP only in a state of persistent pain and/or stress and not under normal conditions is comparable to observations for other CeA systems including CGRP, CRH and mGluR regulated-functions (8, 49-51). A majority of PBn neurons projecting to the CeLC coexpress PACAP and CGRP, and from recent work, the two peptides may have similar or complementary roles in transmitting nociceptive signals to the amygdala (31, 52). The mechanisms through which CCI-induced CeA PACAP may result in anxiety-like behaviors is not clear but may involve the potentiation of basolateral amygdala (BLA) excitatory postsynaptic transmission to the CeL (53). Similarly LPBn PACAP projections to the BNST may not only have anxiogenic but hyperalgesic attributes by interactions with CRH systems (38). Hence, CCI-induced LPBn PACAP expression and release could heighten nociceptive hypersensitivity and anxiety-like behaviors via multiple mechanisms with projections facilitating BLA to CeL neurotransmission, modulating descending inhibitory signals, altering BNST function or enhancing CeLC nociceptive signals to the substantia innominata dorsalis for anxiety, aversion and fear responding (54).
ERK activation is a central means of nociceptive signaling in a variety of pain models. The ability for MEK inhibition to attenuate CeA PACAP-stimulated pERK and c-Fos in parallel with blockade of PACAP-induced thermal sensitivity demonstrated that PACAP/PAC1 receptor-mediated ERK signaling is requisite for CeA nociceptive hypersensitivity responses. Although there are several mechanisms by which PAC1 receptors can activate ERK, the recent observations that PAC1 receptor can participate in endosomal MEK signaling implicated a key mechanism for prolonged intracellular ERK signaling. PACAP induced PAC1 receptor internalization into endosomes and PAC1 receptor antagonism blocked concomitantly both receptor internalization and ERK activation in vitro. As with MEK inhibitors, Pitstop 2, an inhibitor of clathrin mediated endocytosis also blocked PACAP-mediated CeA pERK and c-Fos levels and attenuated PACAP-mediated nociceptive hypersensitivity responses. These results may be one demonstration of how GPCR internalization and endosomal signaling may be relevant in physiological processes and in particular nociceptive mechanisms.
In summary, our results demonstrate that spino-parabrachioamygdaloid PACAP expression and signaling are augmented in neuropathic pain and that this heightened expression may contribute to adverse pain- and stress-related behaviors. While clinical data have placed emphasis on the dysregulation of inhibitory pathways as mechanisms underlying pain-associated psychopathologies, maladaptations from ascending activating pathways including neurophenotypically plastic PACAPergic system may be contributory to that process. CeA PAC1 receptor antagonism or inhibition of downstream endosomal ERK signaling can block PACAP- and CCI-induced nociceptive hypersensitivity and associated anxiety-like responses. As PACAP receptor antagonism during CCI advancement can still ameliorate the adverse neuropathic pain and behavioral responses, these observations suggest that interventions in PACAP signaling during the progression of pain and associated behavioral responses may have therapeutic utility in improving disorder outcomes.
Supplementary Material
Acknowledgments
We thank R. L. Parsons for critical discussions of the data in the manuscript and A. B. Howard for assistance in statistical data analyses. This work was supported in part by National Institutes of Health grants MH-97988 and MH-072088 (SEH) and MH096764 (KJR). Portions of the work were also supported by funds from the Center of Biomedical Research Excellence (COBRE) in Neuroscience at the University of Vermont (National Institute of Health NIGMS P30 GM103498/NCRR P30 RR032135.
Footnotes
Financial Disclosures: All authors report no biomedical financial interests or potential conflicts of interest.
Author Contributions: G. M., V. M and K. M. B. conceived the project, analyzed the data and wrote the manuscript. G. M. and L. M. executed the experiments; M. A. V., J. A. W., S. E. H and K. J. R. assisted in the data analyses, and provided critical experimental guidance, comments and editing of the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Asmundson GJ, Katz J. Understanding the co-occurrence of anxiety disorders and chronic pain: state-of-the-art. Depress Anxiety. 2009;26:888–901. doi: 10.1002/da.20600. [DOI] [PubMed] [Google Scholar]
- 2.Scioli-Salter ER, Forman DE, Otis JD, Gregor K, Valovski I, Rasmusson AM. The shared neuroanatomy and neurobiology of comorbid chronic pain and PTSD: therapeutic implications. Clin J Pain. 2015;31:363–374. doi: 10.1097/AJP.0000000000000115. [DOI] [PubMed] [Google Scholar]
- 3.Veinante P, Yalcin I, Barrot M. The amygdala between sensation and affect: a role in pain. Journal of Molecular Psychiatry. 2013;1:9. doi: 10.1186/2049-9256-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernard JF, Bester H, Besson JM. Involvement of the spino-parabrachio - amygdaloid and -hypothalamic pathways in the autonomic and affective emotional aspects of pain. Prog Brain Res. 1996;107:243–255. doi: 10.1016/s0079-6123(08)61868-3. [DOI] [PubMed] [Google Scholar]
- 5.Gauriau C, Bernard JF. Pain pathways and parabrachial circuits in the rat. Exp Physiol. 2002;87:251–258. doi: 10.1113/eph8702357. [DOI] [PubMed] [Google Scholar]
- 6.Rouwette T, Vanelderen P, Roubos EW, Kozicz T, Vissers K. The amygdala, a relay station for switching on and off pain. Eur J Pain. 2012;16:782–792. doi: 10.1002/j.1532-2149.2011.00071.x. [DOI] [PubMed] [Google Scholar]
- 7.Todd AJ. Neuronal circuitry for pain processing in the dorsal horn. Nat Rev Neurosci. 2010;11:823–836. doi: 10.1038/nrn2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neugebauer V, Li W, Bird GC, Bhave G, Gereau RWt. Synaptic plasticity in the amygdala in a model of arthritic pain: differential roles of metabotropic glutamate receptors 1 and 5. J Neurosci. 2003;23:52–63. doi: 10.1523/JNEUROSCI.23-01-00052.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bird GC, Lash LL, Han JS, Zou X, Willis WD, Neugebauer V. Protein kinase A-dependent enhanced NMDA receptor function in pain-related synaptic plasticity in rat amygdala neurones. J Physiol. 2005;564:907–921. doi: 10.1113/jphysiol.2005.084780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carrasquillo Y, Gereau RWt. Activation of the extracellular signal-regulated kinase in the amygdala modulates pain perception. J Neurosci. 2007;27:1543–1551. doi: 10.1523/JNEUROSCI.3536-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu YC, Chen YZ, Wei YY, He XT, Li X, Hu W, et al. Neurochemical properties of the synapses between the parabrachial nucleus-derived CGRP-positive axonal terminals and the GABAergic neurons in the lateral capsular division of central nucleus of amygdala. Mol Neurobiol. 2015;51:105–118. doi: 10.1007/s12035-014-8713-x. [DOI] [PubMed] [Google Scholar]
- 12.Ren W, Neugebauer V. Pain-related increase of excitatory transmission and decrease of inhibitory transmission in the central nucleus of the amygdala are mediated by mGluR1. Mol Pain. 2010;6:93. doi: 10.1186/1744-8069-6-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han JS, Adwanikar H, Li Z, Ji G, Neugebauer V. Facilitation of synaptic transmission and pain responses by CGRP in the amygdala of normal rats. Mol Pain. 2010;6:10. doi: 10.1186/1744-8069-6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vaudry D, Falluel-Morel A, Bourgault S, Basille M, Burel D, Wurtz O, et al. Pituitary adenylate cyclase-activating polypeptide and its receptors: 20 years after the discovery. Pharmacol Rev. 2009;61:283–357. doi: 10.1124/pr.109.001370. [DOI] [PubMed] [Google Scholar]
- 15.Ressler KJ, Mercer KB, Bradley B, Jovanovic T, Mahan A, Kerley K, et al. Post-traumatic stress disorder is associated with PACAP and the PAC1 receptor. Nature. 2011;470:492–497. doi: 10.1038/nature09856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Almli LM, Mercer KB, Kerley K, Feng H, Bradley B, Conneely KN, et al. ADCYAP1R1 genotype associates with post-traumatic stress symptoms in highly traumatized African-American females. Am J Med Genet B Neuropsychiatr Genet. 2013;162B:262–272. doi: 10.1002/ajmg.b.32145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jovanovic T, Norrholm SD, Davis J, Mercer KB, Almli L, Nelson A, et al. PAC1 receptor (ADCYAP1R1) genotype is associated with dark-enhanced startle in children. Mol Psychiatry. 2013;18:742–743. doi: 10.1038/mp.2012.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uddin M, Chang SC, Zhang C, Ressler K, Mercer KB, Galea S, et al. Adcyap1r1 genotype, posttraumatic stress disorder, and depression among women exposed to childhood maltreatment. Depress Anxiety. 2013;30:251–258. doi: 10.1002/da.22037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pohlack ST, Nees F, Ruttorf M, Cacciaglia R, Winkelmann T, Schad LR, et al. Neural mechanism of a sex-specific risk variant for posttraumatic stress disorder in the type I receptor of the pituitary adenylate cyclase activating polypeptide. Biol Psychiatry. 2015;78:840–847. doi: 10.1016/j.biopsych.2014.12.018. [DOI] [PubMed] [Google Scholar]
- 20.Hashimoto H, Shintani N, Tanaka K, Mori W, Hirose M, Matsuda T, et al. Altered psychomotor behaviors in mice lacking pituitary adenylate cyclase-activating polypeptide (PACAP) Proc Natl Acad Sci USA. 2001;98:13355–13360. doi: 10.1073/pnas.231094498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Girard BA, Lelievre V, Braas KM, Razinia T, Vizzard MA, Ioffe Y, et al. Noncompensation in peptide/receptor gene expression and distinct behavioral phenotypes in VIP- and PACAP-deficient mice. J Neurochem. 2006;99:499–513. doi: 10.1111/j.1471-4159.2006.04112.x. [DOI] [PubMed] [Google Scholar]
- 22.Hattori S, Takao K, Tanda K, Toyama K, Shintani N, Baba A, et al. Comprehensive behavioral analysis of pituitary adenylate cyclase-activating polypeptide (PACAP) knockout mice. Front Behav Neurosci. 2012;6:58. doi: 10.3389/fnbeh.2012.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Otto C, Martin M, Wolfer DP, Lipp HP, Maldonado R, Schutz G. Altered emotional behavior in PACAP-type-I-receptor-deficient mice. Brain Res Mol Brain Res. 2001;92:78–84. doi: 10.1016/s0169-328x(01)00153-x. [DOI] [PubMed] [Google Scholar]
- 24.Stroth N, Eiden LE. Stress hormone synthesis in mouse hypothalamus and adrenal gland triggered by restraint is dependent on pituitary adenylate cyclase-activating polypeptide signaling. Neuroscience. 2010;165:1025–1030. doi: 10.1016/j.neuroscience.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsukiyama N, Saida Y, Kakuda M, Shintani N, Hayata A, Morita Y, et al. PACAP centrally mediates emotional stress-induced corticosterone responses in mice. Stress. 2011;14:368–375. doi: 10.3109/10253890.2010.544345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Botz B, Imreh A, Sandor K, Elekes K, Szolcsanyi J, Reglodi D, et al. Role of pituitary adenylate-cyclase activating polypeptide and Tac1 gene derived tachykinins in sensory, motor and vascular functions under normal and neuropathic conditions. Peptides. 2013;43:105–112. doi: 10.1016/j.peptides.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 27.Jongsma H, Pettersson LM, Zhang Y, Reimer MK, Kanje M, Waldenstrom A, et al. Markedly reduced chronic nociceptive response in mice lacking the PAC1 receptor. Neuroreport. 2001;12:2215–2219. doi: 10.1097/00001756-200107200-00034. [DOI] [PubMed] [Google Scholar]
- 28.Hammack SE, Cheung J, Rhodes KM, Schutz KC, Falls WA, Braas KM, et al. Chronic stress increases pituitary adenylate cyclase-activating peptide (PACAP) and brain-derived neurotrophic factor (BDNF) mRNA expression in the bed nucleus of the stria terminalis (BNST): roles for PACAP in anxiety-like behavior. Psychoneuroendocrinology. 2009;34:833–843. doi: 10.1016/j.psyneuen.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roman CW, Lezak KR, Hartsock MJ, Falls WA, Braas KM, Howard AB, et al. PAC1 receptor antagonism in the bed nucleus of the stria terminalis (BNST) attenuates the endocrine and behavioral consequences of chronic stress. Psychoneuroendocrinology. 2014;47:151–165. doi: 10.1016/j.psyneuen.2014.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lezak KR, Roman CW, Braas KM, Schutz KC, Falls WA, Schulkin J, et al. Regulation of bed nucleus of the stria terminalis PACAP expression by stress and corticosterone. J Mol Neurosci. 2014;54:477–484. doi: 10.1007/s12031-014-0269-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Missig G, Roman CW, Vizzard MA, Braas KM, Hammack SE, May V. Parabrachial nucleus (PBn) pituitary adenylate cyclase activating polypeptide (PACAP) signaling in the amygdala: implication for the sensory and behavioral effects of pain. Neuropharmacology. 2014;86:38–48. doi: 10.1016/j.neuropharm.2014.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang YZ, Hannibal J, Zhao Q, Moller K, Danielsen N, Fahrenkrug J, et al. Pituitary adenylate cyclase activating peptide expression in the rat dorsal root ganglia: up-regulation after peripheral nerve injury. Neuroscience. 1996;74:1099–1110. doi: 10.1016/0306-4522(96)00168-6. [DOI] [PubMed] [Google Scholar]
- 33.Pettersson LM, Dahlin LB, Danielsen N. Changes in expression of PACAP in rat sensory neurons in response to sciatic nerve compression. Eur J Neurosci. 2004;20:1838–1848. doi: 10.1111/j.1460-9568.2004.03644.x. [DOI] [PubMed] [Google Scholar]
- 34.Carrasquillo Y, Gereau RWt. Hemispheric lateralization of a molecular signal for pain modulation in the amygdala. Mol Pain. 2008;4:24. doi: 10.1186/1744-8069-4-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ji G, Neugebauer V. Hemispheric lateralization of pain processing by amygdala neurons. J Neurophysiol. 2009;102:2253–2264. doi: 10.1152/jn.00166.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ji RR, G RW, 4th, Malcangio M, Strichartz GR. MAP kinase and pain. Brain Res Rev. 2009;60:135–148. doi: 10.1016/j.brainresrev.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng SJ, Chen CC, Yang HW, Chang YT, Bai SW, Chen CC, et al. Role of extracellular signal-regulated kinase in synaptic transmission and plasticity of a nociceptive input on capsular central amygdaloid neurons in normal and acid-induced muscle pain mice. J Neurosci. 2011;31:2258–2270. doi: 10.1523/JNEUROSCI.5564-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tran L, Schulkin J, Greenwood-Van Meerveld B. Importance of CRF receptor-mediated mechanisms of the bed nucleus of the stria terminalis in the processing of anxiety and pain. Neuropsychopharmacology. 2014;39:2633–2645. doi: 10.1038/npp.2014.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barrie AP, Clohessy AM, Buensuceso CS, Rogers MV, Allen JM. Pituitary adenylyl cyclase-activating peptide stimulates extracellular signal-regulated kinase 1 or 2 (ERK1/2) activity in a Ras-independent, mitogen-activated protein kinase/ERK kinase 1 or -dependent manner in PC12 cells. J Biol Chem. 1997;272:19666–19671. doi: 10.1074/jbc.272.32.19666. [DOI] [PubMed] [Google Scholar]
- 40.Bouschet T, Perez V, Fernandez C, Bockaert J, Eychene A, Journot L. Stimulation of the ERK pathway by GTP-loaded Rap1 requires the concomitant activation of Ras, protein kinase C, and protein kinase A in neuronal cells. J Biol Chem. 2003;278:4778–4785. doi: 10.1074/jbc.M204652200. [DOI] [PubMed] [Google Scholar]
- 41.May V, Lutz E, MacKenzie C, Schutz KC, Dozark K, Braas KM. Pituitary adenylate cyclase-activating polypeptide (PACAP)/PAC1HOP1 receptor activation coordinates multiple neurotrophic signaling pathways: Akt activation through phosphatidylinositol 3-kinase and vesicle endocytosis for neuronal survival. J Biol Chem. 2010;285:9749–9761. doi: 10.1074/jbc.M109.043117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.May V, Buttolph TR, Girard BM, Clason TA, Parsons RL. PACAP-induced ERK activation in HEK cells expressing PAC1 receptors involves both receptor internalization and PKC signaling. Am J Physiol Cell Physiol. 2014;306:C1068–C1079. doi: 10.1152/ajpcell.00001.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jean-Alphonse F, Hanyaloglu AC. Regulation of GPCR signal networks via membrane trafficking. Mol Cell Endocrinol. 2011;331:205–214. doi: 10.1016/j.mce.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 44.Luttrell LM, Gesty-Palmer D. Beyond desensitization: phyisiological relevance of arrestin-dependent signaling. Pharmacol Rev. 2010;62:305–330. doi: 10.1124/pr.109.002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moller K, Reimer M, Ekblad E, Hannibal J, Fahrenkrug J, Kanje M, et al. The effects of axotomy and preganglionic denervation on the expression of pituitary adenylate cyclase activating peptide (PACAP), galanin and PACAP type 1 receptors in the rat superior cervical ganglion. Brain Res. 1997;775:166–182. doi: 10.1016/s0006-8993(97)00923-2. [DOI] [PubMed] [Google Scholar]
- 46.Alba-Delgado C, Llorca-Torralba M, Horrillo I, Ortega JE, Mico JA, Sanchez-Blazquez P, et al. Chronic pain leads to concomitant noradrenergic impairment and mood disorders. Biol Psychiatry. 2013;73:54–62. doi: 10.1016/j.biopsych.2012.06.033. [DOI] [PubMed] [Google Scholar]
- 47.Hammack SE, May V. Pituitary adenylate cyclase activating polypeptide in stress-related disorders: data convergence from animal and human studies. Biol Psychiatry. 2014;78:167–177. doi: 10.1016/j.biopsych.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lehmann ML, Mustafa T, Eiden AM, Herkenham M, Eiden LE. PACAP-deficient mice show attenuated corticosterone secretion and fail to develop depressive behavior during chronic social defeat stress. Psychoneuroendocrinology. 2013;38:702–715. doi: 10.1016/j.psyneuen.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Han JS, Li W, Neugebauer V. Critical role of calcitonin gene-related peptide 1 receptors in the amygdala in synaptic plasticity and pain behavior. J Neurosci. 2005;25:10717–10728. doi: 10.1523/JNEUROSCI.4112-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fu Y, Neugebauer V. Differential mechanisms of CRF1 and CRF2 receptor functions in the amygdala in pain-related synaptic facilitation and behavior. J Neurosci. 2008;28:3861–3876. doi: 10.1523/JNEUROSCI.0227-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Adwanikar H, Ji G, Li W, Doods H, Willis WD, Neugebauer V. Spinal CGRP1 receptors contribute to supraspinally organized pain behavior and pain-related sensitization of amygdala neurons. Pain. 2007;132:53–66. doi: 10.1016/j.pain.2007.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Han S, Soleiman MT, Soden ME, Zweifel LS, Palmiter RD. Elucidating an Affective Pain Circuit that Creates a Threat Memory. Cell. 2015;162:363–374. doi: 10.1016/j.cell.2015.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cho JH, Zushida K, Shumyatsky GP, Carlezon WAJ, Meloni EG, Bolshakov VY. Pituitary adenylate cyclase-activating polypeptide induces postsynaptically expressed potentiation in the intra-amygdala circuit. J Neurosci. 2012;32:14165–14177. doi: 10.1523/JNEUROSCI.1402-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bourgeais L, Gauriau C, Bernard JF. Projections from the nociceptive area of the central nucleus of the amygdala to the forebrain: a PHA-L study in the rat. Eur J Neurosci. 2001;14(2):229–255. doi: 10.1046/j.0953-816x.2001.01640.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.