

Abstract
Although experimental studies suggest that low and oscillatory wall shear stress (WSS) promotes plaque transformation to a more vulnerable phenotype, this relationship has not been examined in human atherosclerosis progression. Thus, the aim of this investigation was to examine the association between oscillatory WSS, in combination with WSS magnitude, and coronary atherosclerosis progression. We hypothesized that regions of low and oscillatory WSS will demonstrate progression towards more vulnerable lesions, while regions exposed to low and non-oscillatory WSS will exhibit progression towards more stable lesions. Patients (n = 20) with non-flow-limiting coronary artery disease (CAD) underwent baseline and six-month follow-up angiography, Doppler velocity and radiofrequency intravascular ultrasound (VH-IVUS) acquisition. Computational fluid dynamics models were constructed to compute time-averaged WSS magnitude and oscillatory WSS. Changes in VH-IVUS-defined total plaque and constituent areas were quantified in focal regions (i.e. sectors; n = 14 235) and compared across haemodynamic categories. Compared with sectors exposed to low WSS magnitude, high WSS sectors demonstrated regression of total plaque area (p < 0.001) and fibrous tissue (p < 0.001), and similar progression of necrotic core. Sectors subjected to low and oscillatory WSS exhibited total plaque area regression, while low and non-oscillatory WSS sectors demonstrated total plaque progression (p < 0.001). Furthermore, compared with low and non-oscillatory WSS areas, sectors exposed to low and oscillatory WSS demonstrated regression of fibrous (p < 0.001) and fibrofatty (p < 0.001) tissue and similar progression of necrotic core (p = 0.82) and dense calcium (p = 0.40). Herein, we demonstrate that, in patients with non-obstructive CAD, sectors subjected to low and oscillatory WSS demonstrated regression of total plaque, fibrous and fibrofatty tissue, and progression of necrotic core and dense calcium, which suggest a transformation to a more vulnerable phenotype.
Keywords: atherosclerosis, coronary artery disease, computational fluid dynamics, haemodynamics, intravascular ultrasound, wall shear stress
1. Introduction
Despite genetic and systemic risk factors, coronary atherosclerotic lesions are discretely distributed within the epicardial coronary vessels [1] with a predilection towards focal sites of complex haemodynamics, i.e. regions exposed to low time-averaged magnitude and temporally oscillatory wall shear stress (herein, low and oscillatory WSS) [2,3]. WSS is a spatially localized vector quantity, defined by both magnitude and direction, that is time-varying due to the pulsatile nature of arterial blood flow. Thus, complete evaluation of the local blood flow environment requires characterization of vector magnitudes and directional changes across the cardiac cycle. In vitro studies have demonstrated that low and oscillatory WSS creates a pro-atherogenic environment for lesion development mediated through altered endothelial cell gene expression [4] and function [4,5]. Furthermore, a recent porcine study reported that regions exposed to low and oscillatory WSS resulted in the formation of advanced atherosclerotic plaques, including thin cap fibroatheromas (TCFAs) [6].
Clinical studies have demonstrated plaque progression in coronary segments exposed to low time-averaged WSS magnitude [7,8] and increased plaque vulnerability in regions of high WSS [7]. However, the impact of oscillatory WSS and the co-localization of regions of low and oscillatory WSS has not been investigated in the clinical setting of plaque progression. We hypothesized that co-localized regions of low and oscillatory WSS will exhibit progression of necrotic core with regression of total plaque and fibrous tissue area, suggestive of greater plaque vulnerability, while regions exposed to low and non-oscillatory WSS will demonstrate progression of total plaque and fibrous tissue area, suggestive of progression towards more stable lesions. Accordingly, we employed our recently developed framework [9], which allows for the focal quantification of coronary haemodynamics and plaque progression, to investigate the relationship between oscillatory WSS in combination with WSS magnitude and coronary artery disease (CAD) progression in patients with non-obstructive CAD treated with optimal medical therapy.
2. Material and methods
2.1. Study population and clinical data acquisition
Patients (n = 20) enrolled in a prospective study evaluating the association between WSS and radiofrequency intravascular ultrasound (i.e. virtual histology–intravascular ultrasound; VH-IVUS) defined plaque progression were included in this investigation [7]. Patients presenting with an abnormal non-invasive stress test or stable angina and determined to have a non-obstructive lesion requiring physiological evaluation were enrolled. Patients underwent baseline and six-month follow-up biplane angiographic and IVUS image (phased-array 20 MHz Eagle Eye® Gold Catheter; Volcano Corp., San Diego, CA, USA) acquisition (figure 1). Electrocardiogram-gated greyscale and radiofrequency data were continuously acquired (0.5 mm s−1 motorized pullback) from the distal left anterior descending (LAD) coronary artery up to the guide catheter in the aorta, sampling approximately 60 mm of the proximal vessel and stored for offline analysis. Doppler-derived velocity data were acquired in the left main (LM) coronary arteries with a 0.014″ (0.3556 mm) monitoring guidewire (ComboWire; Volcano Corp.). Lipid profiling was performed at baseline and follow-up, and patients received optimal medical therapy for cardiovascular risk factors, including 80 mg atorvastatin daily. Emory University's Institutional Review Board approved the study and each patient provided informed consent.
Figure 1.

Schematic of the presented study. Multi-modal clinical imaging data and intracoronary haemodynamic measures were collected in the cardiac catheterization laboratory. The imaging data were used to construct the 3D lumen geometry, flow extensions were added and the geometry discretized. Acquired patient-specific haemodynamic data were prescribed as boundary conditions, and pulsatile simulations were performed to evaluate the flow field. These computed data were post-processed to quantify the haemodynamics metrics of interest (e.g. wall shear stress).
2.2. Anatomical reconstruction and computational fluid dynamics modelling
The end-diastolic three-dimensional (3D) coronary geometry was constructed by fusion of angiographic views and lumen contours segmented from IVUS images as previously described [7,9]. Biplane angiographic images of the IVUS catheter in its most distal position in the LAD prior to pullback were acquired. The 3D spatial position of the catheter was determined via back-projection image analysis (QAngio XA 3D RE; Medis, Lieden, The Netherlands) and served as the backbone for vessel reconstruction. An experienced IVUS reader segmented the lumen and external elastic membrane (EEM) contours from each frame of the IVUS pullback (echoPlaque 4.0; INDEC Medical Systems, Santa Clara, CA, USA). Contours were stacked perpendicular to the catheter centreline at specific locations determined by the catheter pullback speed and identified time step of each image (figure 1). As a result of catheter torsion during the pullback, the relative orientation of each contour was quantified by the sequential triangulation algorithm derived from differential geometry [10]. Major coronary branching vessels (e.g. left circumflex, diagonal, septal coronary arteries) were included in the reconstructions through extraction of diameter and orientation information from angiographic and IVUS data. Incorporation of branch vessels in the computational geometry is critical to ensure correct flow rates in distal vessels through branching flow divisions, and to avoid incorrectly high flow rates (and WSS values) in these distal regions [11]. The reconstruction algorithm resulted in an anatomically correct 3D geometry of the indexed coronary artery.
Computational fluid dynamics (CFD) techniques were employed, as described previously in detail [9], to quantify the patient-specific haemodynamic environment and quantify WSS metrics throughout the reconstructed artery (figure 1). Briefly, a surface was fitted to the reconstructed lumen surface (Geomagic Studio 11; Geomagic, Inc., Research Triangle Park, NC, USA) and flow extensions were added to the inlet (one diameter) and outlets (seven diameters). Preliminary investigations confirmed that the inlet extension length allowed viscous effects to slightly develop the spatially uniform velocity profile into a blunted profile at the coronary main body (figure 1), which agrees with experimental observations [3], and that outlet extension length was sufficiently long not to affect the flow field in the region of interest (coronary main body) and for flow to be fully developed at all outlets [7]. The volume was discretized with tetrahedral and prismatic elements, which were located adjacent to the lumen boundary, and the average mesh size was approximately 1.2 million elements per computational domain (ICEM CFD, ANSYS 15; Ansys, Inc., Canonsburg, PA, USA). The computational mesh was imported into a commercial software package (Fluent 15; ANSYS 15) to numerically solve the Navier–Stokes and continuity equations. Patient-specific pulsatile velocity values were extracted from Doppler data acquired in the LM coronary artery and applied as a series of spatially uniform profiles (i.e. plug flow) at the inlet. All outlets were assumed pressure-free (traction-free). Preliminary validation studies were performed to confirm that application of a pressure-free boundary condition reproduced the in vivo haemodynamic environment, as quantified with invasive intracoronary measurements, throughout the coronary territory of interest. Blood was assumed to be an incompressible Newtonian fluid (ρ = 1.06 g cm−3, μ = 3.5 cP). Following solution convergence, flow-field data were post-processed to quantify instantaneous WSS vectors at all surface nodes. Vector WSS magnitudes were temporally averaged across the cardiac cycle (i.e. time-averaged WSS; figure 2). Temporal oscillatory WSS was quantified at time points (n) throughout the cardiac cycle by the WSS angle deviation (WSSAD) [12], which is defined as
where τi and τj are the instantaneous and time-averaged WSS vectors, respectively. At each node, the maximum WSSAD across the cardiac cycle was used to characterize the oscillatory haemodynamic environment (figure 2). Previously analysis demonstrated solution independence from the mesh resolution, as sector time-averaged WSS values were less than 4% when mesh volume size was reduced by 50% (3.75× initial mesh size) [9]. Note that oscillatory shear index (OSI) [2] values were not quantified due to a lack of a predominant axial direction in the coronary arteries and the difficulty in defining a positive flow direction in a model with multiple outlets; however, analogous to OSI, WSSAD quantifies the deviation of an instantaneous WSS vector (or vector components) from the temporal mean vector (WSSAD > 90° ≈ OSI > 0).
Figure 2.

Example time-averaged WSS (magnitude), oscillatory WSS and co-localized regions of low WSS and high oscillatory WSS (i.e. low and oscillatory WSS) distributions. Polar plots indicate the time-varying changes in WSS magnitude and direction throughout the cardiac cycle; 0° indicates the time-averaged WSS vector direction. Note the large variation in instantaneous WSS vector directions (high oscillatory WSS) and low WSS magnitudes in a region immediately near branching vessels (location A), while WSS vectors have a uniform direction (low oscillatory WSS) with instantaneous magnitudes > 50 dynes cm−2 in a straight segment not near a coronary branch (location B).
Given the positive correlation previously reported between low and oscillatory WSS and atherogenesis [2,3,13], haemodynamic data were categorized, based on previous experimental [5,14] and clinical studies [7,8], to identify sectors exposed to this WSS environment. Time-averaged WSS magnitude values were defined as low (<10 dynes cm−2), intermediate (10–25 dynes cm−2) or high (>25 dynes cm−2), and oscillatory WSS (i.e. WSSAD) values were categorized as low (<45°), intermediate (45–90°) or high (>90°). Note that an oscillatory WSS value > 90° indicates that flow reversal occurred during the cardiac cycle (i.e. an instantaneous WSS vector was directed in the opposite direction of the time-averaged WSS vector; figure 2, location A), which is a flow characteristic commonly observed in regions of atherosclerosis development [2], and high oscillatory WSS largely occurs in a low time-averaged WSS environment, where fluid momentum is low and flow directional changes are possible. Finally, sectors were identified that exhibited both low time-averaged WSS magnitude and high oscillatory WSS values (i.e. low and oscillatory WSS; figure 2).
2.3. Focal plaque progression and haemodynamics analysis
VH-IVUS data were derived from offline analysis of each acquired image (echoPlaque 4.0; INDEC Medical Systems). Briefly, VH-IVUS is an imaging technique that uses an automated plaque tissue classification scheme, which identifies fibrous, fibrofatty, necrotic core and dense calcium tissues, to create tissue maps (see figures 1 and 3) from spectral analysis of the IVUS backscatter data [15]. The imaging modality has been validated in ex vivo and in vivo studies [16,17], and employed in several prospective clinical trials to evaluate the prognostic accuracy of VH-IVUS-defined plaque phenotypes for major adverse cardiac events [18,19]. Acquired image data were analysed, in accordance with consensus documents [20], by a single experienced investigator, who demonstrated strong reproducibility for total plaque and necrotic core areas [7,21]. Serial imaging data were co-registered in the axial and circumferential directions to quantify changes in total plaque and constituent (fibrous tissue, fibrofatty tissue, necrotic core and dense calcium) areas. An experienced analyst manually co-registered the baseline and follow-up images in the axial direction with the aid of fiduciary anatomic landmarks [7]. Subsequently, paired axial images were automatically co-registered in the circumferential direction by employing a multi-variate normalized cross-correlation algorithm, which has been verified and validated across multiple lesion types [22]. Images were divided into eight sectors (45° intervals), and VH-IVUS-derived total plaque and constituent areas were quantified in each sector (figure 3a). Area changes were determined by taking the difference in values between corresponding sectors in co-registered images (figure 3b). Progression was defined as a positive change in plaque or constituent area from baseline to follow-up, while regression was defined as a negative change. Note that, because VH-IVUS images are acquired at approximately 0.5 mm intervals, plaque progression in the axial direction, between consecutive images, is not captured. Note, too, that because changes in plaque areas are evaluated in focal regions, which cover approximately one-eighth of the artery circumference, reported values are an order of magnitude less than if evaluated around the complete circumference [7].
Figure 3.
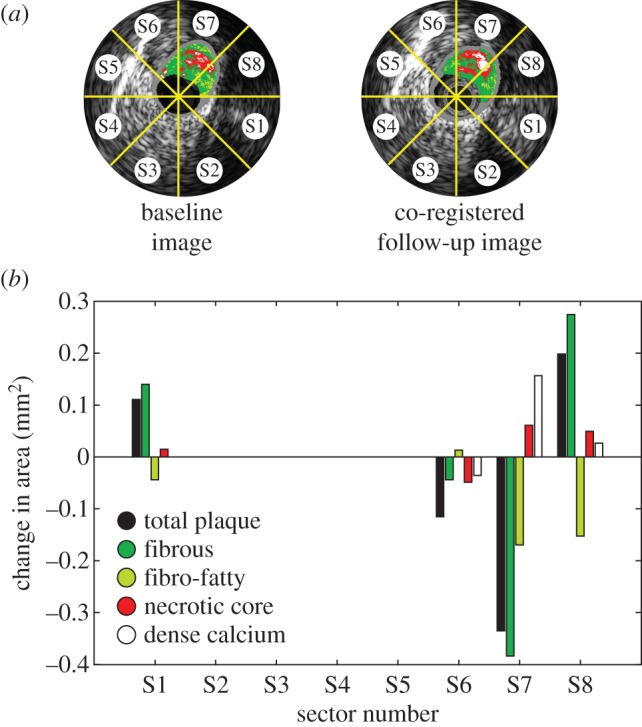
Evaluation of focal coronary artery disease progression. (a) Baseline and co-registered follow-up images divided into focal regions (sectors). (b) Quantification of focal (sector) changes in total plaque and VH-IVUS-defined constituent areas. (Online version in colour.)
The spatial location of each VH-IVUS image was identified in the computational geometry, and values of WSS magnitude and oscillation were extracted at these locations and divided into equivalent sectors (figure 4a). Thus, each sector had one value each for time-averaged WSS magnitude, oscillatory WSS and changes in total plaque and VH-IVUS-derived constituent areas. Sectors that resided within a branching vessel were excluded from the analysis; however, other sectors within that cross-section were included (figure 4b,c). Furthermore, only data in the LAD were included in the analysis, due to the difficulties associated with co-registering VH-IVUS images in the LM coronary artery, and imaging data were excluded if accurate axial and circumferential co-registration could not be performed (e.g. mismatch of baseline and follow-up images acquired between identified landmarks, poor circumferential co-registration correlation value [22]).
Figure 4.

Quantification of the focal haemodynamic environment. (a) WSS data are extracted from the computational geometry at the location of the VH-IVUS images. (b,c) Time-averaged WSS magnitude and oscillatory WSS data are divided up into sectors, and values are averaged within each sector. Sectors with nodes within branching vessels were excluded from the analysis.
2.4. Statistical analysis
Continuous data are reported as mean ± s.d. or median and interquartile range as appropriate. Categorical variables are presented as counts and proportions. To correct for systematic error, introduced due to repeated measures within subjects, a random-effects ANOVA was employed. p-values were adjusted for multiple comparisons with the method of Scheffé. All statistical tests were two-tailed, and p-values were derived from comparisons between haemodynamic categories. p < 0.05 was established as the level of statistical significance. Statistical analyses were performed using the statistical package SPSS (v. 21; IBM Corp., Armonk, NY, USA).
3. Results
Baseline and follow-up data were evaluated in the proximal LAD of 20 patients. No adverse clinical events occurred in the enrolled patient cohort during the study. Baseline demographic and clinical characteristics of the study cohort are presented in table 1. A total of 1840 serial VH-IVUS image pairs (median: 107; Q1–Q3: 63–122 per artery; 31.5–61 mm) were analysed and divided into 14 235 sectors (median: 792; Q1–Q3: 495–955 per artery) to examine the focal relationship between baseline haemodynamics and six-month changes in plaque area and composition. Sectors were distributed across the three time-averaged WSS classifications with 22%, 42% and 36% sectors in the low, intermediate and high time-averaged WSS classifications, respectively (figure 5a), with average values of 6.59 ± 2.00, 16.67 ± 4.21 and 45.24 ± 20.55 dynes cm−2, respectively (p < 0.0001, table 2). Sectors were largely exposed to low oscillatory WSS with 83%, 7% and 10% of sectors in the low, intermediate and high oscillatory WSS categories, respectively (figure 5a). Furthermore, low time-averaged WSS magnitude co-localized with high oscillatory WSS (i.e. low and oscillatory WSS) in 7% of sectors and low oscillatory WSS (i.e. low and non-oscillatory WSS) in 13% of sectors (figure 5b). Baseline VH-IVUS image characteristics stratified by haemodynamic classifications are presented in tables 2 and 3.
Table 1.
Patient demographic and clinical characteristics. Continuous data are reported as median (interquartile range). CAD, coronary artery disease.
| characteristic | all patients (n = 20) |
|---|---|
| age (years) | 54 (46–68) |
| male, n (%) | 13 (65) |
| white, n (%) | 14 (70) |
| body mass index (kg m−1) | 30 (27–36) |
| hypertension, n (%) | 14 (70) |
| current smoking, n (%) | 5 (25) |
| diabetes mellitus, n (%) | 7 (35) |
| family history of CAD, n (%) | 8 (40) |
| previous myocardial infarction, n (%) | 2 (10) |
| coronary flow reserve | 2.35 (2.03–2.59) |
| fractional flow reserve | 0.90 (0.82–0.96) |
| baseline lipid profile | |
| total cholesterol (mg dl−1) | 186.0 (168.0–212.5) |
| triglycerides (mg dl−1) | 115.5 (83.5–158.8) |
| high-density lipoprotein (mg dl−1) | 39.5 (33.3–52.8) |
| low-density lipoprotein (mg dl−1) | 118.5 (105.3–140.5) |
| C-reactive protein (mg l−1) | 2.7 (1.5–7.2) |
| follow-up lipid profile | |
| total cholesterol (mg dl−1) | 139.0 (124.3–151.3) |
| triglycerides (mg dl−1) | 107.0 (75.8–138.8) |
| high-density lipoprotein (mg dl−1) | 42.5 (31.3–57.3) |
| low-density lipoprotein (mg dl−1) | 70.5 (54–87.5) |
Figure 5.
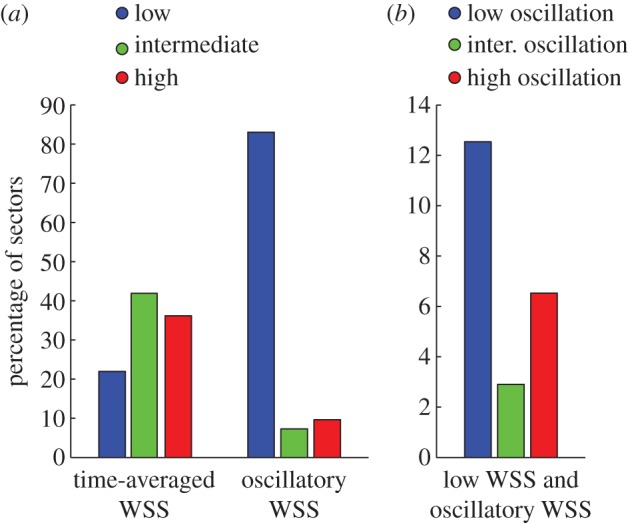
Distribution of sectors across haemodynamic classifications. (a) Time-averaged WSS and oscillatory WSS categories. (b) Co-localization of low time-averaged WSS and oscillatory WSS categories. (Online version in colour.)
Table 2.
Baseline VH-IVUS data stratified by haemodynamic classifications. Data are reported as mean ± s.d. WSS, wall shear stress.
| low WSS | intermediate WSS | high WSS | p-valuea | |
|---|---|---|---|---|
| sectors, n | 3127 | 5964 | 5144 | |
| WSS magnitude (dynes cm−2) | 6.59 ± 2.00 | 16.67 ± 4.21 | 45.24 ± 20.55 | <0.0001 |
| oscillatory WSS (°) | 56.14 ± 56.99 | 23.12 ± 33.18 | 11.65 ± 17.66 | <0.0001 |
| plaque area (mm2) | 0.40 ± 0.53 | 0.43 ± 0.58 | 0.49 ± 0.66 | 0.11 |
| fibrous area (mm2) | 0.25 ± 0.34 | 0.26 ± 0.36 | 0.28 ± 0.38 | 0.52 |
| fibrofatty area (mm2) | 0.05 ± 0.11 | 0.04 ± 0.01 | 0.06 ± 0.14 | 0.02 |
| necrotic core (mm2) | 0.07 ± 0.15 | 0.09 ± 0.17 | 0.11 ± 0.18 | 0.08 |
| dense calcium (mm2) | 0.03 ± 0.11 | 0.04 ± 0.01 | 0.05 ± 0.12 | 0.13 |
aDerived from comparison across three WSS categories.
Table 3.
Baseline VH-IVUS data stratified by oscillatory WSS classifications in low time-averaged WSS magnitude sectors. Data are reported as mean ± s.d. WSS, wall shear stress.
| low WSS magnitude and low oscillatory WSS | low WSS magnitude and intermediate oscillatory WSS | low WSS magnitude and high oscillatory WSS | p-valuea | |
|---|---|---|---|---|
| sectors, n | 1785 | 413 | 929 | |
| WSS (dynes cm−2) | 6.98 ± 1.86 | 6.17 ± 2.05 | 6.02 ± 2.08 | <0.0001 |
| oscillatory WSS (°) | 12.42 ± 10.96 | 67.53 ± 13.23 | 135.08 ± 25.62 | <0.0001 |
| plaque area (mm2) | 0.25 ± 0.40 | 0.45 ± 0.56 | 0.66 ± 0.64 | 0.001 |
| fibrous area (mm2) | 0.17 ± 0.26 | 0.25 ± 0.33 | 0.41 ± 0.42 | <0.0001 |
| fibrofatty area (mm2) | 0.03 ± 0.09 | 0.05 ± 0.16 | 0.07 ± 0.13 | 0.01 |
| necrotic core (mm2) | 0.04 ± 0.10 | 0.09 ± 0.16 | 0.13 ± 0.20 | 0.21 |
| dense calcium (mm2) | 0.01 ± 0.05 | 0.06 ± 0.15 | 0.05 ± 0.15 | 0.60 |
aDerived from comparison across three haemodynamic categories.
3.1. High time-averaged wall shear stress magnitude leads to total plaque area regression and necrotic core progression
Baseline time-averaged WSS magnitude greatly influenced total plaque area progression and changes in VH-IVUS-derived plaque constituent areas over the six-month follow-up. Sectors subjected to low WSS magnitude demonstrated a trend towards an increase in total plaque area (0.007 ± 0.010 mm2), which was significantly different from the progression patterns observed in areas of intermediate (−0.032 ± 0.006 mm2; p < 0.001) and high WSS (−0.028 ± 0.007 mm2; p < 0.001), both of which exhibited a decrease in total plaque area (p = 0.64; figure 6a). Evaluation of changes in plaque constituent areas revealed that sectors exposed to low WSS magnitude exhibited a significant increase in necrotic core (p < 0.001) and dense calcium (p < 0.05) area compared with areas of intermediate WSS (figure 6b). However, no significant change in fibrous tissue area was observed in the low WSS group, while significant regression occurred in the intermediate WSS group (p < 0.001). Comparison of changes in constituent areas between intermediate and high WSS magnitude sectors revealed greater progression of necrotic core (p < 0.001) and dense calcium (p < 0.001) in sectors exposed to high WSS. Similar to sectors exposed to low WSS, high WSS sectors demonstrated necrotic core progression (0.008 ± 0.004 versus 0.015 ± 0.004 mm2; p < 0.05); however, sectors subjected to high WSS demonstrated greater regression of fibrous tissue than low WSS sectors (−0.031 ± 0.006 versus −0.002 ± 0.007 mm2; p < 0.001).
Figure 6.

Changes in plaque area in low, intermediate and high time-averaged WSS sectors over six months. (a) Total plaque area. (b) VH-IVUS-derived plaque constituents. Sectors exposed to intermediate or high WSS were associated with a decrease in total plaque area, while sectors exposed to high WSS sectors demonstrated an increase in necrotic core and dense calcium. Error bars are 95% CIs. p < 0.05: low versus intermediate (*), intermediate versus high (#) and low versus high WSS (†).
3.2. Coincidence of low and oscillatory wall shear stress promotes total plaque area regression and necrotic core progression
In a low time-averaged WSS magnitude environment, the presence or absence of oscillatory WSS resulted in distinct VH-IVUS-defined plaque progression patterns over six months. Sectors exposed to low and oscillatory WSS demonstrated total plaque area regression (−0.026 ± 0.020 mm2), while areas of low and non-oscillatory WSS exhibited an increase in total plaque area (0.028 ± 0.010 mm2; p < 0.001; figure 7). Examination of changes in VH-IVUS-defined constituent areas revealed that low and oscillatory WSS was associated with a significant decrease in fibrous (p < 0.001) and fibrofatty (p < 0.001) tissue areas when compared with sectors subjected to low and non-oscillatory WSS (figure 7), with similar progression of necrotic core (p = 0.82) and dense calcium (p = 0.40). Figure 8 presents representative IVUS images that include sectors exposed to baseline haemodynamic values across the haemodynamic classifications, as well as the co-registered follow-up image and the sector progression values.
Figure 7.
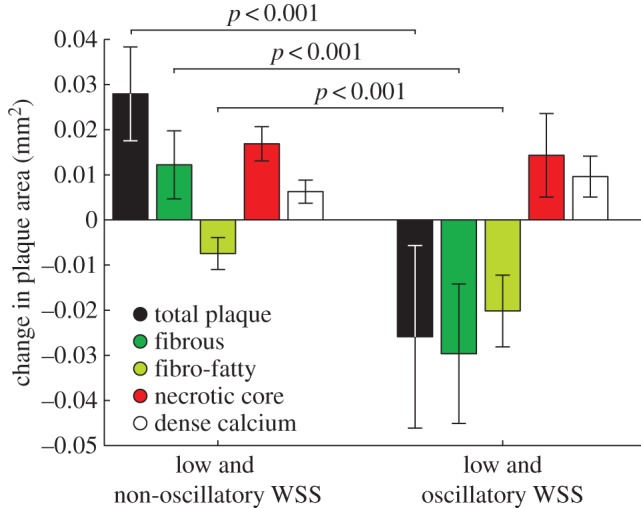
Changes in total plaque and VH-IVUS derived plaque constituent areas in sectors subjected to low and non-oscillatory (low time-averaged WSS, low oscillatory WSS) or low and oscillatory (low time-averaged WSS, high oscillatory WSS) WSS. Notably, focal regions of low and oscillatory WSS demonstrated regression of total plaque, fibrous and fibrofatty tissue area, and progression of necrotic core and dense calcium, suggestive of a transformation to a more vulnerable phenotype. Error bars are 95% CIs.
Figure 8.

Example IVUS images at baseline and follow-up demonstrating focal changes in plaque area (greyscale IVUS) and constituents (VH-IVUS) stratified by haemodynamic categories. Sectors exposed to low time-averaged WSS and oscillatory WSS or high time-averaged WSS exhibited a decrease in total plaque area with progression of necrotic core and dense calcium tissue.
4. Discussion
This is the first clinical study to investigate the effect of low and oscillatory WSS on plaque progression and transformation in patients with CAD treated with optimal medical therapy. We observed that sectors exposed to low and oscillatory WSS were associated with regression of total plaque, fibrous and fibrofatty tissue area, but progression of necrotic core and dense calcium, whereas sectors subjected to low and non-oscillatory WSS demonstrated progression of total plaque, fibrous tissue, necrotic core and calcium and regression of fibrofatty tissue (figure 7). Similarly, high time-averaged WSS magnitude demonstrated plaque regression, as well as progression of necrotic core and dense calcium (figure 6).
Our results are consistent with previous clinical studies reporting that baseline low WSS magnitude promotes total plaque area progression and high WSS leads to plaque area reduction [7,8,23]. However, these studies employed spatially averaged haemodynamic data, either around the lumen circumference or within a coronary segment, thus limiting the range of WSS values evaluated and the focal nature of these data. Employing a developed framework that allows for the examination of the focal association between haemodynamics and VH-IVUS-defined plaque progression (figures 3 and 4), our observations confirm that WSS magnitude modulates changes in total plaque size and VH-IVUS-defined constituent areas. Further, we now demonstrate for the first time in humans that oscillatory WSS significantly affects CAD progression patterns. Notably, our data indicate that WSS magnitude and oscillation may act cooperatively in affecting coronary lesion progression. Specifically, in areas of low WSS we observed necrotic core progression and total plaque size regression or progression depending on the presence or absence of oscillatory WSS, respectively. Thus, these data suggest that a haemodynamic environment characterized by either low and oscillatory WSS or high time-averaged WSS magnitude is associated with transformation towards a more vulnerable plaque phenotype [24].
In addition to the compelling experimental evidence linking haemodynamics to atherogenesis and early lesion formation [25], the data also suggest a role of WSS in atherosclerosis progression and plaque vulnerability. In a pro-atherogenic region, low time-averaged WSS has been linked to increased expression of matrix metalloproteinases (MMPs) [26], changes in endothelial cell morphology [27], and vascular smooth muscle cell (VSMC) migration and proliferation [28]. A study that used a perivascular cast to induce altered haemodynamics in a mouse model of atherosclerosis demonstrated a vulnerable plaque phenotype in areas of lowered WSS and more stable lesions in areas of oscillatory, but not lowered, WSS [29], which contrasts the presented results; however, limitations exist in studying atherosclerosis in murine models [30]. In diabetic, hypercholesterolaemic pigs, arterial segments exposed to persistently low WSS magnitude showed increased plaque size, decreased intimal VSMC content, increased expression of collagenases and collagenolytic activity and thinning of the fibrous cap [31]. These observations are consistent with data from the present study, as we observed total plaque progression in areas of low WSS. However, the role of oscillatory WSS could not be determined in that study [31], as the computational methods were limited by a steady flow assumption. Thus, it is possible that lesions in two different haemodynamic environments, low WSS and low and oscillatory WSS, were collectively analysed. More recently, it was observed in proprotein convertase subtilisin/kexin type 9 (PCSK9) ‘gain-of-function’, hypercholesterolaemic minipigs that regions of low and multi-directional WSS, which was quantified by transverse WSS (transWSS) [32], led to the formation of advanced coronary lesions, including TCFAs [6], which supports the clinical observations presented here. Furthermore, the study [6] showed that transWSS alone led to distinct plaque phenotypes, highlighting the role of WSS magnitude, oscillatory WSS and their combination in atherosclerosis progression and plaque vulnerability. We note that transWSS and WSSAD are analogous haemodynamic parameters in identifying focal regions where there exist large deviations of instantaneous WSS vectors from the main flow direction. Finally, our observation that sectors exposed to high WSS magnitude demonstrated progression patterns towards lesions with high-risk features is supported by experimental data that we have previously discussed in detail [7]. It has been shown that high WSS promotes increased production of plasmin, transforming growth factor-β, nitric oxide, bone morphogenic protein-4 and MMPs [33–35], each of which contribute to increased lesion vulnerability.
It cannot be overemphasized that CAD progression, vulnerable plaque formation and plaque rupture are focal pathophysiological episodes. Indeed, atherosclerotic lesions are classified by focal morphological and histological features [36] and plaque ruptures are a local disruption of a thin fibrous cap overlying a large necrotic core [37]. Thus, examination of variables that influence these events should be evaluated at an equivalent scale. We acknowledge that previous investigations have provided a critical understanding of the role of WSS in the natural history of coronary atherosclerosis; however, nearly all studies evaluating the relationship between WSS and CAD progression spatially averaged the data over the lumen circumference [7], axial ‘strips’ [38] or coronary segments [8], all of which reduce data width and the spatial dependence of the associations [39].
We have previously observed a threefold reduction in the range of time-averaged WSS values when data are averaged around the circumference versus evaluation in focal regions, and considerable differences between the two analysis techniques in the association of WSS and VH-IVUS-defined plaque progression [9]. In the current investigation, averaging time-averaged WSS and oscillatory WSS data around the circumference yielded no segments of low and oscillatory WSS, which highlights the advantage of our analysis framework to capture local flow features (figure 2). Our current data indicate that CAD progression and plaque phenotypic transformation, which were evaluated at a focal level, are associated with the baseline haemodynamic environment that was quantified at the same length scale. The novel approach presented herein provides motivation for future investigations that seek to identify asymptomatic coronary lesions that precede rapid lumen obstruction or an acute thrombotic event.
As coronary plaque rupture is the underlying aetiology for a majority of acute coronary syndromes [37], early identification of these high-risk plaques is of significant clinical importance. Adverse clinical outcomes have been individually associated with plaque burden, composition and phenotype; vessel remodelling patterns; and the surrounding haemodynamic environment. Studies have shown that the combination of distinct lesion characteristics demonstrates higher predictive value than a single characteristic in identifying lesions at greatest risk for future cardiac events [40]. However, to date, the required tools to identify focal WSS patterns at an individual coronary lesion have been absent. As a result, the complexity of the haemodynamic environment that includes focal regions of high WSS (plaque throat) juxtaposed to focal regions of low and oscillatory WSS (downstream of plaque), both of which are linked to plaque-destabilizing pathobiological mechanisms [35], cannot be accurately captured and characterized in humans. Our present findings demonstrate that WSS magnitude and oscillation are critical in identifying progression patterns of increased vulnerability and suggest that identification of these unique WSS patterns, in conjunction with other lesion characteristics, will afford increased predictive value and clinical prognostication of high-risk coronary lesions.
4.1. Study limitations
The limitations of this study should be noted. First, the limited study size of patients with stable, non-obstructive CAD treated with aggressive medical therapy resulted in no clinical events in the study cohort during the investigation. Nevertheless, our novel analysis techniques afforded the statistical power to detect significant changes in total plaque and constituent areas across the haemodynamic categories. Second, we used serial radiofrequency intravascular ultrasound imaging (i.e. VH-IVUS) to quantify CAD progression. Although one study questioned the accuracy of VH-IVUS in identifying necrotic core size [41], several identified flaws limit the clinical significance of the conclusions [42]. Furthermore, the PROSPECT study identified VH-IVUS-defined TCFAs in combination with greyscale as an independent predictor of plaque rupture [18]. Third, we acknowledge that arterial remodelling, which is defined as the change in EEM area from baseline to follow-up, is an important component of CAD progression and clinical manifestation of the disease [43]; however, as this study was focused on the focal association between haemodynamics and CAD progression, we chose not to analyse remodelling or other circumferentially defined parameters that confound the focal analysis. Fourth, there are limitations in our computational methods that should be acknowledged. Blood was assumed to be an incompressible Newtonian fluid; however, the Reynolds (Re) numbers in these models were moderate (Re ≈ 250–400), where the non-Newtonian behaviour of blood is minimal [44], and an exploratory study in one of our models resulted in minimal differences (less than 1%) in time-averaged WSS values when incorporating a non-Newtonian fluid model (Carreau model [45]). Also, we assumed a rigid wall and neglected coronary motion. We acknowledge that these are a significant approximation of in vivo conditions; yet, the lack of a validated fluid–solid interaction computational framework to quantify the 3D coronary artery mechanical environment and processing techniques to accurately quantify coronary motion from cine angiography (currently) restricts inclusion of these advanced modelling techniques. Finally, the results presented do not establish a causal relationship between coronary haemodynamics, CAD progression and plaque vulnerability in humans. Experimental models are better suited to advance mechanistic understanding of the role of haemodynamics in atherosclerosis progression, and larger clinical trials deploying such sophisticated methodology for evaluation of coronary haemodynamics are challenging, but warranted.
5. Conclusion
We demonstrate that both WSS magnitude and oscillation are closely associated with coronary plaque progression over six months in patients with non-obstructive CAD treated with optimal medical therapy. Using an analysis framework to examine the focal association between coronary haemodynamics and CAD progression, we report that, in a low WSS environment, focal regions (sectors) subjected to an oscillatory WSS exhibit regression of total plaque area, while non-oscillatory WSS sectors demonstrate total plaque progression. Furthermore, sectors exposed to low and oscillatory WSS exhibit a decrease in fibrous and fibrofatty tissue and an increase in necrotic core and dense calcium, implying a phenotypic transformation towards a more vulnerable phenotype [24]. Future clinical studies that rigorously evaluate the haemodynamic environment and CAD progression at the focal level will provide significant insight on the prognostic utility of WSS in the early identification of high-risk coronary lesions.
Acknowledgement
We thank the Emory interventional cardiology fellows and catheterization staff for participation in the study performance.
Ethics
This study was approved by the Institutional Review Board at Emory University, and all patients provided informed consent.
Authors' contributions
L.H.T. developed and performed the computations, performed the data analysis and prepared the manuscript. D.S.M. developed the computations, advised on data analysis, and provided feedback and suggestions on the manuscript. P.E. assisted in clinical data collection, advised on data analysis, and provided feedback and suggestions on the manuscript. M.C.M. assisted in clinical data collection and provided feedback and suggestions on the manuscript. J.N.O. and D.P.G. advised on computational model development, data analysis, and provided feedback and suggestions on the manuscript. H.S. collected the clinical data, advised on the data analysis, and provided feedback and suggestions on the manuscript.
Competing interests
We have no competing interests.
Funding
This research was supported by the American Heart Association (L.H.T.: 11POST7210012, D.S.M.: 13POST17110030), Georgia Research Alliance (D.P.G.), Pfizer Pharmaceuticals (H.S.) and Volcano Corp. (H.S.).
References
- 1.Grottum P, Svindland A, Walloe L. 1983. Localization of atherosclerotic lesions in the bifurcation of the main left coronary artery. Atherosclerosis 47, 55–62. ( 10.1016/0021-9150(83)90071-0) [DOI] [PubMed] [Google Scholar]
- 2.Ku DN, Giddens DP, Zarins CK, Glagov S. 1985. Pulsatile flow and atherosclerosis in the human carotid bifurcation. Positive correlation between plaque location and low oscillating shear stress. Arteriosclerosis 5, 293–302. ( 10.1161/01.ATV.5.3.293) [DOI] [PubMed] [Google Scholar]
- 3.He X, Ku DN. 1996. Pulsatile flow in the human left coronary artery bifurcation: average conditions. J. Biomech. Eng. 118, 74–82. ( 10.1115/1.2795948) [DOI] [PubMed] [Google Scholar]
- 4.Garcia-Cardena G, Comander J, Anderson KR, Blackman BR, Gimbrone MA Jr. 2001. Biomechanical activation of vascular endothelium as a determinant of its functional phenotype. Proc. Natl Acad. Sci. USA 98, 4478–4485. ( 10.1073/pnas.071052598) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou J, Li YS, Chien S. 2014. Shear stress-initiated signaling and its regulation of endothelial function. Arterioscler. Thromb. Vasc. Biol. 34, 2191–2198. ( 10.1161/ATVBAHA.114.303422) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pedrigi RM, et al. 2015. Inducing persistent flow disturbances accelerates atherogenesis and promotes thin cap fibroatheroma development in D374Y-PCSK9 hypercholesterolemic minipigs. Circulation 132, 1003–1012. ( 10.1161/CIRCULATIONAHA.115.016270) [DOI] [PubMed] [Google Scholar]
- 7.Samady H, Eshtehardi P, McDaniel MC, Suo J, Dhawan SS, Maynard C, Timmins LH, Quyyumi AA, Giddens DP. 2011. Coronary artery wall shear stress is associated with progression and transformation of atherosclerotic plaque and arterial remodeling in patients with coronary artery disease. Circulation 124, 779–788. ( 10.1161/CIRCULATIONAHA.111.021824) [DOI] [PubMed] [Google Scholar]
- 8.Stone PH, et al. 2012. Prediction of progression of coronary artery disease and clinical outcomes using vascular profiling of endothelial shear stress and arterial plaque characteristics: the PREDICTION Study. Circulation 126, 172–181. ( 10.1161/CIRCULATIONAHA.112.096438) [DOI] [PubMed] [Google Scholar]
- 9.Timmins LH, Molony DS, Eshtehardi P, McDaniel MC, Oshinski JN, Samady H, Giddens DP. 2015. Focal association between wall shear stress and clinical coronary artery disease progression. Ann. Biomed. Eng. 43, 94–106. ( 10.1007/s10439-014-1155-9) [DOI] [PubMed] [Google Scholar]
- 10.Wahle A, Prause PM, DeJong SC, Sonka M. 1999. Geometrically correct 3-D reconstruction of intravascular ultrasound images by fusion with biplane angiography—methods and validation. IEEE Trans. Med. Imaging 18, 686–699. ( 10.1109/42.796282) [DOI] [PubMed] [Google Scholar]
- 11.Molony DS, Timmins LH, Rasoul-Arzrumly E, Samady H, Giddens DP. 2014. Investigation of the influence of side-branches on wall shear stress in coronary arteries reconstructed from intravascular ultrasound. In Computational biomechancis for medicine: fundamental science and patient-specific applications (ed. Doyle B.). New York, NY: Springer. [Google Scholar]
- 12.Timmins LH, Gupta D, Corban MT, Molony DS, Oshinski JN, Samady H, Giddens DP. 2015. Co-localization of disturbed flow patterns and oclusive cardiac allograft vasculopathy lesion formation in heart transplant patients. Cardiovasc. Eng. Technol. 6, 25–35. ( 10.1007/s13239-014-0198-2) [DOI] [PubMed] [Google Scholar]
- 13.Moore JE Jr, Xu C, Glagov S, Zarins CK, Ku DN. 1994. Fluid wall shear stress measurements in a model of the human abdominal aorta: oscillatory behavior and relationship to atherosclerosis. Atherosclerosis 110, 225–240. ( 10.1016/0021-9150(94)90207-0) [DOI] [PubMed] [Google Scholar]
- 14.Malek AM, Izumo S. 1996. Mechanism of endothelial cell shape change and cytoskeletal remodeling in response to fluid shear stress. J. Cell Sci. 109, 713–726. [DOI] [PubMed] [Google Scholar]
- 15.Nair A, Kuban BD, Tuzcu EM, Schoenhagen P, Nissen SE, Vince DG. 2002. Coronary plaque classification with intravascular ultrasound radiofrequency data analysis. Circulation 106, 2200–2206. ( 10.1161/01.cir.0000035654.18341.5e) [DOI] [PubMed] [Google Scholar]
- 16.Nair A, Margolis MP, Kuban BD, Vince DG. 2007. Automated coronary plaque characterisation with intravascular ultrasound backscatter: ex vivo validation. EuroIntervention 3, 113–120. [PubMed] [Google Scholar]
- 17.Nasu K, et al. 2006. Accuracy of in vivo coronary plaque morphology assessment: a validation study of in vivo virtual histology compared with in vitro histopathology. J. Am. Coll. Cardiol. 47, 2405–2412. ( 10.1016/j.jacc.2006.02.044) [DOI] [PubMed] [Google Scholar]
- 18.Stone GW, et al. 2011. A prospective natural-history study of coronary atherosclerosis. N. Engl. J. Med. 364, 226–235. ( 10.1056/NEJMoa1002358) [DOI] [PubMed] [Google Scholar]
- 19.Calvert PA, et al. 2011. Association between IVUS findings and adverse outcomes in patients with coronary artery disease: the VIVA (VH-IVUS in vulnerable atherosclerosis) study. JACC Cardiovasc. Imaging 4, 894–901. ( 10.1016/j.jcmg.2011.05.005) [DOI] [PubMed] [Google Scholar]
- 20.Mintz GS, Garcia-Garcia HM, Nicholls SJ, Weissman NJ, Bruining N, Crowe T, Tardif JC, Serruys PW. 2011. Clinical expert consensus document on standards for acquisition, measurement and reporting of intravascular ultrasound regression/progression studies. EuroIntervention 6, 1123–1130, 1129 ( 10.4244/EIJV6I9A195) [DOI] [PubMed] [Google Scholar]
- 21.Eshtehardi P, et al. 2012. Association of coronary wall shear stress with atherosclerotic plaque burden, composition, and distribution in patients with coronary artery disease. J. Am. Heart Assoc. 1, e002543 ( 10.1161/JAHA.112.002543) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Timmins LH, Suever JD, Eshtehardi P, McDaniel MC, Oshinski JN, Samady H, Giddens DP. 2013. Framework to co-register longitudinal virtual histology-intravascular ultrasound data in the circumferential direction. IEEE Trans. Med. Imaging 32, 1989–1996. ( 10.1109/TMI.2013.2269275) [DOI] [PubMed] [Google Scholar]
- 23.Stone PH, et al. 2003. Effect of endothelial shear stress on the progression of coronary artery disease, vascular remodeling, and in-stent restenosis in humans: in vivo 6-month follow-up study. Circulation 108, 438–444. ( 10.1161/01.CIR.0000080882.35274.AD) [DOI] [PubMed] [Google Scholar]
- 24.Finn AV, Nakano M, Narula J, Kolodgie FD, Virmani R. 2010. Concept of vulnerable/unstable plaque. Arterioscler. Thromb. Vasc. Biol. 30, 1282–1292. ( 10.1161/ATVBAHA.108.179739) [DOI] [PubMed] [Google Scholar]
- 25.Chiu JJ, Chien S. 2011. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol. Rev. 91, 327–387. ( 10.1152/physrev.00047.2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deguchi JO, Aikawa E, Libby P, Vachon JR, Inada M, Krane SM, Whittaker P, Aikawa M. 2005. Matrix metalloproteinase-13/collagenase-3 deletion promotes collagen accumulation and organization in mouse atherosclerotic plaques. Circulation 112, 2708–2715. ( 10.1161/CIRCULATIONAHA.105.562041) [DOI] [PubMed] [Google Scholar]
- 27.Himburg HA, Grzybowski DM, Hazel AL, LaMack JA, Li XM, Friedman MH. 2004. Spatial comparison between wall shear stress measures and porcine arterial endothelial permeability. Am. J. Physiol. Heart Circ. Physiol. 286, H1916–H1922. ( 10.1152/ajpheart.00897.2003) [DOI] [PubMed] [Google Scholar]
- 28.Bentzon JF, Weile C, Sondergaard CS, Hindkjaer J, Kassem M, Falk E. 2006. Smooth muscle cells in atherosclerosis originate from the local vessel wall and not circulating progenitor cells in ApoE knockout mice. Arterioscler. Thromb. Vasc. Biol. 26, 2696–2702. ( 10.1161/01.ATV.0000247243.48542.9d) [DOI] [PubMed] [Google Scholar]
- 29.Cheng C, Tempel D, van Haperen R, van der Baan A, Grosveld F, Daemen MJ, Krams R, de Crom R. 2006. Atherosclerotic lesion size and vulnerability are determined by patterns of fluid shear stress. Circulation 113, 2744–2753. ( 10.1161/CIRCULATIONAHA.105.590018). [DOI] [PubMed] [Google Scholar]
- 30.Seok J, et al. 2013. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl Acad. Sci. USA 110, 3507–3512. ( 10.1073/pnas.1222878110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koskinas KC, et al. 2013. Thin-capped atheromata with reduced collagen content in pigs develop in coronary arterial regions exposed to persistently low endothelial shear stress. Arterioscler. Thromb. Vasc. Biol. 33, 1494–1504. ( 10.1161/ATVBAHA.112.300827) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peiffer V, Sherwin SJ, Weinberg PD. 2013. Computation in the rabbit aorta of a new metric—the transverse wall shear stress—to quantify the multidirectional character of disturbed blood flow. J. Biomech. 46, 2651–2658. ( 10.1016/j.jbiomech.2013.08.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Casey PJ, Dattilo JB, Dai G, Albert JA, Tsukurov OI, Orkin RW, Gertler JP, Abbott WM. 2001. The effect of combined arterial hemodynamics on saphenous venous endothelial nitric oxide production. J. Vasc. Surg. 33, 1199–1205. ( 10.1067/mva.2001.115571) [DOI] [PubMed] [Google Scholar]
- 34.Kenagy RD, Fischer JW, Davies MG, Berceli SA, Hawkins SM, Wight TN, Clowes AW. 2002. Increased plasmin and serine proteinase activity during flow-induced intimal atrophy in baboon PTFE grafts. Arterioscler. Thromb. Vasc. Biol. 22, 400–404. ( 10.1161/hq0302.105376) [DOI] [PubMed] [Google Scholar]
- 35.Slager CJ, Wentzel JJ, Gijsen FJ, Thury A, van der Wal AC, Schaar JA, Serruys PW. 2005. The role of shear stress in the destabilization of vulnerable plaques and related therapeutic implications. Nat. Clin. Pract. Cardiovasc. Med. 2, 456–464. ( 10.1038/ncpcardio0298) [DOI] [PubMed] [Google Scholar]
- 36.Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. 2000. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 20, 1262–1275. ( 10.1161/01.ATV.20.5.1262) [DOI] [PubMed] [Google Scholar]
- 37.Virmani R, Burke AP, Farb A, Kolodgie FD. 2006. Pathology of the vulnerable plaque. J. Am. Coll. Cardiol. 47, C13–C18. ( 10.1016/j.jacc.2005.10.065) [DOI] [PubMed] [Google Scholar]
- 38.Wentzel JJ, Janssen E, Vos J, Schuurbiers JC, Krams R, Serruys PW, de Feyter PJ, Slager CJ. 2003. Extension of increased atherosclerotic wall thickness into high shear stress regions is associated with loss of compensatory remodeling. Circulation 108, 17–23. ( 10.1161/01.CIR.0000078637.21322.D3) [DOI] [PubMed] [Google Scholar]
- 39.Peiffer V, Sherwin SJ, Weinberg PD. 2013. Does low and oscillatory wall shear stress correlate spatially with early atherosclerosis? A systematic review. Cardiovasc. Res. 99, 242–250. ( 10.1093/cvr/cvt044) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Corban MT, et al. 2014. Combination of plaque burden, wall shear stress, and plaque phenotype has incremental value for prediction of coronary atherosclerotic plaque progression and vulnerability. Atherosclerosis 232, 271–276. ( 10.1016/j.atherosclerosis.2013.11.049) [DOI] [PubMed] [Google Scholar]
- 41.Thim T, Hagensen MK, Wallace-Bradley D, Granada JF, Kaluza GL, Drouet L, Paaske WP, Botker HE, Falk E. 2010. Unreliable assessment of necrotic core by virtual histology intravascular ultrasound in porcine coronary artery disease. Circ. Cardiovasc. Imaging 3, 384–391. ( 10.1161/CIRCIMAGING.109.919357) [DOI] [PubMed] [Google Scholar]
- 42.Stone GW, Mintz GS. 2010. Letter by Stone and Mintz regarding article, ‘unreliable assessment of necrotic core by virtual histology intravascular ultrasound in porcine coronary artery disease’. Circ. Cardiovasc. Imaging 3, e4; author reply e5 ( 10.1161/CIRCIMAGING.110.958553) [DOI] [PubMed] [Google Scholar]
- 43.Glagov S, Weisenberg E, Zarins CK, Stankunavicius R, Kolettis GJ. 1987. Compensatory enlargement of human atherosclerotic coronary arteries. N. Engl. J. Med. 316, 1371–1375. ( 10.1056/NEJM198705283162204) [DOI] [PubMed] [Google Scholar]
- 44.Ku DN. 1997. Blood flow in arteries. Annu. Rev. Fluid Mech. 29, 399–434. ( 10.1146/annurev.fluid.29.1.399) [DOI] [Google Scholar]
- 45.Cho YI, Kensey KR. 1991. Effects of the non-Newtonian viscosity of blood on flows in a diseased arterial vessel. Part 1: steady flows. Biorheology 28, 241–262. [DOI] [PubMed] [Google Scholar]