

Abstract
Background
Gene prediction is one of the most important steps in the genome annotation process. A large number of software tools and pipelines developed by various computing techniques are available for gene prediction. However, these systems have yet to accurately predict all or even most of the protein-coding regions. Furthermore, none of the currently available gene-finders has a universal Hidden Markov Model (HMM) that can perform gene prediction for all organisms equally well in an automatic fashion.
Results
We present an automated gene prediction pipeline, Seqping that uses self-training HMM models and transcriptomic data. The pipeline processes the genome and transcriptome sequences of the target species using GlimmerHMM, SNAP, and AUGUSTUS pipelines, followed by MAKER2 program to combine predictions from the three tools in association with the transcriptomic evidence. Seqping generates species-specific HMMs that are able to offer unbiased gene predictions. The pipeline was evaluated using the Oryza sativa and Arabidopsis thaliana genomes. Benchmarking Universal Single-Copy Orthologs (BUSCO) analysis showed that the pipeline was able to identify at least 95% of BUSCO’s plantae dataset. Our evaluation shows that Seqping was able to generate better gene predictions compared to three HMM-based programs (MAKER2, GlimmerHMM and AUGUSTUS) using their respective available HMMs. Seqping had the highest accuracy in rice (0.5648 for CDS, 0.4468 for exon, and 0.6695 nucleotide structure) and A. thaliana (0.5808 for CDS, 0.5955 for exon, and 0.8839 nucleotide structure).
Conclusions
Seqping provides researchers a seamless pipeline to train species-specific HMMs and predict genes in newly sequenced or less-studied genomes. We conclude that the Seqping pipeline predictions are more accurate than gene predictions using the other three approaches with the default or available HMMs.
Keywords: Gene prediction, Gene model, Species specific HMM
Background
Rapid and cost-effective next-generation sequencing (NGS) technologies produce large volumes of DNA sequencing data in large-scale genome projects. These advances enabled the research community to sequence many plant genomes and transcriptomes. After the assembly process, the next critical step is annotation of these newly sequenced genomes. Experimental methods for gene validation, biological interpretation and annotation are costly, time-consuming, and labor intensive. Hence, there is a pressing need to develop accurate and fast tools to analyze genomic sequences, especially to identify genes and determine their functions. Many computational tools had been developed with intent to solve the gene finding problem. Protein coding genes are commonly predicted using Hidden Markov Model (HMM) approach [1–5], Conditional Random Field [6], Support Vector Machine [7], Neural Network [8, 9], or by combining multiple predictions from various programs [10, 11]. However, gene finders are often trained using known gene models and this leads to biases in gene structure [12–14]. None of these systems incorporates a flexible, universal gene model that can perform gene prediction for a wide range of species. The process is more complex for plants due to its typically large genome size, short exons bordered by large introns, highly repetitive sequences, and alternative spliced transcripts. Currently available gene finders do not accurately predict most of the protein-coding regions [15], and predicting the complete set of an organism’s protein-coding genes remains a significant challenge.
Recently developed automatic pipeline, such as SnowyOwl [16] and CodingQuarry [17] is designed and optimized for fungal genomes, while BRAKER1 [18] is generally for eukaryotic genomes. The main goal of our work was to develop a versatile gene prediction pipeline that could be applied to any newly (even partially) sequenced plant genome. In order to address these issues, we combined existing gene-finders with self-trained HMMs constructed from a training set of the same species to predict gene models. Our program automates and streamlines the gene prediction process by preparing the training dataset, building species-specific HMMs, predicting gene models and compiles the relevant information for the gene models.
Methods
The scripts run on Linux platform in Bash shell and require some preinstalled software like BLAST+ 2.2.30 [19], CD-HIT 4.5.4 [20], Splign 1.39.8 [21], GlimmerHMM 3.0 [5, 22], AUGUSTUS 2.6.1 [23], SNAP [4], MAKER 2.10 [24, 25], and EMBOSS 6.4.0 [26].
Scripting
UNIX based Bash and Perl scripting was used in the current work. “seqping.sh” is the main script that executes a sequence of commands, including invoking other scripts written in Bash and Perl. The pipeline is shown in Fig. 1. We divided the task into seven stages: (1) setting up the working directories, (2) preparation of the training set, (3) GlimmerHMM [5, 22] training, (4) AUGUSTUS [23] training, (5) SNAP [4] training, (6) MAKER2 [24, 25] prediction, and (7) results filtering. Seqping supports multiple processors analysis, as well as job submission to Sun Grid Engine (SGE) or Portable Batch System (PBS) job schedulers. The script’s optimized parameters provide an automated and efficient tool for filtering and structural annotation of gene predictions.
Fig. 1.
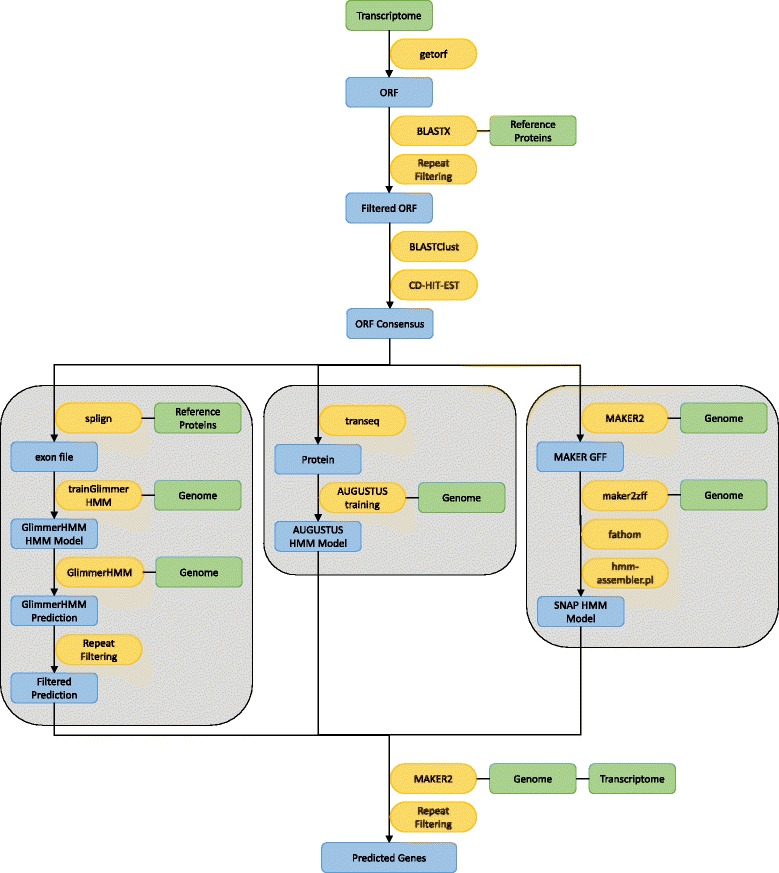
The Seqping self-training gene prediction pipeline. Green boxes indicate input sequences in FASTA format, yellow boxes indicate software or processes, and blue boxes indicate intermediate output files
Program input
The user is prompt to submit the respective species’ (1) transcriptome and (2) genome sequence in FASTA format. A reference protein file containing full length protein sequences selected from the NCBI Protein Database [27] is required for validation and annotation of the gene predictions. We selected only proteins from the phylum Magnoliophyta (flowering plants) and excluded hypothetical, ribosomal, mitochondrial and chloroplast proteins. TIGR Plant Repeat [28] and RepBase [29] sequences were combined into a database for TBLASTX filtering while HMM profiles from Gypsy Database [30] were used for HMMER [31] hmmsearch filtering.
Preparation of training dataset
Transcriptome from the organism of interest is used to generate the training set. Seqping extracts open reading frames (ORFs), sized between 500 and 5000 nucleotides, from the transcriptome using getorf tool from the EMBOSS package [26]. Next, ORFs with reference proteins support (BLASTX E-value < E-10) are clustered using BLASTClust and CD-HIT-EST [20] tools with stringent parameters. Transcripts that have similarity to repeats are removed (TBLASTX against TIGR Plant Repeat [28] and RepBase [29] with E-value < 1E-10, and hmmsearch against Gypsy Database [30] with E-value < 1E-5). The remaining sequences are used as the training set to develop species-specific HMMs for gene prediction.
In the next step, the program aligns the training set to the genome using Splign and Compart tools [21]. The aligned training set and corresponding genomic sequences are used to train GlimmerHMM [5, 22]. Then a custom Perl script is used to convert the Splign output into an exon file, and trainGlimmerHMM is activated to generate a HMM model. Gene prediction by GlimmerHMM is executed using the newly generated species-specific HMM, followed by filtering of repeats. To generate HMM for AUGUSTUS [23], the training set is translated into protein sequences using EMBOSS’s transeq. A different HMM is produced using the AUGUSTUS-specific training script that can be found in the AUGUSTUS package. In order to build the HMM for SNAP [4], Seqping runs a basic MAKER2 [24] prediction using DNA and protein sequences from the training set. The SNAP HMM model is finally produced by fathom and hmm-assembler scripts from the SNAP package.
Program output
The output is stored in a user-defined directory. The self-trained HMM models and gene prediction outputs are located in several sub-directories labeled by the names of the respective gene-finding modules. MAKER2, which is the final tool to combine all models (GlimmerHMM’s prediction, AUGUSTUS’s HMM and SNAP’s HMM) and evidences (transcriptome data and NCBI Protein Database), generate the list of predicted genes in GFF format, as well as predicted genes and proteins sequences in FASTA format. The list of output directories in a tree-like format is shown in Fig. 2. A comprehensive log file is generated as the pipeline is executed.
Fig. 2.
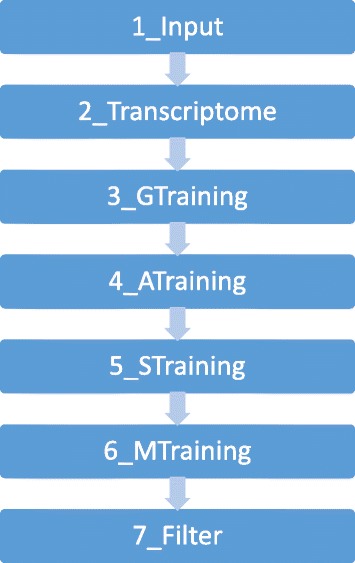
The list of output directories from the seven stages in Seqping
Comparison of gene prediction tools
To demonstrate the effectiveness of the Seqping pipeline, the rice (Oryza sativa ssp. japonica) and Arabidopsis thaliana genomes were used. Benchmarking Universal Single-Copy Orthologs (BUSCO) [32] analysis of the predicted genes were tested using the 956 plantae BUSCO profiles. The predicted gene models were also compared to manually curated gene sets for both organisms. A total of 24,229 complete genes of O. sativa ssp. japonica from RefSeq were used as the reference set to calculate sensitivity (Sn) and specificity (Sp). For A. thaliana, annotations from TAIR10 [33] was used to compare the performance of Seqping. Sn and Sp were calculated as described by Burset and Guigo [34] using GenomeTools [35] gt-eval. Sn and Sp are defined as and where TP, FN and FP is the number of true positives, false negatives, and false positives, respectively. Accuracy (Acc) is defined as the average of Sn and Sp, [34]. Comparison was done at CDS, exon and single-nucleotide levels.
Materials
Twelve rice chromosomes were obtained from the MSU Rice Genome Annotation Project release 7 [36]. The transcriptome set contained assembled transcripts from three RNA-Seq projects in NCBI BioProject: PRJNA79825, PRJDA67119, and PRJNA80103. A total of 175,251 assembled transcripts were used as input for the pipeline. The contigs N50 and mean length are 1693 and 956 respectively. For A. thaliana gene prediction, the genome and transcriptome data were downloaded from TAIR10 [33] genome release (1,476,275 ESTs and 77,415 cDNAs).
Results and discussion
Transcriptome data is a key source of experimental evidence for genome annotation, since it reflects the genes that are expressed in specific cell types or conditions [37]. Mapping of a large number of full-length transcripts greatly improves identification of the exon structures of eukaryotic genes [38–40]. It also allows identification of alternative splicing [40, 41], and accurate prediction of transcription start sites and promoters [42, 43]. Incorporation of transcriptome data into the gene prediction pipeline is a feasible and cost-effective approach for annotation of newly sequenced or less-studied genomes [17, 40, 44], in spite of existing computational challenges and complexity of higher eukaryotic genomes [45].
HMMs form the basis for most currently used gene finders. HMMs for gene prediction contain probabilistic state models for different functional parts of the genomic sequence, such as translational and splicing signals and coding regions, depending on the base frequency. The three main gene finders: GlimmerHMM, AUGUSTUS, and SNAP, have pre-build HMM models for several model species in their software packages, but the available existing HMMs may not be suitable for highly complex plant genomes. The prior probabilities calculated for HMM in other species difficultly identify the genes in targeted plant genome. Species-specific HMMs are required to find both novel and well-characterized genes. Seqping performs species-specific HMM training for three programs: GlimmerHMM, AUGUSTUS and SNAP, and uses MAKER2 to combine the predictions in order to take advantages of the different algorithms used by the respective programs. MAKER2 uses the GFF3 file of GlimmerHMM, AUGUSTUS HMM, and SNAP HMM, in addition with the transcriptome data, to generate the final set of predicted genes. All the transcriptome and gene models available are also used by MAKER2 to generate a quality metric called Annotation Edit Distance (AED) for each gene model, in which AED score of 0 is the best-supported gene models.
Seqping also filter repetitive sequences, since these sequences are mainly represented by noncoding sequences. In plants, repetitive sequences may account for up to 90% of the genome [46]. These repeats may also create challenges during the automatic gene finding process. Filtering of repetitive sequences is implemented in several stages in the pipeline, namely during the selection of ORFs for the training set, GlimmerHMM gene prediction and MAKER2 gene prediction. The presence of repetitive sequences is identified by comparison to the TIGR Plant Repeat [28], RepBase [29], and Gypsy Database [30].
Oryza sativa gene prediction
The pipeline was first tested in rice (O. sativa ssp. japonica). It took ~100 h to execute the gene prediction pipeline on the Linux SGE cluster with 9 nodes (8 CPUs per node). The rice transcripts were treated as described in the Training Set Preparation section, producing 11,729 putative full-length ORFs that were then used for HMM training. The Seqping pipeline, using the new HMMs and transcriptome data identified 24,009 highly confidence rice genes. BUSCO analysis, which is a benchmarking tool to determine the completeness of genome assemblies and annotations, revealed that Seqping was able to identify 95.92% of the highly conserved plant genes (Table 1). This was the best performance, followed by MAKER2 (92.26%), GlimmerHMM (91.53%) and Augustus (88.70%).
Table 1.
Accuracy of four methods of gene prediction using the O. sativa genome
| Seqpinga | MAKER2b | GlimmerHMMc | AUGUSTUSd, e | ||
|---|---|---|---|---|---|
| BUSCO | 95.92% | 92.26% | 91.53% | 88.70% | |
| CDS structure | Sn | 0.6175 | 0.5193 | 0.4394 | 0.4717 |
| Sp | 0.5120 | 0.4922 | 0.2774 | 0.3008 | |
| Acc | 0.5648 | 0.5058 | 0.3584 | 0.3863 | |
| Exon structure | Sn | 0.4820 | 0.4028 | 0.3089 | - |
| Sp | 0.4116 | 0.3880 | 0.2129 | - | |
| Acc | 0.4468 | 0.3954 | 0.2609 | - | |
| Nucleotide Level | Sn | 0.6906 | 0.5950 | 0.6597 | 0.6581 |
| Sp | 0.6484 | 0.6680 | 0.4381 | 0.3698 | |
| Acc | 0.6695 | 0.6315 | 0.5489 | 0.5140 | |
aSeqping: Trained using rice transcriptome; bMAKER2: SNAP’s rice HMM, AUGUSTUS’s maize model and rice transcriptome; cGlimmerHMM: Trained using rice transcriptome; dAUGUSTUS: maize model
eUsing the available maize models, AUGUSTUS does not predict exon structure
It also had the highest Sn, Sp and Acc for three comparison levels (CDS, exon, single-nucleotide), with the exception of the Sp at the nucleotide level, in which it scored the second highest score of 0.6484 after MAKER2 (0.6680). This shows that the Seqping pipeline was able to produce the most precise rice models compared to MAKER2, GlimmerHMM and AUGUSTUS. It also indicates that optimization of parameters to train the gene finders in Seqping was an important step to enable the gene prediction software to accurately identify gene structure. The predicted rice genes from Seqping were also independently verified using a different approach. Comparison of the Seqping models to the MSU annotation using ParsEval [47] yielded 87.70% shared gene loci. These results indicate that Seqping had the best prediction for the rice genome.
Arabidopsis thaliana gene prediction
Using the Seqping pipeline, a total of 25,829 putative full-length ORFs were identified and used for the HMM training. The pipeline identified 21,229 highly confidence genes. BUSCO analysis showed that AUGUSTUS was able to identify the highest number (98.64%) of conserved orthologs. This was followed by GlimmerHMM (98.12%). Seqping was ranked as the third, with 96.44% identified. Nevertheless, it was also still able to identify more than 95% of the orthologs available.
To compare the performance of the four programs, TAIR10 [33] A. thaliana annotations were used as the reference gene set (Table 2). Overall, the Sn for CDS structure was much lower compared to rice as the annotations from TAIR10 covers many alternative splicing forms. Seqping had the best Sn at the exon level and Sp at the nucleotide level, while MAKER2 performed better in Sp at the CDS and exon levels. GlimmerHMM achieved the highest Sn for nucleotide structure. Augustus was able to predict the best Sn at CDS structure. Nevertheless, Seqping had the best overall Acc at all three levels. This shows that while each tool was sacrificing either Sn or Sp, Seqping was able to balance the predictions by using a combination of the tools.
Table 2.
Accuracy of four methods of gene prediction using the A. thaliana genome
| Seqpinga | MAKER2b | GlimmerHMMc, e | AUGUSTUSd | ||
|---|---|---|---|---|---|
| BUSCO | 96.44% | 94.14% | 98.12% | 98.64% | |
| CDS Structure | Sn | 0.2749 | 0.0738 | 0.2804 | 0.3075 |
| Sp | 0.8867 | 0.8877 | 0.7515 | 0.7527 | |
| Acc | 0.5808 | 0.4808 | 0.5160 | 0.5301 | |
| Exon structure | Sn | 0.4596 | 0.1207 | - | 0.4155 |
| Sp | 0.7313 | 0.7457 | - | 0.5373 | |
| Acc | 0.5955 | 0.4332 | - | 0.4764 | |
| Nucleotide Level | Sn | 0.7929 | 0.1932 | 0.9350 | 0.9634 |
| Sp | 0.9748 | 0.9750 | 0.8150 | 0.7974 | |
| Acc | 0.8839 | 0.5841 | 0.8750 | 0.8804 | |
aSeqping: Trained using A. thaliana transcriptome; bMAKER2: SNAP’s A. thaliana HMM, AUGUSTUS’s A. thaliana model and A. thaliana transcriptome; cGlimmerHMM: A. thaliana model; dAUGUSTUS: A. thaliana model
eUsing the available A. thaliana model, GlimmerHMM does not predict exon structure
Conclusions
The Seqping pipeline predictions are more accurate compared to the other three approaches with the default or available HMMs. We demonstrated that integration of multiple tools result in higher quality gene predictions in both dicotyledon and monocotyledon plants. By training species-specific HMMs, Seqping provides an effective, organism independent, gene prediction tool for non-model plant species. Expectedly, the performance is influenced by the quality of the transcriptome and genome sequences of the target species. The pipeline is most suitable for used in newly sequenced or less-studied plant genomes.
Acknowledgements
The authors would like to thank MPOB for permission to publish this manuscript. We wish to thank the members of MPOB’s Bioinformatics Unit for their valuable thoughts and assistance. We appreciate the unwavering support from Datuk Dr. Choo Yuen May, Director General of MPOB.
Declarations
This article has been published as part of BMC Bioinformatics Vol 18 Suppl 1, 2017: Selected articles from BGRS\SB-2016: bioinformatics. The full contents of the supplement are available online at http://bmcbioinformatics.biomedcentral.com/articles/supplements/volume-18-supplement-1.
Funding
This work (including the publication costs) was funded by the Malaysian Palm Oil Board. Dr. Tatarinova was supported by the NSF Awards #1456634 and #1622840.
Availability of data and materials
Project name: Seqping
Project home page: https://sourceforge.net/projects/seqping/
Archived version: seqping_0.1.45
Operating system: Linux platform
Programming language: Bash, Perl
Other requirements: BLAST 2.2.25, CD-HIT 4.5.4, Splign 1.39.8, Exonerate 2.2, GlimmerHMM 3.0, AUGUSTUS 2.6.1, MAKER 2.10, EMBOSS 6.4.0, Cufflinks 2.0.2
License: GNU General Public License
Any restrictions to use by non-academics: None
Authors’ contributions
KLC, MH, MFR and ETLL conceptualized the research project; KLC wrote the code; RR, MH, and MFR evaluated the performance; TVT and ETLL supervised the work; KLC, ETLL and TVT wrote the paper. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Contributor Information
Kuang-Lim Chan, Email: chankl@mpob.gov.my.
Rozana Rosli, Email: lizana@mpob.gov.my.
Tatiana V. Tatarinova, Email: tatiana.tatarinova@usc.edu
Michael Hogan, Email: mhogan@oriongenomics.com.
Mohd Firdaus-Raih, Email: firdaus@mfrlab.org.
Eng-Ti Leslie Low, Email: lowengti@mpob.gov.my.
References
- 1.Stanke M, Waack S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics. 2003;19(SUPPL 2):ii215–25. doi: 10.1093/bioinformatics/btg1080. [DOI] [PubMed] [Google Scholar]
- 2.Salamov AA, Solovyev VV. Ab initio gene finding in Drosophila genomic DNA. Genome Res. 2000;10:516–22. doi: 10.1101/gr.10.4.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ter-Hovhannisyan V, Lomsadze A, Chernoff YO, Borodovsky M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008;18:1979–90. doi: 10.1101/gr.081612.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Korf I. Gene finding in novel genomes. BMC Bioinformatics. 2004;5:59. doi: 10.1186/1471-2105-5-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Majoros WHH, Pertea M, Salzberg SLL. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics. 2004;20:2878–9. doi: 10.1093/bioinformatics/bth315. [DOI] [PubMed] [Google Scholar]
- 6.DeCaprio D, Vinson JP, Pearson MD, Montgomery P, Doherty M, Galagan JE. Conrad: gene prediction using conditional random fields. Genome Res. 2007;17:1389–98. doi: 10.1101/gr.6558107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schweikert G, Zien A, Zeller G, Behr J, Dieterich C, Cheng SO, Philips P, De Bona F, Hartmann L, Bohlen A, Krüger N, Sonnenburg S, Rätsch G. mGene: accurate SVM-based gene finding with an application to nematode genomes. Genome Res. 2009;19:2133–43. doi: 10.1101/gr.090597.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ying XU, Mural RJ, Ralph Einstein J, Shah MB, Uberbacher EC. GRAIL: a multi-agent neural network system for gene identification. Proc IEEE. 1996;84:1544–51. doi: 10.1109/5.537117. [DOI] [Google Scholar]
- 9.Snyder EE, Stormo GD. Identification of protein coding regions in genomic DNA. J Mol Biol. 1995;248:1–18. doi: 10.1006/jmbi.1995.0198. [DOI] [PubMed] [Google Scholar]
- 10.Allen JE, Salzberg SL. JIGSAW: integration of multiple sources of evidence for gene prediction. Bioinformatics. 2005;21:3596–603. doi: 10.1093/bioinformatics/bti609. [DOI] [PubMed] [Google Scholar]
- 11.Cantarel BL, Korf I, Robb SMC, Parra G, Ross E, Moore B, Holt C, Alvarado AS, Yandell M. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008;18:188–96. doi: 10.1101/gr.6743907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seaver SMD, Gerdes S, Frelin O, Lerma-Ortiz C, Bradbury LMT, Zallot R, Hasnain G, Niehaus TD, El Yacoubi B, Pasternak S, Olson R, Pusch G, Overbeek R, Stevens R, de Crécy-Lagard V, Ware D, Hanson AD, Henry CS. High-throughput comparison, functional annotation, and metabolic modeling of plant genomes using the PlantSEED resource. Proc Natl Acad Sci U S A. 2014;111:9645–50. doi: 10.1073/pnas.1401329111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goel N, Singh S, Aseri TC, Goel N, Singh S, Aseri TC. A review of soft computing techniques for gene prediction. ISRN Genomics. 2013;2013:1–8. doi: 10.1155/2013/191206. [DOI] [PubMed] [Google Scholar]
- 14.Goel N, Singh S, Aseri TC. A comparative analysis of soft computing techniques for gene prediction. Anal Biochem. 2013;438:14–21. [DOI] [PubMed]
- 15.Goodswen SJ, Kennedy PJ, Ellis JT. Evaluating high-throughput ab initio gene finders to discover proteins encoded in eukaryotic pathogen genomes missed by laboratory techniques. PLoS One. 2012;11:7. [DOI] [PMC free article] [PubMed]
- 16.Reid I, O’Toole N, Zabaneh O, Nourzadeh R, Dahdouli M, Abdellateef M, Gordon PM, Soh J, Butler G, Sensen CW, Tsang A. SnowyOwl: accurate prediction of fungal genes by using RNA-Seq and homology information to select among ab initio models. BMC Bioinformatics. 2014;15:229. doi: 10.1186/1471-2105-15-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Testa AC, Hane JK, Ellwood SR, Oliver RP. CodingQuarry: highly accurate hidden Markov model gene prediction in fungal genomes using RNA-seq transcripts. BMC Genomics. 2015;16:170. doi: 10.1186/s12864-015-1344-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoff KJ, Lange S, Lomsadze A, Borodovsky M, Stanke M. BRAKER1: unsupervised RNA-seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics. 2015;32:767–9. doi: 10.1093/bioinformatics/btv661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28:3150–2. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kapustin Y, Souvorov A, Tatusova T, Lipman D. Splign: algorithms for computing spliced alignments with identification of paralogs. Biol Direct. 2008;3:20. doi: 10.1186/1745-6150-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Allen JE, Majoros WH, Pertea M, Salzberg SL. JIGSAW, GeneZilla, and GlimmerHMM: puzzling out the features of human genes in the ENCODE regions. Genome Biol. 2006;7(Suppl 1):S9.1–13. doi: 10.1186/gb-2006-7-s1-s9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stanke M, Diekhans M, Baertsch R, Haussler D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics. 2008;24:637–44. doi: 10.1093/bioinformatics/btn013. [DOI] [PubMed] [Google Scholar]
- 24.Holt C, Yandell M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics. 2011;12:491. [DOI] [PMC free article] [PubMed]
- 25.Campbell MS, Law M, Holt C, Stein JC, Moghe GD, Hufnagel DE, Lei J, Achawanantakun R, Jiao D, Lawrence CJ, Ware D, Shiu S-H, Childs KL, Sun Y, Jiang N, Yandell M. MAKER-P: a tool kit for the rapid creation, management, and quality control of plant genome annotations. Plant Physiol. 2014;164:513–24. doi: 10.1104/pp.113.230144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–7. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 27.Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35(Database issue):D61–5. [DOI] [PMC free article] [PubMed]
- 28.Ouyang S, Buell CR. The TIGR Plant Repeat Databases: a collective resource for the identification of repetitive sequences in plants. Nucleic Acids Res. 2004;32(Database issue):D360–3. doi: 10.1093/nar/gkh099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–7. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 30.Llorens C, Futami R, Covelli L, Domínguez-Escribá L, Viu JM, Tamarit D, Aguilar-Rodríguez J, Vicente-Ripolles M, Fuster G, Bernet GP, Maumus F, Munoz-Pomer A, Sempere JM, Latorre A, Moya A. The Gypsy Database (GyDB) of mobile genetic elements: release 2.0. Nucleic Acids Res. 2011;39(Database issue):D70–4. doi: 10.1093/nar/gkq1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mistry J, Finn RD, Eddy SR, Bateman A, Punta M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013;41(12):e121. [DOI] [PMC free article] [PubMed]
- 32.Sima FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;2015:1–3. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 33.Lamesch P, Berardini TZ, Li D, Swarbreck D, Wilks C, Sasidharan R, Muller R, Dreher K, Alexander DL, Garcia-Hernandez M, Karthikeyan AS, Lee CH,Nelson WD, Ploetz L, Singh S, Wensel A, Huala E. The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res. 2012;D1:40. [DOI] [PMC free article] [PubMed]
- 34.Burset M, Guigó R. Evaluation of gene structure prediction programs. Genomics. 1996;34:353–67. doi: 10.1006/geno.1996.0298. [DOI] [PubMed] [Google Scholar]
- 35.Gremme G, Steinbiss S, Kurtz S. Genome tools: a comprehensive software library for efficient processing of structured genome annotations. IEEE/ACM Trans Comput Biol Bioinform. 2013;10:645–56. doi: 10.1109/TCBB.2013.68. [DOI] [PubMed] [Google Scholar]
- 36.Kawahara Y, de la Bastide M, Hamilton JP, Kanamori H, McCombie WR, Ouyang S, Schwartz DC, Tanaka T, Wu J, Zhou S, Childs KL, Davidson RM, Lin H, Quesada-Ocampo L, Vaillancourt B, Sakai H, Lee SS, Kim J, Numa H, Itoh T, Buell CR, Matsumoto T. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice (N Y) 2013;6:4. doi: 10.1186/1939-8433-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zickmann F, Lindner MS, Renard BY. GIIRA - RNA-Seq driven gene finding incorporating ambiguous reads. Bioinformatics. 2014;30:606–13. doi: 10.1093/bioinformatics/btt577. [DOI] [PubMed] [Google Scholar]
- 38.Haas BJ, Volfovsky N, Town CD, Troukhan M, Alexandrov N, Feldmann KA, Flavell RB, White O, Salzberg SL. Full-length messenger RNA sequences greatly improve genome annotation. Genome Biol. 2002;3:RESEARCH0029. doi: 10.1186/gb-2002-3-6-research0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alexandrov NN, Brover VV, Freidin S, Troukhan ME, Tatarinova TV, Zhang H, Swaller TJ, Lu Y-PP, Bouck J, Flavell RB, Feldmann KA. Insights into corn genes derived from large-scale cDNA sequencing. Plant Mol Biol. 2009;69:179–94. doi: 10.1007/s11103-008-9415-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Z, Zhang Z, Yan P, Huang S, Fei Z, Lin K. RNA-Seq improves annotation of protein-coding genes in the cucumber genome. BMC Genomics. 2011;12:540. doi: 10.1186/1471-2164-12-540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alexandrov NN, Troukhan ME, Brover VV, Tatarinova T, Flavell RB, Feldmann KA. Features of Arabidopsis genes and genome discovered using full-length cDNAs. Plant Mol Biol. 2006;60:69–85. doi: 10.1007/s11103-005-2564-9. [DOI] [PubMed] [Google Scholar]
- 42.Tatarinova T, Brover V, Troukhan M, Alexandrov N. Skew in CG content near the transcription start site in Arabidopsis thaliana. Bioinformatics. 2003;19(Suppl 1):i313–4. doi: 10.1093/bioinformatics/btg1043. [DOI] [PubMed] [Google Scholar]
- 43.Troukhan M, Tatarinova T, Bouck J, Flavell RB, Alexandrov NN. Genome-wide discovery of cis-elements in promoter sequences using gene expression. OMICS. 2009;13:139–51. doi: 10.1089/omi.2008.0034. [DOI] [PubMed] [Google Scholar]
- 44.Ahmad T, Sablok G, Tatarinova TV, Xu Q, Guo WW. Evaluation of codon biology in citrus and Poncirus trifoliata based on genomic features and frame corrected expressed sequence tags. DNA Res. 2013;20:135–50. doi: 10.1093/dnares/dss039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Steijger T, Abril JF, Engström PG, Kokocinski F, Akerman M, Alioto T, Ambrosini G, Antonarakis SE, Behr J, Bertone P, Bohnert R, Bucher P, Cloonan N, Derrien T, Djebali S, Du J, Dudoit S, Gerstein M, Gingeras TR, Gonzalez D, Grimmond SM, Guigó R, Habegger L, Harrow J, Hubbard TJ, Iseli C, Jean G, Kahles A, Lagarde J, Leng J, et al. Assessment of transcript reconstruction methods for RNA-seq. Nat Methods. 2013;10:1177–84. doi: 10.1038/nmeth.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mehrotra S, Goyal V. Repetitive sequences in plant nuclear DNA: types, distribution, evolution and function. Genomics Proteomics Bioinformatics. 2014;12(4):164–71. [DOI] [PMC free article] [PubMed]
- 47.Standage DS, Brendel VP. ParsEval: parallel comparison and analysis of gene structure annotations. BMC Bioinformatics. 2012;13:187. doi: 10.1186/1471-2105-13-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Project name: Seqping
Project home page: https://sourceforge.net/projects/seqping/
Archived version: seqping_0.1.45
Operating system: Linux platform
Programming language: Bash, Perl
Other requirements: BLAST 2.2.25, CD-HIT 4.5.4, Splign 1.39.8, Exonerate 2.2, GlimmerHMM 3.0, AUGUSTUS 2.6.1, MAKER 2.10, EMBOSS 6.4.0, Cufflinks 2.0.2
License: GNU General Public License
Any restrictions to use by non-academics: None