

Abstract
Background
Female moths synthesize species-specific sex pheromone components and release them to attract male moths, which depend on precise sex pheromone chemosensory system to locate females. Two types of genes involved in the sex pheromone biosynthesis and degradation pathways play essential roles in this important moth behavior. To understand the function of genes in the sex pheromone pathway, this study investigated the genome-wide and digital gene expression of sex pheromone biosynthesis and degradation genes in various adult tissues in the diamondback moth (DBM), Plutella xylostella, which is a notorious vegetable pest worldwide.
Results
A massive transcriptome data (at least 39.04 Gb) was generated by sequencing 6 adult tissues including male antennae, female antennae, heads, legs, abdomen and female pheromone glands from DBM by using Illumina 4000 next-generation sequencing and mapping to a published DBM genome. Bioinformatics analysis yielded a total of 89,332 unigenes among which 87 transcripts were putatively related to seven gene families in the sex pheromone biosynthesis pathway. Among these, seven [two desaturases (DES), three fatty acyl-CoA reductases (FAR) one acetyltransferase (ACT) and one alcohol dehydrogenase (AD)] were mainly expressed in the pheromone glands with likely function in the three essential sex pheromone biosynthesis steps: desaturation, reduction, and esterification. We also identified 210 odorant-degradation related genes (including sex pheromone-degradation related genes) from seven major enzyme groups. Among these genes, 100 genes are new identified and two aldehyde oxidases (AOXs), one aldehyde dehydrogenase (ALDH), five carboxyl/cholinesterases (CCEs), five UDP-glycosyltransferases (UGTs), eight cytochrome P450 (CYP) and three glutathione S-transferases (GSTs) displayed more robust expression in the antennae, and thus are proposed to participate in the degradation of sex pheromone components and plant volatiles.
Conclusions
To date, this is the most comprehensive gene data set of sex pheromone biosynthesis and degradation enzyme related genes in DBM created by genome- and transcriptome-wide identification, characterization and expression profiling. Our findings provide a basis to better understand the function of genes with tissue enriched expression. The results also provide information on the genes involved in sex pheromone biosynthesis and degradation, and may be useful to identify potential gene targets for pest control strategies by disrupting the insect-insect communication using pheromone-based behavioral antagonists.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3592-y) contains supplementary material, which is available to authorized users.
Keywords: Pheromone-biosynthesis enzymes, Pheromone-degrading enzymes, Detoxification, Carboxyl/Cholinesterase, Aldehyde oxidase, Fatty acyl reductase, Desaturase
Background
In moths, reproductive isolation relies heavily on mature female adults producing and releasing species-specific sex pheromone components to attract conspecific males. Adult male antennae on the other hand specifically perceive these sex pheromone components from a distance resulting in the successful location of the female to initiate and complete mating behavior [1]. In lepidopterans, type I sex pheromone components are fatty acid derivatives with 0–4 double bond containing carbon chains of varying lengths (C10-C18) and an oxygenated functional group that can be an aldehyde, alcohol or an acetate ester. Type II sex pheromone components are hydrocarbons or epoxides [2, 3]. In female moths, the type I sex pheromones are synthesized de novo and the genes involved in the biosynthesis are regulated in a specialized manner in their sex pheromone glands (PGs). Sex pheromone biosynthesis employs a modified fatty-acid biosynthesis pathway that includes various processes such as acetylation, desaturation, chain shortening, reduction, and oxidation, either separately or in combination [2, 4].
Type I sex pheromone biosynthesis pathway in moths initiates in PG by the release of pheromone biosynthesis activating neuropeptide (PBAN) from the subesophageal ganglion, which then circulates unbound to the PG where it binds to the pheromone biosynthesis activating neuropeptide receptor (PBANr) [5]. The resulting signal, then triggers several enzyme pathways starting with acetyl-CoA carboxylase (ACC) that catalyzes the conversion of acetyl-CoA to malonyl-CoA and finally resulting in the synthesis of pheromone precursors (mostly 14, 16 or 18 carbon saturated fatty acids) [4]. Later, double bonds are introduced into the pheromone precursors at specific positions (Δ5 [6], Δ6 [7], Δ9 [8–12], Δ10 [13, 14], Δ11 [9, 15, 16] and Δ14 [17, 18]) by different desaturases (DESs), and the chain is shortened by a modified β-oxidation pathway [19]. Finally, the unsaturated pheromone precursors are modified to form various functional groups such as aldehyde, alcohol or acetate ester, by three enzymes, aldehyde reductase, fatty acyl reductase and acetyltransferase, respectively [2, 20–22]. Some of these enzymes have been shown to have substrate specificity. For example, pgFARs from Spodoptera exigua have substrate preference for C14 and C16 fatty acids [23].
Males on the other hand, have evolved a sensitive antennal olfaction system to trace even very low amounts of sex pheromone components released from the female located at a long distance [24]. These sex pheromone components enter the male antennal pores and are transported by pheromone binding proteins (PBPs) [25–27] through the antennal sensillum, and activate membrane-bound pheromone receptors (PRs) [28–30]. After PR activation, the pheromone molecules must be rapidly degraded to release and refresh the PR. Depending on the functional group on a pheromone, whether an aldehyde, alcohol or ester, degradation of pheromones involves specific enzymes in the pheromone degradation pathway. Aldehyde oxidase (AOX) is an antennal enzyme that catalyzes the oxidation of aldehyde sex pheromones to carboxylic acids, which were biochemically characterized in the antennae of Manduca sexta [31], Antheraea polyphemus and Bombyx mori [32]. Thus far, only one insect AOX (AtraAOX2) from Amyelois transitella has been functionally characterized to hydrolyze aldehyde sex pheromone components and some plant volatiles in vitro [33]. However, a number of putative AOX genes have been reported in lepidopterans at the nucleic acid level [34–36]. Aldehyde-degradation involves the NAD(P)− dependent aldehyde dehydrogenase (ALDH) and AR enzymes that convert sex pheromones to their corresponding acids and alcohols, respectively [37–39]. Antennal carboxyl/cholinesterase (CCE) hydrolyzes ester sex pheromone components. In previous studies, we functionally characterized five carboxyl/cholinesterases (CXEs) belonging to the CCE family from S. exigua and Spodoptera litura; in vitro expressed SexiCXE10 specifically degraded ester plant volatiles and three other CCEs (SexiCXE4, SexiCXE13 and SlitCXE13) had dual roles, i.e., they hydrolyzed sex pheromone components as well as plant volatiles [40–43]. Other members of the multi-function enzyme families include CYPs [44, 45], UGTs [46] and GSTs [47], which are also considered as odorant-degrading enzymes (ODEs) or pheromone-degrading enzymes (PDEs).
The diamondback moth (DBM), Plutella xylostella (Lepidoptera: Plutellidae), is a notorious pest of cruciferous vegetables worldwide. The major known pheromone components extracted from female DBM moth PGs include three different functional groups that were type I sex pheromone components; (Z)-11-hexadecenal [Z11-16:Ald] [48], (Z)-11-hexadecenyl acetate [Z11-16:Ac] [49–52] and (Z)-11-hexadecenol [Z11-16:OH] [49–52]. These studies also revealed that both female and male DBM need precise regulation of sex pheromone biosynthesis that facilitates their communication through the various structural components. In recent years, sex pheromone reception has been studied extensively because of its potential use as green targets for environmentally friendly pest control measures. Male DBMs have PBPs that are designed for sex pheromone reception. Thus far, three PBPs have been cloned and characterized in P. xylostella. These PBPs robustly bound all three sex pheromone components (Z11-16:Ald, Z11-16:OH and Z11-16) [53] thereby enhancing the electrophysiological responses of DBM to sex pheromone components in vitro [54]. In the recently sequenced DBM genome [54], two gene sequences likely to be odorant/pheromone-degrading enzymes such as CYPs [55] and GSTs [56] were identified. However, the tissue specific expression of these genes are not known. To identify additional genes involved in the DBM pheromone biosynthesis and degradation, in this study, we combined transcriptome based digital gene expression (DGE) and genome mapping. By using these methods, we identified a total of 299 genes belonging to 14 gene families potentially involved in the sex pheromone biosynthesis and degradation pathways. The tissue specific expression pattern of these genes was also confirmed qPCR.
Results and discussion
Overview of the transcriptomes
Six transcriptomes from male antennae (m_Ant), female antennae (f_Ant), legs (L), heads (H), abdomens (AB) and female pheromone glands (PG) were sequenced using the Illumina HiSeq 4000 platform (Illumina, Tianjin, China) and assembled with Trinity (version r20140413p1). To obtain an accurate expression pattern, we removed as much of the ovipositor as possible from the females due to its adherence to the pheromone glands (Fig. 1). Illumina sequencing yielded 5.96–6.84 giga bases (Gb) for each of the six transcriptomes. We first mapped the transcriptome reads to the DBM reference genome (Genome assembly version 2, http://iae.fafu.edu.cn/DBM/) (Table 1) and obtained the following mapping rates that were favourable for annotation: 42.29% (legs), 39.6% (male antennae), 38.65% (female antennae), 42.27% (heads), 43.7% (pheromone glands) and 41.65% (abdomens). Then, we supplemented the original genome annotation file (18,105 genes Genome assembly version 2, http://iae.fafu.edu.cn/DBM/) and found 3,839 novel genes (Additional file 1: Table S1). Further to obtain more unigenes, we also performed a de novo assembly of the six transcriptomes that yielded unigenes with a median length of 334 nt with the longest unigene being 28,680 nt in length. Finally, these reads were assembled into 125,385 transcripts and 89,332 unigenes, with N50 lengths of 1,831 and 1,499 nt, respectively (Table 2).
Fig. 1.
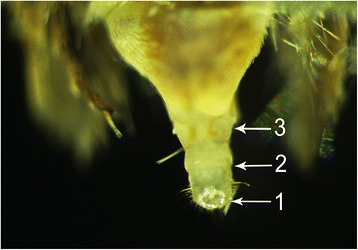
Dissection of P. xylostella female sex pheromone glands. The pheromone glands from female P. xylostella were squeezed out from the abdomen using forceps (the gland is similarly inflated when the female calls). The abdomen of P. xylostella was cut at the sclerotized cuticle from the 8th abdominal segment, and the sclerotized cuticle was removed before immersing the glands in liquid nitrogen. 1: Sclerotized ovipositor valves; 2: Pheromone gland; 3: Sclerotized cuticle that was removed
Table 1.
Summary of P. xylostella transcriptome mapping to the reference genome
| Sample_name | L | M_Ant | F_Ant | H | PG | AB |
|---|---|---|---|---|---|---|
| Total reads | 53439024 | 54660234 | 50948026 | 51117268 | 47655468 | 54606672 |
| Total mapped | 22601392 (42.29%) | 21647440 (39.6%) | 19691333 (38.65%) | 21606597 (42.27%) | 20826116 (43.7%) | 22742109 (41.65%) |
| Multiple mapped | 2634934 (4.93%) | 2802997 (5.13%) | 2425347 (4.76%) | 2059342 (4.03%) | 2283645 (4.79%) | 2736184 (5.01%) |
| Uniquely mapped | 19966458 (37.36%) | 18844443 (34.48%) | 17265986 (33.89%) | 19547255 (38.24%) | 18542471 (38.91%) | 20005925 (36.64%) |
| Read-1 | 10216135 (19.12%) | 9618895 (17.6%) | 8799050 (17.27%) | 9995946 (19.55%) | 9468501 (19.87%) | 10206183 (18.69%) |
| Read-2 | 9750323 (18.25%) | 9225548 (16.88%) | 8466936 (16.62%) | 9551309 (18.69%) | 9073970 (19.04%) | 9799742 (17.95%) |
| Reads map to ‘ + ’ | 9980188 (18.68%) | 9391804 (17.18%) | 8600732 (16.88%) | 9758204 (19.09%) | 9242300 (19.39%) | 9976030 (18.27%) |
| Reads map to ‘-’ | 9986270 (18.69%) | 9452639 (17.29%) | 8665254 (17.01%) | 9789051 (19.15%) | 9300171 (19.52%) | 10029895 (18.37%) |
| Non-splice reads | 13506179 (25.27%) | 12550858 (22.96%) | 11270056 (22.12%) | 10945264 (21.41%) | 12244204 (25.69%) | 13297124 (24.35%) |
| Splice reads | 6460279 (12.09%) | 6293585 (11.51%) | 5995930 (11.77%) | 8601991 (16.83%) | 6298267 (13.22%) | 6708801 (12.29%) |
Table 2.
Summary of P. xylostella de novo transcriptome assembly
| Tissues | F_Ant | M_Ant | L | H | AB | PG |
|---|---|---|---|---|---|---|
| Total size (Gb) | 6.36 | 6.84 | 6.68 | 6.38 | 5.96 | 6.82 |
| GC content | 47.26 | 45.98 | 47.46 | 54.59 | 47.77 | 45.01 |
| Number of transcripts | 125,385 | |||||
| Total unigene count | 89,332 | |||||
| Genes with homologues in NR | 26,657 | |||||
| Total transcript nucleotides | 113,082,390 | |||||
| Total unigene nucleotides | 65,915,278 | |||||
| N50 transcript length | 1831 nt | |||||
| N50 unigene length | 1499 nt | |||||
| Longest unigene length | 28,680 nt | |||||
| Median unigene length | 334 nt | |||||
BLASTx searches of all 89,332 unigenes showed that 29.84% were homologous to proteins in several insect genome databases (Bombyx mori, Danaus plexippus, Acyrthosiphon pisum, Anopheles gambiae, Apis mellifera, Drosophila melanogaster, Tribolium castaneum, Lucilia cuprina, Rhodnius prolixus and Solenopsis invicta, http://ensemblgenomes.org/info/genomes) with a cut-off E-value of 10−5. The highest homology (31.2%) was with B. mori sequences, followed by sequences from D. plexippus (27.8%), A. gambiae (12.6%), D. melanogaster (9.7%), and T. castaneum (5.3%) (Additional file 2: Figure S1a).
Then, we used Blast2GO to annotate unigenes into functional groups based on Gene ontology (GO). The GO annotations were used to classify the transcripts into functional groups according to specific GO categories. In the molecular function category, the genes expressed were mostly enriched for catalytic activity (e.g., hydrolase and oxidoreductase) and binding (e.g., nucleotide, ion, and odorant binding). In the biological process category, the most common were the cellular and metabolic processes. In the cellular component category, the most represented were the following two terms: cell (GO:0005623) and cell part (GO:0044464) (Additional file 2: Figure S1b).
The putative pathways of pheromone biosynthesis and degradation are shown in Fig. 2. We identified a number of genes related to the pathways by using the BLASTx search. Below, we describe the specific tissue expression patterns of these genes using qPCR.
Fig. 2.
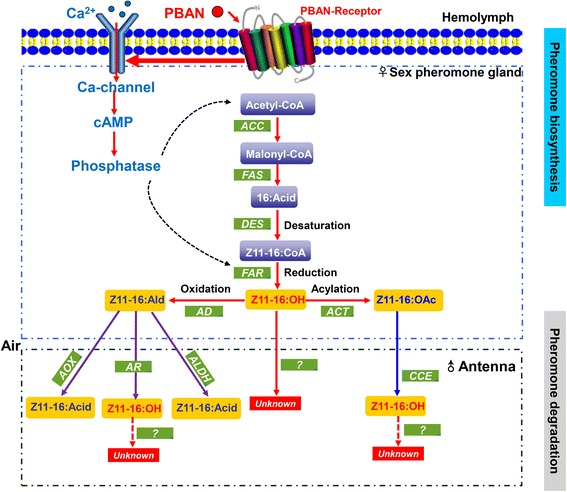
Proposed biosynthetic pathway of sex pheromone production in female DBM moths (adapted from [2, 16, 77])
Genes involved in the sex pheromone biosynthesis pathway
Pheromone biosynthesis activating neuropeptide receptor (PBANr)
Insect pheromone biosynthesis is regulated by PBANr that binds to PBAN released from the suboesophagal ganglion in the brain [5]. To date, no more than four alternative splice transcripts such as isoforms As, A, B and C with differences in their C-terminal sequences have been reported in one insect species [57]. B and C typically consist of the YXXΦ endosomal sorting motif and the complete seven transmembrane regions [58]. Although all four PBANrs are expressed dominantly in PGs, only PBANR-B and PBANR-C have been identified as intracellularly localized with some cell surface localization. PBANR-As and PBANR-A on the other hand, were completely intracellular, suggesting that PBANR-B and PBANR-C could be principal receptors participating in the PBAN signal reception [57]. In this study, we found two additional alternative splice transcripts (Px002138 and c63176_g2; see Additional file 3: Table S2) that possessed C-termini different from DBM PBANr genes previously described in the genome [59, 60] (Additional file 4: Figure S2). We named them as PluxyPBANR-C, −B and -A. Multiple amino acid sequence alignment of these PBANr isoforms showed the presence of typical PBANr sequence features (Additional file 4: Figure S2). We further analyzed the tissue expression pattern of PluxyPBANR-C and -B by comparing their FPKM values (Additional file 3: Table S2 and Fig. 3) in the transcriptomes from the different tissues. The results showed at least 3- and 5-fold higher numbers for PluxyPBANR-C and -B transcripts, respectively in PG than in other tissues studied, and is consistent with reports from other moths such as Agrotis ipsilon [61], B. mori [62] and Heliothis virescens [63].
Fig. 3.
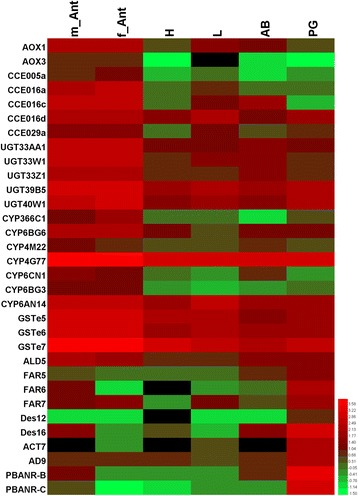
Heat map illustration of the tissue expression profile of sex pheromone biosynthesis and degradation genes based on FPKM values. m_Ant, male antennae; f_Ant, female antennae; H, Head; L, Leg; AB, Abdomen; PG, female pheromone gland. Gene names are indicated on the left. Scale Bar on bottom right indicates the degree of expression
Acetyl CoA Carboxylase (ACC) [EC:6.4.1.-]
The first step in moth pheromone biosynthesis is the ATP-dependent carboxylation of acetyl-CoA to malonyl-CoA by ACC [4]. This is a rate-limiting step in fatty acid biosynthesis [51]. In our transcriptome data set, seven ACC transcripts were identified (Additional file 3: Table S2) with all showing robust expression in the abdomen followed by PG (Additional file 3: Table S2). Among these, PxACC3, PxACC4 and PxACC5 showed higher expression than the others in PGs. In addition, homology analysis revealed that PxACC3 shared 73% amino acid identity with the ACC homolog from Papilio xuthus (Genbank XP_013176189.1), 70% with Helicoverpa assulta (Genbank (AKD01722.1) and 66% with D. plexippus (Genbank EHJ72299.1). PxACC4 is a full-length ACC, and the predicted protein from this transcript shared a high amino acid identity (83%) with the ACC from Agrotis segetum (AID66639.1) [64]. Similar to A. segetum ACC, PxACC4 also had dominant expression in the abdomen as well as in PG but without significant difference. PxACC5 also shared a high amino acid sequence identity (83%) with ACC from B. mori (XP_004930758.1) indicating a similar function for ACC homologs.
Fatty acid synthase (FAS) [EC:2.3.1.-]
In moths, saturated fatty acid is produced from malonyl-CoA and NADPH in a reaction catalyzed by FAS [4]. In this study, we found seven putative transcripts with high amino acid identities to known FAS genes (PxFAS1-PxFAS8) (Additional file 3: Table S2). Only one partial FAS transcript was found in the genome annotation, two novel transcripts were identified via genome mapping and four new FAS transcripts were found among the unigenes after de novo assembly. None of the seven PxFAS genes showed dominant expression in PGs (Additional file 3: Table S2). Five of the PxFASs displayed abdomen biased expression, unlike the two homologs, FAS from A. ipsilon (AGR49310.1) and A. segetum (AID66645.1) that shared 71 and 72% amino acid identity with PxFAS5 and PxFAS1, respectively and were expressed higher in PGs than in the abdomen [61, 64].
Fatty Acid Transport Protein (FATP) [EC:6.2.1.-]
The fatty acid transport protein (FATP) has been functionally characterized by in vitro expression and knock down assay in B. mori [65] and Eilema japonica [66]. These studies showed that FATP catalyzes the ATP-dependent esterification of extracellular long-chain fatty acids in sex PGs to produce the corresponding acyl-CoA derivatives. Four PxFATPs unigenes were found in this study; PxFATP1 shared high amino acid identity (74%) with FATP from E. japonica (BAJ33523.1); PxFATP2 and 4 shared 58 and 81% amino acid identity, respectively with FATP2 and FATP3 from Sesamia inferens [36]. All three PxFATPs displayed ubiquitous expression (Additional file 3: Table S2) indicating the lack of specialized function in the sex pheromone biosynthesis pathway. PxFATP3 was not detected in any tissue in our transcriptome data, and thus maybe a pseudogene (Additional file 3: Table S2).
Desaturase (DES) [EC:1.14.19.-]
Typically moth pheromones contain double bonds, which require desaturases to introduce the bonds into specific locations in the fatty acyl carbon chains [15, 67, 68]. Depending on the locations of the double bonds introduced, DES can be classified into the following major groups: Δ5 [6], Δ6 [7], Δ9 [8–12], Δ10 [13, 14], Δ11 [9, 15, 16] and Δ14 [17, 18]. The major sex pheromone components in DBM are Z11-16:Ald, Z11-16:OAc and Z11-16:OH [48, 51, 52]. A single Z11-16:Ald can trigger copulation behaviour in male moths [48, 69]. Thus, Δ11 desaturase is expected to play a key role in this pheromone component biosynthesis. Investigation of the presence of desaturases in our transcriptome data set using genome mapping and de novo transcriptome assembly revealed 16 PxylDes transcripts (Additional file 3: Table S2). FPKM and qPCR analysis revealed that PxylDes12 and 16 were expressed abundantly in PG (Additional file 3: Table S2, Figs. 3 and 4). In addition, phylogenetic analysis showed that PxylDes12, 14, 15 and 16 clustered into the Δ11 clade, which also had the SinfDes4 homolog from S. inferens [36] (Fig. 5). Thus, we propose that the two PxDes may be involved in the desaturation of Z11-16:CoA, the major sex pheromone component in DBM. Ten additional PxylDes were assigned to other desaturase groups and also did not have PG biased expression. These genes may also be involved in other physiological functions rather than sex pheromone component desaturation.
Fig. 4.
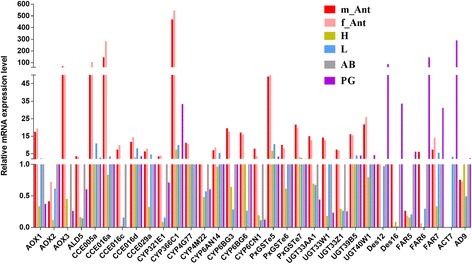
Relative expression levels of sex pheromone biosynthesis and degradation gene transcripts with antennae or pheromone gland-biased expression in different adult tissues. m_Ant, male antennae; f_Ant, female antennae; H, Head; L, Leg; AB, Abdomen; PG, female pheromone gland
Fig. 5.
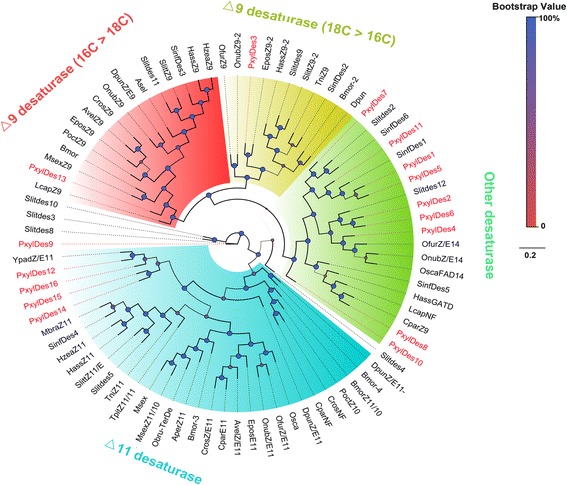
Phylogenetic tree of typical insect DES genes. PxylDES are highlighted in red. Species abbreviations: Bmor. B. mori; Slit. Spodoptera litura; Sinf. Sesamia inferens; Tni. T. ni; Ofur. O. furnacalis; Onub. O. nubilalis; Osca. O. scapulalis; Hass. H. assulta; Lcap. L. capitella; Cpar. C. parallela; Dpun. D. punctatus; Cros. C. rosaceana; Obru. O. brumata; Aper. A. pernyi; Msex. M. sexta; Tpit. T. pityocampa; Hzea. H. zea; Mbra. M. brassicae; Ypad. Y. padellus; Poct. P. octo; Epos. E. postvittana; Avel. A. velutinana; and Asel. A. selenaria cretacea
Fatty Acyl Reductase (FAR) [EC:1.2.1.-]
In DBM, Z11-16: OH is a main sex pheromone component [51, 52], which is produced by the reduction of fatty acyl precursor, Z11-16:CoA, in an NADPH dependent reaction [22, 70–72]. In this study, we identified in total, seven putative FAR transcripts, named PxFAR1-7 (Additional file 3: Table S2). Among them, PxFAR6 and PxFAR7, clustered with the pgFAR clade (Fig. 6). All FARs in this group displayed PG dominant expression, including the first identified pgFAR from B. mori [70], SlitFAR3 from S. litura [73] and SinfFAR2 from S. inferens [36]. Together, the FPKM value and qPCR analysis showed that among the seven PxFARs, PxFAR6 had the highest expression (20-fold higher) in PGs when compared to the other tested tissues (Figs. 3 and 5). Therefore, PxFAR6 may likely play a major role in the conversion of Z11-16:CoA to Z11-16:OH in DBM. In addition, we found another FAR predominantly expressed in PG; PxFAR5 that did not cluster with the pgFAR group. It shared high amino acid identity (71%) with YevoFARI from Yponomeuta evonymellus (ADD62438.1). A previous report on the PG specific expression of YevoFARI determined by qPCR analysis was also not clear [22]. The expression and functional analyses of this clade need further investigation.
Fig. 6.
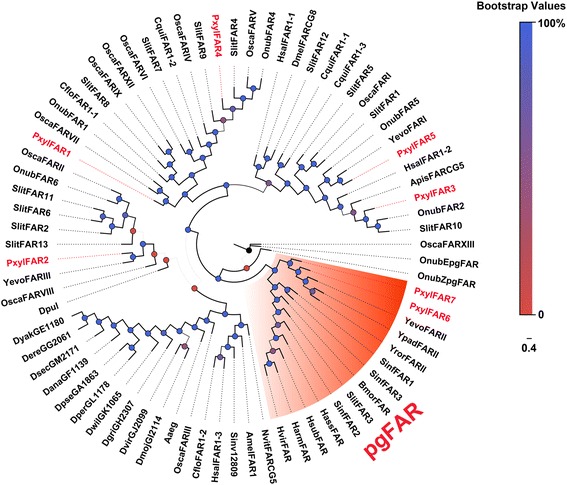
Phylogenetic tree of typical insect FAR genes. PxFARs are highlighted in red. Species abbreviations: Bmor. B. mori; Cqui. C.ulex quinquefasciatus; Yevo. Y. evonymellus; Ypad. Y. padellus; Yror. Y. rorrellus; Hass. H. assulta; Harm. H. armigera; Hsub. H. subflexa; Hvir. H. virescens; Nvit. N. vitripennis; Amel. A. mellifera; Sinv. S. invicta; Hsal. H. saltator; Cflo. C. floridanus; Osca. O. scapulalis; Aaeg. Aedes aegypti; Dmoj. D. mojavensis; Dvir. D. virilis; Dgri. D. grimshawi; Dwii. D. willistoni; Dper. D. persimilis; Dpse. D. pseudoobscura; Dana. D. ananassae; Dsec. D. sechellia; Dere. D. erecta; Dyak. D. yakuba; Dpul. D. pulex; Dmel. D. melanogaster and Apis. A. pisum
Acetyltransferase (ACT)
ACT is known to convert fatty acid alcohol to the corresponding ester in plants [74–76]. However, no ACT related to sex pheromone component biosynthesis has been thus far identified at the molecular level. In this study, we identified 21 ACT transcripts, PxylACT1-22, by homolog searches using sequences (BLASTp) obtained from our previous studies as query; SlitACTs from S. litura [73], SinfACTs from S. inferens [36] and AsegACTs from A. segetum [64] (Additional file 3: Table S2). We found that only PxACT7 displayed PG abundant expression via FPKM and qPCR analysis (Additional file 3: Table S2, Figs. 3 and 4). Further, BLASTx showed that PxACT7 shared a low amino acid identity (39%) with AsegACT (AIN34700.1) [64], which was shown to lack esterification activity by using in vitro yeast expression system and GC-MS analysis [65]. Not surprisingly, AsegACT was not abundant in PG, and its FPKM value in PG was almost equal to that in the abdomen [15]. Three ACTs from Agrotis ipsilon, ATF-KC007357, ATF-KC007358 and ATF-KC007360, were mainly expressed in PG than the rest of the body [61]. Their homologs in DBM are PxACT17, 19 and 14, which shared 65, 75 and 91% amino acid identities, respectively. However, none of these displayed PG dominant expression (Additional file 3: Table S2).
Alcohol dehydrogenase (AD)
Alcohol dehydrogenase converts alcohol compounds to corresponding aldehydes. However, no alcohol dehydrogenase has been identified at the molecular level from insect PGs although biochemical studies have shown the presence of alcohol dehydrogenses in the PGs of several species such as Heliothis virescens [77], Helicoverpa armigera [78] and H. assulta [78]. Z11-16:Ald was found to be a major sex pheromone in DBM, indicating that AD plays a crucial role in its biosynthesis. In this study, we conducted a BLASTp analysis by using previously identified SinfADs from S. inferens [36], AipsADs from A. ipsilon [61], CpomADs from Cydia pomonella [79], HassADs from H. assulta [78], HarmADs from H. armigera [78] and HvirAD from H. virescens [77]. We found in total, 22 ADs in DBM, named PxylAD1-22 (Additional file S3: Table S2). The FPKM values of these 22 PxylADs revealed that only one AD (PxylAD9) had PG dominant expression. qPCR assay also confirmed this (Figs. 3 and 4) and showed at least 2-fold higher expression in PG than in other tested tissues (Fig. 4). We also found several homologs of previously identified ADs from H. armigera (HarmAD5) and H. assulta (HassAD6) that had PG biased expression [78]. In our study, PxylAD15 shared the highest amino acid identity 70.2% with HassAD6, but PxylAD15 had not PG predominant expression. But, PG-enriched PxylAD9 shared only 25.1% amino acid identity with HassAD6.
Sex pheromone and other odorant-degrading enzyme genes
Aldehyde oxidase (AOX)
Aldehyde oxidase is considered as a sensillum enzyme that degrades redundant aldehyde odorants (including aldehyde sex pheromone components) to corresponding acids without the need for coenzymes [31–33]. We found three full-length AOXs in the DBM transcriptomes via genome mapping. These were named PxylAOX1, PxylAOX2 and PxylAOX3 (Additional file 3: Table S2), all containing the typical conserved motifs: a FAD-binding domain, two putative iron-sulfur (2Fe-2S) redox centers, and a molybdenum-cofactor (MoCo)-binding site. One 2Fe-2S redox center contains four conserved cysteine residues that coordinate with iron. PxylAOX1 and PxylAOX3 likely originated from a gene duplication event because they were located in the same scaffold (scaffold_267). Based on FPKM values and qPCR analysis, PxylAOX1 and PxylAOX3 were identified to have antennae enriched expression without sex-bias; PxylAOX1 expression in the antennae was more than 60-fold higher and PxylAOX3 expression was 40-fold higher than in other tissues. In addition, PxylAOX1 expression was higher than PxylAOX3 (Additional file 3: Table S2, Figs. 3 and 4), while PxylAOX2 showed ubiquitous expression and therefore may perform a more generalized function. We also noticed a putative AOX identified in the DBM genome (ID: CCG014884.1) [54]. This sequence was identified as a xanthine dehydrogenase (XDH) member by BLASTx search. A phylogenetic tree constructed by using known insect AOXs and XDH as an out group (Fig. 7) showed the distribution of insect AOXs into three major groups; group 1 consisted of dipteran AOX, group 2 had PxylAOX2 and PxylAOX3, and group 3 had AtraAOX2 from A. transitella and their homologs, which were shown to degrade the aldehyde sex pheromone component, Z11Z13–16Ald and several aldehyde plant volatiles [33], and SinfAOX2 from S. inferens with antennae dominant expression [36]. PxylAOX1 clustered with group 3 along with BmorAOX1 [80], BmorAOX2 [80], SinfAOX1 [36], SinfAOX2 [81], and MbraAOX [31], all of which have antennae enriched expression pattern. However, like other AOXs, PxylAOX1 and 3 did not show sex biased expression suggesting that they may be specifically involved in aldehyde plant volatiles degradation rather than sex pheromone component pathways.
Fig. 7.
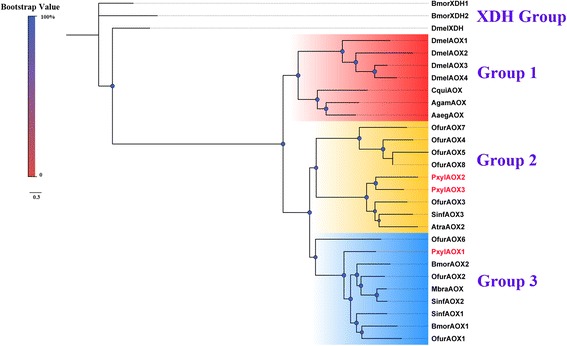
Phylogenetic tree of typical insect AOX genes. PxylAOXs are highlighted in red. Species abbreviations: Dmel, D. melanogaster; Bmor, B. mori; Cqui, C quinquefasciatus; Agam, A. gambiae; Aaeg, A. aegypti; Ofur, Ostrinia. furnacalis; Sinf, S. inferens; Atra, A. transitella and Mbra. Mamestra. brassicae. Scale bar on top left indicates the color code for bootstrap values in the phylogenetic tree
Aldehyde dehydrogenase (ALDH) and Aldehyde reductase (AR)
Unlike AOX, both ALDH and AR are thought to be intracellular and NAD(P)− dependent enzymes that catalyze the oxidation and reduction of a broad spectrum of aldehyde substrates to their corresponding acids and alcohols, respectively. Five PxylALDHs (named PxylALDH1-5) were identified from the DBM transcriptome data set. Among them, only PxylALDH4 was mainly expressed in the antennae as determined by the FPKM value and qPCR analysis (Additional file 3: Table S2). The expression level of PxylALDH4 in PG was at least 2-fold higher than in other tested tissues, indicating its likely function in odorant-degradation. Similar to AOX, this gene also showed no sex-biased expression suggesting that it is involved in plant volatiles degradation. A previous study showed that one ALDH, AaALDH3, oxidizes farnesal into farnesoic acid in Aedes aegypti [39]. But this gene only shared 11% amino acid identity with PxylALDH4, suggesting that they could act on different substrates. The ARs in general, include four reductase superfamilies; medium-chain dehydrogenases/reductases (MDR), short chain dehydrogenases/reductases (SDR), aldo-keto reductases (AKR) and NADB (NAD+ binding) reductases. Through motif and BLAST analyses, we found in total, 15 ARs among which were six MDRs (PxylAR2, 3, 5, 6, 14 and 15), eight AKRs (1, 7, 8, 9, 10, 11, 12 and 13) and one NADB (PxylAD4). However, no SDR was found (Additional file 3: Table S2). In B. mori [82] and H. armigera [38], two AKRs have been characterized in vitro; these were reductases that converted the sex pheromone components, E10,Z12-16:Ald (B. mori), Z9-16:Ald (H. armigera) and Z11-16:Ald (H. armigera) to corresponding alcohols. FPKM values did not show AR expression specific to the antennae. Two AKRs shared the most amino acid identities with PxylARs; these were PxylAD9 (59%) and PxylAD7 (37%), but PxylAD9 and PxylAD7 displayed ubiquitous expression, indicating their involvement in other physiological functions instead of olfaction alone.
Carboxyl/cholinesterase (CCE)
Insect CCE is a superfamily, which participates in many physiological processes and contains various substrates [83]. Some of them are secretory enzymes that refresh ORs by clearing redundant esterase odorants around them. Besides Z11-16:Ald, Z11-16:Ac is another major sex pheromone component reported in P. xylostella [48]. We found 49 CCEs in the DBM transcriptome that was mapped to the reference genome (Additional file 3: Table S2). This total number is lower than 69 identified in B. mori [84, 85], but higher than 39 found in H. armigera [86]. A phylogenetic tree of known insect CCEs is shown in Fig. 8. These CCEs were named from CCE001-30 sub-groups based on the B. mori [84, 85] and H. armigera [86] 13 major clade nomenclature system. By FPKM values and qPCR analysis, five CCEs were found to have antennae enriched expression (Additional file 3: Table S2, Figs. 3 and 4); PxCCE005a, 016a, 016c, 016d and 029a. These were distributed into three clades. PxCCE016a, 016c and 016d were located in the same sub-clade containing most general intracellular enzymes with dietary detoxification functions or ester odorant degradation functions such as the two CCEs with antennae dominant expression, SlCXE10 [87] and SexiCXE10 [40], that degrade ester plant volatiles. Unlike these two CCEs, PxCCE016a, 016c and 016d had no obvious sex biased expression. Other CCEs involved in non-olfactory functions have been described in some previous studies [88–90], We found the homologs of these genes among the PxCCEs, and they did not show antennae dominant expression, thus they may not participate in olfaction. For instance, in Group E, the pheromone degrading esterase, ApolPDE from A. polyphemus [91], PjapPDE from Popillia japonica [92], SexiCXE13/SlitCXE13 from S. exigua and S. litura [43], and DmelEst6 from D. melanogaster [93] were able to hydrolyze ester sex pheromone components. These genes showed abundant expression in the antennae except SexiCXE13 and SlitCXE13. PxCCE026a was their closest homolog, but it showed ubiquitous expression by FPKM analysis. Moreover, PxCCE016b also showed ubiquitous expression. A recent study showed that PxCCE016b was involved in chlorantraniliprole resistance; its increased expression in resistant strains enabled better degradation of ester pesticides [94]. The four 016 sub-group CCEs are derived from the same ancestor because they clustered in the same clade in the phylogenetic tree and may have evolved to perform various physiological functions based on the difference in tissue expression patterns between PxCCE016b and three other PxCCE016 CCEs. PxCCE029a and PxCCE005a were in the uncharacterized functional clade. The 029 clade CCEs lacked one or more of the enzyme catalytic active site residues, but had another active site called carboxyl/cholinesterase-like adhesion molecules including transmembrane and binding domains [95]. Thus, PxCCE029a may be involved in neuro/developmental functions. We also investigated the intron-exon organization in PxCCEs (Fig. 9). The average intron size was 1290 nt, which was slightly lower than 1372 in B. mori [85]. The longest intron was between exons 2 and 3 in PxCCE027b and comprised 10,014 nt, similar to that observed in BmCCE027b [85]. The shortest intron was in PxCCE028b between exons 8 and 9, and contained only 33 nt. Further, the approximate distribution of introns is shown in Fig. 9, which shows the positions of introns in PxCCEs by multiple sequence alignment. We also collected and marked the splice sites into three phases according to their positions in the codons. We noticed the presence of two highly conserved intron insertion sites and the splice site phase was also conserved. The two most conserved introns were a phase 2 intron around position 1500 present in 42 CCEs (Fig. 9, red arrow) and a phase 0 intron around position 500 in 37 CCEs (Fig. 9, gray arrow). The two conserved intron insertion sites and splice site phases were identical to that reported in B. mori CCEs [85], indicating that these sites were likely conserved since the early stage of CCE evolution. In addition, several clade specific conserved sites were also observed and were similar to the clade-specific conservation of intron phase and position reported in P. xylostella GSTs [56]. All CCEs in clade 020 possessed two conserved introns, phase 2 introns at positions 700 and 800 (Fig. 9, blue and dark brown brackets, respectively). Notably, two intron insertions and phase splice sites were conserved in two distant clades, 024–026 and 030 (Fig. 9, light brown and gray brackets, respectively). These conserved introns were also observed in clades 020, 024, 026 and 030 in B. mori and the neuroligins of D. melanogaster and A. mellifera [85].
Fig. 8.
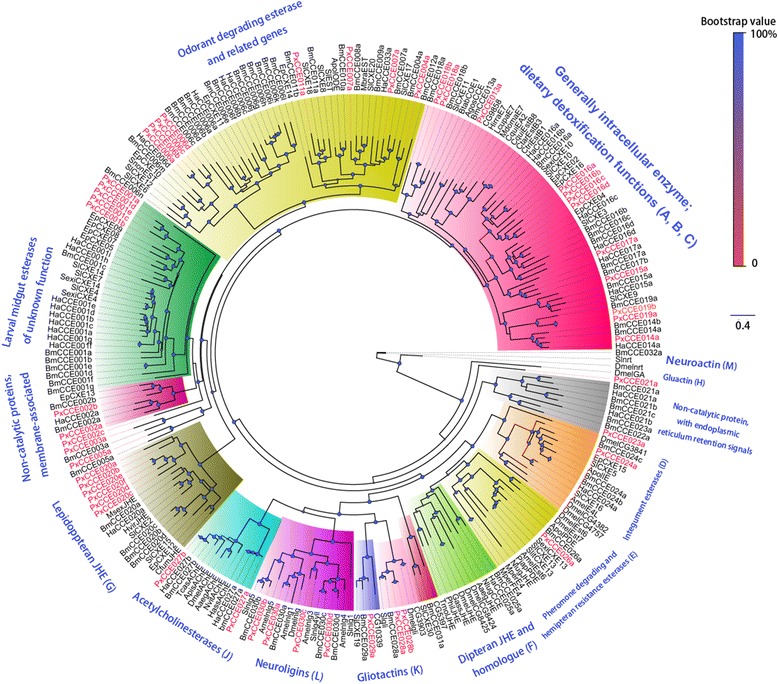
Phylogenetic tree of typical insect CCE genes. PxCCEs are highlighted in red. Species abbreviations: Dmel. D. melanogaster; Bmor. B. mori; Agos. Aphis gossypii; Hirr. Haematobia irritans; Lcup. Lucilia cuprina; Mdom. Musca domestica; Cqui. Culex quinquefasciatus; Ctar. Culex tarsalis; Ctri. Culex tritaeniorhynchus; Ha. H. armigera; Sexi. Spodoptera exigua; Sl. S. littoralis; Slit. Spodoptera litura; Ep. Epiphyas postvittana; Apol. Antheraea polyphemus; Amel. Apis mellifera; Pjap. Popillia japonica; Nlug. Nilaparvata lugens; Mper. Myzus persicae; Aaeg. Aedes aegypti; Gass. Gryllus assimilis; Phil. Psacothea hilaris; Tmol. Tenebrio molitor; Apis. Acyrthosiphon pisum; Tcas. Tribolium castaneum; Hass. Heliothis assulta; Nvit. Nasonia vitripennis; Cfum. Choristoneura fumiferana; Hvir. Heliothis virescens; Mbra. M. brassicae and Msex. Manduca sexta
Fig. 9.
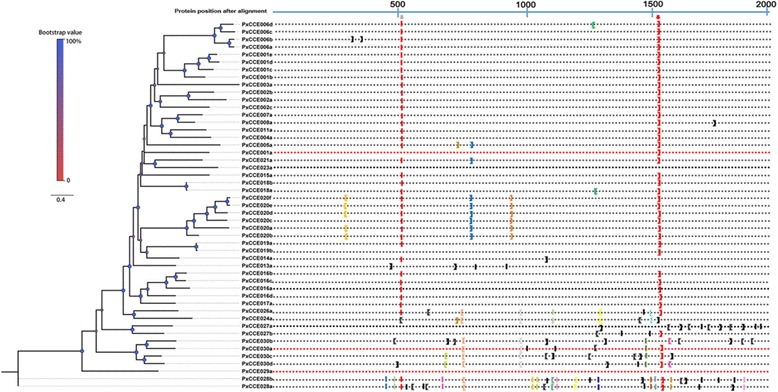
Phylogenetic tree and intron positions of PxCCE genes. The nomenclatures of clades are based on previously published data. The intron positions in sequences are shown as (|) for a phase 0 intron, ([) for a phase 1 intron and (]) for a phase 2 intron. Arrows indicate the most conserved intron insertion site in PxCCEs. Same color brackets indicate the same intron insertion site among different genes
UDP-glycosyltransferases (UGT)
UGT is a superfamily of detoxifying enzymes that improve the excretion of certain molecules by transferring glycosyl residues from nucleotide sugars; for example, hydrophobic molecules are thus converted to hydrophilic molecules for easy excretion. Typical examples of such molecules in insects are odorant molecules [46, 96, 97]. In the present study, 26 UGTs were found via genome mapping and transcriptome assembly of all six transcriptomes from DBM (Additional file 3: Table S2). All PxUGTs presented the classic UGT signature motif (N-terminal substrate binding site and N-glycosylation sites) similar to that described for other insect UGTs [46, 97, 98]. These UGT sequences have been submitted to the UGT Nomenclature Committee (http://prime.vetmed.wsu.edu/resources/udp-glucuronsyltransferase-homepage) for nomenclature assignments [97] (the sub-group names are shown in Additional file 3: Table S2). To further uncover the putative functions of PxUGTs, a phylogenetic tree was constructed by using UGTs from S. littoralis [46], B. mori [98, 99] and H. armigera [98] (Fig. 10). To date, there are 14 UGT families identified in lepidopterans [98]. PxUGTs were distributed within 11 sub-families; none were in UGT34, 47 and 48 sub-families. UGT33 and UGT40 sub-families had the most members in lepidopteran species [46, 98]. Similarly, most PxUGTs were clustered in UGT33 (n = 10) and UGT40 (n = 4) sub-families. In the UGT33 sub-family, BmUGT33D8 [99] and one UGT from M. sexta (AI234470.1) [100] displayed antennae enriched expression. By using FPKM values and qPCR analysis, we also identified three UGT33 family members among PxUGTs (PxUGT33AA1, PxUGT33W1 and PxUGT33Z1) that displayed antennae dominant expression (Additional file 3: Table S2, Figs. 3 and 5) indicating their olfactory function. Recently, two more UGTs from UGT40 and 46 sub-families, SlUGT40R3 and SlUGT46A6 from S. littoralis, showed abundant expression in the male antennae after exposure of moths to the sex pheromone component, Z9E11-14:Ac or an ester plant volatile, Z3-6:Ac. This suggested that the two UGTs may be odorant-degrading enzymes. We also found a UGT40 member in DBM, PxUGT40W1, which displayed the highest expression in antennae than in other tissues. However, there was no significant difference in the expression of PxUGT40W1 between male and female antennae, thus indicating that it may be involved in the removal of plant volatiles rather than sex pheromone components. In addition, PxUGT39B5 of the UGT39 sub-family also had an antennae enriched expression pattern indicating its olfactory function. However, the functions of its homologs have not been reported yet.
Fig. 10.
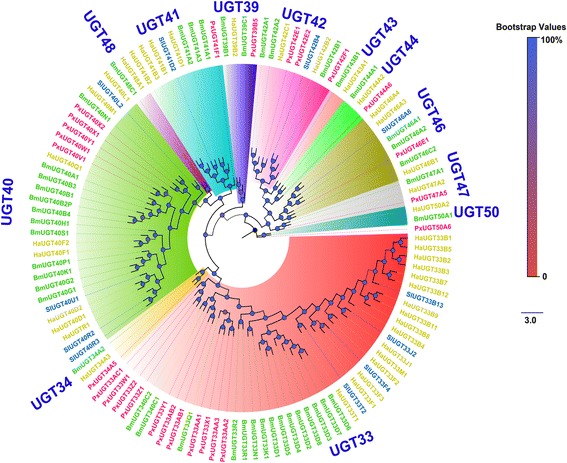
Phylogenetic tree of typical insect UGT genes. PxUGTs are highlighted in red. Species abbreviations: Bm. B. mori; Sl. S. littoralis; Ha. H. armigera
Cytochrome P450
Cytochrome P450 is a multi-functional enzyme in organisms, and is involved in detoxification, resistance, and odorant clearance [44, 101]. A previous study annotated 85 P450 genes (PxCYPs) in DBM but the tissue specific expression pattern was investigated only in the larval stages but not in adult tissues [55]. In the present study, the 85 PxCYPs were analyzed in various adult tissue transcriptomes and validated via qPCR (Additional file 3: Table S2, Figs. 3 and 4). We found that eight PxCYPs had predominant expression in the antennae of both sexes, and belonged to three CYP sub-families, CYP3: PxCYP321E1, 366C1; CYP4: PxCYP 4G77, 4 M22, and CYP6: PxCYP6AN14, 6BG3, 6BG6 and 6CN1 (Additional file 3: Table S2, Figs. 3 and 4). One CYP3 member, CYP345E2, which is an antenna-specific CYP from the pine beetle, D. ponderosae, was shown to catalyze the oxidation of monoterpene volatiles from pine hosts [44]. In our study, we found 2 CYP345E2 homologs in DBM; PxCYP321E1 and PxCYP366C1, both with higher expression in the antennae. In addition, PxCYP366C1 had the highest expression in the antennae when compared to other candidate odorant-degrading CYPs. This gene was also reported as highly expressed in adult heads from different developmental stages [55], indicating that PxCYP366C1 plays a vital role in specific olfactory behaviors of DBM adults; an example of such behavior could be locating oviposition sites. Some members of the CYP4 sub-family were also identified to be olfactory related. CYP4AW1 in P. diversa was reported to be involved in pheromone inactivation by analyzing its antennae specificity and through electrophysiological studies [45]. In DBM, we identified 2 CYP4 genes; PxCYP4G77 and 4 M22 both with robust expression in the antennae. A previous study reported the high expression of PxCYP4M22 in an insecticide-resistance strain than in the susceptible strain [55]. Together with its tissue specific expression pattern, we propose that PxCYP4M22 may participate in insecticide detoxification in the antennae of the insecticide-resistance strain. In the CYP6 sub-family, no member has been reported to have odorant clearance function. However, several genes have been reported to have olfactory organ specific expression; for example CYP6W1 [102] and CYP6A20 [103] in D. melanogaster, and CYP6B48, CYP6B42 and CYP6AE49 expressed in the antennae of both sexes and male proboscis in S. littoralis [104]. We found four CYP6 members in DBM (PxCYP6AN14, 6BG3, 6BG6 and 6CN1) that were abundant in the antennae. PxCYP6BG3 and 6BG6 had high expression in the larvae than in adults [55], suggesting that the two CYP genes may be involved in odorant clearance in larvae.
Glutathione S-Transferase (GSTs)
The functions of GSTs in many insect physiological functions such as insecticide resistance [56] and detoxification [105, 106] have been well-documented. In this study, we identified in total, 25 PxGSTs from the transcriptomes of DBM. In comparison with a previous study [56] we identified three new GSTs, PxGSTe6, PxGSTe7 and PxGSTu3 in DBM (Additional file 3: Table S2). Since PxGSTu3 was not detected in the transcriptomes of all tested tissues we presume that this may be a pseudogene. One olfactory-specific GST from M. sexta named, GSTmsolf1, was reported to degrade the plant volatile, trans-2-hexenal [47]. When we used the deduced amino acid sequence of this protein as query in a BLASTp search, we found that it belonged to the GST sub-family Delta. However, all three antennae enriched PxGSTs we identified in this study belonged to the GST sub-family Epsilon (Additional file 3: Table S2, Figs. 3 and 4). We identified three new GSTs; PxGSTe6, PxGSTe7 and PxGSTe5. Among these, PxGSTe5 had the highest expression and was consistent with a previous report that showed robust expression of PxGSTe5 in adults [56]. This suggested that PxGSTe5 could participate in odorant degradation in the adult stage.
Conclusions
Based on massive Illumina sequencing, in this study we sequenced the transcriptomes from six tissues of the diamondback moth. Genome mapping and de novo assembly of our DBM transcriptome data provides a reference gene set for sex pheromone biosynthesis pathway and degradation genes in this notorious agricultural pest. Further analysis of their tissue specific expression pattern via digital gene expression and qPCR validation revealed the identification of the following genes in the pheromone biosynthesis and degradation pathways. A) In the sex pheromone biosynthesis gene category, we identified two PBAN receptor isoforms with PG biased expression, 7 ACCs, 8 FASs, 4 FATPs, 16 DESs, 7 FARs, 21 ACTs and 22 ADs. Two PG abundant PxDESs, PxDES12 and 16, could be involved in Δ11 desaturation. Three possible candidate pgFARs, PxFAR5, 6 and 7, were also identified to have PG dominant expression and could participate in the reduction of Z11-16:acid to Z11-16:OH. B) In the odorant/sex pheromone-degrading gene category, we identified three AOXs, five ALDHs, 15 ARs, 49 CCEs, 27 UGTs, 85 P450s and 26 GSTs. Among these genes, two AOXs, one ALDH, five CCE, five UGTs, eight P450s and three GSTs displayed antennae dominant expression, and were proposed to be involved in sex pheromone components and plant volatiles degradation. Together, our data provides a strong reference for the genes involved in sex pheromone biosynthesis and degradation pathways, and will be a useful gene set for future gene function studies. This data set may also provide potential gene targets to design pest control strategies based on disrupting pheromone-based behaviors of insect pests.
Methods
Insect samples
The 5th instar larvae of DBMs were purchased from an insect rearing facility in Henan province, China. The rearing conditions were 27 °C, 14 L : 10D photoperiod and 65% ± 5% relative humidity (RH). We collected approximately 300 antennae, 300 heads, 300 legs, and 100 abdomens from 3-day old adults of both sexes (male/female = 1:1), and 100 female sex pheromone glands for transcriptome sequencing. Three biological replicates of these samples were also collected for qPCR. Ovipositors that were adhered to the abdomens were removed as much as possible to avoid tissue contamination. The tissue samples were stored in liquid nitrogen at −80 °C until further use.
cDNA library construction and Illumina sequencing
Total RNA was extracted using TRIzol reagent (Invitrogen Carlsbad, CA, USA) according to the manufacturer’s protocol. RNA degradation and contamination was monitored on 1% agarose gels. RNA purity was checked using the NanoPhotometer spectrophotometer (IMPLEN, CA, USA). RNA concentration was measured using Qubit RNA Assay Kit in Qubit 2.0 Fluorometer (Life Technologies, CA, USA). RNA integrity was assessed using the RNA Nano 6000 Assay Kit of the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA). The cDNA library construction and Illumina sequencing of the samples were performed by Novogene Bioinformatics Technology Co. Ltd, Beijing, China. A total amount of 3 μg RNA was used as input material for the RNA sample preparations. Sequencing libraries were generated using NEBNext Ultra RNA Library Prep Kit for Illumina (NEB, USA) following manufacturer’s recommendations and index codes were added to assign sequences to each sample. Briefly, mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. Fragmentation was carried out using divalent cations under elevated temperature in the NEBNext First Strand Synthesis Reaction Buffer (5×). First strand cDNA was synthesized using random hexamer primer and M-MuLV Reverse Transcriptase (RNase H-). Second strand cDNA synthesis was subsequently performed using DNA polymerase I and RNase H. Remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. After adenylation of 3′ ends of DNA fragments, NEBNext Adaptor with hairpin loop structure were ligated to prepare for hybridization. In order to select cDNA fragments that were 150–200 bp in length, the library fragments were purified with the AMPure XP system (Beckman Coulter, Beverly, USA). Thereafter, 3 μL USER Enzyme (NEB, USA) was added to size-selected, adaptor-ligated cDNA and incubated at 37 °C for 15 min followed by 5 min at 95 °C. Then, PCR was performed with Phusion high-fidelity DNA polymerase, universal PCR primers, and index (X) primer. Finally, the PCR products were purified (AMPure XP system) and the library quality was assessed on an Agilent Bioanalyzer 2100 system.
De novo assembly of short reads and gene annotation
Raw data (raw reads) in fastq format were first processed through in-house perl scripts. In this step, clean data (clean reads) were obtained by removing the adapter sequences, reads containing poly-N, and low quality reads from the raw data. Simultaneously, Q20, Q30, GC-content, and sequence duplication level of the clean data were calculated. All downstream analyses were based on clean data with high quality. De novo transcriptome assembly was performed using the short reads assembly program Trinity (version: r20140413p1) with min_kmer_cov set to 2 by default and all other parameters were set to default values. The overlap settings used for the assembly were 30 bp and 80% similarity, and all the other parameters were set to their default values.
Unigenes >150 bp were aligned by BLASTx with protein databases, including Nr, Swiss-Prot, KEGG, and COG (e-value < 10–5), to identify proteins with high sequence identity and to assign putative functional annotations. Next, we used the Blast2GO program (version: b2g4pipe_v2.5, e-value = 1.0E-6) (https://www.blast2go.com/) to obtain GO annotations of the unigenes and we obtained the GO functional classifications using the WEGO software (http://wego.genomics.org.cn/cgi-bin/wego/index.pl).
Reads mapping to the reference genome
Reference genome and gene model annotation files were downloaded directly from the DBM genome website: http://iae.fafu.edu.cn/DBM/. Index of the reference genome was built using Bowtie v2.2.3 and paired-end clean reads were aligned to the reference genome using TopHat v2.0.12. We selected TopHat as the mapping tool to generate a database of splice junctions based on the gene model annotation file to yield a better mapping result compared to other non-splice mapping tools.
Quantification of gene expression level
HTSeq v0.6.1 was used to count the read numbers mapped to each gene. Then, FPKM of each gene was calculated based on the length of the gene and reads count mapped to this gene. FPKM, which is the expected number of Fragments Per Kilobase of transcript sequence per Millions base pairs sequenced, considers the effect of sequencing depth and gene length for the reads count at the same time, and is currently the most commonly used method for estimating gene expression levels [107].
Differential expression analysis
Prior to differential gene expression analysis, for each sequenced library, the read counts were adjusted by the edgeR program package through one scaling normalized factor. Differential expression analysis of two samples was performed using the DEGSeq R package (1.20.0). The P values were adjusted using the Benjamini & Hochberg method. Corrected P-value of 0.005 and log2 (Fold change) of 1 (for de novo assembly: P value was adjusted using q value. qValue < 0.005 & |log2(foldchange)| > 1 was set as the threshold for significant differential expression).
Novel transcripts prediction
The Cufflinks v2.1.1 Reference Annotation Based Transcript (RABT) assembly method was used to construct and identify both known and novel transcripts from TopHat alignment results.
Phylogenetic tree
Amino acid sequences of selected AOX, CCE, FAR and DES were aligned with MAFFT (E-INS-I parameter). Thereafter, PhyML 3.1 with WAG substitution model was used to construct a maximum likelihood phylogenetic tree using Bayesian analysis. The deduced protein sequences used in the phylogenetic tree are listed in Additional file 5. Finally, the trees were viewed and group edited with FigTree v1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/).
RNA extraction and cDNA synthesis
Total RNA was extracted using EasyPure RNA Kit (TransGen Biotech, Beijing, China) following the manufacturer’s instructions, in which DNase digestion was included to avoid the genomic DNA contamination. RNA quality was checked with a spectrophotometer (NanoDrop 2000, Thermo Fisher Scientific, USA). The single-stranded cDNA templates were synthesized from 1 μg total RNA from various tissue samples using the PrimeScriptRT Master Mix (TaKaRa, Dalian, China) at 42 °C for 1 h. The reaction was terminated by heating at 70 °C for 15 min.
Quantitative real time PCR and data analysis
qPCRs were performed for each sample using an iCycle iQ (Bio-Rad, CA, USA) according to the minimum information for publication of quantitative Real-Time PCR Experiments [108]. Gene-specific primers were designed by Beacon Designer 7.6 (PREMIER Biosoft International, CA, USA) and are listed in Additional file 6: Table S3. The mRNA levels were measured in three technical replicates from each of the three biological replicates by qPCR using TransStart Tip Green qPCR SuperMix, as described by the manufacturer (TransGen Biotech, Beijing, China). The mRNA levels were quantified using ribosomal protein L8 (RPL8) as the reference gene [56]. Means and standard errors were calculated based on at least two biological replicates. Relative expression level of the mRNAs for genes was calculated according to the 2−ΔΔCq method. Relative fold-changes in the different tissues were calculated and normalized based on the transcript levels in the bodies.
Acknowledgements
We thank Zhao-Qun Li (Tea Research Institute, Chinese Academy of Agricultural Science, China) for taking pictures of the DBM female sex pheromone gland, Michael Court (UGT nomenclature committee) for the nomenclature of P. xylostella UGTs, Martine Maïbèche-Coisne (Université Pierre et Marie Curie, France) for help collecting the B. mori and H. armigera UGT sequences and Ya-Nan Zhang (Huaibei Normal University, China) for critique of an earlier version of the manuscript.
Funding
This research was supported by the National Natural Science Foundation of China (Grant Nos. 31401750 and 31360528 for PH and JW), Natural Science Foundation of Guizhou Province of China (Grant No. QKH-J [2014]2062 for PH), Scientific Research Foundation of Guizhou University, China (RJH Project Grant No. 2013–28 for PH), Major Program of Innovation Research Team of Educational Commission of Guizhou Province of China (Grant No. QJH-KY[2016]113 for MH), Science and Technology Platform for Talent of Guizhou Province (Grant No. QKH [2016]5608 for MH and XF-T), Science and Technology Project of Guizhou Province (QKH Major Project, Grant No. QKH (2012) 6012 for MH) and the National Natural Science Foundation of China (Grant No. 21262009 for WM-X). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and materials
The datasets of DBM transcriptomes during the current study are available in the NCBI SRA database (http://trace.ncbi.nlm.nih.gov/Traces/sra/. Accession numbers: SRR3955374; SRR3972213; SRR3972295; SRR3972298; SRR3972299 and SRR3972322). This Transcriptome Shotgun Assembly of genes have been deposited at DDBJ/EMBL/GenBank under the accession number GEYN00000000, these assembled genes are also listed in Additional file 7, the data evaluation of P. xylostella de novo tissue transcriptomes are in Additional file 8 and the detailed annotation information of de novo assembly are listed in Additional file 9: Table S4.
Authors’ contributions
PH conceived and designed the experiments; XL-W and LH-Z prepared the DBM samples, PH and JW collected and evaluated the transcriptome data, PH, YF-Z and LH-Z analysed the odorant degradation genes, WM-X and MH analysed the sex pheromone biosynthesis genes, PH, DY-H and YF-Z performed the qPCR assay of the odorant degradation genes and analysed the data, MH, XF-T and WM-X performed the qPCR assay of the sex pheromone biosynthesis genes and analysed the data. PH, YF-Z and JW constructed the phylogenetic trees, PH, XL-W and MH prepared the initial draft of the manuscript and YF-Z, DY-H, JW and WM-X participating in modifying the final version. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- ACC
Acetyl CoA Carboxylase
- ACT
Acetyltransferase
- ACT
Acetyltransferase
- AD
Alcohol dehydrogenase
- AKR
Aldo-keto reductase
- ALDH
Aldehyde dehydrogenase
- AOX
Aldehyde oxidase
- AR
Aldehyde reductase
- CCE
Carboxyl/cholinesterase
- cDNA
Complementary DNA
- CYP
Cytochrome P450
- DBM
Diamondback moth
- DES
Desaturase
- FAR
Fatty acyl-CoA reductases
- FAS
Fatty acid synthase
- FATP
Fatty Acid Transport Protein
- FPKM
Fragments per kilobase per million mapped fragments
- GO
Gene ontology
- GST
Glutathione S-transferase
- MDR
Medium-chain dehydrogenases/reductase
- PBANr
Pheromone biosynthesis activating neuropeptide receptor
- PBP
Pheromone binding protein
- PR
Pheromone receptor
- qPCR
Quantitative real time qPCR
- SDR
Short chain dehydrogenases/reductase
- UGT
UDP-glycosyltransferases
Additional files
The genome location of novel genes. (XLS 1969 kb)
Annotation summaries of DBM unigenes. (A) Species distribution of unigenes with the best hit annotation terms in the non-redundant (Nr) database. (B) Gene ontology (GO) classifications of DBM unigenes. (TIF 817 kb)
Information of the pheromone biosynthesis and degradation genes in DBM. (XLSX 4618 kb)
Multiple sequence alignment of PBANr variants from different insect species. The seven predicted TM domains are lined. Location of the YXXΦ endosomal sorting motif is highlighted in red. Pluxy, Plutella xylostella, Bommo, B. mori, Helar, Psese, Pseudaletia separate, Helvi, H. virescens, Chisu, Chilo suppressalis, Ostnu, Ostrinia nubilalis, Helze, Helicoverpa zea (ZIP 4167 kb)
The phylogenetic tree of sex pheromone gene sequences used in this study. (XLSX 26 kb)
Sequences of primers used in qPCR. (TXT 647 kb)
cDNA sequences of assembled genes. (XLS 38124 kb)
The data evaluation of P. xylostella de novo tissue transcriptomes. (DOCX 15 kb)
The detailed annotation information of de novo assembly. (RAR 9052 kb)
Contributor Information
Peng He, Email: phe1@gzu.edu.cn.
Yun-Fei Zhang, Email: zhangyunfei@caas.cn.
Duan-Yang Hong, Email: m18285185142@163.com.
Jun Wang, Email: fccjwang@126.com.
Xing-Liang Wang, Email: wxl@njau.edu.cn.
Ling-Hua Zuo, Email: zlh123@ruc.edu.cn.
Xian-Fu Tang, Email: 535612131@qq.com.
Wei-Ming Xu, Email: wmxu@gzu.edu.cn.
Ming He, Email: hmher@126.com.
References
- 1.Wyatt TD. Fifty years of pheromones. Nature. 2009;457(7227):262–3. doi: 10.1038/457262a. [DOI] [PubMed] [Google Scholar]
- 2.Jurenka R: Insect pheromone biosynthesis. Top Curr Chem. 2004;239:97-132. doi:10.1007/b95450. [DOI] [PubMed]
- 3.Ando T, Inomata S, Yamamoto M. Lepidopteran sex pheromones. Top Curr Chem. 2004;239:51–96. doi: 10.1007/b95449. [DOI] [PubMed] [Google Scholar]
- 4.Volpe JJ, Vagelos PR. Saturated fatty acid biosynthesis and its regulation. Annu Rev Biochem. 1973;42:21–60. doi: 10.1146/annurev.bi.42.070173.000321. [DOI] [PubMed] [Google Scholar]
- 5.Zhu J-W, Millar J, Löfstedt C. Hormonal regulation of sex pheromone biosynthesis in the turnip moth, Agrotis segetum. Arch Insect Biochem Physiol. 1995;30(1):41–59. doi:10.1002/arch.940300104.
- 6.Foster SP, Roelofs WL. Sex pheromone biosynthesis in the tortricid moth, Ctenopseustis herana (Felder & Rogenhofer) Arch Insect Biochem Physiol. 1996;33(2):135–47. doi: 10.1002/(SICI)1520-6327(1996)33:2<135::AID-ARCH4>3.0.CO;2-X. [DOI] [Google Scholar]
- 7.Wang H-L, Liénard MA, Zhao C-H, Wang C-Z, Löfstedt C. Neofunctionalization in an ancestral insect desaturase lineage led to rare Δ6 pheromone signals in the Chinese tussah silkworm. Insect Biochem Mol Biol. 2010;40(10):742–51. doi: 10.1016/j.ibmb.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 8.Albre J, Liénard MA, Sirey TM, Schmidt S, Tooman LK, Carraher C, et al. Sex pheromone evolution is associated with differential regulation of the same desaturase gene in two genera of leafroller moths. PLoS Genet. 2012;8(1):e1002489. doi: 10.1371/journal.pgen.1002489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liénard MA, Lassance J-M, Wang H-L, Zhao C-H, Piškur J, Johansson T, et al. Elucidation of the sex-pheromone biosynthesis producing 5,7-dodecadienes in Dendrolimus punctatus (Lepidoptera: Lasiocampidae) reveals Δ11- and Δ9-desaturases with unusual catalytic properties. Insect Biochem Mol Biol. 2010;40(6):440–52. doi: 10.1016/j.ibmb.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 10.Löfstedt C, Bengtsson M. Sex pheromone biosynthesis of (E, E)-8,10-dodecadienol in codling moth Cydia pomonella involves E9 desaturation. J Chem Ecol. 1988;14(3):903–15. doi: 10.1007/BF01018782. [DOI] [PubMed] [Google Scholar]
- 11.Liénard MA, Strandh M, Hedenström E, Johansson T, Löfstedt C. Key biosynthetic gene subfamily recruited for pheromone production prior to the extensive radiation of Lepidoptera. BMC Evol Biol. 2008;8(1):1–15. doi: 10.1186/1471-2148-8-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinez T, Fabriás G, Camps F. Sex pheromone biosynthetic pathway in Spodoptera littoralis and its activation by a neurohormone. J Biol Chem. 1990;265(3):1381–7. [PubMed] [Google Scholar]
- 13.Hao G, Liu W, O’Connor M, Roelofs WL. Acyl-CoA Z9- and Z10-desaturase genes from a New Zealand leafroller moth species, Planotortrix octo. Insect Biochem Mol Biol. 2002;32(9):961–6. doi:10.1016/S0965-1748(01)00176-X. [DOI] [PubMed]
- 14.Foster SP, Roelofs WL. Sex pheromone biosynthesis in the leafroller moth Planotortix excessana by Δ10 desaturation. Arch Insect Biochem Physiol. 1988;8(1):1–9. doi: 10.1002/arch.940080102. [DOI] [Google Scholar]
- 15.Ding B-J, Carraher C, Löfstedt C. Sequence variation determining stereochemistry of a Δ11 desaturase active in moth sex pheromone biosynthesis. Insect Biochem Mol Biol. 2016;74:68–75. doi: 10.1016/j.ibmb.2016.05.002. [DOI] [PubMed] [Google Scholar]
- 16.Ki M, Suzuki MG, Hull JJ, Kurata R, Takahashi S, Yamamoto M, et al. Involvement of a bifunctional fatty-acyl desaturase in the biosynthesis of the silkmoth, Bombyx mori, sex pheromone. Proc Natl Acad Sci U S A. 2004;101(23):8631–6. doi: 10.1073/pnas.0402056101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roelofs WL, Liu W, Hao G, Jiao H, Rooney AP, Linn CE. Evolution of moth sex pheromones via ancestral genes. Proc Natl Acad Sci U S A. 2002;99(21):13621–6. doi: 10.1073/pnas.152445399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao C, Löfstedt C, Wang X. Sex pheromone biosynthesis in the Asian corn borer Ostrinia furnacalis (II): Biosynthesis of (E)- and (Z)-12-tetradecenyl acetate involves Δ14 desaturation. Arch Insect Biochem Physiol. 1990;15(1):57–65. doi: 10.1002/arch.940150106. [DOI] [Google Scholar]
- 19.Wang H-L, Zhao C-H, Wang C-Z. Comparative study of sex pheromone composition and biosynthesis in Helicoverpa armigera, H. assulta and their hybrid. Insect Biochem Mol Biol. 2005;35(6):575–83. doi: 10.1016/j.ibmb.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 20.Carot-Sans G, Muñoz L, Piulachs MD, Guerrero A, Rosell G. Identification and characterization of a fatty acyl reductase from a Spodoptera littoralis female gland involved in pheromone biosynthesis. Insect Mol Biol. 2015;24(1):82–92. doi: 10.1111/imb.12138. [DOI] [PubMed] [Google Scholar]
- 21.Lassance JM, Lienard MA, Antony B, Qian S, Fujii T, Tabata J, et al. Functional consequences of sequence variation in the pheromone biosynthetic gene pgFAR for Ostrinia moths. Proc Natl Acad Sci U S A. 2013;110(10):3967–72. doi: 10.1073/pnas.1208706110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lienard MA, Hagstrom AK, Lassance JM, Lofstedt C. Evolution of multicomponent pheromone signals in small ermine moths involves a single fatty-acyl reductase gene. Proc Natl Acad Sci U S A. 2010;107(24):10955–60. doi: 10.1073/pnas.1000823107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Antony B, Ding BJ, Moto K, Aldosari SA, Aldawood AS. Two fatty acyl reductases involved in moth pheromone biosynthesis. Sci Rep. 2016;6:29927. doi: 10.1038/srep29927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vogt RG. Molecular basis of pheromone detection in insects. In: Gilbert LI, Iatro K, Gill S, editors. Comprehensive insect physiology, biochemistry, pharmacology and molecular biology. Volume 3. Endocrinology. London: Elsevier; 2005. pp. 753–804. [Google Scholar]
- 25.Liu N-Y, Yang F, Yang K, He P, Niu X-H, Xu W, et al. Two subclasses of odorant-binding proteins in Spodoptera exigua display structural conservation and functional divergence. Insect Mol Biol. 2015;24(2):167–82. doi:10.1111/imb.12143. [DOI] [PubMed]
- 26.Liu N-Y, He P, Dong S-L. Binding properties of pheromone-binding protein 1 from the common cutworm Spodoptera litura. Comp Biochem Physiol B Biochem Mol Biol. 2012;161(4):295–302. doi:10.1016/j.cbpb.2011.11.007. [DOI] [PubMed]
- 27.Vogt RG, Grosse-Wilde E, Zhou J-J. The lepidoptera odorant binding protein gene family: gene gain and loss within the GOBP/PBP complex of moths and butterflies. Insect Biochem Mol Biol. 2015;62:142–53. doi: 10.1016/j.ibmb.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 28.Chang H-T, Liu Y, Yang T, Pelosi P, Dong S-L, Wang G-R. Pheromone binding proteins enhance the sensitivity of olfactory receptors to sex pheromones in Chilo suppressalis. Sci Rep. 2015;5:13093. doi: 10.1038/srep13093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y-N, Zhang J, Yan S-W, Chang H-T, Liu Y, Wang G-R, et al. Functional characterization of sex pheromone receptors in the purple stem borer, Sesamia inferens (Walker) Insect Mol Biol. 2014;23(5):611–20. doi: 10.1111/imb.12109. [DOI] [PubMed] [Google Scholar]
- 30.Leal WS. Odorant reception in insects: roles of receptors, binding proteins, and degrading enzymes. Annu Rev Entomol. 2013;58:373–91. doi: 10.1146/annurev-ento-120811-153635. [DOI] [PubMed] [Google Scholar]
- 31.Rybczynski R, Reagan J, Lerner MR. A pheromone-degrading aldehyde oxidase in the antennae of the moth Manduca sexta. J Neurosci. 1989;9(4):1341–53. doi: 10.1523/JNEUROSCI.09-04-01341.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rybczynski R, Vogt RG, Lerner MR. Antennal-specific pheromone-degrading aldehyde oxidases from the moths Antheraea polyphemus and Bombyx mori. J Biol Chem. 1990;265(32):19712–5. [PubMed] [Google Scholar]
- 33.Choo Y-M, Pelletier J, Atungulu E, Leal WS. Identification and characterization of an antennae-specific aldehyde oxidase from the navel orangeworm. PLoS One. 2013;8(6):e67794. doi: 10.1371/journal.pone.0067794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang B, Ozaki K, Ishikawa Y, Matsuo T. Identification of candidate odorant receptors in Asian corn borer Ostrinia furnacalis. PLoS One. 2015;10(3):e0121261. doi: 10.1371/journal.pone.0121261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xia Y-H, Zhang Y-N, Hou X-Q, Li F, Dong S-L. Large number of putative chemoreception and pheromone biosynthesis genes revealed by analyzing transcriptome from ovipositor-pheromone glands of Chilo suppressalis. Sci Rep. 2015;5:7888. doi: 10.1038/srep07888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y-N, Xia Y-H, Zhu J-Y, Li S-Y, Dong S-L. Putative pathway of sex pheromone biosynthesis and degradation by expression patterns of genes identified from female pheromone gland and adult antenna of Sesamia inferens (Walker). J Chem Ecol. 2014;40(5):439–51. doi:10.1007/s10886-014-0433-1. [DOI] [PubMed]
- 37.Leal JFM, Barbancho M. Aldehyde dehydrogenase (ALDH) activity in Drosophila melanogaster adults: evidence for cytosolic localization. Insect Biochem Mol Biol. 1993;23(5):543–7. doi: 10.1016/0965-1748(93)90027-P. [DOI] [PubMed] [Google Scholar]
- 38.Guo H, Del Corso A, Huang L-Q, Mura U, Pelosi P, Wang C-Z. Aldehyde reductase activity in the antennae of Helicoverpa armigera. Insect Mol Biol. 2014;23(3):330–40. doi: 10.1111/imb.12084. [DOI] [PubMed] [Google Scholar]
- 39.Rivera-Perez C, Nouzova M, Clifton ME, Garcia EM, Leblanc E, Noriega FG. Aldehyde dehydrogenase 3 converts farnesal into farnesoic acid in the corpora allata of mosquitoes. Insect Biochem Mol Biol. 2013;43(8):675–82. doi: 10.1016/j.ibmb.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He P, Zhang Y-N, Yang K, Li Z-Q, Dong S-L. An antenna-biased carboxylesterase is specifically active to plant volatiles in Spodoptera exigua. Pestic Biochem Physiol. 2015;123:93–100. doi: 10.1016/j.pestbp.2015.03.009. [DOI] [PubMed] [Google Scholar]
- 41.He P, Zhang Y-N, Li Z-Q, Yang K, Zhu J-Y, Liu S-J, et al. An antennae-enriched carboxylesterase from Spodoptera exigua displays degradation activity in both plant volatiles and female sex pheromones. Insect Mol Biol. 2014;23(4):475–86. doi: 10.1111/imb.12095. [DOI] [PubMed] [Google Scholar]
- 42.He P, Zhang J, Li Z-Q, Zhang Y-N, Yang K, Dong S-L. Functional characterization of an antennal esterase from the noctuid moth, Spodoptera exigua. Arch Insect Biochem Physiol. 2014;86(2):85–99. doi: 10.1002/arch.21164. [DOI] [PubMed] [Google Scholar]
- 43.He P, Li Z-Q, Liu C-C, Liu S-J, Dong S-L. Two esterases from the genus Spodoptera degrade sex pheromones and plant volatiles. Genome. 2014;57(4):201–8. doi: 10.1139/gen-2014-0041. [DOI] [PubMed] [Google Scholar]
- 44.Keeling CI, Henderson H, Li M, Dullat HK, Ohnishi T, Bohlmann J. CYP345E2, an antenna-specific cytochrome P450 from the mountain pine beetle, Dendroctonus ponderosae Hopkins, catalyses the oxidation of pine host monoterpene volatiles. Insect Biochem Mol Biol. 2013;43(12):1142–51. doi: 10.1016/j.ibmb.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 45.Maïbèche-Coisne M, Nikonov AA, Ishida Y, Jacquin-Joly E, Leal WS. Pheromone anosmia in a scarab beetle induced by in vivo inhibition of a pheromone-degrading enzyme. Proc Natl Acad Sci U S A. 2004;101(31):11459–64. doi: 10.1073/pnas.0403537101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bozzolan F, Siaussat D, Maria A, Durand N, Pottier MA, Chertemps T, et al. Antennal uridine diphosphate (UDP)-glycosyltransferases in a pest insect: diversity and putative function in odorant and xenobiotics clearance. Insect Mol Biol. 2014;23(5):539–49. doi: 10.1111/imb.12100. [DOI] [PubMed] [Google Scholar]
- 47.Rogers ME, Jani MK, Vogt RG. An olfactory-specific glutathione-S-transferase in the sphinx moth Manduca sexta. J Exp Biol. 1999;202(Pt 12):1625–37. doi: 10.1242/jeb.202.12.1625. [DOI] [PubMed] [Google Scholar]
- 48.Tamaki YZ. Z-11-hexadecenal and Z-11-hexadecenyl acteact :sex pheromone components of the diamonback moth (Lepideptera :Plutellidae) Appl Entomol Zool. 1977;12:208–10. [Google Scholar]
- 49.Môttus E, Nômm V, Williams IH, Liblikas I. Optimization of Pheromone Dispensers for Diamondback Moth Plutella xylostella. J Chem Ecol. 1997;23(9):2145–59. doi: 10.1023/B:JOEC.0000006435.13481.ba. [DOI] [Google Scholar]
- 50.Chisholm MD, Steck WF, Underhill EW, Palaniswamy P. Field trapping of diamondback moth Plutella xylostella using an improved four-component sex attractant blend. J Chem Ecol. 1983;9(1):113–8. doi: 10.1007/BF00987775. [DOI] [PubMed] [Google Scholar]
- 51.Koshihara T, Yamada H. Attractant activity of the female Sex pheromone of diamondback moth, Plutella xylostella (L.), and analogue. Jap J Appl Entomol Zool. 1980;24(1):6–12. doi: 10.1303/jjaez.24.6. [DOI] [Google Scholar]
- 52.Ando T, Koshihara T, Yamada H, Vu MH, Takahashi N, Tamaki Y. Electroantennogram activities of Sex pheromone analogues and their synergistic effect on field attraction in the diamondback moth. Appl Entomol Zool. 1979;14(3):362–4. [Google Scholar]
- 53.Sun M-J, Liu Y, Wang G-R. Expression patterns and binding properties of three pheromone binding proteins in the diamondback moth, Plutella xyllotella. J Insect Physiol. 2013;59(1):46–55. doi: 10.1016/j.jinsphys.2012.10.020. [DOI] [PubMed] [Google Scholar]
- 54.You M-S, Yue Z, He W-Y, Yang X-H, Yang G, Xie M, et al. A heterozygous moth genome provides insights into herbivory and detoxification. Nat Genet. 2013;45:220–5. doi:10.1038/ng.2524. [DOI] [PubMed]
- 55.Yu L-Y, Tang W-Q, He W-Y, Ma X-L, Vasseur L, Baxter SW, et al. Characterization and expression of the cytochrome P450 gene family in diamondback moth, Plutella xylostella (L.). Sci Rep. 2015;5:8952. doi:10.1038/srep08952. [DOI] [PMC free article] [PubMed]
- 56.You Y-C, Xie M, Ren N-N, Cheng X-M, Li J-Y, Ma X-L, et al. Characterization and expression profiling of glutathione S-transferases in the diamondback moth, Plutella xylostella (L.). BMC Genomics. 2015;16(1):152. doi:10.1186/s12864-015-1343-5. [DOI] [PMC free article] [PubMed]
- 57.Lee JM, Hull JJ, Kawai T, Goto C, Kurihara M, Tanokura M, et al. Re-evaluation of the PBAN receptor molecule: characterization of PBANR variants expressed in the pheromone glands of moths. Front Endocrinol (Lausanne) 2012;3:6. doi: 10.3389/fendo.2012.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hull JJ, Ohnishi A, Matsumoto S. Regulatory mechanisms underlying pheromone biosynthesis activating neuropeptide (PBAN)-induced internalization of the Bombyx mori PBAN receptor. Biochem Biophys Res Commun. 2005;334(1):69–78. doi: 10.1016/j.bbrc.2005.06.050. [DOI] [PubMed] [Google Scholar]
- 59.Lee D-W, Kim Y, Koh Y-H. RNA interference of PBAN receptor suppresses expression of two fatty acid desaturases in female Plutella xylostella. J Asia-Pacific Entomol. 2011;14(4):405–10. doi: 10.1016/j.aspen.2011.05.004. [DOI] [Google Scholar]
- 60.Lee D-W, Boo K-S. Molecular characterization of pheromone biosynthesis activating neuropeptide from the diamondback moth, Plutella xylostella (L.) Peptides. 2005;26(12):2404–11. doi: 10.1016/j.peptides.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 61.Gu S-H, Wu K-M, Guo Y-Y, Pickett JA, Field LM, Zhou J-J, et al. Identification of genes expressed in the sex pheromone gland of the black cutworm Agrotis ipsilon with putative roles in sex pheromone biosynthesis and transport. BMC Genomics. 2013;14(1):636. doi: 10.1186/1471-2164-14-636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hull JJ, Ohnishi A, Moto K, Kawasaki Y, Kurata R, Suzuki MG, et al. Cloning and characterization of the pheromone biosynthesis activating neuropeptide receptor from the silkmoth, Bombyx mori: significance of the carboxyl terminus in receptor internalization. J Biol Chem. 2004;279(49):51500–7. doi: 10.1074/jbc.M408142200. [DOI] [PubMed] [Google Scholar]
- 63.Kim Y-J, Nachman RJ, Aimanova K, Gill S, Adams ME. The Pheromone Biosynthesis Activating Neuropeptide (PBAN) receptor of Heliothis virescens: identification, functional expression, and structure-activity relationships of ligand analogs. Peptides. 2008;29(2):268–75. doi: 10.1016/j.peptides.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ding B-J, Löfstedt C. Analysis of the agrotis segetum pheromone gland transcriptome in the light of Sex pheromone biosynthesis. BMC Genomics. 2015;16(1):711. doi: 10.1186/s12864-015-1909-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ohnishi A, Hashimoto K, Imai K, Matsumoto S. Functional characterization of the Bombyx mori Fatty Acid Transport Protein (BmFATP) within the silkmoth pheromone gland. J Biol Chem. 2009;284(8):5128–36. doi: 10.1074/jbc.M806072200. [DOI] [PubMed] [Google Scholar]
- 66.Qian S, Fujii T, Ito K, Nakano R, Ishikawa Y. Cloning and functional characterization of a fatty acid transport protein (FATP) from the pheromone gland of a lichen moth, Eilema japonica, which secretes an alkenyl sex pheromone. Insect Biochem Mol Biol. 2011;41(1):22–8. doi:10.1016/j.ibmb.2010.09.008. [DOI] [PubMed]
- 67.Hagström A, Wang H-L, Lienard MA, Lassance JM, Johansson T, Löfstedt C. A moth pheromone brewery: production of (Z)-11-hexadecenol by heterologous co-expression of two biosynthetic genes from a noctuid moth in a yeast cell factory. Microb Cell Fact. 2013;12:125. doi: 10.1186/1475-2859-12-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fujii T, Suzuki MG, Katsuma S, Ito K, Rong Y, Matsumoto S, et al. Discovery of a disused desaturase gene from the pheromone gland of the moth Ascotis selenaria, which secretes an epoxyalkenyl sex pheromone. Biochem Biophys Res Commun. 2013;441(4):849–55. doi: 10.1016/j.bbrc.2013.10.143. [DOI] [PubMed] [Google Scholar]
- 69.Sakurai T, Mitsuno H, Haupt SS, Uchino K, Yokohari F, Nishioka T, et al. A single sex pheromone receptor determines chemical response specificity of sexual behavior in the silkmoth Bombyx mori. PLoS Genet. 2011;7(6):e1002115. doi: 10.1371/journal.pgen.1002115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Moto K, Yoshiga T, Yamamoto M, Takahashi S, Okano K, Ando T et al. Pheromone gland-specific fatty-acyl reductase of the silkmoth, Bombyx mori. Proc Natl Acad Sci U S A. 2003;100. doi:10.1073/pnas.1531993100. [DOI] [PMC free article] [PubMed]
- 71.Antony B, Fujii T, Moto K, Matsumoto S, Fukuzawa M, Nakano R, et al. Pheromone-gland-specific fatty-acyl reductase in the adzuki bean borer, Ostrinia scapulalis (Lepidoptera: Crambidae) Insect Biochem Mol Biol. 2009;39(2):90–5. doi: 10.1016/j.ibmb.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 72.Hagström ÅK, Liénard MA, Groot AT, Hedenström E, Löfstedt C. Semi–selective fatty acyl reductases from four heliothine moths influence the specific pheromone composition. PLoS One. 2012;7(5):e37230. doi: 10.1371/journal.pone.0037230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang Y-N, Zhu X-Y, Fang L-P, He P, Wang Z-Q, Chen G, et al. Identification and expression profiles of sex pheromone biosynthesis and transport related genes in Spodoptera litura. PLoS One. 2015;10(10):e0140019. doi: 10.1371/journal.pone.0140019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Beekwilder J, Alvarez-Huerta M, Neef E, Verstappen FWA, Bouwmeester HJ, Aharoni A. Functional characterization of enzymes forming volatile esters from strawberry and banana. Plant Physiol. 2004;135(4):1865–78. doi: 10.1104/pp.104.042580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.D’Auria JC. Acyltransferases in plants: a good time to be BAHD. Curr Opin Plant Biol. 2006;9(3):331–40. doi: 10.1016/j.pbi.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 76.Kalscheuer R, Stoveken T, Luftmann H, Malkus U, Reichelt R, Steinbuchel A. Neutral lipid biosynthesis in engineered Escherichia coli: jojoba oil-like wax esters and fatty acid butyl esters. Appl Environ Microbiol. 2006;72(2):1373–9. doi: 10.1128/AEM.72.2.1373-1379.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vogel H, Heidel AJ, Heckel DG, Groot AT. Transcriptome analysis of the sex pheromone gland of the noctuid moth Heliothis virescens. BMC Genomics. 2010;11(1):1–21. doi: 10.1186/1471-2164-11-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li Z-Q, Zhang S, Luo J-Y, Wang C-Y, Lv L-M, Dong S-L, et al. Transcriptome comparison of the sex pheromone glands from two sibling Helicoverpa species with opposite sex pheromone components. Sci Rep. 2015;5. doi:10.1038/srep09324. [DOI] [PMC free article] [PubMed]
- 79.Huang X, Liu L, Su X, Feng J. Identification of biotransformation enzymes in the antennae of codling moth Cydia pomonella. Gene. 2016;580(1):73–9. doi: 10.1016/j.gene.2016.01.008. [DOI] [PubMed] [Google Scholar]
- 80.Pelletier J, Bozzolan F, Solvar M, Francois MC, Jacquin-Joly E, Maïbèche-Coisne M. Identification of candidate aldehyde oxidases from the silkworm Bombyx mori potentially involved in antennal pheromone degradation. Gene. 2007;404(1–2):31–40. doi: 10.1016/j.gene.2007.08.022. [DOI] [PubMed] [Google Scholar]
- 81.Rund SS, Bonar NA, Champion MM, Ghazi JP, Houk CM, Leming MT, et al. Daily rhythms in antennal protein and olfactory sensitivity in the malaria mosquito Anopheles gambiae. Sci Rep. 2013;3:2494. doi: 10.1038/srep02494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yamamoto K, Higashiura A, Suzuki M, Shiotsuki T, Sugahara R, Fujii T, et al. Structural characterization of an aldo-keto reductase (AKR2E5) from the silkworm Bombyx mori. Biochem Biophys Res Commun. 2016;474(1):104–10. doi: 10.1016/j.bbrc.2016.04.079. [DOI] [PubMed] [Google Scholar]
- 83.Oakeshott JG, Claudianos C, Campbell PM, Newcomb RD, Russell RJ. Biochemical genetics and genomics of insect esterases. Insect pharmacology: channels, receptors, toxins and enzymes. London: Elsevier; 2005. pp. 309–81. [Google Scholar]
- 84.Tsubota T, Shiotsuki T. Genomic and phylogenetic analysis of insect carboxyl/cholinesterase genes. J Pestic Sci. 2010;35(3):310–4. doi: 10.1584/jpestics.R10-04. [DOI] [Google Scholar]
- 85.Tsubota T, Shiotsuki T. Genomic analysis of carboxyl/cholinesterase genes in the silkworm Bombyx mori. BMC Genomics. 2010;11(1):377. doi: 10.1186/1471-2164-11-377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Teese MG, Campbell PM, Scott C, Gordon KHJ, Southon A, Hovan D, et al. Gene identification and proteomic analysis of the esterases of the cotton bollworm, Helicoverpa armigera. Insect Biochem Mol Biol. 2010;40(1):1–16. doi: 10.1016/j.ibmb.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 87.Durand N, Carot-Sans G, Chertemps T, Bozzolan F, Party V, Renou M, et al. Characterization of an antennal carboxylesterase from the pest moth Spodoptera littoralis degrading a host Plant Odorant. PLoS One. 2010;5(11):e15026. doi: 10.1371/journal.pone.0015026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Newcomb RD, Campbell PM, Ollis DL, Cheah E, Russell RJ, Oakeshott JG. A single amino acid substitution converts a carboxylesterase to an organophosphorus hydrolase and confers insecticide resistance on a blowfly. Proc Natl Acad Sci U S A. 1997;94(14):7464–8. doi: 10.1073/pnas.94.14.7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Poupardin R, Reynaud S, Strode C, Ranson H, Vontas J, David J-P. Cross-induction of detoxification genes by environmental xenobiotics and insecticides in the mosquito Aedes aegypti: Impact on larval tolerance to chemical insecticides. Insect Biochem Mol Biol. 2008;38(5):540–51. doi: 10.1016/j.ibmb.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 90.Campbell PM, Robin DQ, Court LN, Dorrian SJ, Russell RJ, Oakeshott JG. Developmental expression and gene/enzyme identifications in the alpha esterase gene cluster of Drosophila melanogaster. Insect Mol Biol. 2003;12(5):459–71. doi: 10.1046/j.1365-2583.2003.00430.x. [DOI] [PubMed] [Google Scholar]
- 91.Ishida Y, Leal WS. Rapid inactivation of a moth pheromone. Proc Natl Acad Sci U S A. 2005;102(39):14075–9. doi: 10.1073/pnas.0505340102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ishida Y, Leal WS. Chiral discrimination of the Japanese beetle sex pheromone and a behavioral antagonist by a pheromone-degrading enzyme. Proc Natl Acad Sci U S A. 2008;105(26):9076–80. doi: 10.1073/pnas.0802610105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chertemps T, Francois A, Durand N, Rosell G, Dekker T, Lucas P, et al. A carboxylesterase, Esterase-6, modulates sensory physiological and behavioural response dynamics to pheromone in Drosophila. BMC Biol. 2012;10(1):56. doi: 10.1186/1741-7007-10-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hu Z-D, Feng X, Lin Q-S, Chen H-Y, Li Z-Y, Yin F, et al. cDNA cloning and characterization of the carboxylesterase pxCCE016b from the diamondback moth, Plutella xylostella L. J Integr Agr. 2016;15(5):1059–68. doi: 10.1016/S2095-3119(15)61278-3. [DOI] [Google Scholar]
- 95.Gilbert MM, Auld VJ. Evolution of clams (cholinesterase-like adhesion molecules): structure and function during development. Front Biosci. 2005;10:2177–92. doi: 10.2741/1689. [DOI] [PubMed] [Google Scholar]
- 96.Krempl C, Sporer T, Reichelt M, Ahn S-J, Heidel-Fischer H, Vogel H, et al. Potential detoxification of gossypol by UDP-glycosyltransferases in the two Heliothine moth species Helicoverpa armigera and Heliothis virescens. Insect Biochem Mol Biol. 2016 doi: 10.1016/j.ibmb.2016.02.005. [DOI] [PubMed] [Google Scholar]
- 97.Mackenzie PI, Owens IS, Burchell B, Bock KW, Bairoch A, Belanger A, et al. The UDP glycosyltransferase gene superfamily: recommended nomenclature update based on evolutionary divergence. Pharmacogenetics. 1997;7(4):255–69. doi: 10.1097/00008571-199708000-00001. [DOI] [PubMed] [Google Scholar]
- 98.Ahn SJ, Vogel H, Heckel DG. Comparative analysis of the UDP-glycosyltransferase multigene family in insects. Insect Biochem Mol Biol. 2012;42(2):133–47. doi: 10.1016/j.ibmb.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 99.Huang F-F, Chai C-L, Zhang Z, Liu Z-H, Dai F-Y, Lu C, et al. The UDP-glucosyltransferase multigene family in Bombyx mori. BMC Genomics. 2008;9:563. doi:10.1186/1471-2164-9-563. [DOI] [PMC free article] [PubMed]
- 100.Robertson HM, Martos R, Sears CR, Todres EZ, Walden KK, Nardi JB. Diversity of odourant binding proteins revealed by an expressed sequence tag project on male Manduca sexta moth antennae. Insect Mol Biol. 1999;8(4):501–18. doi: 10.1046/j.1365-2583.1999.00146.x. [DOI] [PubMed] [Google Scholar]
- 101.Liu N-N, Li M, Gong Y-H, Liu F, Li T. Cytochrome P450s – Their expression, regulation, and role in insecticide resistance. Pestic Biochem Physiol. 2015;120:77–81. doi:10.1016/j.pestbp.2015.01.006. [DOI] [PubMed]
- 102.Wang Q, Hasan G, Pikielny CW. Preferential expression of biotransformation enzymes in the olfactory organs of Drosophila melanogaster, the antennae. J Biol Chem. 1999;274(15):10309–15. doi: 10.1074/jbc.274.15.10309. [DOI] [PubMed] [Google Scholar]
- 103.Wang L-M, Dankert H, Perona P, Anderson DJ. A common genetic target for environmental and heritable influences on aggressiveness in Drosophila. Proc Natl Acad Sci U S A. 2008;105(15):5657–63. doi:10.1073/pnas.0801327105. [DOI] [PMC free article] [PubMed]
- 104.Pottier MA, Bozzolan F, Chertemps T, Jacquin-Joly E, Lalouette L, Siaussat D, et al. Cytochrome P450s and cytochrome P450 reductase in the olfactory organ of the cotton leafworm Spodoptera littoralis. Insect Mol Biol. 2012;21(6):568–80. doi: 10.1111/j.1365-2583.2012.01160.x. [DOI] [PubMed] [Google Scholar]
- 105.Zou X-P, Xu Z-B, Zou H-W, Liu J-S, Chen S-N, Feng Q-L, et al. Glutathione S-transferase SlGSTE1 in Spodoptera litura may be associated with feeding adaptation of host plants. Insect Biochem Mol Biol. 2016;70:32–43. doi:10.1016/j.ibmb.2015.10.005. [DOI] [PubMed]
- 106.Sun X-Q, Zhang M-X, Yu J-Y, Jin Y, Ling B, Du J-P, et al. Glutathione S-Transferase of brown planthoppers (Nilaparvata lugens) is essential for their adaptation to gramine-containing host plants. PLoS One. 2013;8(5):e64026. doi: 10.1371/journal.pone.0064026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25-R. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55(4):611–22. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets of DBM transcriptomes during the current study are available in the NCBI SRA database (http://trace.ncbi.nlm.nih.gov/Traces/sra/. Accession numbers: SRR3955374; SRR3972213; SRR3972295; SRR3972298; SRR3972299 and SRR3972322). This Transcriptome Shotgun Assembly of genes have been deposited at DDBJ/EMBL/GenBank under the accession number GEYN00000000, these assembled genes are also listed in Additional file 7, the data evaluation of P. xylostella de novo tissue transcriptomes are in Additional file 8 and the detailed annotation information of de novo assembly are listed in Additional file 9: Table S4.