

Abstract
Autophagy is an important homeostatic cellular process encompassing a number of consecutive steps indispensable for degrading and recycling cytoplasmic materials. Basically autophagy is an adaptive response that under stressful conditions guarantees the physiological turnover of senescent and impaired organelles and, thus, controls cell fate by various cross-talk signals. Diabetic retinopathy (DR) is a serious microvascular complication of diabetes and accounts for 5% of all blindness. Although, various metabolic disorders have been linked with the onset of DR, due to the complex character of this multi-factorial disease, a connection between any particular defect and DR becomes speculative. Diabetes increases inflammation, advanced glycation end products (AGEs) and oxidative stress in the retina and its capillary cells. Particularly, a great number of evidences suggest a mutual connection between oxidative stress and other major metabolic abnormalities implicated in the development of DR. In addition, the intricate networks between autophagy and apoptosis establish the degree of cellular apoptosis and the progression of DR. Growing data underline the crucial role of reactive oxygen species (ROS) in the activation of autophagy. Depending on their delicate balance both redox signaling and autophagy, being detrimental or beneficial, retain opposing effects. The molecular mechanisms of autophagy are very complex and involve many signaling pathways cooperating at various steps. This review summarizes recent advances of the possible molecular mechanisms in autophagic process that are involved in pathophysiology of DR. In-depth analysis on the molecular mechanisms leading to autophagy in the retinal pigment epithelial (RPE) will be helpful to plan new therapies aimed at preventing or improving the progression of DR.
Keywords: Damage-regulated autophagy modulator, Diabetic retinopathy, MTOR deregulation, MTORC1, UPR, XBP1
1. INTRODUCTION
Autophagy is a membrane-trafficking trial, which permits to remove cytoplasmic proteins and dysfunctional organelles to the lysosome for degradation. It is a cellular machinery genetically programmed in which the numerous components implicated are highly conserved from yeast to man.
Autophagy can be regarded as a metabolic process occurring at a basal level mainly to maintain homeostatic function during protein and organelle turnover. After various pathophysiological stress conditions such as hypoxia, growth factor removal, nutrient deprivation, or after enhanced release of ROS it can be up-regulated to supply the increased demand for intracellular nutrients and energy, autophagy allows the degradation and recycling of cellular elements, such as longstanding proteins and superfluous or damaged organelles, macromolecules and invading microorganisms to preserve intracellular homeostasis [1]. Macroautophagy or
autophagy, microautophagy, which employs the invagination of lysosomal membranes for the sequestration and digestion of cytoplasmic components [1] and chaperone- mediated autophagy, in which cytosolic chaperones vehicle cytoplasmic elements through lysosomal membranes [1], are the autophagic pathways known in mammalian cells. The autophagic process consists in the removal of cytoplasmic proteins and organelles’ debris within autophagosomes. These organelles then coalesce with the lysosome and release its contents for possible degradation and are afterwards recycled for successive cellular processes [2]. There are two forms of autophagy: non-selective autophagy and cargo-specific autophagy. In the first, nutrients deficiency triggers autophagy as a medium of acquiring vital metabolic components from inside of the cell for usual cellular preservation and healing and to generate energy [1, 3]. In the second, autophagy is employed by a nutrient-rich cell to eliminate either useless or impaired organelles or protein clusters that could be deleterious for the cell. Non-selective autophagy consents to the cells the reutilization of basic structural constituents such as proteins, carbo-hydrates and lipids, and to re-allocate them in periods of nutrients shortage. Cargo-specific autophagy arises when the autophagy is committed particularly at an organelle or substrate. Ribophagy (ribosomes removal), xenophagy (intracellular pathogens removal), pexophagy (peroxisomes elimination) and aggrephagy (aggregate-prone proteins clearance), mitophagy (mitochondria elimination) belong to this category [4-9]. Remarkably, mitophagy removes damaged mitochondria and their lethal content to protect the cell acting as another option to apoptosis (programmed cell death). Mitochondria are competent to trigger apoptosis through the production of ROS and cytochrome C, which aid to activate apoptosis. In addition, mitochondria (being actively involved in mitophagy) allow the cell to gap the To gap is a scientific terminology to indicate that the cell intervals the cycle in order to repair the damages. cycle and recover impaired mitochondria that might alternatively end in cell death. Since it is subject to the availability of adequate amounts of Adenosine triphosphate (ATP) autophagy is a process energy-demanding [7, 10]. Nutrient shortage [11], hypoxia [12] ischemia/reperfusion injury [13], oxidative stress, cellular accumulation of ROS and other cellular stress responses [14] play a critical role in the triggering of autophagy. An efficient autophagic system is fundamental for maintaining a healthy cellular environment. However, in mammalian cells the inhibition of autophagy result in increased level of apoptosis [8]. Therefore, the cells are well-endowed to control any type of damage using both apoptosis and autophagy as a standby machinery [15]. Generally, the mitochondrial fusion is sufficient for minor injuries, nevertheless when that process operates ineffectively to heal the lesion, the cell would undergo to apoptosis to renovate the tissue or organism [16]. The rapid progress on understanding the mechanisms involved in autophagy has disclosed that it plays a crucial role in the pathogenesis of various metabolic and age-related diseases. Recently an emerging role of autophagy in diabetes mellitus has been reported [17]. Moreover, autophagy is involved significantly in the progression of Diabetic retinopathy (DR). DR is one of the most common eye diseases leading to vision loss in world and is the consequence of the numerous pathogenic complications triggered by hyperglycemia and defects of insulin signaling pathways, inducing retinal microvascular defects and neuroretinal dysfunction and degeneration. So far, the molecular mechanisms originating the pathogenesis of DR are not entirely clarified and, so, there is no effective treatment or prevention. Hyperglycemia is the fundamental causative factor in the pathogenesis of diabetic retinopathy determining a series of reactions which include among others the altered cellular signal transduction. Cellular damage induced in endothelial and pericytes results in an imbalance in the regulatory mechanisms of flow control with subsequent hypoxia and retinal edema. Increasing evidence suggests that DR related factors including nutrient stress, oxidative stress, hypoxia, endoplasmic reticulum stress are strictly associated with the generation of autophagic process [18]. Similarly, autophagy and its consequences on cellular and mitochondrial function have important effects on the pathobiology of retinal diseases such as diabetic retinopathy. The role of autophagy as a pro-survival or death mechanism is a highly controversial subject [19]. In this review, we want to disclose whether alterations in autophagic function are detrimental and can cause the increase of noxious elements, thus exacerbating the deleterious processes of DR. The knowledge of the molecular mechanisms underlying this process can be helpful for the prevention or treatment of this disease.
2. MOLECULAR MECHANISMS INVOLVED IN DIABETIC RETINOPATHY
During the last twenty years DR has become one of the most difficult health problems in the world. Retinal pigment epithelial (RPE) cells are the most important cells involved in DR. In diabetic patients retinal blood vessels and neurons are persistently injured by inflammation, ROS and endoplasmic reticulum (ER) stress. Therefore, in the initiation of the disease and in the progression of diabetic complications including DR the chronic hyperglycemia connected with oxidative stress and low-grade inflammation play crucial roles [20, 21] For a long time, DR has been regarded as a microvascular disease characterized by vessel basement membrane thickening, blood retinal barrier breakdown, capillary cell death, acellular capillary, neovascularization, and retinal detachment [22]. In the 19th century, clinical features of DR first detected by ophthalmoscopy retinal vascular abnormalities including microaneurysm, intraretinal hemorrhages, and cottonwool spots [23]. With the progression of the disease, the loss of blood vessels induces retinal ischemia, which may be accompanied by venous beading, loops, and intraretinal microvascular abnormalities (IRMA). These alterations can result in retinal neovascularization (a characteristic of proliferative diabetic retinopathy), fibrosis, and retinal detachment. It has been estimated that half of the patients with untreated proliferative retinopathy will lose their sight within five years [24]. Therefore, it is imperative to disclose the mechanism of DR in order to determine more specific and effective therapeutic trials to prevent it. Recently, it has been reported that DR can be regarded as a neurovascular disease affecting both blood vessel and neuroglia [25]. An important characteristic of DR is the break of the blood-retinal barrier (BRB), which can occur at any stage of the disease, passing from macular edema and exudation (a non-proliferative phase) to extremely permeable neovasculature (a proliferative phase) [26]. Although BRB damage principally involves disturbed function of tight junctions between vascular endothelial cells (inner BRB) and retinal pigment epithelial cells (outer BRB), emerging investigations suggest that other neural and vascular cells (e.g., glial cells and pericytes) are important in the normal maintaining BRB function. [26]. In addition, neuronal and glial cells release metabolites such as lactate and nitric oxide (NO), which set retinal blood flow to consent the metabolic process essentials for retinal tissue. Alterations in the interplay between neural and vascular cells contribute to vascular malfunction in the pathogenesis of DR [23]. In DR, mitochondrial dysfunction is supposed to be a consequence of oxidative injuries within the cell [27]. The oxidative injury in diabetic tissue can result from augmented levels of ROS or a reduced capacity of the retina to challenge the oxidative stress [18]. This concept is sustained by the finding that in diabetic rat lower retinal levels of free radical scavengers such as glutathione peroxidase and ascorbic acid was correlated with increased oxidative damage [28]. These effects have been also observed in rodent models of diabetes in which mitochondria in the retina are undamaged and functional two-month post- diabetes induction, but display dysfunction six months post-induction [29]. Mitochondrial DNA is originally safeguarded by short- lived compensatory mechanisms which in the end become overwhelmed by the consequences of persistent hyperglycemia [30]. Therefore, in early stages of DR mitochondria is unsusceptible to superoxide damage, but becomes damaged after a prolonged period of disease. Evidence suggests that basal autophagy has anti-inflammatory effects by suppressing unscheduled inflammasome activation [31], whereas induced autophagy promotes inflammasome secretion of cytokines, such as IL-1 [32]. The history of DR indicates that both chronic inflammatory and oxidative stress appear to be effective in the progression of DR [33]. In fact, oxidative stress is increased in retinal capillary cells (both endothelial cells and pericytes) treated with high-glucose (HG) medium [34], and also in other nonvascular retinal cells, including Müller cells and photoreceptors [35]. Experimental studies in streptozotocin-induced diabetic rats demonstrate differential expression of 20 genes of the transforming growth factor (TGF)-β signaling cascade, in addition to genes involved in oxidative stress, inflammation, vascular remodeling, and apoptosis suggesting that retinal microangiopathy in diabetic rats is the product of several molecular pathways that are interconnected but not of equal pathogenic importance [36]. In the diabetic retina, an advanced glycation end product (AGEs) modifies proteins promoting oxidative stress and increase inflammatory cytokines that alter vascular function [37]. In DR, activation of tumor necrosis alpha (TNFα), mitochondrial injury by oxidative stress and endoplasmic reticulum (ER) stress are the main responsible factors of the cellular impairment. TNFα has been found increased in the vitreous of diabetic patients [38] and in diabetic rat retinas [39] and through protein kinase C (PKC) and nuclear factor- kappa B (NF-κB) signals impairs the tight junction complex and alters retinal endothelial cell permeability [40]. Microglial-mediated release of TNFα and interleukin 1beta (IL1β) is a mechanism by which a pro-inflammatory environment is present in the diabetic retina and plays a part to the development of experimental DR. In vitro studies demonstrate that ARPE-19 cells respond to high glucose with an increase in autophagy. The 3-methyladenine (3-MA) inhibits occurrence of autophagy and induces the collection of damaged-mitochondria- producing-ROS, the activation of NOD-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome, and subsequently, causes IL1β secretion [41]. Lipid-soluble tetracycline, class of antibiotics that reduce TNFα and NF-κB, inhibits downstream inflammatory mediators and pro-apoptotic signals resulting from triggered retinal microglial cells [42]. The transcription factor NF-κB is one of the principal inflammatory regulators that mediate the release of cytokines and other chemotactic factors involved in inflammation. A localized inflammatory process in the retina is fundamental to the onset of DR. This inflammatory process results in a local increase of IL- 1β, cytokines, inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), prostaglandin 2 (PGE2), vascular endothelial growth factor (VEGF), NF-κB, caspases, the adhesion molecule intercellular adhesion molecule (ICAM-1), and augmented permeability and leukostasis in the retina [43]. The microangiopathy evolving in DR is associated to localized inflammation. The retinal vessels of TNFα deficient mice show a reduction of leucocytosis indicating that this pro-inflammatory cytokine contributes to the leukostasis caused by platelet-activating factor, IL-1β, and VEGF [44]. Validation that leukostasis in DR is associated to oxidant stress and other downstream mediators arises from the finding that alphalipoic acid abolishes augmentations in leukocyte adhesion while other mechanisms, connected to PKC pathways, are accountable for hemodynamic changes occurring along with leukostasis [45]. The elevated circulating amounts of polymorphonuclear leukocytes in the retinal microvasculature contribute to progressive microangiopathy including vascular occlusion and zones of nonperfusion that could improve the susceptibility of retina to hypoxia. In addition, the pathological neovascularization arising in DR entails of the inflammatory response induction and leukocyte adhesion to the vessel wall mediated by VEGF-164 isoform [46]. VEGF is a chemotactic factor for monocytes and upregulates intercellular adhesion molecule -1(ICAM-1) expression, promoting leukostasis [47]. This inflammatory environment seems to be essential for the onset and the evolution of DR pathogenesis. The activation of oxidative stress mechanisms induces the mitochondria to produce superoxide in endothelial cells, elicits inflammatory mediators and alters angiogenesis [48]. Poly (ADP- ribose) polymerase (PARP) is involved in oxidative-stress pathways initiated during DR. In diabetic animal models, PARP is connected to hypoxia-induced VEGF overexpression, and PARP inhibitors are capable to avoid VEGF overexpression by a post-translational mechanism [49]. Increased levels of PARP are also involved in the manifestation of early stage diabetic microangiopathy, including cellularity and pericytes degeneration. Oxidative stress is involved in autophagy of retinal pericytes by the induction of highly-oxidized glycated low-density lipoprotein (HOG-LDL) [50]. Furthermore, lipid peroxidation induces oxidative stress leading to RPE cell death [51]. Alternatively, the elevated expression of the CCAAT/enhancer-binding protein (C/EBP) homologous protein growth arrest and DNA damage-inducible gene 153 (CHOP/GADD153) in retinas of diabetic rats and in human retinal capillary endothelial cells (HRCECs) cultured under hyperglycemic conditions could facilitate the initial development of DR through ER stress [52]. Moreover, ROS can indirectly promote the nuclear translocation of NF-κB via the degradation of the negative regulator inhibitor of kappa B alpha (IκBα) in the cytoplasm. Into the nucleus, NF-κB controls the expression of the genes regulatory of the inflammatory response by the binding to the DNA Fig. (2) [53]. The suggested mechanism occurs via the activation of NF-κB and as a consequence of initiating downstream effectors such as ICAM-1 which induces the leukostasis [54]. Since the pericytes of diabetics display augmented levels of NF-κB, it is reasoned that hyperglycemia triggers NF-κB and induces apoptosis of retinal pericytes [55]. The death of pericytes is one of the first histopathological lesions and distinguishing mark of DR [56]. Additionally, high glucose level modulates TGFβ signals in mesenchymal cells linked to Ca(2+)/ PKC/MAPKs as well as PI3K/Akt/mTOR signal pathways [57]. The correlation among TGFβ, pericytes, and the preservation of quiescent retinal endothelial cells has been also explored [58]. A subgroup of pericytes expresses TGFβ1, and cross-talk signaling with the endothelial cell increases the expression of vascular endothelial growth factor receptor 1 (VEGFR1) on endothelium protecting the vasculature from oxidative injury [59]. The contribution of mTOR signaling in pericytes could affect the autophagic mechanisms, since impairing pericytes biology could have a substantial impact during the initial subclinical stages of DR.
Fig. (2).
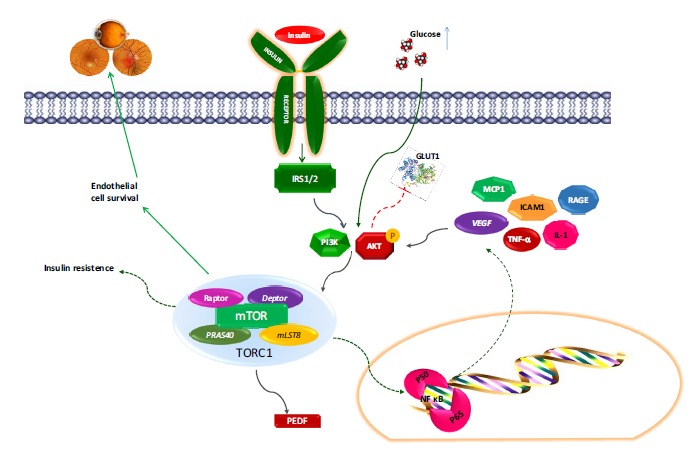
Mechanistic target of rapamycin (mTOR) pathway in autophagy and diabetic retinopathy.
3. MOLECULAR MECHANISM OF AUTOPHAGY
Autophagy is involved in numerous neurodegenerative disorders. Neural tissues, such as retina, are fully reliant on glucose for usual metabolic activity. In both type I and II diabetes, maintenance of blood glucose level is an essential process to prevent secondary long-term microvascular complications, including neuropathy and retinopathy [60]. The molecular machinery of autophagy is very complex, engages various signaling pathways that, in turn, might cooperate at multiple stages. In the course of autophagy, the microtubule-associated protein 1 light chain 3 (LC3-I) is transformed by the addition of a group of phosphatidyl- ethanolamine (LC3-II) which permits the combination of the protein to autophagosome membranes. Similarly, sequestosome 1 (p62/SQSTM1), hired to the auto-phagosomal membrane by interaction with LC3, participate to the autophagic process [61]. Defect in autophagy elicits p62 overexpression [62], whereas the autophagy triggered by the degradation of p62 prevents tumorigenesis [63]. Both apoptotic and autophagic mechanisms adopt common pathways with several proteins such as BCL2 (B/cell CLL/ lymphoma 2) family protein which play crucial role in the regulation of both these processes [64-65]. BCL2 after binding BECN1 (Beclin 1, autophagy-related) disturbs the formation of the class III phosphatidylinositol 3-kinase (PtdIns3K) complex, preventing the autophagic process. Besides to BECN1, ER- located BCL2 may avoid autophagy by regulating free ER Ca2+ homeostasis [66]. Thus, both pathways can co-occur in the same cell [67].
Autophagy is initially triggered in the dendrites of retinal ganglion cells to protect the cell. Subsequently, autophagy is principally activated in the cytoplasm to elicit cell death. BECN1, belonging to class III PtdIns3K complexes, plays a part to the initial autophagic vesicles development [68], which are mainly localized at the ganglion cell layer (GCL) in the retina [69]. It has been reported that a low glucose in 661W photoreceptor cells, inducing a reduction of BCL2 and BCL-XL and leading to the expression of the active pro-apoptotic BAX, could disturb the autophagic process [70]. The mechanistic target of rapamycin (mTOR) is one of the principal regulators of autophagy and it’s important because play a key role in this mechanism. In physiological conditions mTOR prevents autophagy, whereas in the course of nutrient deficiency Class I phosphoinositide 3-kinase induces autophagy, principally modulating TOR activity in response to insulin-like and other growth factors [71]. The AMP activated protein kinase (AMPK) is an intracellular nutrient sensor, which reacting to energy reduction induces the autophagic process. Depending on the stimulus or the cellular system, the activated-autophagy could show contrasting aspects, either harmful or beneficial. Not only hyperglycemia, but also hypoglycemia, could be detrimental for the retina and can lead to autophagy [72]. As validated in 661W photoreceptor cells, in which low levels of glucose exposure resulted in the activation of the caspase 3 signaling cascade, the reduction of glutathione (GSH) level and retinal cell death. Further, was found that low glucose activated apoptotic process via the BCL2/BAX pathway and autophagy via the AMPK/RAPTOR/mTOR pathway. In addition, low glucose produces an altered autophagosome/lysosome fusion through a reduction of the lysosome-associated membrane protein type 2α (LAMP2α) protein expression [73]. LAMP2α, belonging to the lysosomal membrane, plays a crucial role in the activity of the chaperone-mediated autophagic (CMA) pathway [74] and autophagosomes maturation [75]. It has been demonstrated that LAMP2 deficient mice exhibit a proliferation of autophagic vacuoles [76]. The reduction of autophagosome/lysosome fusion via LAMP2α expression decrease elicit an increase of LC3-II and p62 proteins, two markers of autophagosomes accumulation, suggesting that LAMP2 reduction might be involved in the altered process of autophagosome/lysosome fusion. Autophagy suppression, either by 3- methyladenine (3-MA) or by specific knock-down of either ATG5 or ATG7 trigger a reduction of low glucose-induced LC3-II accumulation and sensitize the cells to low glucose by induction of caspase 3 activity and cell death Fig. (1). The equilibrium between apoptosis and autophagy appears crucial for 661W cells survival in low energy conditions. The increase of autophagy represents a survival response to preserve vital functions of cells, which are neutralized by a lysosomal fusion defect. Modulation of both apoptosis and autophagy pathways might be important to avoid diabetic retinopathy [77]. Interestingly, a cone-rod dystrophy characterized by loss of photoreceptor and RPE cells was reported in a patient carrying a LAMP2 mutation [78]. Additionally, an involvement of LAMP2 has been reported in Danon disease, where it induced autophagic vacuolar myopathy in muscles [79] and retinopathy [80]. Additional in vivo investigations should be performed to improve the knowledge on the detailed involvement of LAMP2 in diabetic side effects and in hypoglycemic-induced retinal cell death.
Fig. (1).
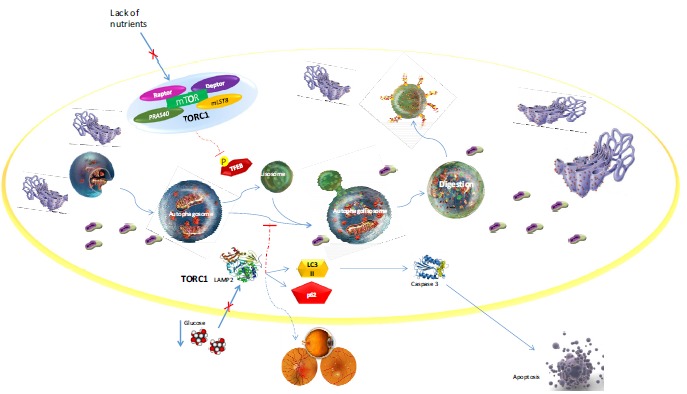
Molecular mechanism of autophagy in the occurrence of Diabetic Retinopathy.
3.1. Role of Mechanistic Target of Rapamycin (mTOR) Pathway in Autophagy and Diabetic Retinopathy
The mTOR, also termed the mammalian target of rapamycin, is a cytoplasmic kinase that controls cell growth and metabolism as reaction to mitogens (such as IGF-I and VEGF, nutrients (amino acids, glucose and fatty acids), hormones including insulin and cytokines [81]. mTOR is a 289-kDa serine/threonine protein kinase. It is encoded by a single gene FRAP1 [82]. Signal and activation due to the increased expression of adenovirus-mediated phosphatases that inhibit Akt phosphorylation also disturb angiogenesis. Therefore, all those growth factors exhibiting a role in the progression of vasculopathies typical of human proliferative diabetic retinopathy are associated to the PI3K/ Akt/mTOR signaling pathway for the modulation of their expression and activity. The mTOR pathway is also involved in other disorders of the retina. The dedifferentiation of RPE and consequent photoreceptor degeneration is associated with mTOR activation [83-95]. In murine model the suppression of mTOR pathway inhibits RPE dedifferentiation and preservation of photoreceptor functionality [96]. Some of the mTOR inhibitors, such as Rapamycin, show an immuno-suppressive action, which on the one hand can have an unfavorable side effect, but, on the other hand it can be beneficial and helpful to control the pro-inflammatory phenotype that occurs in diabetes.
3.2. HIF-1α, VEGF, and mTOR Deregulation in Diabetic Retinopathy
The enhanced level of insulin leads to IR, avoidable by rapamycin [71]. High glucose (HG) concentrations activate mTOR Fig. (2). By stabilizing glucose concentrations, insulin therapy may deactivate mTOR. Conversely, hyperactivation of mTOR results in IR, [97, 98] mTOR activates S6 kinase (S6K), which phosphorylating and degrading the insulin receptor substrate 1/2 (IRS1/2) impairs insulin signaling. Further, mTOR by inducing growth factor receptor- bound protein 10 causes IR [99-100] Fig. (3). Amino acids administration triggers mTOR/S6K1, which in turn causes a feedback IR in human skeletal muscle [101-102]. Oral rapamycin reduced mTOR activation, inhibiting IR induced by nutrients [103]. Similarly, TNFα and other pro-inflammatory cytokines trigger mTOR which leads damage in insulin signaling Fig. (3). The investigation on the mechanisms implicated in the manifestation of DR worsening indicates that the process arises from hypoxia produced by an impaired hemodynamic and low-glucose availability in the retina [104]. The mechanism by which low calorie diet reduces IR may be explained by the observation that dietary restriction deactivates mTOR [86]. Interestingly, VEGF production in retinal pigment epithelial cells is induced by the insulin/mTOR pathway [105-108], these evidences explain why hyperactivation of mTOR is involved in DR. It is well kwon that abnormal expansion of small blood vessels (angiogenesis or neovascularization) plays a part to retinopathy. VEGF induces angiogenesis and leads to BRB collapse [109, 110]. Moreover, insulin and IGF-1 are implicated in angiogenesis and DR [107, 111-113]. This observation explains why increased insulin treatment may exacerbate DR [111-114]. It has been reported that Rapamycin inhibits insulin-induced hypoxia-inducible factor-1 (HIF-1) and retinal cells senescence [108,115] and destroys retinal and choroidal neovascularization in mice [116]. The hypoxia is worsened by an acute decrease of free glucose due to the “tight” glucose control. Intensive reduction of glucose by insulin could induce inadequate amount of glucose essential for the metabolic demand of the retina. Contemporarily, the massive insulin treatment could elicit HIF-α expression via PI3K-dependent pathway [117]. Ischemia causes the proliferative stage of DR, in which the hypoxia intensifies the proliferative component of angiogenesis. Further, the signaling pathway of mTOR enhances mitogen-stimulated vascular cell proliferation and angiogenesis as reaction to hypoxia [118]. Tissue hypoxia controls HIF-1α hydroxylation and modulates its protein levels and activity [119]. HIF-1α is one of the major regulators of VEGF expression Fig. (3). The binding of HIF-1α to the VEGF hypoxia-responsive elements promoter induces signaling via MAPK, PI3K, and JNK pathways with a consequential production of various growth factors and genes including VEGF, VEGF flt-1 receptor, PDGF, βFGF, IGF-1, angiopoietin 2 and nitric oxide synthases, that are recognized inducers of angiogenesis Fig. (3). It has been found that in ocular tissue, the up-regulation of VEGF expression induced activating the PI3K/Akt/mTOR pathway and post-transcriptional activation of HIF- α mediated the proangiogenic effects of IGF-1 [120]. Moreover, it has been reported that mTOR pathway influences the mechanism by which growth factor, such as IGF-1, can display conflicting pleiotropic effects in an HIF- 1α -dependent manner [121]. In fact, IGF-1 can facilitate VEGF expression as well as stress and cytokine-induced VEGF production dependently or independently of HIF- 1α [127, 128] Fig. (3). Additionally, in the retina of transgenic mice overexpressing IGF-1 have been observed vascular modifications similar to that observed in human DR [124]. The increase of calcium levels may be one of the causes inducing hyperglycemia, which in turn activates retinal Müller cells and angiogenesis in patients with DR, as calcium contributes to HG-induced expression of the major angiogenic factors HIF-1α and VEGF in retinal Müller cells. This reaction is mediated by activation of the CaMKII-CREB signaling cascade, which through HG may be a potential mechanism responsible of the pathogenesis of DR. On the contrary, the inhibition of this signal may be a beneficial new therapeutic approach on the prevention of visual deficiency and blindness in DR patients [125]. The Src kinase signal contributes to VEGF-mediated retinal vascular availability and BRB collapse detected in diabetes [126]. VEGF along with PKC activation cooperate to enhance permeability of the endothelium in diabetes. In fact, VEGF induces the phosphorylation of the tight-junction complex protein occludin via a PKC-dependent pathway [126] Fig. (3). Additionally, the finding that VEGF immunoreactivity is linked to vascular leakage of macromolecules in human diabetic retinas [127] support the crucial contribution of VEGF in this process. Furthermore, chimeric antibodies that prevent VEGF bioavailability (“VEGF-trap”) decrease vascularization [128]. In diabetic patients increased VEGF levels promote acute breakdown of the BRB and the clinical appearances including retinal edema and exudates. The BRB collapse explains, in part, the clinical manifestations of “early worsening” effect in patients with minimal to moderate retinopathy [129]. The recognitions that oxygen levels regulate mTOR function and that mTOR is implicated in hypoxia- facilitated vasoproliferative responses propose a quite novel downstream functional connection between hypoxia and mitogenic signaling engaged in proliferation of vascular cells [130]. Overall, these remarks indicate that PI3K/Akt/mTOR inhibition signal would be suitable to accomplish the evolution of the proliferative stages of DR where hypoxia-driven vasoproliferative mechanisms prevail in the evolution of vasculopathies [131]. The mTOR inhibitors could be able to inhibit the onset of the “early worsening” avoiding the BRB collapse through the control of HIF-1α-mediated downstream activation of growth factors, such as the transcriptional regulation of retinal VEGF. The effectiveness of this treatment could prevent the progression of irreparable structural injury of the retinal microvasculature and could restrain forthcoming detrimental processes and possibly defer or avoid the evolution of retinal micro-angiopathies.
Fig. (3).
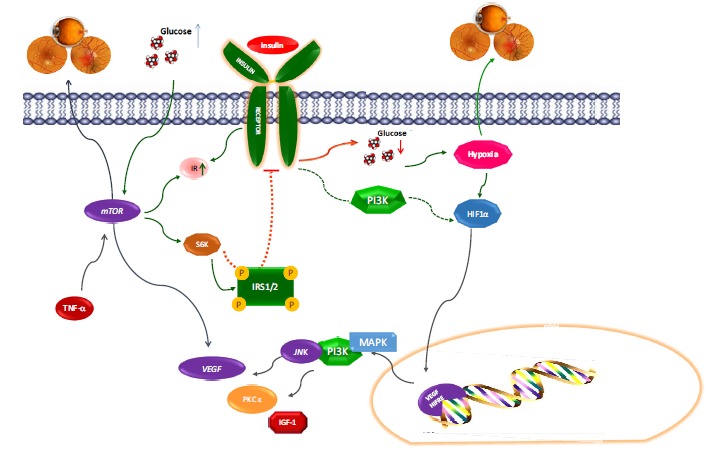
HIF-1α, VEGF, and mTOR deregulation in Diabetic Retinopathy.
3.3. mTOR Complex 1 (mTORC1) Regulates Trans-cription Factors Involved in DR
Nutrients and growth factors such as insulin or insulin-like growth factor 1 are the best described cellular modulators of the mTOR complex 1 (mTORC1) activation. The mTORC1 activation elicits cell growth and proliferation by inducing protein synthesis, lipid metabolism and biogenesis, and by decreasing autophagy [132]. In addition, several factors such
as oxygen, energy, inflammation, Wnt signaling and phosphatidic acid have been recognized as modulators of mTORC1 [85]. In its active form, mTORC1 phosphorylates the eukaryotic translation initiation factor 4E (eIF4E) binding protein 1 (4E-BP1) and S6 kinase 1 (S6K1), which, consecutively, induce protein synthesis [133]. Additionally, by the phosphorylation of other effectors, mTORC1 induces lipid biogenesis and metabolism and inhibits autophagy [85] Fig. (4). Similarly, mTORC1 activity is very sensitive to the macrolide rapamycin. Once, rapamycin binds the 12 kDa FK506-binding protein (FKBP12), interacts with mTORC1 kinase and inhibits mTORC1 activity and interrupts gene transcription suppressing protein synthesis [134]. In the last few years, a number of evidences indicated that mTORC1 is required in lipid biogenesis by modulating the expression of numerous lipogenic genes. The sterol-regulatory- element-binding proteins (SREBPs) are basic helix-loop-helix (bHLH) transcription factors involved in lipid homeostasis. SREBPs regulate the expression of lipogenic genes, and therefore are fundamental in lipid synthesis [135]. SREBP family encompasses three members SREBP1a and SREBP1c (currently named SREBP1) and SREBP2. In particular, SREBP1 is involved in insulin-mediated fatty acid synthesis, while SREBP2 principally modulates cholesterol biosynthesis [135]. It has been shown that insulin induces SREBP1 expression and cleavage, thus, the mature form of SREBP1 translocate into the nuclei and control gene expression. The inhibition of mTOR signal decreases the expression of both SREBPs mRNA and protein [136]. Some investigations have indicated that mTORC1 controls transcription of SREBPs by a mechanism operating independently to mTORC1 substrate S6K1. Moreover, mTORC1 promotes the nuclear accumulation of the mature and active form of SREBPs [137] Fig. (4) by a mechanism that involves Lipin 1, a phosphatidic acid phosphatase that also acts as a transcriptional coactivator [138]. In particular, the active mTORC1 phosphorylates Lipin 1 leading to its elimination from the nucleus. On the contrary, if mTORC1 is inhibited, Lipin 1 accumulates in the nucleus and inducing the bind of SREBPs to the nuclear matrix hampers their binding to target genes [137] Fig. (4). Studies have revealed the expression of SREBP also in the retina. In diabetes the altered circadian pattern of SREBP could cause retinal metabolic defects triggering the onset of DR. The evidence showing that mTORC1 controls SREBPs at various levels indicates that lipid synthesis regulation is closely related to nutrient and growth factor pathways to guarantee cellular homeostasis and suggests investigating the role of these factors in DR [133]. It has been reported that the mTORC1–4E-BP axis modulates also the translation of peroxisome proliferator activated receptor γ (PPARγ), a member of the nuclear receptor superfamily. PPARγ is a ligand-activated transcription factor essential for the regulation of several physiological processes including the control of genes expression involved in fatty acid synthesis, uptake and esterification [138] and the control of CCAAT/enhancer-binding protein α (C/EBPα) and delta (C/ EBPδ) that are molecules needed for the activation of the adipogenic cascade [139]. It has been reported that mTORC1 helps the transactivation capability of PPARγ [140], possibly, by SREBP1 activation, which induces the endogenous PPARγ ligands production [141] (Fig. 4). Although mTORC1 is a crucial regulator of PPARγ, its activity must be strongly controlled to achieve the adipogenic pathway in physiological conditions. Studies in recent years have revealed that mTORC1 during adipogenesis is involved in numerous processes, such as mitotic clonal expansion, epigenetic regulation, unfolded protein response, and autophagy. Conversely to mTORC1, the overexpression of DEPTOR, an endogenous and partial inhibitor of mTOR signaling, does not enhance adipose tissue accumulation in vivo [130] and adipogenesis in vitro. In fact, it was demonstrated that DEPTOR induces adipogenesis by inhibiting the negative effect of mTORC1 on insulin signaling, which stimulates the pro-adipogenic action of Akt.
Fig. (4).
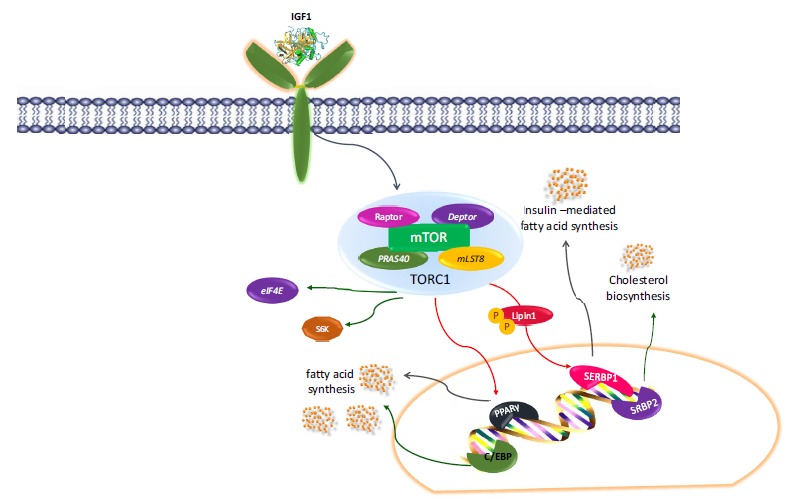
Transcription factors involved in DR regulated by mTOR complex.
3.4. Control of Lysosome Mediated by the mTORC1
Lysosomes are organelles that play a part to cellular homeostasis by controlling a large number of physiological processes, such as lipid homeostasis, energy metabolism, cellular clearance and pathogen defense [142]. Lysosomes are also indispensable for the activation of mTORC1 by nutrients. Lysosomal dysfunction can give rise to health problems by decreasing the cellular competence of remove organelles, protein aggregates, and debris. This lysosomal aptitude deteriorates over time, and is associated to the onset of age-related diseases and aging [143].
The βHLH leucine zipper transcription factor EB (TFEB) is a regulator of lysosome functions including their adaptation to environmental signals. As reaction to food shortage or lysosomal dysfunction, TFEB positively regulates the expression of lysosomal hydrolases, lysosomal membrane proteins and components of the VATPase complex [144]. Moreover, it induces autophagosomes development and their fusion with the lysosome [145]. Overall, these processes help the cells to survive in stressful conditions by increasing their energy production from the degradation of cellular elements. mTORC1 has been recognized as an important regulator of TFEB function [146]. In fact, in the presence of nutrients it promotes TFEB phosphorylation at the lysosome surface. The phosphorilation induces the binding of TFEB to 14-3-3 proteins and prevents its transport into the nucleus. On the contrary, the mTORC1 damage decrease TFEB phosphorylation and its binding to 14-3-3 proteins, which promptly elicits TFEB accumulation in the nucleus, where it organizes the development of lysosomal and autophagic areas (Fig. 1). Since mTORC1 suppression facilitates lysosomal biogenesis and autophagy, trials designed at inhibiting mTORC1 could be beneficial treatments to improve age-related diseases and aging [147].
4. UPR and XBP1 and DR
As previously mentioned malfunction of the endoplasmic reticulum (ER), or ER stress, is involved in the pathogenesis of diabetes and its complications [148,149]. The ER is the essential cellular organelle answerable for protein folding and maturation. To assure the correct protein folding, the ER has sophisticated machinery to recognize irregular proteins and target them for refolding or clearance [150]. This process is recognized as the unfolded protein response (UPR). The UPR regulates the course of protein synthesis, protein folding, and degradation to assure proteostasis, that is essential for cell survival and activity. The UPR pathways are begun by three ER stress sensors situated on the ER membrane, specifically, IRE1, ATF6, and PERK. In diabetic retinal cells have been identify all three UPR devices, including the PERK/ATF4 signaling pathway, which has been broadly analyzed in the progress of DR. The three UPR pathways could be stimulated individually and independently of each other and behave with different roles in inducing downstream target genes and regulate various physiological processes. X-box binding protein 1 (XBP1) is a main transcription factor in the central UPR pathway and controls a set of genes implicated in cellular metabolism, redox state, autophagy, inflammation, cell survival, and vascular function. Activation of XBP1 is crucial for preserving ER function and dysregulated XBP1 expression/activity has been associated to apoptosis, cell death, and insulin resistance in diseased conditions [151]. XBP1 is a basic-region leucine zipper protein in the cAMP binding protein/activating transcription factor (CREB/ATF) family of transcription factors and regulates a subcategory of UPR target genes [152]. It is expressed ubiquitously in adult tissues, activated by IRE1 in the course of ER stress. Deletion or downregulation of XBP1 exacerbates retinal cell sensitivity to apoptosis [153], inflammation [154], and oxidative stress [155]. In quiescent cells, the ER luminal domain of these proteins binds to a chaperone molecule known as the glucose-regulated protein 78 (GRP78, also famous as immunoglobulin binding protein, BiP) and the binding preclude their activation. About ER stress, GRP78 is sequestered from the sensors and links to unfolded/misfolded proteins to make easy their refolding. The dissociation of GRP78 results in activation of the ER stress sensors and afterward activates the UPR [151]. So far the exact function of UPR and XBP1 in the DR it is still unclear, therefore further experimental studies are required in order to explore its role in autophagy and in the pathogenesis of DR in order to develop new potential therapeutic target.
5. DRAM2 GENE
Recently have been identified new autophagy regulators, which include the Damage-Regulated Autophagy Modulator (DRAM). DRAM is a novel p53-induced transmembrane protein. The DRAM expression is necessary for p53-mediated apoptosis [156, 157]. Since p53 expression is stimulated by several cellular stresses including genotoxic stress, DRAM is considered an important molecule connecting p53 and autophagy [158]. Furthermore, c-Jun NH2 Terminal Kinase (JNK) activation increases the DRAM expression to arouse autophagy and apoptosis [159, 161]. In humans, 5 additional proteins display significant homology to DRAM, including damage-regulated autophagy modulator 2 (DRAM2), which showing 45% identity and 67% conservation when compared to DRAM, is highly homologue to DRAM [162, 163]. For this tight homology DRAM and DRAM2 share common features. Both DRAM and DRAM2 are mainly located in lysosomes and include six putative transmembrane domains [162, 163]. Further, their expression is usually down-regulated in tumors [158, 163]. Recently it has been reported that like to DRAM, DRAM2 is implicated in autophagy induction. DRAM2 overexpression leads to cytoplasmic GFP-LC3 scatter, and increases the level of endogenous LC3-II [164]. In addition, the silencing of endogenous DRAM2 affects starvation -induced autophagy and an efficient autophagosome formation [165, 166]. Autophagy develops in the retinal pigment epithelium, because the necessity of a constant renewal of the outer segments of the photoreceptors resulting from damage induced by light daily. Immunohistochemical analysis displayed that DRAM2 is confined to photoreceptor inside the segments and to the apical surface of retinal pigment epithelial cells where likely participate in the process of photoreceptor regeneration and recycling to maintain visual function. Therefore, it has been supposed in the absence of properly functioning gene DRAM2, the autophagy and the renewal of photoreceptors decreases with the thinning of photoreceptor cells [166].
6. CONCLUDING REMARKS
Drugs inducing pro-autophagic or autophagy process could be evaluated as new therapeutic strategies reducing retinal cells death. TZDs, synthetic PPARγ agonists, elicit anti-inflammatory, antiatherogenic, neuroprotective, and antioxidative effects [167]. Therefore, they could be used in the treatment of diabetic microvascular complications such as DR. Both in vitro and in vivo experiments showed that TZDs may induce retinal microcirculatory stability [168], reduce pathological retinal microvessel formation [169], inhibit the fibrotic change of RPE cells [170], as well as avoid retinal neuronal injury [171] in DR. TZDs may inhibit the progression of DR [172]. In fact, troglitazone and rosiglitazone could prevent the proliferation of retinal endothelial cell and tube formation induced by VEGF. Moreover, it has been reported that rosiglitazone may linger the onset of proliferative DR [173]. However, since some investigations revealed that TZDs increased the risk of macular edema, the association between TZDs and DME is still debatable [174].
Treatments with pioglitazone [175] and troglitazone [176] augmented significantly VEGF expression in diabetic patients increasing the risk of diabetic macular edema (DME) and DR evolution. In contrast, some authors did not detected fluid retention in the macula or subclinical DME under TZDs treatment [177, 178]. Recent studies related to PPARγ activation and its mechanism of action addressed to identify valid therapeutic approach for DR have been performed mixing herbal or traditional medicine [179]. It has been found that plants such as Astragalus membranaceus, Pueraria thomsonii [180], Swietenia mahagoni [181], Korean red ginseng [182], Dan-shao-hua-xian formula [183], and Turmeric [184], are potential modulator of DR through PPARγ activation and, thus could be used to avoid DR evolution. Since, fenofibrate an agonist of peroxisome proliferator-activated receptor (PPAR) α is able to reduce levels of serum lipids, its effect was evaluated in DR treatment and it was reported that fenofibrate reduces DME and proliferative diabetic retinopathy (PDR) by 30% [184]. Additional evidence showed that the combination of fenofibrate with simvastatin is more effective, in the evolution of DR, than the treatment with placebo plus simvastatin [185]. This positive result is linked to the potential mechanisms on the BRB and not to the quantitative variations in serum lipids [186]. Further investigation on the influence of fenofibrate on survival signaling or stress in retinal tissues treated in experimental conditions reproducing the diabetic environment demonstrated that fenofibric acid inhibits the detrimental effects of hypoxia and/or of HG levels [187]. The extensive research on the functional role of mTOR pathways involving a variety of regulators of cellular survival essential to the initiation and progression of DR strongly indicate that mTOR is an interesting therapeutic target for DR. The inhibitors of mTOR appear to delay or inhibit the evolution of retinal microangiopathies avoiding the BRB collapse by controlling the activation of growth factors HIF-α-mediated. With the evolution of DR in proliferative stages, the inhibition of PI3K/Akt/mTOR pathway could be beneficial to suppress neovascularization by inhibition of growth factors, restraining the inflammation, promoting apoptosis of nascent vessels and preventing angiogenesis. So far the best detected mTOR complex inhibitor is rapamycin, “a macrolide antifungal compound produced by the soil bacterium Streptomyces hygroscopicus found in Rapa Nui [134]. It has been reported that in mice with laser-induced choroidal neovascularization and in oxygen-induced retinopathy the Rapamycin administration induces anti-angiogenesis [188]. Nevertheless, several clinical and preclinical studies demonstrate that mTOR inhibitors that are beneficial for diabetes display adverse effects [189]. The side effects present in many organs after mTOR inhibitors administered for systemic exposure, including cutaneous lesions and oral ulcerations [190], metabolic [191], hematological alterations [192], renal toxicities [193], high incidence of reversible infertility [194] showed different occurrence and duration.
Rapamycin, consequently to the feedback activation of Akt via TORC2, has displayed a paradoxical augmentation of VEGF and Flt-1 protein levels as reaction to pathway inhibition. This consequence could be problematic for the longstanding treatment of DR. Nevertheless, it has been developed efficacious therapies avoiding the limits mTOR inhibitors. For example, selective and powerful mTOR inhibitors suppressing both mTORC1 and mTORC2 show extraordinary efficacy in avoiding the feedback-loop activation. Furthermore, the new generation mTOR inhibitors do not exert this negative feedback. The new drugs embrace highly specific mTOR inhibitors, dual PI3K/mTOR inhibitors [87], and AKT inhibitors possessing ATP-competitive or ATP-independent allosteric modulators [88]. Moreover, Green Tea [195] Epigallocatechin Gallate (EGCG) [195], and Ginkgo Biloba [196] natural mTOR inhibitors display protective effects in DR, which seems to be principally mediated by their powerful antioxidative properties. In addition, the polyphenol resveratrol has mTOR-modulating properties, is cytoprotective and suppress VEGF production in human retinal ARPE-19 cells [197]. Interestingly, more recent investigation reports that Alpha Lipoic Acid (LA) regulates high glucose-induced mesangial cell dysfunction by modulating mTOR/4E-BP1/p70S6K signaling. This study suggests a possible application of LA in the regulation of diabetes-induced mesangial cell proliferation and matrix expansion in vivo [198]. The mTOR inhibitors combined with anti- inflammatory compounds are beneficial to contain early hemodynamic alterations in the retina and ocular angiogenesis. The mTOR inhibitors are suitable to treat both early and advanced manifestations of DR. The second-generation mTOR inhibitors should accomplish several basic criteria against neovascularization by precise mechanism, inhibit the angiogenic process of the disease, validate specificity and selectivity for atypical vessels, achieve a formulation non-toxic at long-term administration, stabilize or avoid additional decline of vision, and prevent late-stage complications of DR such as detachment and scarring. Therefore, is important to identify the activating factors contributing to the microangiopathy detected in progressive diabetic retinopathy to achieve a clear understanding of the autophagic and apoptotic molecular mechanisms. In addition, it is imperative that emerging therapeutic device targeting UPR or mTOR pathways should be carefully considered in the setting of their molecular pathways operant in DR, such as mechanism of action, stage of evolution of the disease, and the crucial time of pharmacological treatment. Further developments in advanced therapies in this field would improve patients’ management and prevent the occurrence and progression of DR.
ACKNOWLEDGEMENTS
This work was supported by grants from the PON 01_02464.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
REFERENCES
- 1.Cao J., Ying M., Xie N., Lin G., Dong R., Zhang J., Yan H., Yang X., He Q., Yang B. The oxidation states of DJ-1 dictate the cell fate in response to oxidative stress triggered by 4-hpr: autophagy or apoptosis? Antioxid. Redox Signal. 2014;21(10):1443–1459. doi: 10.1089/ars.2013.5446. [http://dx.doi.org/10.1089/ars.2013.5446]. [PMID: 24392637]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maiese K. Novel applications of trophic factors, Wnt and WISP for neuronal repair and regeneration in metabolic disease. Neural Regen. Res. 2015;10(4):518–528. doi: 10.4103/1673-5374.155427. [http://dx.doi.org/10.4103/1673-5374.155427]. [PMID: 26170801]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vakifahmetoglu-Norberg H., Xia H.G., Yuan J. Pharmacologic agents targeting autophagy. J. Clin. Invest. 2015;125(1):5–13. doi: 10.1172/JCI73937. [http://dx.doi.org/10.1172/JCI73937]. [PMID: 25654545]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wirawan E., Vanden Berghe T., Lippens S., Agostinis P., Vandenabeele P. Autophagy: for better or for worse. Cell Res. 2012;22(1):43–61. doi: 10.1038/cr.2011.152. [http://dx.doi.org/10.1038/cr.2011.152]. [PMID: 21912435]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Müller M., Reichert A.S. Mitophagy, mitochondrial dynamics and the general stress response in yeast. Biochem. Soc. Trans. 2011;39(5):1514–1519. doi: 10.1042/BST0391514. [http://dx.doi.org/10.1042/BST0391514]. [PMID: 21936844]. [DOI] [PubMed] [Google Scholar]
- 6.Tanida I. Autophagosome formation and molecular mechanism of autophagy. Antioxid. Redox Signal. 2011;14(11):2201–2214. doi: 10.1089/ars.2010.3482. [http://dx.doi.org/10.1089/ars.2010.3482]. [PMID: 20712405]. [DOI] [PubMed] [Google Scholar]
- 7.Oku M., Sakai Y. Peroxisomes as dynamic organelles: autophagic degradation. FEBS J. 2010;277(16):3289–3294. doi: 10.1111/j.1742-4658.2010.07741.x. [http://dx.doi.org/10.1111/j.1742-4658.2010.07741.x]. [PMID: 20629742]. [DOI] [PubMed] [Google Scholar]
- 8.Knodler L.A., Celli J. Eating the strangers within: host control of intracellular bacteria via xenophagy. Cell. Microbiol. 2011;13(9):1319–1327. doi: 10.1111/j.1462-5822.2011.01632.x. [http://dx.doi.org/10.1111/j.1462-5822.2011.01632.x]. [PMID: 21740500]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mijaljica D., Prescott M., Devenish R.J. The intriguing life of autophagosomes. Int. J. Mol. Sci. 2012;13(3):3618–3635. doi: 10.3390/ijms13033618. [http://dx.doi.org/10.3390/ijms13033618]. [PMID: 22489171]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Plomp P.J., Gordon P.B., Meijer A.J., Høyvik H., Seglen P.O. Energy dependence of different steps in the autophagic-lysosomal pathway. J. Biol. Chem. 1989;264(12):6699–6704. [PMID: 2708336]. [PubMed] [Google Scholar]
- 11.Russell F.D., Hamilton K.D. Nutrient deprivation increases vulnerability of endothelial cells to proinflammatory insults. Free Radic. Biol. Med. 2014;67:408–415. doi: 10.1016/j.freeradbiomed.2013.12.007. [http://dx.doi.org/10.1016/ j.freeradbiomed.2013.12.007]. [PMID: 24334251]. [DOI] [PubMed] [Google Scholar]
- 12.Cimini S., Rizzardini M., Biella G., Cantoni L. Hypoxia causes autophagic stress and derangement of metabolic adaptation in a cell model of amyotrophic lateral sclerosis. J. Neurochem. 2014;129(3):413–425. doi: 10.1111/jnc.12642. [http://dx.doi.org/10.1111/jnc.12642]. [PMID: 24359187]. [DOI] [PubMed] [Google Scholar]
- 13.Hariharan N., Zhai P., Sadoshima J. Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid. Redox Signal. 2011;14(11):2179–2190. doi: 10.1089/ars.2010.3488. [http://dx.doi.org/10.1089/ars. 2010.3488]. [PMID: 20812860]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Essick E.E., Sam F. Oxidative stress and autophagy in cardiac disease, neurological disorders, aging and cancer. Oxid. Med. Cell. Longev. 2010;3(3):168–177. doi: 10.4161/oxim.3.3.2. [http://dx.doi.org/10.4161/oxim. 3.3.12106]. [PMID: 20716941]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boya P., González-Polo R.A., Casares N., Perfettini J.L., Dessen P., Larochette N., Métivier D., Meley D., Souquere S., Yoshimori T., Pierron G., Codogno P., Kroemer G. Inhibition of macroautophagy triggers apoptosis. Mol. Cell. Biol. 2005;25(3):1025–1040. doi: 10.1128/MCB.25.3.1025-1040.2005. [http://dx.doi.org/10.1128/MCB.25.3.1025-1040.2005]. [PMID: 15657430]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kubli D.A., Gustafsson Å.B. Mitochondria and mitophagy: the yin and yang of cell death control. Circ. Res. 2012;111(9):1208–1221. doi: 10.1161/CIRCRESAHA.112.265819. [http://dx.doi.org/10.1161/CIRCRESAHA.112.265819]. [PMID: 23065344]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maiese K. New Insights for Oxidative Stress and Diabetes Mellitus. Longev. 2015 doi: 10.1155/2015/875961. 2015, 875961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin W.J., Kuang H.Y. Oxidative stress induces autophagy in response to multiple noxious stimuli in retinal ganglion cells. Autophagy. 2014;10(10):1692–1701. doi: 10.4161/auto.36076. [http://dx.doi.org/10.4161/ auto.36076]. [PMID: 25207555]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Z.F., Li Y.B., Han J.Y., Wang J., Yin J.J., Li J.B., Tian H. The double-edged effect of autophagy in pancreatic beta cells and diabetes. Autophagy. 2011;7(1):12–16. doi: 10.4161/auto.7.1.13607. [http://dx.doi.org/10.4161/auto.7.1.13607]. [PMID: 20935505]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frey T., Antonetti D.A. Alterations to the blood-retinal barrier in diabetes: cytokines and reactive oxygen species. Antioxid. Redox Signal. 2011;15(5):1271–1284. doi: 10.1089/ars.2011.3906. [http://dx.doi.org/10.1089/ars. 2011.3906]. [PMID: 21294655]. [DOI] [PubMed] [Google Scholar]
- 21.Vincent J.A., Mohr S. Inhibition of caspase-1/interleukin-1β signaling prevents degeneration of retinal capillaries in diabetes and galactosemia. Diabetes. 2007;56(1):224–230. doi: 10.2337/db06-0427. [http://dx.doi.org/10.2337/db06-0427]. [PMID: 17192486]. [DOI] [PubMed] [Google Scholar]
- 22.Cheung N., Mitchell P., Wong T.Y. Diabetic retinopathy. Lancet. 2010;376(9735):124–136. doi: 10.1016/S0140-6736(09)62124-3. [http://dx.doi.org/10.1016/S0140-6736(09)62124-3]. [PMID: 20580421]. [DOI] [PubMed] [Google Scholar]
- 23.Antonetti D.A., Klein R., Gardner T.W. Diabetic retinopathy. N. Engl. J. Med. 2012;366(13):1227–1239. doi: 10.1056/NEJMra1005073. [http://dx.doi.org/10. 1056/NEJMra1005073]. [PMID: 22455417]. [DOI] [PubMed] [Google Scholar]
- 24.Ferris F.L. III Results of 20 years of research on the treatment of diabetic retinopathy. Prev. Med. 1994;23(5):740–742. doi: 10.1006/pmed.1994.1127. [http://dx. doi.org/10.1006/pmed.1994.1127]. [PMID: 7845951]. [DOI] [PubMed] [Google Scholar]
- 25.Tang J., Kern T.S. Inflammation in diabetic retinopathy. Prog. Retin. Eye Res. 2011;30(5):343–358. doi: 10.1016/j.preteyeres.2011.05.002. [http://dx.doi.org/10.1016/ j.preteyeres.2011.05.002]. [PMID: 21635964]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pournaras C.J., Rungger-Brändle E., Riva C.E., Hardarson S.H., Stefansson E. Regulation of retinal blood flow in health and disease. Prog. Retin. Eye Res. 2008;27(3):284–330. doi: 10.1016/j.preteyeres.2008.02.002. [http://dx.doi.org/10.1016/j.preteyeres.2008.02.002]. [PMID: 18448380]. [DOI] [PubMed] [Google Scholar]
- 27.Jarrett S.G., Lin H., Godley B.F., Boulton M.E. Mitochondrial DNA damage and its potential role in retinal degeneration. Prog. Retin. Eye Res. 2008;27(6):596–607. doi: 10.1016/j.preteyeres.2008.09.001. [http://dx.doi.org/10.1016/ j.preteyeres.2008.09.001]. [PMID: 18848639]. [DOI] [PubMed] [Google Scholar]
- 28.Altomare E., Grattagliano I., Vendemaile G., Micelli-Ferrari T., Signorile A., Cardia L. Oxidative protein damage in human diabetic eye: evidence of a retinal participation. Eur. J. Clin. Invest. 1997;27(2):141–147. doi: 10.1046/j.1365-2362.1997.780629.x. [http://dx.doi.org/10.1046/j.1365-2362.1997.780629.x]. [PMID: 9061308]. [DOI] [PubMed] [Google Scholar]
- 29.Santos J.M., Tewari S., Goldberg A.F., Kowluru R.A. Mitochondrial biogenesis and the development of diabetic retinopathy. Free Radic. Biol. Med. 2011;51(10):1849–1860. doi: 10.1016/j.freeradbiomed.2011.08.017. [http://dx.doi.org/10.1016/j.freeradbiomed.2011.08.017]. [PMID: 21911054]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Santos J.M., Tewari S., Kowluru R.A. A compensatory mechanism protects retinal mitochondria from initial insult in diabetic retinopathy. Free Radic. Biol. Med. 2012;53(9):1729–1737. doi: 10.1016/j.freeradbiomed.2012.08.588. [http://dx.doi.org/10.1016/j.freeradbiomed.2012.08.588]. [PMID: 22982046]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou R., Yazdi A.S., Menu P., Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. doi: 10.1038/nature09663. [http://dx.doi.org/10.1038/nature09663]. [PMID: 21124315]. [DOI] [PubMed] [Google Scholar]
- 32.Dupont N., Jiang S., Pilli M., Ornatowski W., Bhattacharya D., Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 2011;30(23):4701–4711. doi: 10.1038/emboj.2011.398. [http://dx.doi.org/10.1038/emboj.2011.398]. [PMID: 22068051]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Joussen A.M., Poulaki V., Le M.L., Koizumi K., Esser C., Janicki H., Schraermeyer U., Kociok N., Fauser S., Kirchhof B., Kern T.S., Adamis A.P. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004;18(12):1450–1452. doi: 10.1096/fj.03-1476fje. [PMID: 15231732]. [DOI] [PubMed] [Google Scholar]
- 34.Cui Y., Xu X., Bi H., Zhu Q., Wu J., Xia X., Ren Q. Ho, P.C. Expression modification of uncoupling proteins and MnSOD in retinal endothelial cells and pericytes induced by high glucose: the role of reactive oxygen species in diabetic retinopathy. Exp. Eye Res. 2006;83(4):807–816. doi: 10.1016/j.exer.2006.03.024. [http://dx.doi.org/10.1016/j.exer.2006. 03.024]. [PMID: 16750827]. [DOI] [PubMed] [Google Scholar]
- 35.Abrahan C.E., Insua M.F., Politi L.E., German O.L., Rotstein N.P. Oxidative stress promotes proliferation and dedifferentiation of retina glial cells in vitro. J. Neurosci. Res. 2009;87(4):964–977. doi: 10.1002/jnr.21903. [http://dx.doi.org/10.1002/jnr.21903]. [PMID: 18855938]. [DOI] [PubMed] [Google Scholar]
- 36.Gerhardinger C., Dagher Z., Sebastiani P., Park Y.S., Lorenzi M. The transforming growth factor-beta pathway is a common target of drugs that prevent experimental diabetic retinopathy. Diabetes. 2009;58(7):1659–1667. doi: 10.2337/db08-1008. [http://dx.doi.org/10.2337/db08-1008]. [PMID: 19401417]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coucha M., Elshaer S.L., Eldahshan W.S., Mysona B.A., El-Remessy A.B. Molecular mechanisms of diabetic retinopathy: potential therapeutic targets. Middle East Afr. J. Ophthalmol. 2015;22(2):135–144. doi: 10.4103/0974-9233.154386. [http://dx.doi.org/10.4103/0974-9233.154386]. [PMID: 25949069]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abu el Asrar A.M., Maimone D., Morse P.H., Gregory S., Reder A.T. Cytokines in the vitreous of patients with proliferative diabetic retinopathy. Am. J. Ophthalmol. 1992;114(6):731–736. doi: 10.1016/s0002-9394(14)74052-8. [http://dx.doi.org/10.1016/S0002-9394(14)74052-8]. [PMID: 1463043]. [DOI] [PubMed] [Google Scholar]
- 39.Joussen A.M., Poulaki V., Mitsiades N., Kirchhof B., Koizumi K., Döhmen S., Adamis A.P. Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-alpha suppression. FASEB J. 2002;16(3):438–440. doi: 10.1096/fj.01-0707fje. [PMID: 11821258]. [DOI] [PubMed] [Google Scholar]
- 40.Aveleira C.A., Lin C.M., Abcouwer S.F., Ambrósio A.F., Antonetti D.A. TNF-α signals through PKCζ/NF-κB to alter the tight junction complex and increase retinal endothelial cell permeability. Diabetes. 2010;59(11):2872–2882. doi: 10.2337/db09-1606. [http://dx.doi.org/10.2337/db09-1606]. [PMID: 20693346]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi H., Zhang Z., Wang X., Li R., Hou W., Bi W., Zhang X. Inhibition of autophagy induces IL-1β release from ARPE-19 cells via ROS mediated NLRP3 inflammasome activation under high glucose stress. Biochem. Biophys. Res. Commun. 2015;463(4):1071–1076. doi: 10.1016/j.bbrc.2015.06.060. [http://dx.doi.org/10.1016/j.bbrc.2015.06.060]. [PMID: 26102024]. [DOI] [PubMed] [Google Scholar]
- 42.Krady J.K., Basu A., Allen C.M., Xu Y., LaNoue K.F., Gardner T.W., Levison S.W. Minocycline reduces proinflammatory cytokine expression, microglial activation, and caspase-3 activation in a rodent model of diabetic retinopathy. Diabetes. 2005;54(5):1559–1565. doi: 10.2337/diabetes.54.5.1559. [http://dx.doi.org/10.2337/diabetes.54.5.1559]. [PMID: 15855346]. [DOI] [PubMed] [Google Scholar]
- 43.Perez V.L., Caspi R.R. Immune mechanisms in inflammatory and degenerative eye disease. Trends Immunol. 2015;36(6):354–363. doi: 10.1016/j.it.2015.04.003. [http://dx.doi.org/10.1016/j.it.2015.04.003]. [PMID: 25981967]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vinores S.A., Xiao W.H., Shen J., Campochiaro P.A. TNF-α is critical for ischemia-induced leukostasis, but not retinal neovas- cularization nor VEGF-induced leakage. J. Neuroimmunol. 2007;182(1-2):73–79. doi: 10.1016/j.jneuroim.2006.09.015. [http://dx.doi.org/10.1016/j.jneuroim.2006.09.015]. [PMID: 17107717]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abiko T., Abiko A., Clermont A.C., Shoelson B., Horio N., Takahashi J., Adamis A.P., King G.L., Bursell S.E. Characterization of retinal leukostasis and hemodynamics in insulin resistance and diabetes: role of oxidants and protein kinase-C activation. Diabetes. 2003;52(3):829–837. doi: 10.2337/diabetes.52.3.829. [http://dx.doi.org/10. 2337/diabetes.52.3.829]. [PMID: 12606527]. [DOI] [PubMed] [Google Scholar]
- 46.Ishida S., Usui T., Yamashiro K., Kaji Y., Amano S., Ogura Y., Hida T., Oguchi Y., Ambati J., Miller J.W., Gragoudas E.S., Ng Y.S., D’Amore P.A., Shima D.T., Adamis A.P. VEGF164-mediated inflammation is required for pathological, but not physiological, ischemia-induced retinal neovascularization. J. Exp. Med. 2003;198(3):483–489. doi: 10.1084/jem.20022027. [http://dx.doi.org/10.1084/jem. 20022027]. [PMID: 12900522]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Usui T., Ishida S., Yamashiro K., Kaji Y., Poulaki V., Moore J., Moore T., Amano S., Horikawa Y., Dartt D., Golding M., Shima D.T., Adamis A.P. VEGF164(165) as the pathological isoform: differential leukocyte and endothelial responses through VEGFR1 and VEGFR2. Invest. Ophthalmol. Vis. Sci. 2004;45(2):368–374. doi: 10.1167/iovs.03-0106. [http://dx.doi.org/10.1167/iovs.03-0106]. [PMID: 14744874]. [DOI] [PubMed] [Google Scholar]
- 48.Giacco F., Brownlee M. Oxidative stress and diabetic complications. Circ. Res. 2010;107(9):1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [http://dx.doi.org/10.1161/ CIRCRESAHA.110.223545]. [PMID: 21030723]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Obrosova I.G., Minchenko A.G., Frank R.N., Seigel G.M., Zsengeller Z., Pacher P., Stevens M.J., Szabó C. Poly(ADP-ribose) polymerase inhibitors counteract diabetes- and hypoxia-induced retinal vascular endothelial growth factor overexpression. Int. J. Mol. Med. 2004;14(1):55–64. [PMID: 15202016]. [PubMed] [Google Scholar]
- 50.Fu D., Wu M., Zhang J., Du M., Yang S., Hammad S.M., Wilson K., Chen J., Lyons T.J. Mechanisms of modified LDL-induced pericyte loss and retinal injury in diabetic retinopathy. Diabetologia. 2012;55(11):3128–3140. doi: 10.1007/s00125-012-2692-0. [http://dx.doi.org/10.1007/ s00125-012-2692-0]. [PMID: 22935961]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qin Y., Zhu M., Qu X., Xu G., Yu Y., Witt R.E., Wang W. Regional macular light sensitivity changes in myopic Chinese adults: an MP1 study. Invest. Ophthalmol. Vis. Sci. 2010;51(9):4451–4457. doi: 10.1167/iovs.09-4642. [http://dx.doi.org/10.1167/iovs.09-4642]. [PMID: 20357202]. [DOI] [PubMed] [Google Scholar]
- 52.Li B., Wang H.S., Li G.G., Zhao M.J., Zhao M.H. The role of endoplasmic reticulum stress in the early stage of diabetic retinopathy. Acta Diabetol. 2011;48(2):103–111. doi: 10.1007/s00592-009-0170-z. [http://dx.doi.org/10.1007/s00592-009-0170-z]. [PMID: 19924374]. [DOI] [PubMed] [Google Scholar]
- 53.Ito C.Y., Kazantsev A.G., Baldwin A.S., Jr Three N.F. -κ B sites in the I κ B-α promoter are required for induction of gene expression by TNF α. Nucleic Acids Res. 1994;22(18):3787–3792. doi: 10.1093/nar/22.18.3787. [http://dx.doi.org/10.1093/nar/22.18.3787]. [PMID: 7937093]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zheng L., Szabó C., Kern T.S. Poly(ADP-ribose) polymerase is involved in the development of diabetic retinopathy via regulation of nuclear factor-kappaB. Diabetes. 2004;53(11):2960–2967. doi: 10.2337/diabetes.53.11.2960. [http://dx.doi.org/10.2337/diabetes.53.11.2960]. [PMID: 15504977]. [DOI] [PubMed] [Google Scholar]
- 55.Romeo G., Liu W.H., Asnaghi V., Kern T.S., Lorenzi M. Activation of nuclear factor-kappaB induced by diabetes and high glucose regulates a proapoptotic program in retinal pericytes. Diabetes. 2002;51(7):2241–2248. doi: 10.2337/diabetes.51.7.2241. [http://dx.doi.org/10.2337/ diabetes.51.7.2241]. [PMID: 12086956]. [DOI] [PubMed] [Google Scholar]
- 56.Robison W.G., Jr Prevention of diabetes-related retinal microangiopathy with aldose reductase inhibitors. Adv. Exp. Med. Biol. 1988;246:365–372. doi: 10.1007/978-1-4684-5616-5_45. [http://dx.doi.org/10.1007/978-1-4684-5616-5_45]. [PMID: 3150650]. [DOI] [PubMed] [Google Scholar]
- 57.Ryu J.M., Lee M.Y., Yun S.P., Han H.J. High glucose regulates cyclin D1/E of human mesenchymal stem cells through TGF-β1 expression via Ca2+/PKC/MAPKs and PI3K/Akt/mTOR signal pathways. J. Cell. Physiol. 2010;224(1):59–70. doi: 10.1002/jcp.22091. [PMID: 20232305]. [DOI] [PubMed] [Google Scholar]
- 58.D’Amore P.A. Mechanisms of retinal and choroidal neovas- cularization. Invest. Ophthalmol. Vis. Sci. 1994;35(12):3974–3979. [PMID: 7525506]. [PubMed] [Google Scholar]
- 59.Shih S.C., Ju M., Liu N., Mo J.R., Ney J.J., Smith L.E. Transforming growth factor β1 induction of vascular endothelial growth factor receptor 1: mechanism of pericyte-induced vascular survival in vivo. Proc. Natl. Acad. Sci. USA. 2003;100(26):15859–15864. doi: 10.1073/pnas.2136855100. [http://dx.doi.org/10.1073/pnas.2136855100]. [PMID: 14657382]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414(6865):813–820. doi: 10.1038/414813a. [http://dx.doi.org/10.1038/414813a]. [PMID: 11742414]. [DOI] [PubMed] [Google Scholar]
- 61.Moscat J., Diaz-Meco M.T. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137(6):1001–1004. doi: 10.1016/j.cell.2009.05.023. [http://dx.doi.org/10.1016/j.cell.2009.05.023]. [PMID: 19524504]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rusten T.E., Stenmark H. p62, an autophagy hero or culprit? Nat. Cell Biol. 2010;12(3):207–209. doi: 10.1038/ncb0310-207. [http://dx.doi.org/10.1038/ ncb0310-207]. [PMID: 20190829]. [DOI] [PubMed] [Google Scholar]
- 63.Mathew R., Karp C.M., Beaudoin B., Vuong N., Chen G., Chen H.Y., Bray K., Reddy A., Bhanot G., Gelinas C., Dipaola R.S., Karantza-Wadsworth V., White E. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137(6):1062–1075. doi: 10.1016/j.cell.2009.03.048. [http://dx.doi.org/10.1016/j.cell.2009.03.048]. [PMID: 19524509]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maiuri M.C., Zalckvar E., Kimchi A., Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007;8(9):741–752. doi: 10.1038/nrm2239. [http://dx.doi.org/10. 1038/nrm2239]. [PMID: 17717517]. [DOI] [PubMed] [Google Scholar]
- 65.Eisenberg-Lerner A., Bialik S., Simon H.U., Kimchi A. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009;16(7):966–975. doi: 10.1038/cdd.2009.33. [http://dx.doi.org/10.1038/cdd.2009.33]. [PMID: 19325568]. [DOI] [PubMed] [Google Scholar]
- 66.Høyer-Hansen M., Bastholm L., Szyniarowski P., Campanella M., Szabadkai G., Farkas T., Bianchi K., Fehrenbacher N., Elling F., Rizzuto R., Mathiasen I.S., Jäättelä M. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol. Cell. 2007;25(2):193–205. doi: 10.1016/j.molcel.2006.12.009. [http://dx.doi.org/10.1016/j.molcel.2006.12.009]. [PMID: 17244528]. [DOI] [PubMed] [Google Scholar]
- 67.Kunchithapautham K., Rohrer B. Apoptosis and autophagy in photoreceptors exposed to oxidative stress. Autophagy. 2007;3(5):433–441. doi: 10.4161/auto.4294. [http://dx.doi.org/10.4161/auto.4294]. [PMID: 17471016]. [DOI] [PubMed] [Google Scholar]
- 68.Guillot-Sestier M.V., Sunyach C., Druon C., Scarzello S., Checler F. The alpha-secretase-derived N-terminal product of cellular prion, N1, displays neuroprotective function in vitro and in vivo. J. Biol. Chem. 2009;284(51):35973–35986. doi: 10.1074/jbc.M109.051086. [http://dx.doi.org/10.1074/jbc.M109.051086]. [PMID: 19850936]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Russo R., Berliocchi L., Adornetto A., Varano G.P., Cavaliere F., Nucci C., Rotiroti D., Morrone L.A., Bagetta G., Corasaniti M.T. Calpain-mediated cleavage of Beclin-1 and autophagy deregulation following retinal ischemic injury in vivo. Cell Death Dis. 2011;2:e144. doi: 10.1038/cddis.2011.29. [http://dx.doi.org/10.1038/cddis.2011.29]. [PMID: 21490676]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Balmer D., Emery M., Andreux P., Auwerx J., Ginet V., Puyal J., Schorderet D.F., Roduit R. Autophagy defect is associated with low glucose-induced apoptosis in 661W photoreceptor cells. PLoS One. 2013;8(9):e74162. doi: 10.1371/journal.pone.0074162. [http://dx.doi.org/10.1371/journal.pone. 0074162]. [PMID: 24066113]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Levine B., Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. doi: 10.1016/j.cell.2007.12.018. [http://dx.doi.org/10.1016/j.cell.2007. 12.018]. [PMID: 18191218]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Emery M., Schorderet D.F., Roduit R. Acute hypoglycemia induces retinal cell death in mouse. PLoS One. 2011;6(6):e21586. doi: 10.1371/journal.pone.0021586. [http://dx.doi.org/10.1371/journal.pone.0021586]. [PMID: 21738719]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Balmer D., Emery M., Andreux P., Auwerx J., Ginet V., Puyal J., Schorderet D.F., Roduit R. Autophagy defect is associated with low glucose-induced apoptosis in 661W photoreceptor cells. PLoS One. 2013;8(9):e74162. doi: 10.1371/journal.pone.0074162. [http://dx.doi.org/10.1371/journal.pone. 0074162]. [PMID: 24066113]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cuervo A.M., Dice J.F. Regulation of lamp2a levels in the lysosomal membrane. Traffic. 2000;1(7):570–583. doi: 10.1034/j.1600-0854.2000.010707.x. [http://dx.doi.org/10.1034/j.1600-0854.2000.010707.x]. [PMID: 11208145]. [DOI] [PubMed] [Google Scholar]
- 75.Saftig P., Beertsen W., Eskelinen E-L. LAMP-2: a control step for phagosome and autophagosome maturation. Autophagy. 2008;4(4):510–512. doi: 10.4161/auto.5724. [http://dx.doi.org/10.4161/auto.5724]. [PMID: 18376150]. [DOI] [PubMed] [Google Scholar]
- 76.Tanaka Y., Guhde G., Suter A., Eskelinen E.L., Hartmann D., Lüllmann-Rauch R., Janssen P.M., Blanz J., von Figura K., Saftig P. Accumulation of autophagic vacuoles and cardio- myopathy in LAMP-2-deficient mice. Nature. 2000;406(6798):902–906. doi: 10.1038/35022595. [http://dx.doi.org/10.1038/35022595]. [PMID: 10972293]. [DOI] [PubMed] [Google Scholar]
- 77.Balmer D., Emery M., Andreux P., Auwerx J., Ginet V., Puyal J., Schorderet D.F., Roduit R. Autophagy defect is associated with low glucose-induced apoptosis in 661W photoreceptor cells. PLoS One. 2013;8(9):e74162. doi: 10.1371/journal.pone.0074162. [http://dx.doi.org/10.1371/journal.pone. 0074162]. [PMID: 24066113]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Thiadens A.A., Slingerland N.W., Florijn R.J., Visser G.H., Riemslag F.C., Klaver C.C. Cone-rod dystrophy can be a manifestation of Danon disease. Graefes Arch. Clin. Exp. Ophthalmol. 2012;250(5):769–774. doi: 10.1007/s00417-011-1857-8. [http://dx.doi.org/10.1007/ s00417-011-1857-8]. [PMID: 22290069]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang Z., Vatta M. Danon disease as a cause of autophagic vacuolar myopathy. Congenit. Heart Dis. 2007;2(6):404–409. doi: 10.1111/j.1747-0803.2007.00132.x. [http://dx.doi.org/10.1111/j.1747-0803.2007.00132.x]. [PMID: 18377432]. [DOI] [PubMed] [Google Scholar]
- 80.Schorderet D.F., Cottet S., Lobrinus J.A., Borruat F.X., Balmer A., Munier F.L. Retinopathy in Danon disease. Arch. Ophthalmol. 2007;125(2):231–236. doi: 10.1001/archopht.125.2.231. [http://dx.doi.org/10.1001/archopht.125.2.231]. [PMID: 17296900]. [DOI] [PubMed] [Google Scholar]
- 81.Zoncu R., Efeyan A., Sabatini D.M. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011;12(1):21–35. doi: 10.1038/nrm3025. [http://dx.doi.org/10.1038/nrm3025]. [PMID: 21157483]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Maiese K. Cutting through the complexities of mTOR for the treatment of stroke. Curr. Neurovasc. Res. 2014;11(2):177–186. doi: 10.2174/1567202611666140408104831. [http://dx.doi.org/10.2174/1567202611666140408104831]. [PMID: 24712647]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Neasta J., Barak S., Hamida S.B., Ron D. mTOR complex 1: a key player in neuroadaptations induced by drugs of abuse. J. Neurochem. 2014;130(2):172–184. doi: 10.1111/jnc.12725. [http://dx.doi.org/10.1111/jnc. 12725]. [PMID: 24666346]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Maiese K., Chong Z.Z., Shang Y.C., Wang S. mTOR: on target for novel therapeutic strategies in the nervous system. Trends Mol. Med. 2013;19(1):51–60. doi: 10.1016/j.molmed.2012.11.001. [http://dx.doi.org/10.1016/j.molmed. 2012.11.001]. [PMID: 23265840]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Laplante M., Sabatini D.M. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–293. doi: 10.1016/j.cell.2012.03.017. [http://dx.doi.org/10.1016/j.cell.2012.03.017]. [PMID: 22500797]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang G.B., Ni Y.L., Zhou X.P., Zhang W.F. The AKT/mTOR pathway mediates neuronal protective effects of erythropoietin in sepsis. Mol. Cell. Biochem. 2014;385(1-2):125–132. doi: 10.1007/s11010-013-1821-5. [http://dx. doi.org/10.1007/s11010-013-1821-5]. [PMID: 24057122]. [DOI] [PubMed] [Google Scholar]
- 87.Wang H., Zhang Q., Wen Q., Zheng Y., Lazarovici P., Jiang H., Lin J., Zheng W. Proline-rich Akt substrate of 40kDa (PRAS40): a novel downstream target of PI3k/Akt signaling pathway. Cell. Signal. 2012;24(1):17–24. doi: 10.1016/j.cellsig.2011.08.010. [http://dx.doi.org/10.1016/j.cellsig.2011.08.010]. [PMID: 21906675]. [DOI] [PubMed] [Google Scholar]
- 88.Chong Z.Z., Kang J.Q., Maiese K. Erythropoietin is a novel vascular protectant through activation of Akt1 and mitochondrial modulation of cysteine proteases. Circulation. 2002;106(23):2973–2979. doi: 10.1161/01.cir.0000039103.58920.1f. [http://dx.doi.org/10.1161/01.CIR.0000039103.58920. 1F]. [PMID: 12460881]. [DOI] [PubMed] [Google Scholar]
- 89.Shang Y.C., Chong Z.Z., Wang S., Maiese K. Prevention of β-amyloid degeneration of microglia by erythropoietin depends on Wnt1, the PI 3-K/mTOR pathway, Bad, and Bcl-xL. Aging (Albany, N.Y.) 2012;4(3):187–201. doi: 10.18632/aging.100440. [http://dx.doi.org/10.18632/ aging.100440]. [PMID: 22388478]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sanghera K.P., Mathalone N., Baigi R., Panov E., Wang D., Zhao X., Hsu H., Wang H., Tropepe V., Ward M., Boyd S.R. The PI3K/Akt/mTOR pathway mediates retinal progenitor cell survival under hypoxic and superoxide stress. Mol. Cell. Neurosci. 2011;47(2):145–153. doi: 10.1016/j.mcn.2011.03.010. [http://dx.doi.org/10.1016/j.mcn.2011.03.010]. [PMID: 21463685]. [DOI] [PubMed] [Google Scholar]
- 91.Huang Q., Sheibani N. High glucose promotes retinal endothelial cell migration through activation of Src, PI3K/Akt1/eNOS, and ERKs. Am. J. Physiol. Cell Physiol. 2008;295(6):C1647–C1657. doi: 10.1152/ajpcell.00322.2008. [http://dx.doi.org/10.1152/ajpcell.00322.2008]. [PMID: 18945941]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kim D.I., Lim S.K., Park M.J., Han H.J., Kim G.Y., Park S.H. The involvement of phosphatidylinositol 3-kinase /Akt signaling in high glucose-induced downregulation of GLUT-1 expression in ARPE cells. Life Sci. 2007;80(7):626–632. doi: 10.1016/j.lfs.2006.10.026. [http://dx.doi.org/10.1016/j.lfs.2006.10.026]. [PMID: 17141276]. [DOI] [PubMed] [Google Scholar]
- 93.Adini I., Rabinovitz I., Sun J.F., Prendergast G.C., Benjamin L.E. RhoB controls Akt trafficking and stage-specific survival of endothelial cells during vascular development. Genes Dev. 2003;17(21):2721–2732. doi: 10.1101/gad.1134603. [http://dx.doi.org/10.1101/gad.1134603]. [PMID: 14597666]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang B., Atherton P., Patel R., Manning G., Donnelly R. Antiangiogenic effects and transcriptional regulation of pigment epithelium-derived factor in diabetic retinopathy. Microvasc. Res. 2010;80(1):31–36. doi: 10.1016/j.mvr.2010.02.012. [http://dx.doi.org/10.1016/j.mvr.2010.02.012]. [PMID: 20219495]. [DOI] [PubMed] [Google Scholar]
- 95.Cai J., Ahmad S., Jiang W.G., Huang J., Kontos C.D., Boulton M., Ahmed A. Activation of vascular endothelial growth factor receptor-1 sustains angiogenesis and Bcl-2 expression via the phosphatidylinositol 3-kinase pathway in endothelial cells. Diabetes. 2003;52(12):2959–2968. doi: 10.2337/diabetes.52.12.2959. [http://dx.doi.org/10.2337/ diabetes.52.12.2959]. [PMID: 14633857]. [DOI] [PubMed] [Google Scholar]
- 96.Zhao C., Yasumura D., Li X., Matthes M., Lloyd M., Nielsen G., Ahern K., Snyder M., Bok D., Dunaief J.L., LaVail M.M., Vollrath D. mTOR-mediated dedifferentiation of the retinal pigment epithelium initiates photoreceptor degeneration in mice. J. Clin. Invest. 2011;121(1):369–383. doi: 10.1172/JCI44303. [http://dx.doi.org/10.1172/ JCI44303]. [PMID: 21135502]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shah O.J., Wang Z., Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr. Biol. 2004;14(18):1650–1656. doi: 10.1016/j.cub.2004.08.026. [http://dx.doi.org/10.1016/j.cub.2004.08.026]. [PMID: 15380067]. [DOI] [PubMed] [Google Scholar]
- 98.Barbour L.A., McCurdy C.E., Hernandez T.L., Friedman J.E. Chronically increased S6K1 is associated with impaired IRS1 signaling in skeletal muscle of GDM women with impaired glucose tolerance postpartum. J. Clin. Endocrinol. Metab. 2011;96(5):1431–1441. doi: 10.1210/jc.2010-2116. [http://dx.doi.org/10.1210/jc.2010-2116]. [PMID: 21289241]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hsu P.P., Kang S.A., Rameseder J., Zhang Y., Ottina K.A., Lim D., Peterson T.R., Choi Y., Gray N.S., Yaffe M.B., Marto J.A., Sabatini D.M. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science. 2011;332(6035):1317–1322. doi: 10.1126/science.1199498. [http://dx. doi.org/10.1126/science.1199498]. [PMID: 21659604]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yu Y., Yoon S.O., Poulogiannis G., Yang Q., Ma X.M., Villén J., Kubica N., Hoffman G.R., Cantley L.C., Gygi S.P., Blenis J. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science. 2011;332(6035):1322–1326. doi: 10.1126/science.1199484. [http://dx.doi.org/10.1126/science.1199484]. [PMID: 21659605]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tremblay F., Brûlé S., Hee Um S., Li Y., Masuda K., Roden M., Sun X.J., Krebs M., Polakiewicz R.D., Thomas G., Marette A. Identification of IRS-1 Ser-1101 as a target of S6K1 in nutrient- and obesity-induced insulin resistance. Proc. Natl. Acad. Sci. USA. 2007;104(35):14056–14061. doi: 10.1073/pnas.0706517104. [http://dx.doi.org/10.1073/pnas. 0706517104]. [PMID: 17709744]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tremblay F., Krebs M., Dombrowski L., Brehm A., Bernroider E., Roth E., Nowotny P., Waldhäusl W., Marette A., Roden M. Overactivation of S6 kinase 1 as a cause of human insulin resistance during increased amino acid availability. Diabetes. 2005;54(9):2674–2684. doi: 10.2337/diabetes.54.9.2674. [http://dx.doi.org/10.2337/diabetes.54.9.2674]. [PMID: 16123357]. [DOI] [PubMed] [Google Scholar]
- 103.Krebs M., Brunmair B., Brehm A., Artwohl M., Szendroedi J., Nowotny P., Roth E., Fürnsinn C., Promintzer M., Anderwald C., Bischof M., Roden M. The Mammalian target of rapamycin pathway regulates nutrient-sensitive glucose uptake in man. Diabetes. 2007;56(6):1600–1607. doi: 10.2337/db06-1016. [http://dx.doi.org/10.2337/db06-1016]. [PMID: 17329620]. [DOI] [PubMed] [Google Scholar]
- 104.Kennedy A., Frank R.N. The influence of glucose concentration and hypoxia on VEGF secretion by cultured retinal cells. Curr. Eye Res. 2011;36(2):168–177. doi: 10.3109/02713683.2010.521968. [http://dx.doi.org/10.3109/02713683. 2010.521968]. [PMID: 21158590]. [DOI] [PubMed] [Google Scholar]
- 105.Forsythe J.A., Jiang B.H., Iyer N.V., Agani F., Leung S.W., Koos R.D., Semenza G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996;16(9):4604–4613. doi: 10.1128/mcb.16.9.4604. [http://dx.doi.org/10. 1128/MCB.16.9.4604]. [PMID: 8756616]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Vinores S.A., Xiao W.H., Aslam S., Shen J., Oshima Y., Nambu H., Liu H., Carmeliet P., Campochiaro P.A. Implication of the hypoxia response element of the Vegf promoter in mouse models of retinal and choroidal neovascularization, but not retinal vascular development. J. Cell. Physiol. 2006;206(3):749–758. doi: 10.1002/jcp.20525. [http://dx.doi.org/10.1002/jcp.20525]. [PMID: 16245301]. [DOI] [PubMed] [Google Scholar]
- 107.Poulaki V., Joussen A.M., Mitsiades N., Mitsiades C.S., Iliaki E.F., Adamis A.P. Insulin-like growth factor-I plays a patho- genetic role in diabetic retinopathy. Am. J. Pathol. 2004;165(2):457–469. doi: 10.1016/S0002-9440(10)63311-1. [http://dx.doi.org/10.1016/S0002-9440(10)63311-1]. [PMID: 15277220]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Demidenko Z.N., Blagosklonny M.V. The purpose of the HIF-1/PHD feedback loop: to limit mTOR-induced HIF-1α. Cell Cycle. 2011;10(10):1557–1562. doi: 10.4161/cc.10.10.15789. [http://dx.doi.org/10.4161/cc.10.10.15789]. [PMID: 21521942]. [DOI] [PubMed] [Google Scholar]
- 109.Poulaki V., Qin W., Joussen A.M., Hurlbut P., Wiegand S.J., Rudge J., Yancopoulos G.D., Adamis A.P. Acute intensive insulin therapy exacerbates diabetic blood-retinal barrier breakdown via hypoxia-inducible factor-1alpha and VEGF. J. Clin. Invest. 2002;109(6):805–815. doi: 10.1172/JCI13776. [http://dx.doi.org/10.1172/JCI0213776]. [PMID: 11901189]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Aiello L.P., Avery R.L., Arrigg P.G., Keyt B.A., Jampel H.D., Shah S.T., Pasquale L.R., Thieme H., Iwamoto M.A., Park J.E. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N. Engl. J. Med. 1994;331(22):1480–1487. doi: 10.1056/NEJM199412013312203. [http://dx.doi.org/10. 1056/NEJM199412013312203]. [PMID: 7526212]. [DOI] [PubMed] [Google Scholar]
- 111.Chantelau E., Meyer-Schwickerath R., Klabe K. Downregulation of serum IGF-1 for treatment of early worsening of diabetic retinopathy: a long-term follow-up of two cases. Ophthalmologica. 2010;224(4):243–246. doi: 10.1159/000260231. [http://dx.doi.org/10.1159/000260231]. [PMID: 19940532]. [DOI] [PubMed] [Google Scholar]
- 112.Smith L.E., Shen W., Perruzzi C., Soker S., Kinose F., Xu X., Robinson G., Driver S., Bischoff J., Zhang B., Schaeffer J.M., Senger D.R. Regulation of vascular endothelial growth factor-dependent retinal neovascularization by insulin-like growth factor-1 receptor. Nat. Med. 1999;5(12):1390–1395. doi: 10.1038/70963. [http://dx.doi.org/10. 1038/70963]. [PMID: 10581081]. [DOI] [PubMed] [Google Scholar]
- 113.Lu M., Amano S., Miyamoto K., Garland R., Keough K., Qin W., Adamis A.P. Insulin-induced vascular endothelial growth factor expression in retina. Invest. Ophthalmol. Vis. Sci. 1999;40(13):3281–3286. [PMID: 10586954]. [PubMed] [Google Scholar]
- 114.Chantelau E., Frystyk J. Progression of diabetic retinopathy during improved metabolic control may be treated with reduced insulin dosage and/or somatostatin analogue administration -- a case report. Growth Horm. IGF Res. 2005;15(2):130–135. doi: 10.1016/j.ghir.2004.12.005. [http://dx.doi.org/10.1016/j.ghir.2004.12.005]. [PMID: 15809016]. [DOI] [PubMed] [Google Scholar]
- 115.Demidenko Z.N., Zubova S.G., Bukreeva E.I., Pospelov V.A., Pospelova T.V., Blagosklonny M.V. Rapamycin decelerates cellular senescence. Cell Cycle. 2009;8(12):1888–1895. doi: 10.4161/cc.8.12.8606. [http://dx.doi.org/10.4161/cc.8.12.8606]. [PMID: 19471117]. [DOI] [PubMed] [Google Scholar]
- 116.Dejneka N.S., Kuroki A.M., Fosnot J., Tang W., Tolentino M.J., Bennett J. Systemic rapamycin inhibits retinal and choroidal neovascularization in mice. Mol. Vis. 2004;10:964–972. [PMID: 15623986]. [PubMed] [Google Scholar]
- 117.Treins C., Giorgetti-Peraldi S., Murdaca J., Semenza G.L., Van Obberghen E. Insulin stimulates hypoxia-inducible factor 1 through a phosphatidylinositol 3-kinase/target of rapamycin-dependent signaling pathway. J. Biol. Chem. 2002;277(31):27975–27981. doi: 10.1074/jbc.M204152200. [http://dx.doi.org/10.1074/jbc.M204152200]. [PMID: 12032158]. [DOI] [PubMed] [Google Scholar]
- 118.Humar R., Kiefer F.N., Berns H., Resink T.J., Battegay E.J. Hypoxia enhances vascular cell proliferation and angiogenesis in vitro via rapamycin (mTOR)-dependent signaling. FASEB J. 2002;16(8):771–780. doi: 10.1096/fj.01-0658com. [http://dx.doi.org/10.1096/fj.01-0658com]. [PMID: 12039858]. [DOI] [PubMed] [Google Scholar]
- 119.Treins C., Giorgetti-Peraldi S., Murdaca J., Monthouël-Kartmann M.N., Van Obberghen E. Regulation of hypoxia-inducible factor (HIF)-1 activity and expression of HIF hydroxylases in response to insulin-like growth factor I. Mol. Endocrinol. 2005;19(5):1304–1317. doi: 10.1210/me.2004-0239. [http://dx.doi.org/10.1210/me.2004-0239]. [PMID: 15695372]. [DOI] [PubMed] [Google Scholar]
- 120.Ren H., Accili D., Duan C. Hypoxia converts the myogenic action of insulin-like growth factors into mitogenic action by differentially regulating multiple signaling pathways. Proc. Natl. Acad. Sci. USA. 2010;107(13):5857–5862. doi: 10.1073/pnas.0909570107. [http://dx.doi.org/10. 1073/pnas.0909570107]. [PMID: 20231451]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Liu Q., Xu Z., Mao S., Chen W., Zeng R., Zhou S., Liu J. Effect of hypoxia on hypoxia inducible factor-1α, insulin-like growth factor I and vascular endothelial growth factor expression in hepatocellular carcinoma HepG2 cells. Oncol. Lett. 2015;9(3):1142–1148. doi: 10.3892/ol.2015.2879. [PMID: 25663870]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Slomiany M.G., Rosenzweig S.A. Hypoxia-inducible factor-1-dependent and -independent regulation of insulin-like growth factor-1-stimulated vascular endothelial growth factor secretion. J. Pharmacol. Exp. Ther. 2006;318(2):666–675. doi: 10.1124/jpet.106.104158. [http://dx.doi.org/10.1124/jpet.106.104158]. [PMID: 16682453]. [DOI] [PubMed] [Google Scholar]
- 123.Treins C., Murdaca J., Obberghen E.V. Giorgetti- Peraldi, S. AMPK activation inhibits the expression of HIF-1a induced by insulin and IGF-1. Biochem. Biophys. Res. Commun. 2006;342:1197–1202. doi: 10.1016/j.bbrc.2006.02.088. [http://dx.doi.org/10.1016/j.bbrc.2006.02.088]. [PMID: 16516166]. [DOI] [PubMed] [Google Scholar]
- 124.Ruberte J., Ayuso E., Navarro M., Carretero A., Nacher V., Haurigot V., George M., Llombart C., Casellas A., Costa C., Bosch A., Bosch F. Increased ocular levels of IGF-1 in transgenic mice lead to diabetes-like eye disease. J. Clin. Invest. 2004;113(8):1149–1157. doi: 10.1172/JCI19478. [http://dx.doi.org/10.1172/JCI19478]. [PMID: 15085194]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Li J., Zhao S.Z., Wang P.P., Yu S.P., Zheng Z., Xu X. Calcium mediates high glucose-induced HIF-1α and VEGF expression in cultured rat retinal Müller cells through CaMKII-CREB pathway. Acta Pharmacol. Sin. 2012;33(8):1030–1036. doi: 10.1038/aps.2012.61. [http://dx.doi.org/10.1038/aps.2012.61]. [PMID: 22796763]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Scheppke L., Aguilar E., Gariano R.F., Jacobson R., Hood J., Doukas J., Cao J., Noronha G., Yee S., Weis S., Martin M.B., Soll R., Cheresh D.A., Friedlander M. Retinal vascular permeability suppression by topical application of a novel VEGFR2/Src kinase inhibitor in mice and rabbits. J. Clin. Invest. 2008;118(6):2337–2346. doi: 10.1172/JCI33361. [PMID: 18483622]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Harhaj N.S., Felinski E.A., Wolpert E.B., Sundstrom J.M., Gardner T.W., Antonetti D.A. VEGF activation of protein kinase C stimulates occludin phosphorylation and contributes to endothelial permeability. Invest. Ophthalmol. Vis. Sci. 2006;47(11):5106–5115. doi: 10.1167/iovs.06-0322. [http://dx.doi.org/10.1167/iovs.06-0322]. [PMID: 17065532]. [DOI] [PubMed] [Google Scholar]
- 128.Saishin Y., Saishin Y., Takahashi K., Takahashi K., Lima Silva R., Hylton D, Rudge J. S., Wiegand S. J. Campochiaro, P. A. VEGF-TRAPR1R2 suppresses choroidal neovascularization and VEGF-induced breakdown of the blood-retinal barrier. J. Cell. Physiol. 2003;195:241–248. doi: 10.1002/jcp.10246. [http://dx.doi.org/10.1002/jcp.10246]. [PMID: 12652651]. [DOI] [PubMed] [Google Scholar]
- 129.Mathews M.K., Merges C., McLeod D.S., Lutty G.A. Vascular endothelial growth factor and vascular permeability changes in human diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 1997;38(13):2729–2741. [PMID: 9418725]. [PubMed] [Google Scholar]
- 130.Humar R., Kiefer F.N., Berns H., Resink T.J., Battegay E.J. Hypoxia enhances vascular cell proliferation and angiogenesis in vitro via rapamycin (mTOR)-dependent signaling. FASEB J. 2002;16(8):771–780. doi: 10.1096/fj.01-0658com. [http://dx.doi.org/10.1096/fj.01-0658com]. [PMID: 12039858]. [DOI] [PubMed] [Google Scholar]
- 131.Jacot J.L., Sherris D. Potential Therapeutic Roles for Inhibition of the PI3K/Akt/mTOR Pathway in the Pathophysiology of Diabetic Retinopathy. J. Ophthalmol. 2011 doi: 10.1155/2011/589813. 2011, 589813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Laplante M., Sabatini D.M. Regulation of mTORC1 and its impact on gene expression at a glance. J. Cell Sci. 2013;126(Pt 8):1713–1719. doi: 10.1242/jcs.125773. [http://dx.doi.org/10.1242/jcs.125773]. [PMID: 23641065]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Blommaart E.F., Luiken J.J., Blommaart P.J., van Woerkom G.M., Meijer A.J. Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J. Biol. Chem. 1995;270(5):2320–2326. doi: 10.1074/jbc.270.5.2320. [http://dx.doi.org/10.1074/jbc.270.5.2320]. [PMID: 7836465]. [DOI] [PubMed] [Google Scholar]
- 134.Bain J., Plater L., Elliott M., Shpiro N., Hastie C.J., McLauchlan H., Klevernic I., Arthur J.S., Alessi D.R., Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem. J. 2007;408(3):297–315. doi: 10.1042/BJ20070797. [http://dx.doi.org/10.1042/ BJ20070797]. [PMID: 17850214]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Horton J.D., Goldstein J.L., Brown M.S. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 2002;109(9):1125–1131. doi: 10.1172/JCI15593. [http://dx.doi.org/10.1172/JCI0215593]. [PMID: 11994399]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Owen J.L., Zhang Y., Bae S.H., Farooqi M.S., Liang G., Hammer R.E., Goldstein J.L., Brown M.S. Insulin stimulation of SREBP-1c processing in transgenic rat hepatocytes requires p70 S6-kinase. Proc. Natl. Acad. Sci. USA. 2012;109(40):16184–16189. doi: 10.1073/pnas.1213343109. [http://dx.doi.org/10.1073/pnas.1213343109]. [PMID: 22927400]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Peterson T.R., Sengupta S.S., Harris T.E., Carmack A.E., Kang S.A., Balderas E., Guertin D.A., Madden K.L., Carpenter A.E., Finck B.N., Sabatini D.M. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146(3):408–420. doi: 10.1016/j.cell.2011.06.034. [http://dx.doi.org/10.1016/j.cell.2011.06.034]. [PMID: 21816276]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rosen E.D., MacDougald O.A. Adipocyte differentiation from the inside out. Nat. Rev. Mol. Cell Biol. 2006;7(12):885–896. doi: 10.1038/nrm2066. [http://dx.doi.org/10.1038/nrm2066]. [PMID: 17139329]. [DOI] [PubMed] [Google Scholar]
- 139.Le Bacquer O., Petroulakis E., Paglialunga S., Poulin F., Richard D., Cianflone K., Sonenberg N. Elevated sensitivity to diet-induced obesity and insulin resistance in mice lacking 4E-BP1 and 4E-BP2. J. Clin. Invest. 2007;117(2):387–396. doi: 10.1172/JCI29528. [http://dx.doi.org/10.1172/JCI29528]. [PMID: 17273556]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kim J.E., Chen J. regulation of peroxisome proliferator-activated receptor-gamma activity by mammalian target of rapamycin and amino acids in adipogenesis. Diabetes. 2004;53(11):2748–2756. doi: 10.2337/diabetes.53.11.2748. [http://dx.doi.org/10.2337/diabetes.53.11.2748]. [PMID: 15504954]. [DOI] [PubMed] [Google Scholar]
- 141.Kim J.B., Wright H.M., Wright M., Spiegelman B.M. ADD1/ SREBP1 activates PPARgamma through the production of endogenous ligand. Proc. Natl. Acad. Sci. USA. 1998;95(8):4333–4337. doi: 10.1073/pnas.95.8.4333. [http://dx.doi.org/10.1073/pnas.95.8.4333]. [PMID: 9539737]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Singh R., Cuervo A.M. Autophagy in the cellular energetic balance. Cell Metab. 2011;13(5):495–504. doi: 10.1016/j.cmet.2011.04.004. [http://dx.doi.org/10. 1016/j.cmet.2011.04.004]. [PMID: 21531332]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Rubinsztein D.C., Mariño G., Kroemer G. Autophagy and aging. Cell. 2011;146(5):682–695. doi: 10.1016/j.cell.2011.07.030. [http://dx.doi.org/10.1016/j.cell.2011. 07.030]. [PMID: 21884931]. [DOI] [PubMed] [Google Scholar]
- 144.Palmieri M., Impey S., Kang H., di Ronza A., Pelz C., Sardiello M., Ballabio A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011;20(19):3852–3866. doi: 10.1093/hmg/ddr306. [http://dx.doi.org/10.1093/hmg/ddr306]. [PMID: 21752829]. [DOI] [PubMed] [Google Scholar]
- 145.Settembre C., Di Malta C., Polito V.A., Garcia Arencibia M., Vetrini F., Erdin S., Erdin S.U., Huynh T., Medina D., Colella P., Sardiello M., Rubinsztein D.C., Ballabio A. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332(6036):1429–1433. doi: 10.1126/science.1204592. [http://dx.doi.org/10.1126/science.1204592]. [PMID: 21617040]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Settembre C., Zoncu R., Medina D.L., Vetrini F., Erdin S., Erdin S., Huynh T., Ferron M., Karsenty G., Vellard M.C., Facchinetti V., Sabatini D.M., Ballabio A. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31(5):1095–1108. doi: 10.1038/emboj.2012.32. [http://dx.doi.org/10.1038/emboj.2012.32]. [PMID: 22343943]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sarkar S., Rubinsztein D.C. Small molecule enhancers of autophagy for neurodegenerative diseases. Mol. Biosyst. 2008;4(9):895–901. doi: 10.1039/b804606a. [http://dx.doi.org/10.1039/b804606a]. [PMID: 18704227]. [DOI] [PubMed] [Google Scholar]
- 148.Ozcan U., Yilmaz E., Ozcan L., Furuhashi M., Vaillancourt E., Smith R.O., Görgün C.Z., Hotamisligil G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313(5790):1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ozcan U., Cao Q., Yilmaz E., Lee A.H., Iwakoshi N.N., Ozdelen E., Tuncman G., Görgün C., Glimcher L.H., Hotamisligil G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306(5695):457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 150.Hotamisligil G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140(6):900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ron D., Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007;8(7):519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 152.Lee A.H., Iwakoshi N.N., Glimcher L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 2003;23(21):7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ryoo H.D., Domingos P.M., Kang M.J., Steller H. Unfolded protein response in a Drosophila model for retinal degeneration. EMBO J. 2007;26(1):242–252. doi: 10.1038/sj.emboj.7601477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Li J., Wang J.J., Yu Q., Wang M., Zhang S.X. Endoplasmic reticulum stress is implicated in retinal inflammation and diabetic retinopathy. FEBS Lett. 2009;583(9):1521–1527. doi: 10.1016/j.febslet.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Zhong Y., Li J., Wang J.J., Chen C., Tran J.T., Saadi A., Yu Q., Le Y.Z., Mandal M.N., Anderson R.E., Zhang S.X. X-box binding protein 1 is essential for the anti-oxidant defense and cell survival in the retinal pigment epithelium. PLoS One. 2012;7(6):e38616. doi: 10.1371/journal.pone.0038616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Crighton D., Wilkinson S., O’Prey J., Syed N., Smith P., Harrison P.R., Gasco M., Garrone O., Crook T., Ryan K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126(1):121–134. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 157.Kerley-Hamilton J.S., Pike A.M., Hutchinson J.A., Freemantle S.J., Spinella M.J. The direct p53 target gene, FLJ11259/DRAM, is a member of a novel family of transmembrane proteins. Biochim. Biophys. Acta. 2007;1769(4):209–219. doi: 10.1016/j.bbaexp.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Crighton D., Wilkinson S., Ryan K.M. DRAM links autophagy to p53 and programmed cell death. Autophagy. 2007;3(1):72–74. doi: 10.4161/auto.3438]. [DOI] [PubMed] [Google Scholar]
- 159.Lorin S., Borges A., Ribeiro Dos Santos L., Souquère S., Pierron G., Ryan K.M., Codogno P., Djavaheri-Mergny M. c-Jun NH2-terminal kinase activation is essential for DRAM-dependent induction of autophagy and apoptosis in 2-methoxyestradiol-treated Ewing sarcoma cells. Cancer Res. 2009;69(17):6924–6931. doi: 10.1158/0008-5472.CAN-09-1270. [DOI] [PubMed] [Google Scholar]
- 160.Lorin S., Borges A., Ribeiro Dos Santos L., Souquère S., Pierron G., Ryan K.M., Codogno P., Djavaheri-Mergny M. c-Jun NH2-terminal kinase activation is essential for DRAM-dependent induction of autophagy and apoptosis in 2-methoxyestradiol-treated Ewing sarcoma cells. Cancer Res. 2009;69(17):6924–6931. doi: 10.1158/0008-5472.CAN-09-1270. [DOI] [PubMed] [Google Scholar]
- 161.Lorin S., Pierron G., Ryan K.M., Codogno P., Djavaheri-Mergny M. Evidence for the interplay between JNK and p53-DRAM signalling pathways in the regulation of autophagy. Autophagy. 2010;6(1):153–154. doi: 10.4161/auto.6.1.10537. [DOI] [PubMed] [Google Scholar]
- 162.O’Prey J., Skommer J., Wilkinson S., Ryan K.M. Analysis of DRAM-related proteins reveals evolutionarily conserved and divergent roles in the control of autophagy. Cell Cycle. 2009;8(14):2260–2265. doi: 10.4161/cc.8.14.9050. [DOI] [PubMed] [Google Scholar]
- 163.Park S.M., Kim K., Lee E.J., Kim B.K., Lee T.J., Seo T., Jang I.S., Lee S.H., Kim S., Lee J.H., Park J. Reduced expression of DRAM2/TMEM77 in tumor cells interferes with cell death. Biochem. Biophys. Res. Commun. 2009;390(4):1340–1344. doi: 10.1016/j.bbrc.2009.10.149. [DOI] [PubMed] [Google Scholar]
- 164.Yoon J.H., Her S., Kim M., Jang I.S., Park J. The expression of damage-regulated autophagy modulator 2 (DRAM2) contributes to autophagy induction. Mol. Biol. Rep. 2012;39(2):1087–1093. doi: 10.1007/s11033-011-0835-x. [DOI] [PubMed] [Google Scholar]
- 165.Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. . EMBO. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Tanida I., Minematsu-Ikeguchi N., Ueno T., Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1(2):84–91. doi: 10.4161/auto.1.2.1697. [DOI] [PubMed] [Google Scholar]
- 167.Calkin A.C., Forbes J.M., Smith C.M., Lassila M., Cooper M.E., Jandeleit-Dahm K.A., Allen T.J. Rosiglitazone attenuates atherosclerosis in a model of insulin insufficiency independent of its metabolic effects. Arterioscler. Thromb. Vasc. Biol. 2005;25(9):1903–1909. doi: 10.1161/01.ATV.0000177813.99577.6b. [DOI] [PubMed] [Google Scholar]
- 168.Ji S., Kronenberg G., Balkaya M., Färber K., Gertz K., Kettenmann H., Endres M. Acute neuroprotection by pioglitazone after mild brain ischemia without effect on long-term outcome. Exp. Neurol. 2009;216(2):321–328. doi: 10.1016/j.expneurol.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 169.Zheng Z., Chen H., Wang H., Ke B., Zheng B., Li Q., Li P., Su L., Gu Q., Xu X. Improvement of retinal vascular injury in diabetic rats by statins is associated with the inhibition of mitochondrial reactive oxygen species pathway mediated by peroxisome proliferator-activated receptor gamma coactivator 1alpha. Diabetes. 2010;59(9):2315–2325. doi: 10.2337/db10-0638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Higuchi A., Ohashi K., Shibata R., Sono-Romanelli S., Walsh K., Ouchi N. Thiazolidinediones reduce pathological neovascularization in ischemic retina via an adiponectin-dependent mechanism. Arterioscler. Thromb. Vasc. Biol. 2010;30(1):46–53. doi: 10.1161/ATVBAHA.109.198465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Hatanaka H., Koizumi N., Okumura N., Kay E.P., Mizuhara E., Hamuro J., Kinoshita S. Epithelial-mesenchymal transition-like phenotypic changes of retinal pigment epithelium induced by TGF-β are prevented by PPAR-γ agonists. Invest. Ophthalmol. Vis. Sci. 2012;53(11):6955–6963. doi: 10.1167/iovs.12-10488. [DOI] [PubMed] [Google Scholar]
- 172.Li P., Xu X., Zheng Z., Zhu B., Shi Y., Liu K. Protective effects of rosiglitazone on retinal neuronal damage in diabetic rats. Curr. Eye Res. 2011;36(7):673–679. doi: 10.3109/02713683.2011.572220. [DOI] [PubMed] [Google Scholar]
- 173.Jiang Y., Thakran S., Bheemreddy R., Ye E.A., He H., Walker R.J., Steinle J.J. Pioglitazone normalizes insulin signaling in the diabetic rat retina through reduction in tumor necrosis factor α and suppressor of cytokine signaling 3. J. Biol. Chem. 2014;289(38):26395–26405. doi: 10.1074/jbc.M114.583880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Murata T., Hata Y., Ishibashi T., Kim S., Hsueh W.A., Murata T., Hata Y., Ishibashi T., Kim S., Hsueh W.A., Law R.E., Hinton D.R. Response of experimental retinal neovascularization to thiazolidinediones. Arch. Ophthalmol. 2001;119(5):709–717. doi: 10.1001/archopht.119.5.709. [DOI] [PubMed] [Google Scholar]
- 175.Shen L.Q., Child A., Weber G.M., Folkman J., Aiello L.P. Rosiglitazone and delayed onset of proliferative diabetic retinopathy. Arch. Ophthalmol. 2008;126(6):793–799. doi: 10.1001/archopht.126.6.793. [DOI] [PubMed] [Google Scholar]
- 176.Idris I., Warren G., Donnelly R. Association between thiazolidinedione treatment and risk of macular edema among patients with type 2 diabetes. Arch. Intern. Med. 2012;172(13):1005–1011. doi: 10.1001/archinternmed.2012.1938. [DOI] [PubMed] [Google Scholar]
- 177.Baba T., Shimada K., Neugebauer S., Yamada D., Hashimoto S., Watanabe T. The oral insulin sensitizer, thiazolidinedione, increases plasma vascular endothelial growth factor in type 2 diabetic patients. Diabetes Care. 2001;24(5):953–954. doi: 10.2337/diacare.24.5.953. [DOI] [PubMed] [Google Scholar]
- 178.Azar S., El-Mollayess G.M., Al Shaar L., Salti H.I., Bashshur Z.F. Impact of thiazolidinediones on macular thickness and volume in diabetic eyes. Can. J. Ophthalmol. 2013;48(4):312–316. doi: 10.1016/j.jcjo.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 179.Zhang S., Gu H., Hu N. Role of Peroxisome Proliferator-Activated Receptor γ in Ocular Disease. J. Ophthalmol. 2015 doi: 10.1155/2015/275435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Asgary S., Naderi G.A., Zadegan N.S., Vakili R. The inhibitory effects of pure flavonoids on in vitro protein glycosylation. J. Herb. Pharmacother. 2002;2(2):47–55. doi: 10.1080/J157v02n02_05. [DOI] [PubMed] [Google Scholar]
- 181.Rodrigues G.A., Maurier-Mah e.F., Shurland D. L., McLaughlin A., Luhrs K., Throo E., Delalonde-Delaunay L., Pallares D., Schweighoffer F., Donello J. Differential effects of PPAR gamma ligands on oxidative stress-induced death of retinal pigmented epithelial cells. Invest. Ophthalmol. Vis. Sci. 2011;52:890–903. doi: 10.1167/iovs.10-5715. [DOI] [PubMed] [Google Scholar]
- 182.Herm R.J. Age-related macular degeneration. N. Engl. J. Med. 2008;359(16):1735–1736. doi: 10.1056/NEJMc081470. [DOI] [PubMed] [Google Scholar]
- 183.Kowluru R.A., Kanwar M. Effects of curcumin on retinal oxidative stress and inflammation in diabetes. Nutr. Metab. (Lond) 2007;4:8. doi: 10.1186/1743-7075-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Keech A.C., Mitchell P., Summanen P.A., O’Day J., Davis T.M., Moffitt M.S., Taskinen M.R., Simes R.J., Tse D., Williamson E., Merrifield A., Laatikainen L.T., d’Emden M.C., Crimet D.C., O’Connell R.L., Colman P.G. Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): a randomised controlled trial. Lancet. 2007;370(9600):1687–1697. doi: 10.1016/S0140-6736(07)61607-9. [DOI] [PubMed] [Google Scholar]
- 185.Chew E.Y., Ambrosius W.T., Davis M.D., Danis R.P., Gangaputra S., Greven C.M., Hubbard L., Esser B.A., Lovato J.F., Perdue L.H., Goff D.C., Jr, Cushman W.C., Ginsberg H.N., Elam M.B., Genuth S., Gerstein H.C., Schubart U., Fine L.J. Effects of medical therapies on retinopathy progression in type 2 diabetes. N. Engl. J. Med. 2010;363(3):233–244. doi: 10.1056/NEJMoa1001288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Simó R., Hernández C. Advances in the medical treatment of diabetic retinopathy. Diabetes Care. 2009;32(8):1556–1562. doi: 10.2337/dc09-0565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Dejneka N.S., Kuroki A.M., Fosnot J., Tang W., Tolentino M.J., Bennett J. Systemic rapamycin inhibits retinal and choroidal neovascularization in mice. Mol. Vis. 2004;10:964–972. [PubMed] [Google Scholar]
- 188.Sánchez-Fructuoso A.I., Ruiz J.C., Pérez-Flores I., Gómez Alamillo C., Calvo Romero N., Arias M. Comparative analysis of adverse events requiring suspension of mTOR inhibitors: everolimus versus sirolimus. Transplant. Proc. 2010;42(8):3050–3052. doi: 10.1016/j.transproceed.2010.07.083. [DOI] [PubMed] [Google Scholar]
- 189.Sonis S., Treister N., Chawla S., Demetri G., Haluska F. Preliminary characterization of oral lesions associated with inhibitors of mammalian target of rapamycin in cancer patients. Cancer. 2010;116(1):210–215. doi: 10.1002/cncr.24696. [DOI] [PubMed] [Google Scholar]
- 190.Soefje S.A., Karnad A., Brenner A.J. Common toxicities of mammalian target of rapamycin inhibitors. Target. Oncol. 2011;6(2):125–129. doi: 10.1007/s11523-011-0174-9. [DOI] [PubMed] [Google Scholar]
- 191.Sofroniadou S., Goldsmith D. Mammalian target of rapamycin (mTOR) inhibitors: potential uses and a review of haematological adverse effects. Drug Saf. 2011;34(2):97–115. doi: 10.2165/11585040-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 192.Hainsworth J.D., Spigel D.R., Burris H.A., III, Waterhouse D., Clark B.L., Whorf R. Phase II trial of bevacizumab and everolimus in patients with advanced renal cell carcinoma. J. Clin. Oncol. 2010;28(13):2131–2136. doi: 10.1200/JCO.2009.26.3152. [DOI] [PubMed] [Google Scholar]
- 193.Boobes Y., Bernieh B., Saadi H., Raafat Al Hakim M., Abouchacra S. Gonadal dysfunction and infertility in kidney transplant patients receiving sirolimus. Int. Urol. Nephrol. 2010;42(2):493–498. doi: 10.1007/s11255-009-9644-8. [DOI] [PubMed] [Google Scholar]
- 194.Mustata G.T., Rosca M., Biemel K.M., Reihl O., Smith M.A., Viswanathan A., Strauch C., Du Y., Tang J., Kern T.S., Lederer M.O., Brownlee M., Weiss M.F., Monnier V.M. Paradoxical effects of green tea (Camellia sinensis) and antioxidant vitamins in diabetic rats: improved retinopathy and renal mitochondrial defects but deterioration of collagen matrix glycoxidation and cross-linking. Diabetes. 2005;54(2):517–526. doi: 10.2337/diabetes.54.2.517. [DOI] [PubMed] [Google Scholar]
- 195.Zhang B., Osborne N.N. Oxidative-induced retinal degeneration is attenuated by epigallocatechin gallate. Brain Res. 2006;1124(1):176–187. doi: 10.1016/j.brainres.2006.09.067. [DOI] [PubMed] [Google Scholar]
- 196.Koh P.O. Gingko biloba extract (EGb 761) prevents cerebral ischemia-induced p70S6 kinase and S6 phosphorylation. Am. J. Chin. Med. 2010;38(4):727–734. doi: 10.1142/S0192415X10008196. [DOI] [PubMed] [Google Scholar]
- 197.Dugas B., Charbonnier S., Baarine M., Ragot K., Delmas D., Ménétrier F., Lherminier J., Malvitte L., Khalfaoui T., Bron A., Creuzot-Garcher C., Latruffe N., Lizard G. Effects of oxysterols on cell viability, inflammatory cytokines, VEGF, and reactive oxygen species production on human retinal cells: cytoprotective effects and prevention of VEGF secretion by resveratrol. Eur. J. Nutr. 2010;49(7):435–446. doi: 10.1007/s00394-010-0102-2. [DOI] [PubMed] [Google Scholar]
- 198.Lv C., Wu C., Zhou Y.H., Shao Y., Wang G., Wang Q.Y. Alpha Lipoic Acid Modulated High Glucose-Induced Rat Mesangial Cell Dysfunction via mTOR/p70S6K/4E-BP1 Pathway. Int. J. Endocrinol. 2014 doi: 10.1155/2014/658589. 2014, 658589. [DOI] [PMC free article] [PubMed] [Google Scholar]