

Abstract
Sulfonamides have been prepared in high yields by the reactions of N-silylamines with sulfonyl chlorides and fluorides. In a competition experiment, the sulfonyl chlorides were found to be far more reactive than sulfonyl fluorides. The chemistry may be used to prepare aliphatic, aromatic, tertiary, secondary, and primary sulfonamides. It may also be done in the absence of solvent and the byproduct trimethylsilyl chloride recovered in good yield. Primary sulfonamides were synthesized from the sulfonyl chloride with aminotriphenyl silane (Ph3SiNH2), a conversion demonstrated with the synthesis of the carbonic anhydrase inhibitor, acetazolamide.
Keywords: Sulfonamides, sulfonyl transfer, N-silylamine, substitution
Introduction
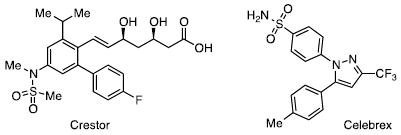
The sulfonamide functional group is a structure with broad importance, as it is found in numerous medicinal agents, dyes/pigments, polymers, and other structures.1 For example, the anti-cholesterol drug Crestor and anti-arthritic drug Celebrex both contain the sulfonamide functional group. Sulfonamides are often prepared by the reactions of sulfonyl chlorides with amines, accompanied by the release of HCl.2 Given the importance of the sulfonamide functional group, convenient methods of preparing this structure are highly desirable. In this manuscript, we report the efficient preparation of sulfonamides from N-silylamines and sulfonyl halides. Our results indicate that sulfonyl chlorides are superior to sulfonyl fluorides in the reaction.
Results and Discussion
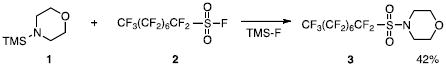
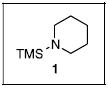
In our initial studies, we sought to react sulfonyl fluorides and N-silylamines. With formation of the sulfonamide, the byproduct of the reaction should be the silyl fluoride. We reasoned that the formation of the fluorine-silicon bond could be a good driving force for the conversion. Our initial experiment involved a reaction between N-(trimethylsilyl)morpholine (1) and the perfluorinated sulfonyl fluoride (2, eq 1). Although the expected sulfonamide (3) was isolated, the yield of the conversion was somewhat disappointing. Other sulfonyl fluorides (4a,6a) were
 |
(1) |
 |
(2) |
 |
(3) |
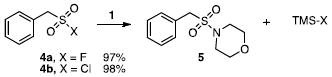
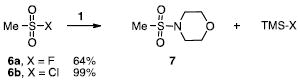
studied and the expected sulfonamide products (5,7) were obtained in fair to good yields (eqs 2-3). Even with added fluoride (20 mol %, Bu4NF), the sulfonyl fluorides exhibited sluggish reactivities. However, the same reactions with analogous sulfonyl chlorides (4b,6b) provide the sulfonamides (5,7) in quantitative yields. The greater reactivity of the sulfonyl chlorides was also observed in a competition experiment. One equivalent of 4a and 4b were reacted with one equivalent of N-(trimethylsilyl)morpholine (1) and product 5 was formed exclusively by reaction of the sulfonyl chloride (4b). Unreacted sulfonyl fluoride (4a) could be recovered quantitatively. These results were somewhat surprising as the formation of the trimethylsilylfluoride – and the strong fluorine-silicon bond – is evidently not the most important factor in determining reaction rates or yields (vide infra).
The N-silylamines 1 and 12 were shown to react with a variety of sulfonyl chlorides to give the respective sulfonamide products (8-11, 13-17; Table 1). Heterocyclic, aromatic, and carbocyclic sulfonamides have all been prepared in quantitative yields. In the optimized reaction conditions, equimolar quantities of N-silylamine and sulfonyl chloride are combined in acetonitrile and the mixture is stirred at reflux for one hour. Removal of the solvent and silyl chloride provides the sulfonamide.
Table 1.
Sulfonamide products (8-17) and yields from N-silylamines 1 and 12.
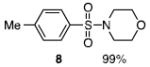
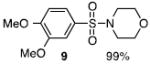
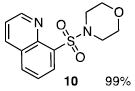
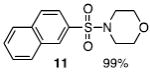
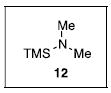
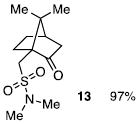
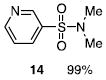
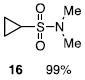
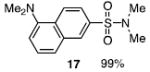
Isolated yield of pure product.
1 mmol N-silylamine, 1 mmol tosyl chloride, and 15 mL CH3CN, reflux 1 hr.
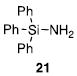
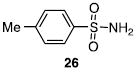
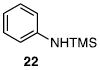
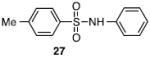
Other N-silylamines were reacted with p-toluenesulfonyl chloride to provide varied sulfonamides (Table 2). The N-silylamines include those from a secondary alkyl amine (18), allyl amine (19), and the imine (20). Primary sulfonamides have found significant use in drug design. In order to prepare the primary sulfonamide (26), aminotriphenylsilane (21) was utilized. The silane 21 itself was prepared from triphenylsilyl chloride and ammonia.3 In the case of sulfonamide 27, this conversion was accomplished without the use of solvent. Thus, the byproduct trimethylsilyl chloride can be distilled off directly and recovered in greater then 75% yield.
Table 2.
Sulfonamide products (23-27) and yields from tosyl chloride and N-silylamines (18-22).
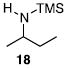
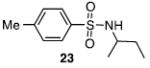
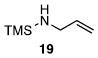
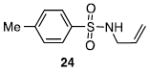
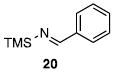
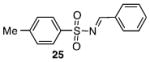
Isolated yield of pure product.
1 mmol N-Silylamine, 1 mmol tosyl chloride, and 15 mL CH3CN, reflux 1 hr.
Neat reaction.
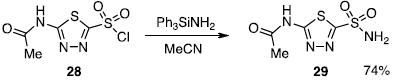
As an application of the chemistry, aminotriphenylsilane (Ph3SiNH2, 21) was also used in the synthesis of the carbonic anhydrase inhibitor, acetazolamide (29; eq 4). Starting from the
 |
(4) |
commercially available 5-amino-1,3,4-thiadiazole-2-thiol, the sulfonyl chloride (28) is prepared in two steps by acetylation of the amino group and oxidation of the thiol group.4 A subsequent reaction between the sulfonyl chloride 28 and Ph3SiNH2 provides acetazolamide (29) in 74% yield.
Regarding the relative reactivities of the sulfonyl fluorides and chlorides in the reactions above, it is known that sulfonyl transfer reactions are considerably faster with sulfonyl chlorides versus sulfonyl fluorides.5 For nitrogen nucleophiles, kCl/kF has been measured to be around 103 to 105 in reactions with benzenesulfonyl halide (acetonitrile/water).5 As noted by Maccarone, these results are clearly related to the relative sulfurhalogen bond strengths,6 where the S-F and S-Cl bonds are respectively 82 and 66 kcal•mol-1.7,8 Previous studies with sulfonyl transfer reactions have suggested a two-step addition-elimination process, SAN, for the substitution mechanism involving nitrogen nulceophiles and sulfonyl halides.6 Other studies have also suggested a one-step, SN2 mechanism for sulfonyl transfer reactions.5
Our own theoretical calculations9 (B3LYP 6-311G(d, p) level) showed the sulfonyl fluoride reaction to be more highly exothermic compared to the same conversion with the sulfonyl chloride (Figure 1). This is likely due to the strong Si-F bond formed in the product mixture. This suggests that sulfonamide formation is a kinetically controlled process involving N-silylamine attack at the sulfur with subsequent elimination of the silyl halide. Assuming the SAN mechanism is operating, the sulfonyl halide (4a,b) reacts with the N-silylamine to give the adduct 30. The reverse reaction is more important with X = F, while the forward reaction is favored with X = Cl. Elimination of the silyl halide gives the sulfonamide 5. Rather than a concerted elimination process, the halide may also dissociate from the sulfur and directly attack the silyl group. This process could occur from the adduct 30, or alternatively, by the SN2-type mechanism where 30 would represent a transition state. Other mechanistic proposals are conceivable, for example a catalytic cycle with halide anion reacting directly with the N-silylamine. Such a pathway is not likely, given that added fluoride does not improve the reaction yields/rates for the conversions involving sulfonyl fluorides.
Figure 1.
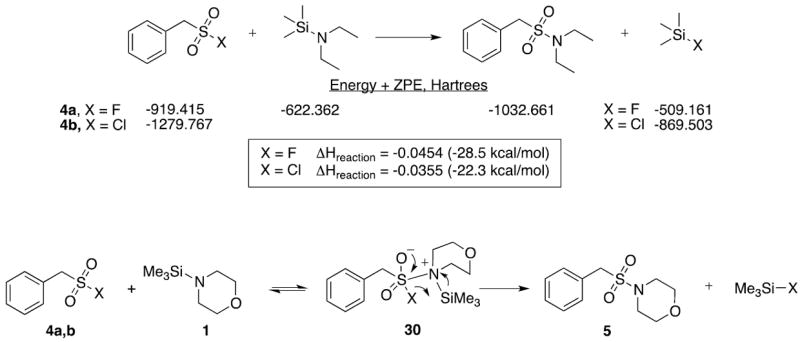
Calculated energies of reactants, products, and the conversion (B3LYP 6-311G (d,p) level), as well as the proposed mechanism of formation for sulfonamide 5.
Conclusions
In summary, we have found a convenient procedure for the synthesis of sulfonamides from N-silylamines and sulfonyl chlorides.10 A “solvent-free” example of the conversion has been demonstrated. Our studies also show that the conversions to sulfonamides may be done with sulfonyl fluorides, although the chemistry is less efficient. A mechanism is proposed involving an addition-elimination process. The methodology compliments an earlier report describing the synthesis of amides with acid chlorides and N-silylamines.11
Supplementary Material
Acknowledgments
We gratefully acknowledge the support of the NIH-National Institute of General Medical Sciences (GM085736-01A1) and the Northern Illinois University for Great Journeys Fellowship (R.R.N.). This manuscript is dedicated to Rachel A. Klumpp on the occasion of her tenth birthday.
Footnotes
Supplementary Material
Supplementary data (characterization data and NMR spectra) and computational methods associated with this article may be found in the online version, at doi:
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.a Wilden JD. J Chem Res. 2010:541. [Google Scholar]; b Bhat MA, Imran M, Khan SA, Siddiqui N. Ind J Pharm Sci. 2005;67:151. [Google Scholar]; c Obreza A, Gobec S. Curr Med Chem. 2004;11:3263. doi: 10.2174/0929867043363659. [DOI] [PubMed] [Google Scholar]; d Scozzafava A, Owa T, Mastrolorenzo A, Supuran CT. Curr Med Chem. 2003;10:925. doi: 10.2174/0929867033457647. [DOI] [PubMed] [Google Scholar]; d Patai S, Rappoport Z, editors. The Chemistry of the Functional Groups: Sulfonic Acids and their Derivatives. Wiley; New York: 1991. [Google Scholar]
- 2.a March J. Advanced Organic Chemistry. 3. Wiley; New York: 1992. p. 499. [Google Scholar]; b de Boer TJ, Backer HJ. Organic Syntheses, Coll. Vol. 4. Wiley; New York: 1963. pp. 943–946. [Google Scholar]; c Doub L. Kirk-Othmer Encyclopedia of Chemical Technology. 2. Wiley-Interscience; Hoboken, New Jersey: 1969. pp. 255–261. [Google Scholar]
- 3.Li Y, Banerjee S, Odom AL. Organometallics. 2005;24:3272. [Google Scholar]
- 4.Roblin RO, Clapp JW. J Am Chem Soc. 1950;72:4890. [Google Scholar]
- 5.Gordon IM, Maskill H. Chem Soc Rev. 1989;18:123. [Google Scholar]
- 6.Maccarone E, Musumarra G, Tomaselli GA. J Org Chem. 1974;39:3286. [Google Scholar]
- 7.Hildenbrand DL. J Phys Chem. 1973;77:897. [Google Scholar]
- 8.Kaufel R, Vahl G, Nunkwitz R, Baumgaertel H. Z Anorg Allg Chem. 1981;481:207. [Google Scholar]
- 9.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision B.01. Gaussian; Wallingford, CT: 2009. [Google Scholar]
- 10.Typical procedure: The sulfonyl chloride (1 mmol) is dissolved in 15 ml acetonitrile and then silylamine (1 mmol) is slowly added. The reaction mixture is refluxed for 1 hour and concentrated with rotary evaporator. If necessary, the product may be further purified by silica gel chromatography (hexane: ethyl acetate).
- 11.Bowser JR, Williams PJ, Kura K. J Org Chem. 1983;48:4111.. For a related preparation of an N-silylated sulfonamide, see: Roy AK. J Am Chem Soc. 1993;115:2598..
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.