

Abstract
A microtiter plate-based screening assay capable of determining the activity and regioselectivity of sialyltransferases was developed. This assay was used to screen two single-site saturation libraries of Pasteurella multocida α2–3-sialyltransferase 1 (PmST1) for α2–6-sialyltransferase activity and total sialyltransferase activity. PmST1 double mutant P34H/M144L was found to be the most effective α2–6-sialyltransferase and displayed 50% reduced donor hydrolysis and 50-fold reduced sialidase activity compared to the wild-type PmST1. It retained the donor substrate promiscuity of the wild-type enzyme and was used in an efficient one-pot multienzyme (OPME) system to selectively catalyze the sialylation of the terminal galactose residue in a multigalactose-containing tetrasaccharide lacto-N-neotetraoside.
Graphical Abstract
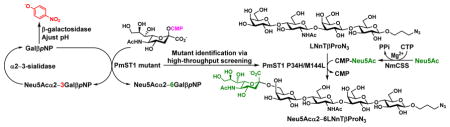
A sialyltransferase mutant for selectively α2–6-sialylating terminal galactose in polyLacNAc-glycan was identified using a novel microtiter plate-based screening.
Introduction
Sialic acid-terminated glycans and glycoconjugates are critical recognition elements involved in a remarkably wide range of pathological and physiological events in human, including cell-cell recognition, inflammation, cancer metastasis, immune regulation, and bacterial and viral infections.1–3 Sialyltransferase-catalyzed reactions with or without in situ enzymatic generation of cytidine 5′-monophosphate (CMP)-sialic acid have been effective approaches to obtain diverse structurally defined sialosides in high yields.2, 3 Of particular note for their low cost and effectiveness are one-pot multienzyme sialylation systems4, 5 combining a sialyltransferase (ST) for the formation of a specific sialyl linkage, a CMP-sialic acid synthetase for the generation of a CMP-sialic acid in situ from CTP and sialic acid, and optionally a sialic acid aldolase for the formation of the sialic acid from its six-carbon monosaccharide precursor N-acetylmannosamine (ManNAc) or derivatives and pyruvate. The flexible substrate specificities of these enzymes have allowed the formation of large libraries of sialosides containing diverse naturally occurring and non-natural modifications at various positions of sialic acid residues. In addition to the diversity of natural sialic acid forms of which more than 50 have been identified,6, 7 sialic acid can be found linked to different monosaccharides in various locations and with a range of sialyl linkages.8 For example, α-linked sialic acid can be found attached to the C3 of a terminal galactose (Gal) residue; to the C6 of a terminal or an internal Gal or N-acetylgalactosamine (GalNAc) residue; to the C6 of an internal glucose (Glc) (e.g. in Neisseria meningitidis serogroup Y capsular polysaccharide) or N-acetyl glucosamine (GlcNAc) (e.g. in milk oligosaccharide disialyllacto-N-tetraose or DSLNT) residue;9, 10 or to the C8 or C9 of another sialic acid residue.2, 3, 11, 12
Several bacterial sialyltransferases have been frequently used for preparative and large-scale synthesis of sialosides.2 Among these enzymes, those from Carbohydrate Active enZyme (CAZy, www.cazy.org) family13, 14 GT80 are of particular interest as several of them have robust heterologous expression in Escherichia coli, high catalytic activity, broad promiscuity toward modified donor substrates and various acceptors, and have been widely used for the synthesis of α2–3- and α2–6-linked sialosides containing different sialic acid structures.15, 16
While the CAZy GT80 family α2–3-sialyltransferases (α2–3STs), such as multifunctional Pasteurella multocida sialyltransferase 1 (PmST1),15 are quite specific in catalyzing α2–3-sialylation to only the terminal galactose residues in galactosides,10, 17–19 the α2–6-sialyltransferase (α2–6ST) activity of multifunctional Photobacterium damselae α2–6ST (Pd2,6ST)20 can catalyze α2–6-sialylation to both terminal and internal galactose or GalNAc residues.10, 12, 16, 17, 21
The lack of selectivity of Pd2,6ST in sialylating terminal or internal Gal was advantageous for one-step sialylation of a tetrasaccharide lacto-N-neotetraose (LNnT) for generating disialyllacto-N-neotetraose DSLNnT, a hexaose mimicking human milk DSLNT.10 Both DSLNnT and DSLNT were effective in protecting neonatal rats from necrotizing enterocolitis in an animal model and could be potentially developed into therapeutic agents.9, 10 This property of Pd2,6ST also allowed facile synthesis of multisialylated poly-N-acetyllactosamine (poly-LacNAc) extensions on O- and N-linked glycans for microarray studies,12, 22 as well as the synthesis of glycans with α2–3-sialylation at the terminal Gal and α2–6-sialylation at the internal Gal.22, 23 However, it generated complications for synthesizing the terminal disialyl tetrasaccharide sequence of gangliosides GD1α, GT1aα, GQ1bα.21 In addition, it would not be suitable for obtaining mono-α2–6-sialylated longer glycans such as α2–6-linked sialyl LNT or sialyl LNnT structures in the milk and on the cell surfaces of human and animals.11, 24
While a human α2–6ST hST6Gal-I12 and a ST6Gal-I purified from rat liver25 were able to catalyze the addition of a sialic acid residue selectively to the terminal Gal on poly-LacNAc-type structures, a bacterial sialyltransferase equivalent has not been identified. Other than unselective α2–6-sialylation of terminal and internal Gal or GalNAc residues by Pd2,6ST,10, 12, 16, 17, 21 several other reported bacterial α2–6STs26–28 including those from GT80 family have not been tested for sialylation of poly-LacNAc-type structures. While hST6Gal-I was able to be expressed in E. coli cells at a level of 0.266 mg purified enzyme per liter culture by N-terminal truncation with fusing to maltose-binding protein (MBP)29 and the expression level was improved to 2 mg purified protein per liter culture by co-expression of multiple chaperon/foldases in the Origami2(DE3) strain,30 the amount was still limited for large-scale synthetic purposes.
The crystal structures have been reported for several GT80 family sialyltransferases, including those preferring the formation of α2–3-31–35 or α2–6-sialyl36, 37 linkage. Comparing the crystal structures and the sialyl linkage specificities of the products formed by PmST1 (PDB ID 2ILV)31, 32 and Photobacterium sp. JT-ISH-224 α2–6ST (Psp2,6ST) (PDB ID 2Z4T)36 indicated that H123 in Psp2,6ST is a key residue determining the α2–6ST specificity of the enzyme while the corresponding P34 in PmST1 is associated with its α2–3ST specificity.36 Indeed, this was also predicted by protein sequence alignment of all characterized GT80 family sialyltransferases and was confirmed by another GT80 family Pasteurella dagmatis α2–3ST (Pd2,3ST) (PDB ID 4V2U)38 for which mutating the corresponding P7 residue to a histidine converted the enzyme from an α2–3ST to an α2–6ST with 95% selectivity for α2–6-sialylation.35 The regio-selectivity for the formation of α2–6-sialyl linkage versus α2–3-sialyl linkage was further enhanced by mutating M117 to alanine. The Pd2,3ST P7H/M117A double mutant had a greater than 99.5% selectivity for α2–6-sialylation. However, each of these mutations resulted in decreased sialylation activity with kcat/KM value dropped from 16 s−1 mM−1 for the wild-type Pd2,3ST to 4.3 s−1 mM−1 and 2.8 s−1 mM−1 for the P7H and P7H/M117A mutants, respectively.
We hypothesized that converting a substrate promiscuous α2–3ST which catalyzes the selective α2–3-sialylation of terminal Gal to an α2–6ST by site-specific mutagenesis might retain its selectivity on sialylating the terminal Gal and substrate promiscuity. PmST1 was chosen as the parent enzyme for engineering. This was because that among all reported GT80 α2–3STs,15, 35, 39–43 PmST1 has the highest expression level in Escherichia coli (100 mg per L culture), the highest measured kinetic constants,15 the largest number of previous mutagenesis studies,31, 38–41, 43–45 and the most complete structural and functional characterization.15, 31, 32 Additionally, PmST1 has been shown to be promiscuous towards a wide range of CMP-sialic acid donors and galactoside acceptors.15, 19, 46–49 PmST1 has multiple functions (Figure 1) including (1) α2–3ST, (2) α2–6ST, (3) α2–3-sialidase, (4) α2–3-trans-sialidase, (5) donor hydrolysis,15, 45 and (6) α2–6-trans-sialidase50 activities activity. As the α2–6ST activity of PmST1 is much weaker than its α2–3ST activity especially at pH≥7.0,15 PmST1 has been broadly used in highly efficient synthesis of α2–3-sialosides with in situ generation of CMP-sialic acids by a CMP-sialic acid synthetase with or without a sialic acid aldolase.2, 51 Furthermore, the product sialyl linkage specificity-switching effect of the P7H mutation for Pd2,3ST has already been confirmed for PmST1 with the corresponding P34H mutation in its trans-sialidase reaction capacity for the formation of α2–6-sialylated products.52
Figure 1.
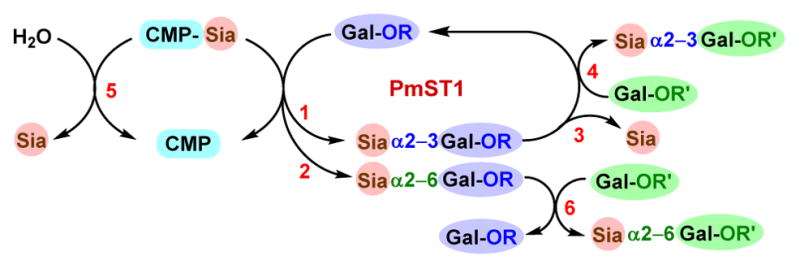
Multiple functions of wild-type PmST1 including (1) α2–3ST, (2) α2–6ST, (3) α2–3-sialidase, (4) α2–3-trans-sialidase, (5) donor hydrolysis, and (6) α2–6-trans-sialidase activities.
Here we report our strategy of converting PmST1 to an α2–6ST with the same selectivity in sialylating the terminal Gal in poly-LacNAc-type structures such as a multigalactose-containing tetrasaccharide lacto-N-neotetraoside Galβ1–4GlcNAcβ1–3Galβ1–4GlcβProN3 (LNnTβProN3). This was accomplished by structure-guided saturation mutagenesis followed by a novel colorimetric screening method in microtiter plates which can be adapted to automation for a high-throughput platform. Various in situ generated CMP-sialic acids containing naturally occurring and non-natural sialic acid forms were shown to be tolerable donor substrates for the mutant, indicating the retaining of the donor substrate promiscuity of the wild-type PmST1 by the mutant. N-Acetylneuraminic acid (Neu5Ac), the most common sialic acid form, was used as a donor precursor for preparative-scale synthesis using a one-pot two-enzyme sialylation system.5 The resulting Neu5Acα2–6LNnT (LSTc) represents a common structure found in human milk11 and on mammalian cell surface.53
Results and discussion
Initially we sought to reproduce the PmST1 mutant homologs for Pd2,3ST P7H and P7H/M117A. The corresponding PmST1 residues P34 and M144 were mutated to generate PmST1 P34H single mutant and P34H/M144A double mutant. Both mutants catalyzed α2–6-sialylation but displayed 2.5- and 6.4-fold, respectively, decreased activity relative to the α2–3ST activity of the wild-type PmST1 (Table 1). A similar loss of activity was reported for the Pd2,3ST P7H single mutant and P7H/M117H double mutant (Table 1).35 Additionally, the PmST1 P34H and P34H/M144A mutants were found to catalyze CMP-Neu5Ac hydrolysis at about 57-fold and nearly 3-fold, respectively, the catalytic efficiency of their α2–6ST activity, hindering their efficiency for the preparation of sialosides. The α2–6-sialidase activities of both mutants were more than 150-fold lower than their corresponding α2–6ST activities, which is a preferable feature for sialoside synthesis.
Table 1.
Apparent kinetic parameters for PmST1 and its mutants. Data from previous reports include PmST1 (aRef15, bRef45), cPd2,3ST,35 Pd2,6ST (dRef54, eRef20) and Psp2,6ST (fRef54, gRef62). ND, not determined. nd, not detected.
| Parent Enzyme |
Mutant | Product sialyl linkage |
Sialyltransferase | CMP-Neu5Ac Hydrolysis | Sialidase Activity | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| kcat (s−1) | KM (mM) |
kcat/KM (s−1 mM−1) |
Specificity (% 6′) |
kcat (s−1) | KM (mM) | kcat/KM (s−1 mM−1) |
kcat (s−1) |
KM (mM) |
kcat/KM (s−1 mM−1) |
|||
| Pd2,3ST | WT | α2–3 | c 24 | c 1.5 | c 16 | c < 0.6 | ND | ND | ND | ND | ND | ND |
| P7H | α2–6 | c 16 | c 3.8 | c 4.3 | c 94.7 | ND | ND | ND | ND | ND | ND | |
| P7H/M117A | α2–6 | c 18 | c 6.4 | c 2.8 | c > 99.6 | ND | ND | ND | ND | ND | ND | |
| PmST1 | WT | α2–3 | a 47 | a 1.4 | a 34 | 3.9 ± 0.8 | b 27 ± 1 | b 1.5 ± 0.2 | b 18 | a 230 | a 24 | a 9.5 |
| P34H | α2–6 | 8.6 ± 1.6 | 0.6 ± 0.4 | 13.5 | 94.0 ± 0.4 | 925.5 ± 66.7 | 1.2 ± 0.5 | 771.3 | 1.2 ± 0.1 | 13.7 ± 1.5 | 0.088 | |
| P34H/M144A | 9.0 ± 1.3 | 1.7 ± 0.5 | 5.3 | 97.9 ± 0.5 | 34.5 ± 0.3 | 2.3 ± 0.1 | 15.1 | 0.51 ± 0.03 | 15.1 ± 1.1 | 0.034 | ||
| P34H/ M144L | 20.0 ± 4.4 | 0.8 ± 0.2 | 26 | 98.7 ± 0.1 | 30.5 ± 0.8 | 3.4 ± 0.1 | 8.9 | 1.14 ± 0.1 | 6.4 ± 0.6 | 0.178 | ||
| P34H/M144V | 5.5 ± 0.4 | 1.4 ± 0.2 | 3.9 | 98.5 ± 0.1 | 62.4 ± 7.6 | 10.5 ± 2.7 | 6.0 | 0.27 ± 0.019 | 14.9 ± 1.3 | 0.018 | ||
| Pd2,6ST | WT | d 2.3 ± 0.1 | d 0.8 ± 0.1 | d 3 | nd | 6.7 ± 1.6 | 44.9 ± 14.2 | 0.15 | e 0.14 ± 0.01 | e 7.6 ± 0.5 | e 0.018 | |
| Psp2,6ST | WT | f 1.7 ± 0.2 | f 0.4 ± 0.1 | f 4.8 | nd | g 4.8 ± 0.5 | g 17 ± 4 | g 0.28 | (7.9 ± 0.3) × 10−3 | 4.7 ± 0.3 | 0.0017 | |
A closer examination of the sequence homology among GT80 STs revealed that while P34 is conserved in α2–3STs, and α2–6STs have a conserved histidine residue at the corresponding site, residues homologous to M144 displayed a greater diversity. Previous mutagenesis studies by our lab and others indicated that M144 of PmST1 and its homologous residues in other GT80 STs are a multifunctional “hotspot,” and that mutations at this position can greatly impact donor hydrolysis activity, sialidase activity, sialyltransferase activity, donor specificity, and acceptor specificity.45, 55 Therefore we hypothesized that screening a PmST1 P34H/M144X saturation library might reveal a more reactive α2–6ST.
In order to identify mutants with improved sialyltransferase activities and with regioselectivity for the formation of either α2–3- or α2–6-sialyl linkage, a novel high-throughput screening assay was developed and used to screen PmST1 P34X and P34H/M144X saturation libraries. To screen each saturation library, a 96-deep well culture plate was inoculated with 88 mutant colonies, 4 wild-type colonies, and 4 empty vector colonies. The libraries were checked by sequencing several colonies from each library as is typical for saturation mutagenesis. No nucleotide sequence bias was detected during this analysis. TopLib analysis56 shows that the likelihood of discovering the best variant from screening 88 wells of an NNK single site saturation library is 96.24%. The lysates from these cultures were used for activity screen in 384-well plates. The screen relied on using sialic acid transferred by the sialyltransferase mutants as a protective group for para-nitrophenyl β-D-galactopyranoside (GalβpNP, 1), a substrate for β-galactosidases which produces a more intense chromophore upon hydrolysis (Figure 2A). On a 384-well microtiter plate, the PmST1 mutants were allowed to sialylate GalβpNP (1), then two aliquots of each reaction were transferred to empty 384-well microtiter plate wells. One of these wells (well A) was treated with β-galactosidase only, while the other (well B) was treated with both β-galactosidase and Streptococcus pneumoniae sialidase SpNanB,57–59 an α2–3-sialyl linkage specific sialidase. In well A, the remaining 1 in the sialylation reaction was hydrolyzed by the excess β-galactosidase added to form para-nitrophenol (pNP). In well B, both remaining 1 and any α2–3-sialoside (2) formed would be hydrolyzed by the combined actions of β-galactosidase and α2–3-specific sialidase SpNanB to form pNP and only the α2–6-sialoside (3) resisted hydrolysis catalyzed by the exoglycosidases and remained in the mixture. Upon quenching with N-cyclohexyl-3-aminopropanesulfonic acid (CAPS) buffer (pH 9.6) to form para-nitrophenolate from pNP and quantification at 405 nm using a plate reader, the extents of total sialylation (from well A) and α2–6-sialylation (from well B) were determined and the α2–3-sialylation could be calculated by the difference between the total and the α2–6-sialylation.
Figure 2.
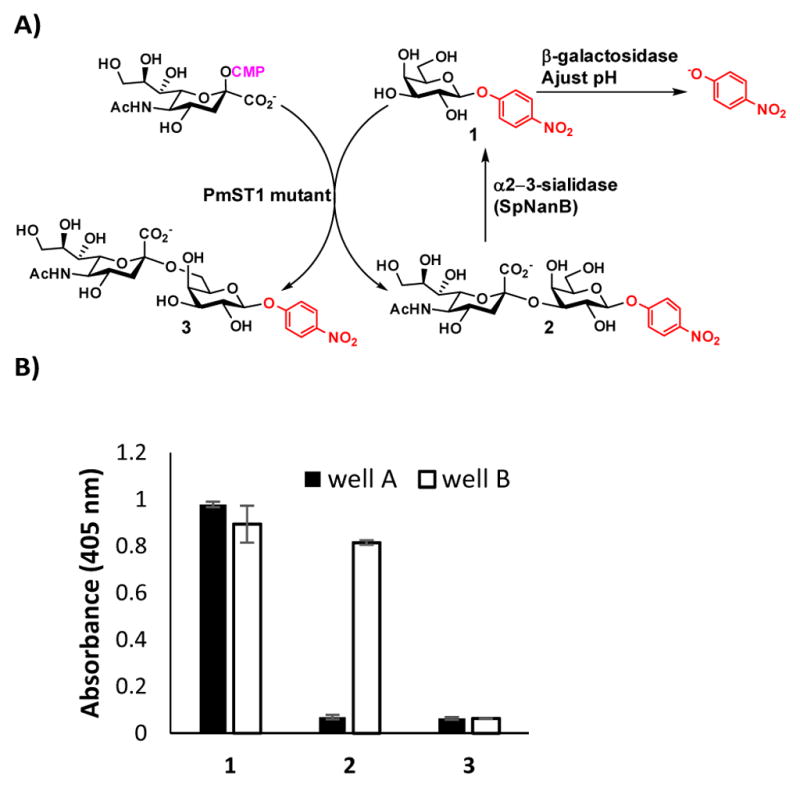
A high-throughput screening assay for identifying PmST1 mutants with improved sialyltransferase activities and with regioselectivity for the formation of either α2–3- or α2–6-sialyl linkage. A) Reaction schemes for the assay and B) Assay validation with starting material GalβpNP (1) and product standards Neu5Acα2–3GalβpNP (2) and Neu5Acα2–6GalβpNP (3) by the addition of β-galactosidase without (in well A, black columns) or with (in well B, white columns) α2–3-sialidase SpNanB.
The assay was validated using synthetic Neu5Acα2–3GalβpNP (2) and Neu5Acα2–6GalβpNP (3) as product standards (Figure 2B). Indeed, treating compound 2 with both β-galactosidase and α2–3-sialidase SpNanB (well B) led to its hydrolysis and a large increase in the A405 nm relative to 2 treated with only β-galactosidase (well A). In comparison, compound 3 was resistant to degradation under both conditions and low signal was observed for both wells. Compound 1 released para-nitrophenolate under both conditions resulting in high A405 nm values for both.
Sialyltransferase activity was tested at pH 6.5. This pH was chosen with the idea that mutants with altered sialidase activity or linkage specificity would be more noticeable, as wild-type PmST1 shows enhanced rates of α2–3-sialidase and α2–6ST activity at low pH. Additionally, this pH was found to be optimal for the degradation of compound 2 in well B. Wells of interest from each plate were re-expressed and re-screened from a single microtiter plate for validation and easy comparison. The combined rescreen data of PmST1 P34X and P34H/M144X libraries are shown in Figure 3. In the P34X library, P34H was identified from two wells as the sole mutant displaying detectable α2–6ST activity. In the P34H/M144X library, enzymes with higher yields in producing α2–6-sialosides than the parent P34H mutant were sequenced and were identified to be mutants with M144 mutated to leucine (L), isoleucine (I), or valine (V). The two wells that displayed the greatest change in absorbance signal were found to harbor P34H/M144L.
Figure 3.
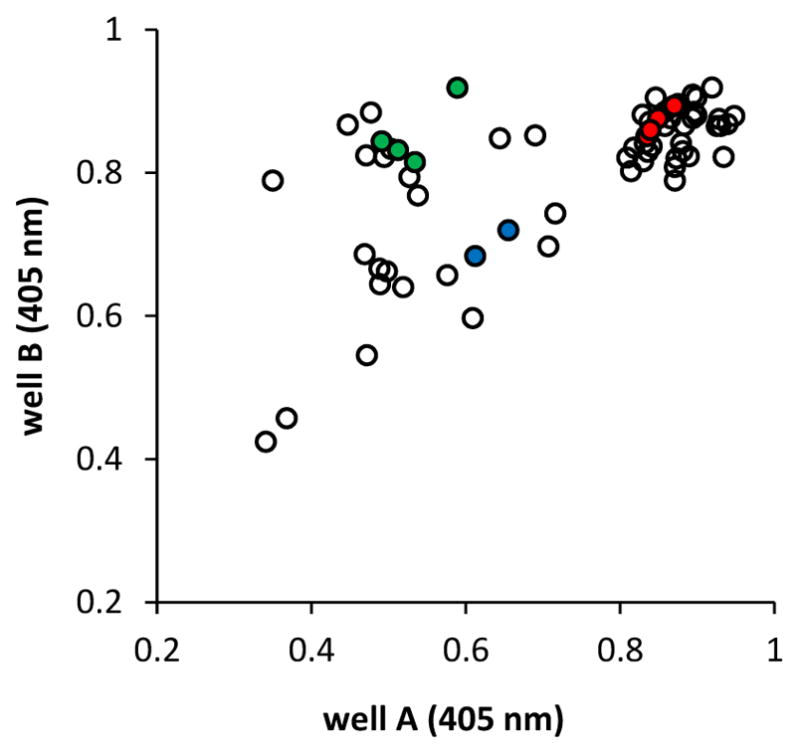
A405 nm data from rescreening PmST1 P34X and P34H/M144X libraries. Empty vector controls are shown in red, wild-type control wells are shown in green, and wells identified to contain the P34H single mutant are shown in blue. Well A was treated with β-galactosidase, and lower absorbance indicates total (both α2–3- and α2–6-) sialylation of the acceptor. Well B was treated with both β-galactosidase and α2–3-sialidase SpNanB, and lower absorbance in both well A and well B indicates α2–6-sialylation of the acceptor.
The two best variants, PmST1 mutants P34H/M144L and P34H/M144V, were purified and their apparent kinetic parameters were measured for sialyltransferase, sialidase, and donor hydrolysis activities. In order to minimize the influence of donor hydrolysis, the rate of which varies between mutants, sialyltransferase kinetics were measured with a constant concentration of CMP-Neu5Ac and varied concentration of an acceptor substrate. To allow comparison to previous kinetic analysis, 4-methylumbelliferyl-β-D-lactoside (LacβMU) was chosen as the acceptor substrate. The choice of an acceptor other than the one used for screening was preferred as it prevented potentially unintentional identification of enzymes that work only specifically for a given acceptor used in the screening.60, 61 The P34H/M144L mutant displayed significantly higher activity (8.7- and 5.4-fold, respectively) than Pd2,6ST or Psp2,6ST,54, 62, 63 the two most characterized and widely used GT80 α2–6STs. Its α2–6ST activity was 76% of the wild-type PmST1 α2–3ST activity. P34H/M144L also displayed 1/3 of the donor hydrolysis activity and 1/53 of the sialidase activity of the wild-type PmST1. In contrast, P34H and P34H/M144A displayed reduced activity consistent with Pd2,3ST P7H and P7H/M117A, although accurate measurement of the P34H ST activity was made impossible by the surprising 43-fold increased donor hydrolysis activity. Using high-performance liquid chromatography (HPLC) to separate α2–3- and α2–6-regioisomers, it was possible to establish the linkage specificity of the mutants. The product α2–6-sialyl linkage specificity of PmST1 P34H was enhanced from 94.0 ± 0.4% to 97.9 ± 0.5% by additional M144A mutation, similarly to the reported shift from 94.7% (P7H single mutant) to 99.6% (P7H/M117A double mutant) for Pd2,3ST (Table 1). To our delight, similar to P34H/M144A, P34H/M144L displayed a high preference for α2–6-sialylation. The P34H/M144V mutant was found to be similarly selective for α2–6-sialylation, though it also displayed decreased α2–6ST activity in a manner similar to P34H/M144A but not P34H/M144L. Nevertheless, P34H/M144V displayed lower donor hydrolysis and sialidase activities than P34H/M144A.
The pH-dependence of the product sialyl linkage specificity was investigated using an HPLC assay in a pH range of 5.0 to 10.5 for both PmST1 P34H/M144L mutant and wild-type PmST1. This assay provided quantitative data instead of qualitative results observed by previous thin-layer chromatography (TLC) assay.15 In addition, instead of using 1:1 ratio of donor versus acceptor in the previous TLC assay,15 a 5:1 ration of donor versus acceptor was used. This was to minimize the interferences by donor hydrolysis and sialidase activities. As shown in Figure S1 (ESI†), PmST1 was found to be a predominant α2–3ST across the entire pH range, although its α2–3-selectivity decreased when the pH was below 7.0. Previously observed linkage specificity switch from α2–3- to α2–6 for PmST1 major sialyl product when the pH changed from a higher (>6.5) to a lower (<6.0) range15 could be explained by the enhanced α2–3-sialidase activity at the lower pH range which caused the hydrolysis of the α2–3-sialyl product rather than a change in sialyltransferase linkage specificity. For the experiments described here, the influence of the sialidase activity of PmST1 was minimized by controlling the amount of enzyme used and using a short reaction time. A similar product linkage selectivity shift observed for PmST1 trans-sialidase reactions was also due to the enhanced α2–3-sialidase activity at low pH which reduced α2–3-sialoside concentration over time.50 In comparison, PmST1 P34H/M144L mutant Figure S2 (ESI†) exhibited a much smaller shift in selectivity across the pH range. The selectivity of α2–6-sialyl product rose from 98.0% at pH 5.0 to 98.9% at pH 10.5. This could be due to the much lower sialidase activity of PmST1 P34H/M144L mutant compared to wild-type PmST1.
It is worth mentioning that the PmST1 mutants studied including P34H, P34H/M144A, P34H/M144V, and P34H/M144L retained a similar expression level as the wild-type enzyme (100 mg L−1 culture) under the same expression conditions. All mutants were routinely expressed at 90–115 mg L−1 culture at 37 °C for 3 h after induction with 100 μM of isopropyl β-D-1-thiogalactopyranoside (IPTG).
The donor substrate specificity of PmST1 P34H/M144L was tested using cytidine 5′-monophosphate-sialic acid (CMP-Sia) and derivatives generated in situ from sialic acids and derivatives or their six-carbon precursors using Neisseria meningitidis CMP-Sia synthetase (NmCSS)64 with or without Pasteurella multocida sialic acid aldolase (PmAldolase)65 in PmST1 P34H/M144L-containing one-pot multienzyme (OPME) systems. Sialic acids and derivatives as well as their precursors used were N-methylglycolylmannosamine (ManNGcOMe, A), mannose (B), 6-azido-6-deoxy-N-acetylmannosamine (ManNAc6N3, C), 4-azido-4-deoxy-N-acetylmannosamine (ManNAc4N3, D), 6-O-methyl-N-acetylmannosamine (ManNAc6OMe, E), and N-azidoacetylmannosamine (ManNAz, F), Neu5Ac (G), N-glycolylneuraminic acid (Neu5Gc, H), 8-O-methyl-N-acetylneuraminic acid (Neu5Ac8OMe, I), and 9-O-acetyl-N-acetylneuraminic acid (Neu5,9Ac2, J). A longer (24 h) reaction time was used for one-pot three-enzyme reactions from sialic acid precursors due to the slower processes by the sialic acid aldolase-catalyzed reactions. In comparison, a shorter (1 h) reaction time was used for one-pot two-enzyme reactions from sialic acid and derivatives. Other than the reaction containing a substrate (Neu5,9Ac2, J) with a labile 9-O-acetyl group which needed to be carried out at a lower pH (7.1) condition, all other reactions were carried out at pH 8.5 which was the optimal for the NmCSS activity without affecting the activities of PmAldolase65 and PmST1 P34H/M144L significantly. Propyl azido β-lactoside (LacβProN3) was used as the acceptor substrate. The formation of the product in each reaction was clearly demonstrated by the electrospray ionization high resolution mass spectrometry (ESI-HRMS) results (Figure S3, ESI†). Therefore, PmST1 P34H/M144L retained the donor substrate promiscuity of the wild-type PmST1 in tolerating CMP-sialic acid and derivatives containing diverse sialic acid forms.
The PmST1 P34H/M144L double mutant was further tested for sialylating LNnTβProN3, a compound containing both terminal and internal Gal residues, using CMP-Neu5Ac as the donor substrate. To our delight, mono-sialylated compound was the major product when 1.0 equivalent of CMP-Neu5Ac was used. In order to confirm the location of the mono-sialylation, a preparative-scale synthesis of monosialylated LNnTβProN3 from LNnTβProN3, 1.0 equivalent of Neu5Ac, and 1.5 equivalent of cytidine 5′-triphosphate (CTP) was carried out using an efficient one-pot two-enzyme sialylation system5 containing Neisseria meningitidis CMP-sialic acid synthetase (NmCSS)64 and PmST1 P34H/M144L mutant. As shown in Figure 4, NmCSS was responsible for converting CTP and Neu5Ac to generate CMP-Neu5Ac in situ which was used as the donor substrate by the PmST1 P34H/M144L mutant. Controlling the amount of enzyme used and the timing of the reaction by monitoring with high-resolution mass spectrometry (HRMS) was important to achieve high-yield production of mono-sialylated product as the major product. Reaction at 37 °C for 1 hour was found to be optimal for the formation of mono-sialylated product. The formation of disialylated product (5% yield) was also observed which was separated from monosialylated product (90% yield) by high performance liquid chromatography (HPLC) purification using a C18 column.
Figure 4.
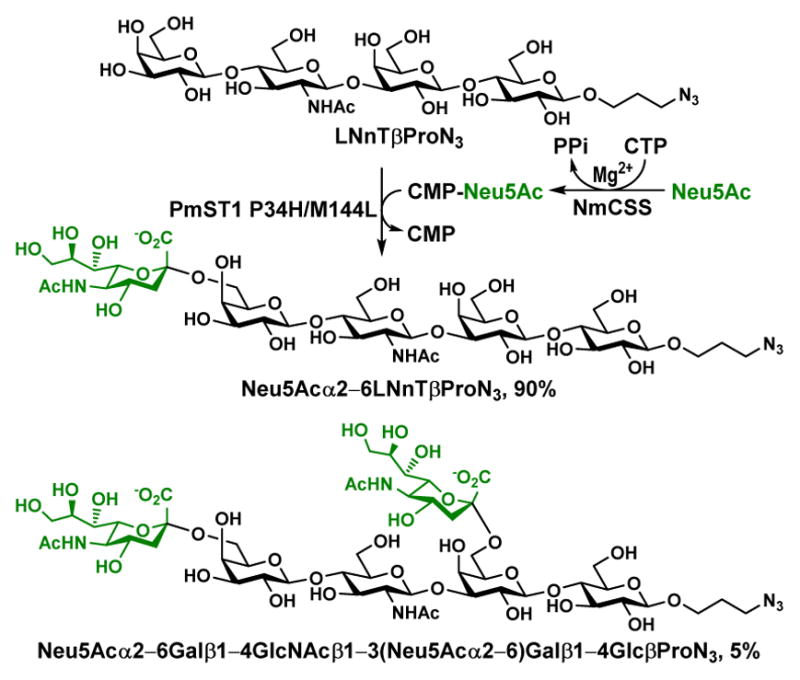
One-pot two-enzyme synthesis of monosialylated Neu5Acα2–6LNnTβProN3.
13C Nuclear magnetic resonance (NMR) data (Table S1, Figure S4–S6 ESI†) confirmed that the attachment of Neu5Ac in the mono-sialylated product was to the C-6 of the terminal Gal residue in LNnTβProN3. Sialylation resulted in a significant downfield shift of the substituted carbon (a downfield shift of 2.31 ppm from 60.91 ppm in the starting tetrasaccharide to 63.22 ppm in the sialylated product) (Table S1, ESI†). This chemical shift change seen in Neu5Acα2–6Gal was consistent with that observed for other glycans containing the same structural component.66, 67 In addition, sialylation led to a significant downfield shift (δ = +2.31 ppm) of C-4 in GlcNAc in the 13C spectra due to the steric interaction between Neu5Ac and GlcNAc in the product.
Conclusions
Regiospecificity is one of the most valued properties of glycosyltransferases, the primary catalysts for the synthesis of carbohydrates. However, changes to this property could not be screened for in a high-throughput fashion with previous glycosyltransferase screening assays. Engineering active, well expressed, and donor promiscuous enzymes for altered linkage regiospecificity is a promising new way to expand the enzyme toolbox for oligosaccharide synthesis. The microtiter plate screening assay demonstrated in this work can be adapted to work for a large variety of glycosyltransferases. With access to suitable exoglycosidases, changes to regioselectivity or stereoselectivity can be determined. The relative simplicity, low cost, and broad applicability of this assay make it a powerful new tool for the field of glycobiology. Using this screening method, a highly active α2–6ST PmST1 mutant P34H/M144L was identified to display 8.7-fold and 5.4-fold higher α2–6ST activity, respectively, than commonly used α2–6STs Pd2,6ST and Psp2,6ST. The identified PmST1 mutant P34H/M144L retained the donor substrate promiscuity of the wild-type enzyme. It allowed highly efficient selectively sialylation of the terminal Gal residue in a multigalactose-containing tetrasaccharide lacto-N-neotetraoside using a one-pot two-enzyme system.
Experimental Section
Materials and methods
Chemicals were purchased and used as received. NMR spectra were recored in the NMR facility of University of California, Davis on a Bruker Avance-800 NMR spectrometer (800 MHz for 1H, 200 MHz for 13C). Chemical shifts are reported in parts per million (ppm) on the δ scale. High resolution (HR) electrospray ionization (ESI) mass spectra were obtained using a Thermo Electron LTQ-Orbitrap Hybrid MS at the Mass Spectrometry Facility in the University of California, Davis. Gel filtration chromatography was performed with a column (100 cm × 2.5 cm) packed with Bio-Gel P-2 Fine resins (Bio-Rad). GalβpNP and sodium pyruvate were from Sigma. N-Acetylneuraminic acid (Neu5Ac) was from Inalco (Italy). Cytosine 5′-triphosphate (CTP) was purchased from Hangzhou Meiya Pharmaceutical Co. Ltd. Recombinant enzymes Neisseria meningitidis CMP-sialic acid synthetase (NmCSS),64 Pasteurella multocida multifunctional α2–3ST 1 (PmST1),15 Photobacterium damselae α2–6ST (Pd2,6ST),16, 54 and Photobacterium species α2–6ST (Psp2,6ST)62 were expressed and purified as described previously. Neu5Acα2–3GalβpNP (2),46 Neu5Acα2–6GalβpNP (3),46 4-methylumbelliferyl-β-D-lactoside (LacβMU),15 and tetrasaccharide LNnTβProN368 were prepared as described previously.
Site-directed saturation mutagenesis
PmST1 point mutant P34H and single-site saturation library P34X were generated with the QuikChange II XL Site-Directed Mutagenesis kit (Agilent Technologies) according to the manufacturer’s instructions using primers P34H_f: ATCACGCTGTATTTAGATCATGCCTCCTTACCGGC, P34H_r: GCCGGTAAGGAGGCATGATCTAAATACAGCGTGAT, P34X_f: GGTCAAAAACAATCACGCTGTATTTAGATNNKGCCTCCTTACCGGCA, and P34X_r: TGCCGGTAAGGAGGCMNNATCTAAATACAGCGTGATTGTTTTTGACC. Single-site saturation library P34H/M144X was generated via the Phusion Site-Directed Mutagenesis Kit (Thermo Scientific) using 5′ phosphorylated primers M144X_f: NNKGAATATGTTGATTTAGAAAAAGAAGAAAATAAAGATATTTCCGC and M144X_r: TGAGCCATCGTCATAAAGATTTAACTGC. The assembled DNA was transformed into E. coli 10 G Electrocompetent Cells (Lucigen). Ten percent of the transformed cells were plated on LB agar plates supplemented with ampicillin in order to determine the number of total transformants. The remaining transformed cells were diluted into fresh LB media (10 g L−1 tryptone, 5 g L−1 yeast extract, and 10 g L−1 NaCl) supplemented with ampicillin, grown overnight at 37 °C 250 rpm, and the plasmid DNA was isolated. This DNA was transformed into homemade chemically competent E. coli BL21(DE3) cells.
Library expression
To each well of a 96-well deep well plate was added LB media (1 mL). For each library, 88 wells were inoculated with colonies from agar plates, 4 wells were inoculated with cells expressing wild-type PmST1, and 4 wells were inoculated with cells containing empty pET22b vector. These plates were cultured for overnight at 37 °C with shaking (350 rpm), and the following day these cultures (20 μL each) were used to inoculate cultures (1 mL each) in another 96-deep well plate. 50% glycerol solution (200 μL) was added to each well on the original plate, and the plate was sealed and stored at -80 °C. The freshly inoculated cells were grown for four hours at 37 °C at 350 rpm before being induced with isopropyl β-D-1-thiogalactopyranoside (IPTG, 100 μM) and grown for another four hours at 37 °C at 350 rpm. The cells were pelleted by centrifugation, resuspended in Tris-HCl (50 mM, pH 8.0) supplemented with 4 mg mL−1 lysozyme, and frozen at -80 °C. After thawing at room temperature, cell debris was pelleted by centrifugation and the lysate was used for the sialyltransferase and sialidase assays.
Sialyltransferase screening
Into every other column and every other row of a clear bottom 384-well microtiter plate was added 20 μL lysate and 20 μL of reaction master mix to final concentrations of GalβpNP (0.4 mM), CMP-Neu5Ac (1.2 mM), Tris-HCl (100 mM, pH 7.5), and MgCl2 (10 mM). After one hour at 37 °C, 15 μL of each reaction was added into two adjacent empty wells. To one of these wells was added β-galactosidase (5 μL, 8 mU μL−1), and to the other of these wells was added β-galactosidase (5 μL, 8 mU μL−1) and NanB (70 μg mL−1). After two hours at 37 °C, CAPS buffer (40 μL, 500 mM, pH 9.6) was added to each well and the absorbances at 405 nm of the samples in the plate were measured using a Synergy HT microplate reader (BioTek).
Overexpression and purification
For the expression of PmST1 and its mutants, 1 L of LB media containing 100 μg mL−1 ampicillin was inoculated with 1 mL of an overnight culture of Escherichia coli BL21(DE3) harboring the appropriate plasmid. This culture was grown at 37 °C with vigorous shaking at 250 rpm until reaching an OD600 of 0.8–1.0, then expression was induced with IPTG to a final concentration of 100 μM and the protein was expressed at 37 °C with vigorous shaking at 250 rpm for another 3 hours. The cells were harvested by centrifugation at 4 °C in a Sorvall Legend RT centrifuge at 4000 rpm for 30 min. The cell pellets were resuspended in Tris-HCl (20 mL, 100 mM, pH 7.5). The cells were broken by sonication with the following method: amplitude at 65%, 10°s pulse on and 20°s pulse off for 18 cycles. Then the cell lysate was centrifuged at 4 °C, 8,000 rpm for 30 min, the supernatant was loaded onto a Ni2+-NTA affinity column at 4 °C that was pre-equilibrated with 6 column volumes of binding buffer (50 mM Tris–HCl buffer, pH 7.5, 10 mM imidazole, 0.5 M NaCl). The column was washed with 10 column volumes of binding buffer and 10 column volumes of washing buffer (50 mM of Tris-HCl buffer, pH 7.5, 50 mM of imidazole, 0.5 M of NaCl) sequentially to wash away the nonspecific binding protein. The target protein was eluted using Tris-HCl buffer (50 mM, pH 7.5) containing 200 mM of imidazole and 0.5 M NaCl. Fractions containing the purified protein were combined and dialyzed against Tris-HCl buffer (20 mM, pH 7.5) supplemented with 10% glycerol. The enzyme solutions were aliquoted, flash frozen in liquid N2, and stored at -20 °C.
Sialyltransferase kinetics
Reactions were performed in duplicate at 37 °C for 10 to 30 minutes with Tris-HCl (100 mM, pH 8.5), MgCl2 (10 mM), CMP-Neu5Ac (5 mM), enzyme (0.0988 μM P34H, 0.06343 μM P34H/M144A, 0.0339 μM P34H/M144V, 0.0169 μM P34H/M144L), and varying concentrations (0.5, 1.0, 2.0, and 5.0 mM) of LacβMU as the acceptor substrate. Reactions were stopped by adding an equal volume of pre-chilled methanol. The mixtures were incubated on ice for 30 min and centrifuged at 13,000 rpm for 5 min. Supernatants were diluted with 25% acetonitrile and analyzed with a Shimadzu LC-6AD system equipped with a membrane online degasser, a temperature control unit, and a fluorescence detector (Shimadzu RF-10AXL). A reverse-phase Premier C18 column (250 mm × 4.6 mm i.d., 5 μm particle size, Shimadzu) protected with a C18 guard column cartridge was used. The fluorophore (MU)-labeled compounds were separated with an isocratic flow of 20% acetonitrile and were detected by excitation at 325 nm and emission at 372 nm. The apparent kinetic parameters were obtained by fitting the experimental data (the average values of duplicate assay results) into the Michaelis–Menten equation using Grafit 5.0.
Donor hydrolysis kinetics
Reactions were performed in duplicate at 37 °C for 10–30 minutes with Tris-HCl (100 mM, pH 8.5), MgCl2 (10 mM), enzyme (0.0011 μM P34H, 0.0339 μM P34H/M144V, 0.07 μM P34H/M144A, 0.0718 μM P34H/M144L, 1.71 μM Psp2,6ST, 0.402 μM Pd2,6ST), and varying concentrations (2.0, 5.0, 10.0, and 20.0 mM) of CMP-Neu5Ac. Reactions were stopped by adding an equal volume of pre-chilled methanol. The mixtures were incubated on ice for 30 min and centrifuged at 13,000 rpm for 5 min. Supernatants were analyzed with a P/ACETM MDQ capillary electrophoresis (CE) system equipped with a UV-Vis detector (Beckman Coulter, Fullerton, CA). The CE procedure utilized a 75 μm i.d. capillary, 25 KV/80 μÅ, 5 s vacuum injections, was monitored at 254 nm, and used sodium tetraborate (25 mM, pH 9.4) buffer as the running buffer. The apparent kinetic parameters were obtained by fitting the experimental data (the average values of duplicate assay results) into the Michaelis–Menten equation using Grafit 5.0.
Sialidase kinetics
Reactions were performed in duplicate at 37 °C for 10 to 30 minutes with Tris-HCl (100 mM, pH 5.5), MgCl2 (10 mM), CMP (0.5 mM), enzyme (3.53 μM P34H/M144A, 1.1 μM P34H, 0.364 μM P34H/M144L, 207.6 μM Psp2,6ST, 33.9 μM P34H/M144V), and varying concentrations (0.5, 1.0, 2.0, and 5.0 mM) of Neu5Acα2–6LacβMU. Reactions were stopped by adding an equal volume of pre-chilled methanol. The mixtures were incubated on ice for 30 min and centrifuged at 13,000 rpm for 5 min. Supernatants were analyzed with a P/ACETM MDQ capillary electrophoresis (CE) system equipped with a UV-Vis detector (Beckman Coulter, Fullerton, CA). The CE procedure utilized a 75 μm i.d. capillary, 25 KV/80 μÅ, 5 s vacuum injections, was monitored at 315 nm, and used sodium tetraborate (25 mM, pH 9.4) buffer as the running buffer. The apparent kinetic parameters were obtained by fitting the experimental data (the average values of duplicate assay results) into the Michaelis–Menten equation using Grafit 5.0.
Product sialyl linkage specificity studies
Reactions were performed in duplicate at 37 °C for 10 minutes in Tris-HCl buffer (100 mM, pH 8.5) containing MgCl2 (10 mM), CMP-Neu5Ac (10 mM), LacβMU (2 mM), and enzyme (0.036 μM PmST1, 0.104 μM PmST1 P34H, 0.104 μM PmST1 P34H/M144A, 0.010 μM PmST1 P34H/M144L, 0.104 μM PmST1 P34H/M144V, 0.082 μM Psp2,6ST, 0.085 μM Pd2,6ST). pH Profiles for sialylation yields and product sialyl linkage percentages were also carried out for PmST1 and PmST1 P34H/M144L mutant with varied pH. Buffers used were 2-(N-morpholino)ethanesulfonic acid (MES) for pH 5.0–6.5, Tris-HCl for pH 7.0–9.0, and CAPS for pH 9.5–10.5. Reactions were stopped by adding an equal volume of pre-chilled methanol. The mixtures were incubated on ice for 30 min and centrifuged at 13,000 rpm for 5 min. Supernatants were analyzed with an Agilent 1290 Infinity II liquid chromatography (LC) system equipped with a UV-Vis detector. A reverse-phase Zorbax Eclipse Plus C18 Rapid Resolution HD column (50 mm × 2.1 mm i.d., 1.8 μm particle size, Agilent) was used. The fluorophore (MU)-labeled compounds were separated with a 2 minute isocratic flow of 9% acetonitrile/91% H2O supplemented with 0.1% TFA and were detected by absorbance at 315 nm.
High resolution mass spectrometry (HRMS)-based donor substrate specificity studies of PmST1 P34H/M144L using one-pot multienzyme sialylation systems
One-pot three-enzyme reactions were carried out at 37 °C for 24 hours in a total volume of 10 μL in Tris-HCl buffer (100 mM, pH 8.5) containing LacβProN3 (1 mM), N-acetylmannosamine (ManNAc), mannose or derivatives as sialic acid precursors (1.5 mM), sodium pyruvate (5 mM), CTP (2 mM), PmAldolase (0.2 μg), NmCSS (0.15 μg), and PmST1 P34H/M144L (0.05 μg). Sialic acid precursors used were ManNGcOMe (A), mannose (B), ManNAc6N3 (C), ManNAc4N3 (D), ManNAc6OMe (E), and ManNAz (F).
One-pot two-enzyme reactions were carried out at 37 °C for 1 hour in a total volume of 10 μL in Tris-HCl buffer (100 mM, pH 8.5; pH 7.5 was used for the reaction containing 9-O-acetyl Neu5Ac) containing LacβProN3 (1 mM), sialic acid or derivatives (1.5 mM), CTP (2 mM), NmCSS (0.15 μg), and PmST1 P34H/M144L (0.05 μg). Sialic acids and derivatives used were Neu5Ac (G), Neu5Gc (H), Neu5Ac8OMe (I), and Neu5,9Ac2 (J).
Samples were analyzed by electrospray ionization (ESI)-HRMS using a Thermo Electron LTQ-Orbitrap Hybrid MS in a negative mode.
One-pot two-enzyme preparative-scale synthesis of sialylated products using PmST1 P34H/M144L in a one-pot two-enzyme system
To prepare sialylated LNnTβProN3, a reaction mixture in a total volume of 4 mL containing Tris-HCl buffer (100 mM, pH 8.5), Galβ1–4GlcNAcβ1–3Galβ1–4GlcβProN3 (LNnTβProN3, 30 mg, 0.038 mmol), Neu5Ac (11 mg, 0.038 mmol), CTP (30 mg, 0.057 mmol), MgCl2 (20 mM), NmCSS (1.2 mg), and PmST1 P34H/M144L (0.02 mg) were incubated in a shaker at 37 °C for 1 h. The reaction was stopped by adding 4 mL of ethanol followed by incubation at 4 °C for 30 min. After centrifugation, the supernatant was concentrated and passed through a Bio-Gel P-2 gel filtration column (water was used as an eluant). Further purification by semi-preparative HPLC using a C18 column provided monosialylated product Neu5Acα2–6Galβ1–4GlcNAcβ1–3Galβ1–4GlcβProN3 as a white powder (37.7 mg, 90%) and disialylated product Neu5Acα2–6Galβ1–4GlcNAcβ1–3(Neu5Acα2–6)Galβ1–4GlcβProN3 (2.7 mg, 5%), also as a white powder.
Neu5Acα2–6Galβ1–4GlcNAcβ1–3Galβ1–4GlcβProN3. 1H NMR (800 MHz, D2O) δ 4.70 (d, J = 8.0 Hz, 1H), 4.46 (d, J = 8.0 Hz, 1H), 4.43 (d, J = 8.0 Hz, 1H), 4.42 (d, J = 8.0 Hz, 1H), 4.14 (d, J = 3.2 Hz, 1H), 3.99–3.52 (m, 25H), 3.44 (t, J = 6.4 Hz, 2H), 3.30 (t, J = 8.8 Hz, 1H), 2.65 (d, J = 12.0 and 4.0 Hz, 1H), 2.04 (s, 3H), 2.01 (s, 3H), 1.89 (m, 2H), 1.70 (t, J = 12.0 Hz, 1H). 13C NMR (200 MHz, D2O) δ 174.81, 174.78, 173.43, 103.33, 102.82, 102.47, 101.98, 100.00, 81.89, 80.30, 78.22, 74.76, 74.65, 74.23, 74.13, 73.56, 72.66, 72.41, 72.29, 72.11, 71.59, 70.60, 69.84, 68.27, 68.23, 68.19, 68.08, 67.24, 63.22, 62.52, 60.84, 59.99, 59.90, 54.80, 51.76, 47.73, 39.94, 28.10, 22.16, 21.91. HRMS (ESI): m/z calculated for C40H67N5O29, m/z [M - H]−, calculated 1080.3843, found 1080.3833.
Neu5Acα2–6Galβ1–4GlcNAcβ1–3(Neu5Acα2–6)Galβ1–4GlcβProN3. 1H NMR (800 MHz, D2O) δ 4.70 (d, J = 8.0 Hz, 1H), 4.48 (d, J = 8.0 Hz, 1H), 4.43 (d, J = 8.0 Hz, 1H), 4.41 (d, J = 8.0 Hz, 1H), 4.16 (d, J = 3.2 Hz, 1H), 3.99–3.51 (m, 32H), 3.45 (t, J = 6.4 Hz, 2H), 3.32 (t, J = 8.8 Hz, 1H), 2.69 (d, J = 12.8 and 4.8 Hz, 1H), 2.65 (d, J = 12.8 and 4.8 Hz, 1H), 2.03 (s, 3H), 2.01 (s, 6H), 1.90 (m, 2H), 1.72 (t, J = 12.0 Hz, 1H), 1.70 (t, J = 12.0 Hz, 1H). 13C NMR (200 MHz, D2O) δ 174.86 (2C), 174.84, 173.52, 173.44, 103.52, 103.20, 102.60, 101.94, 100.25, 100.09, 82.16, 80.57, 79.63, 74.61, 74.58, 74.22, 73.64, 73.22, 72.66, 72.50, 72.49, 72.37, 72.30, 71.71, 71.67, 70.69, 69.62, 68.37, 68.34, 68.32(2C), 68.18, 68.11, 67.28, 63.42, 63.30, 62.61, 62.58, 60.19, 60.11, 54.89, 51.85, 51.74, 47.84, 40.09, 40.04, 28.20, 22.25, 22.01, 21.99. HRMS (ESI): C51H84N6O37, m/z [M/2 - 1]−, calculated 635.2358, found 635.2335.
Supplementary Material
Acknowledgments
This work was supported by US NIH grants R01HD065122 and U01GM120419. Bruker Avance-800 NMR spectrometer was funded by NSF grant DBIO-722538.
H.Y. and X.C. are co-founders of Glycohub, Inc., a company focused on the development of carbohydrate-based reagents, diagnostics, and therapeutics. Glycohub, Inc. played no role in the design, execution, interpretation, or publication of this study.
Footnotes
Electronic Supplementary Information (ESI) available: pH profiles and donor substrate promiscuity studies of PmST1 P34H/M144L. 1H and 13C NMR spectra for Neu5Acα2–6Galβ1–4GlcNAcβ1–3Galβ1–4GlcβProN3 (Neu5Acα2–6LNnTβProN3) and Neu5Acα2–6Galβ1–4GlcNAcβ1–3(Neu5Acα2–6)Galβ1–4GlcβProN3. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Varki A. Nature. 2007;446:1023–1029. doi: 10.1038/nature05816. [DOI] [PubMed] [Google Scholar]
- 2.Li Y, Chen X. Appl Microbiol Biotechnol. 2012;94:887–905. doi: 10.1007/s00253-012-4040-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen X, Varki A. ACS Chem Biol. 2010;5:163–176. doi: 10.1021/cb900266r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu H, Chokhawala HA, Huang S, Chen X. Nat Protoc. 2006;1:2485–2492. doi: 10.1038/nprot.2006.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu H, Chen X. Org Biomol Chem. 2016;14:2809–2818. doi: 10.1039/c6ob00058d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Angata T, Varki A. Chem Rev. 2002;102:439–469. doi: 10.1021/cr000407m. [DOI] [PubMed] [Google Scholar]
- 7.Schauer R. Glycoconj J. 2000;17:485–499. doi: 10.1023/A:1011062223612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cohen M, Varki A. OMICS. 2010;14:455–464. doi: 10.1089/omi.2009.0148. [DOI] [PubMed] [Google Scholar]
- 9.Jantscher-Krenn E, Zherebtsov M, Nissan C, Goth K, Guner YS, Naidu N, Choudhury B, Grishin AV, Ford HR, Bode L. Gut. 2012;61:1417–1425. doi: 10.1136/gutjnl-2011-301404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu H, Lau K, Thon V, Autran CA, Jantscher-Krenn E, Xue M, Li Y, Sugiarto G, Qu J, Mu S, Ding L, Bode L, Chen X. Angew Chem Int Ed. 2014;53:6687–6691. doi: 10.1002/anie.201403588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen X. Adv Carbohydr Chem Biochem. 2015;72:113–190. doi: 10.1016/bs.accb.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nycholat CM, Peng W, McBride R, Antonopoulos A, de Vries RP, Polonskaya Z, Finn MG, Dell A, Haslam SM, Paulson JC. J Am Chem Soc. 2013;135:18280–18283. doi: 10.1021/ja409781c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campbell JA, Davies GJ, Bulone V, Henrissat B. Biochem J. 1997;326(Pt 3):929–939. doi: 10.1042/bj3260929u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coutinho PM, Deleury E, Davies GJ, Henrissat B. J Mol Biol. 2003;328:307–317. doi: 10.1016/s0022-2836(03)00307-3. [DOI] [PubMed] [Google Scholar]
- 15.Yu H, Chokhawala H, Karpel R, Yu H, Wu B, Zhang J, Zhang Y, Jia Q, Chen X. J Am Chem Soc. 2005;127:17618–17619. doi: 10.1021/ja0561690. [DOI] [PubMed] [Google Scholar]
- 16.Yu H, Huang S, Chokhawala H, Sun M, Zheng H, Chen X. Angew Chem Int Ed. 2006;45:3938–3944. doi: 10.1002/anie.200600572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu H, Chen X. Org Lett. 2006;8:2393–2396. doi: 10.1021/ol060736m. [DOI] [PubMed] [Google Scholar]
- 18.Yao W, Yan J, Chen X, Wang F, Cao H. Carbohydr Res. 2015;401:5–10. doi: 10.1016/j.carres.2014.10.017. [DOI] [PubMed] [Google Scholar]
- 19.Huang S, Yu H, Chen X. Sci China Chem. 2011;54:117–128. doi: 10.1007/s11426-010-4175-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng J, Huang S, Yu H, Li Y, Lau K, Chen X. Glycobiology. 2010;20:260–268. doi: 10.1093/glycob/cwp172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meng X, Yao W, Cheng J, Zhang X, Jin L, Yu H, Chen X, Wang F, Cao H. J Am Chem Soc. 2014;136:5205–5208. doi: 10.1021/ja5000609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu Z, Liu Y, Ma C, Li L, Bai J, Byrd-Leotis L, Lasanajak Y, Guo Y, Wen L, Zhu H, Song J, Li Y, Steinhauer DA, Smith DF, Zhao B, Chen X, Guan W, Wang PG. Org Biomol Chem. 2016;14:11106–11116. doi: 10.1039/c6ob01982j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen C, Zhang Y, Xue M, Liu XW, Li Y, Chen X, Wang PG, Wang F, Cao H. Chem Commun. 2015;51:7689–7692. doi: 10.1039/c5cc01330e. [DOI] [PubMed] [Google Scholar]
- 24.Chandrasekaran A, Srinivasan A, Raman R, Viswanathan K, Raguram S, Tumpey TM, Sasisekharan V, Sasisekharan R. Nat Biotechnol. 2008;26:107–113. doi: 10.1038/nbt1375. [DOI] [PubMed] [Google Scholar]
- 25.Blixt O, Brown J, Schur MJ, Wakarchuk W, Paulson JC. J Org Chem. 2001;66:2442–2448. doi: 10.1021/jo0057809. [DOI] [PubMed] [Google Scholar]
- 26.Schur MJ, Lameignere E, Strynadka NC, Wakarchuk WW. Glycobiology. 2012;22:997–1006. doi: 10.1093/glycob/cws071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wakarchuk WW, Gilbert M, Martin A, Wu Y, Brisson JR, Thibault P, Richards JC. Eur J Biochem. 1998;254:626–633. doi: 10.1046/j.1432-1327.1998.2540626.x. [DOI] [PubMed] [Google Scholar]
- 28.Wakarchuk WW, Watson D, St Michael F, Li J, Wu Y, Brisson JR, Young NM, Gilbert M. J Biol Chem. 2001;276:12785–12790. doi: 10.1074/jbc.M011293200. [DOI] [PubMed] [Google Scholar]
- 29.Hidari KI, Horie N, Murata T, Miyamoto D, Suzuki T, Usui T, Suzuki Y. Glycoconj J. 2005;22:1–11. doi: 10.1007/s10719-005-0845-9. [DOI] [PubMed] [Google Scholar]
- 30.Ortiz-Soto ME, Seibel J. PLoS One. 2016;11:e0155410. doi: 10.1371/journal.pone.0155410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ni L, Chokhawala HA, Cao H, Henning R, Ng L, Huang S, Yu H, Chen X, Fisher AJ. Biochemistry. 2007;46:6288–6298. doi: 10.1021/bi700346w. [DOI] [PubMed] [Google Scholar]
- 32.Ni L, Sun M, Yu H, Chokhawala H, Chen X, Fisher AJ. Biochemistry. 2006;45:2139–2148. doi: 10.1021/bi0524013. [DOI] [PubMed] [Google Scholar]
- 33.Kim DU, Yoo JH, Lee YJ, Kim KS, Cho HS. BMB Rep. 2008;41:48–54. doi: 10.5483/bmbrep.2008.41.1.048. [DOI] [PubMed] [Google Scholar]
- 34.Iwatani T, Okino N, Sakakura M, Kajiwara H, Takakura Y, Kimura M, Ito M, Yamamoto T, Kakuta Y. FEBS Lett. 2009;583:2083–2087. doi: 10.1016/j.febslet.2009.05.032. [DOI] [PubMed] [Google Scholar]
- 35.Schmolzer K, Czabany T, Luley-Goedl C, Pavkov-Keller T, Ribitsch D, Schwab H, Gruber K, Weber H, Nidetzky B. Chem Commun. 2015;51:3083–3086. doi: 10.1039/c4cc09772f. [DOI] [PubMed] [Google Scholar]
- 36.Kakuta Y, Okino N, Kajiwara H, Ichikawa M, Takakura Y, Ito M, Yamamoto T. Glycobiology. 2008;18:66–73. doi: 10.1093/glycob/cwm119. [DOI] [PubMed] [Google Scholar]
- 37.Huynh N, Li Y, Yu H, Huang S, Lau K, Chen X, Fisher AJ. FEBS Lett. 2014;588:4720–4729. doi: 10.1016/j.febslet.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmolzer K, Ribitsch D, Czabany T, Luley-Goedl C, Kokot D, Lyskowski A, Zitzenbacher S, Schwab H, Nidetzky B. Glycobiology. 2013;23:1293–1304. doi: 10.1093/glycob/cwt066. [DOI] [PubMed] [Google Scholar]
- 39.Li Y, Sun M, Huang S, Yu H, Chokhawala HA, Thon V, Chen X. Biochem Biophys Res Commun. 2007;361:555–560. doi: 10.1016/j.bbrc.2007.07.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Talafova K, Hrabarova E, Nahalka J. J Biotechnol. 2015;216:116–124. doi: 10.1016/j.jbiotec.2015.09.031. [DOI] [PubMed] [Google Scholar]
- 41.Tsukamoto H, Takakura Y, Yamamoto T. J Biol Chem. 2007;282:29794–29802. doi: 10.1074/jbc.M701907200. [DOI] [PubMed] [Google Scholar]
- 42.Tsukamoto H, Takakura Y, Mine T, Yamamoto T. J Biochem. 2008;143:187–197. doi: 10.1093/jb/mvm208. [DOI] [PubMed] [Google Scholar]
- 43.Takakura Y, Tsukamoto H, Yamamoto T. J Biochem. 2007;142:403–412. doi: 10.1093/jb/mvm147. [DOI] [PubMed] [Google Scholar]
- 44.Sugiarto G, Lau K, Li Y, Khedri Z, Yu H, Le DT, Chen X. Mol Biosyst. 2011;7:3021–3027. doi: 10.1039/c1mb05182b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sugiarto G, Lau K, Qu J, Li Y, Lim S, Mu S, Ames JB, Fisher AJ, Chen X. ACS Chem Biol. 2012;7:1232–1240. doi: 10.1021/cb300125k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chokhawala HA, Yu H, Chen X. Chembiochem. 2007;8:194–201. doi: 10.1002/cbic.200600410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cao H, Muthana S, Li Y, Cheng J, Chen X. Bioorg Med Chem Lett. 2009;19:5869–5871. doi: 10.1016/j.bmcl.2009.08.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Khedri Z, Li Y, Muthana S, Muthana MM, Hsiao CW, Yu H, Chen X. Carbohydr Res. 2014;389:100–111. doi: 10.1016/j.carres.2014.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khedri Z, Muthana MM, Li Y, Muthana SM, Yu H, Cao H, Chen X. Chem Commun. 2012;48:3357–3359. doi: 10.1039/c2cc17393j. [DOI] [PubMed] [Google Scholar]
- 50.Guo Y, Jers C, Meyer AS, Arnous A, Li H, Kirpekar F, Mikkelsen JD. J Biotechnol. 2014;170:60–67. doi: 10.1016/j.jbiotec.2013.11.013. [DOI] [PubMed] [Google Scholar]
- 51.Yu H, Chokhawala HA, Huang S, Chen X. In: Chemical Glycobiology ACS Symposium Series 990. Chen X, Halcomb R, Wang PG, editors. Oxford University Press; 2008. pp. 96–122. [Google Scholar]
- 52.Guo Y, Jers C, Meyer AS, Li H, Kirpekar F, Mikkelsen JD. Enzyme Microb Technol. 2015;78:54–62. doi: 10.1016/j.enzmictec.2015.06.012. [DOI] [PubMed] [Google Scholar]
- 53.Haley SA, O’Hara BA, Nelson CD, Brittingham FL, Henriksen KJ, Stopa EG, Atwood WJ. Am J Pathol. 2015;185:2246–2258. doi: 10.1016/j.ajpath.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun M, Li Y, Chokhawala HA, Henning R, Chen X. Biotechnol Lett. 2008;30:671–676. doi: 10.1007/s10529-007-9588-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Watson DC, Wakarchuk WW, Leclerc S, Schur MJ, Schoenhofen IC, Young NM, Gilbert M. Glycobiology. 2015;25:767–773. doi: 10.1093/glycob/cwv017. [DOI] [PubMed] [Google Scholar]
- 56.Nov Y. Appl Environ Microbiol. 2012;78:258–262. doi: 10.1128/AEM.06265-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu G, Potter JA, Russell RJ, Oggioni MR, Andrew PW, Taylor GL. J Mol Biol. 2008;384:436–449. doi: 10.1016/j.jmb.2008.09.032. [DOI] [PubMed] [Google Scholar]
- 58.Xu G, Kiefel MJ, Wilson JC, Andrew PW, Oggioni MR, Taylor GL. J Am Chem Soc. 2011;133:1718–1721. doi: 10.1021/ja110733q. [DOI] [PubMed] [Google Scholar]
- 59.Tasnima N, Yu H, Li Y, Santra A, Chen X. Org Bomol Chem. 2016;15:160–167. doi: 10.1039/c6ob02240e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aharoni A, Thieme K, Chiu CP, Buchini S, Lairson LL, Chen H, Strynadka NC, Wakarchuk WW, Withers SG. Nat Methods. 2006;3:609–614. doi: 10.1038/nmeth899. [DOI] [PubMed] [Google Scholar]
- 61.Yang G, Rich JR, Gilbert M, Wakarchuk WW, Feng Y, Withers SG. J Am Chem Soc. 2010;132:10570–10577. doi: 10.1021/ja104167y. [DOI] [PubMed] [Google Scholar]
- 62.Ding L, Yu H, Lau K, Li Y, Muthana S, Wang J, Chen X. Chem Commun. 2011;47:8691–8693. doi: 10.1039/c1cc12732b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ding L, Zhao C, Qu J, Li Y, Sugiarto G, Yu H, Wang J, Chen X. Carbohydr Res. 2015;408:127–133. doi: 10.1016/j.carres.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yu H, Yu H, Karpel R, Chen X. Bioorg Med Chem. 2004;12:6427–6435. doi: 10.1016/j.bmc.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 65.Li Y, Yu H, Cao H, Lau K, Muthana S, Tiwari VK, Son B, Chen X. Appl Microbiol Biotechnol. 2008;79:963–970. doi: 10.1007/s00253-008-1506-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Strecker G, Wieruszeski JM, Michalski JC, Montreuil J. Glycoconj J. 1989;6:67–83. doi: 10.1007/BF01047891. [DOI] [PubMed] [Google Scholar]
- 67.Sabesan S, Bock K, Paulson JC. Carbohydr Res. 1991;218:27–54. doi: 10.1016/0008-6215(91)84084-r. [DOI] [PubMed] [Google Scholar]
- 68.Yu H, Zeng J, Li Y, Thon V, Shi B, Chen X. Org Biomol Chem. 2016;14:8586–8597. doi: 10.1039/c6ob01706a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.