

Abstract
Many studies seek to explore the impact of extrinsic soluble factors present in serum, interstitial fluids or cell-conditioned media on cells in vitro. A convenient approach to elucidate the effects of a particular factor is its selective neutralization. However, intrinsic production of soluble factors such as cytokines by the cultured cells is common and can have an impact via autocrine mechanisms. The addition of cytokine-specific neutralizing antibodies leads to neutralization of the targeted factors irrespective of their source and affects paracrine and autocrine effects alike. Thus, neutralization assays are not suitable to irrevocably demonstrate that the examined factors exert their effect via a paracrine mechanism. We were interested in investigating the impact of immunosuppressive factors present in ovarian carcinoma-associated ascites by dissecting paracrine versus autocrine effects of interleukin 10 (IL-10) and prostaglandin E2 (PGE2) on the activation of monocyte-derived dendritic cells (DC). We explored several methods of depletion based on introduction of the neutralizing antibodies bound to beads. Here we describe the pitfalls of the investigated depletion approaches and show the importance of monitoring the presence of residual neutralizing antibodies in the sample upon depletion, which impacts on the suitability of the approach to distinguish paracrine from autocrine effects. Only one of three investigated approaches showed no dislocation of neutralizing antibody from the beads into the sample. This method, which is based on covalently linking antibody to magnetic beads harbouring a reactive group allowed for the complete removal of the investigated factors from ascites and represents an elegant tool to elucidate immunoregulatory or -stimulatory cytokine networks in considerably more depth than the use of neutralizing antibodies in cell cultures alone can contribute.
Keywords: Depletion, Antibodies, Cytokines, Beads, Autocrine, Ascites
1. Introduction
Development of ascites is a hallmark symptom of the late stages of ovarian carcinoma, and soluble tumour-associated factors within can affect disease progression and interfere with anti-tumour vaccination strategies (Giuntoli et al., 2009, Yigit et al., 2011). Studies of cytokines in the tumour environment are frequently conducted in vitro by addition of biological fluids such as malignant ascites or tumour-conditioned media to cell cultures, while functional characterization of individual proteins is attempted by their selective antibody-mediated neutralization. In our laboratory, we investigate how factors within ovarian carcinoma-associated ascites affect Toll-like receptor (TLR)-mediated activation of monocyte-derived DC. Ovarian carcinoma-associated ascites contains a complex mixture of anti- and pro-inflammatory factors which can differ significantly between individual patients and which has the potential to influence the activation of various cell types (Chen et al., 2009, Mustea et al., 2009, Nowak et al., 2010, Yigit et al., 2010). We observed that ascites partially impairs DC activation in response to TLR agonists in vitro (unpublished data) and we decided to selectively neutralize various factors using specific antibodies to identify the mechanism by which ascites reduces DC activation. IL-10 is one well-known immunosuppressive factor present in ovarian carcinoma ascites and has previously been implicated to play an important role in disease progression and prognosis (Punnonen et al., 1998, Santin et al., 2001, Mustea et al., 2006, Zhou et al., 2007, Giuntoli et al., 2009, Yigit et al., 2011, Matte et al., 2012). We therefore considered IL-10 to be a candidate protein potentially important to the immunosuppressive effects of ascites. Tumour cells, but also tumour-associated antigen-presenting cells have been shown to produce IL-10 and are thought to contribute to the IL-10 levels in ascites (Melichar et al., 1998, Loercher et al., 1999, Zhou et al., 2007). Incidentally, DC produce IL-10 upon TLR activation (Jarrossay et al., 2001, Boonstra et al., 2006). IL-10 induction under pro-inflammatory conditions is an important protective mechanism of the immune system preventing excessive and uncontrolled immune activation. Because of the induction of DC-derived IL-10, blocking experiments based on the addition of neutralizing antibodies to DC cultures cannot conclusively attribute the suppression to ascites-derived paracrine IL-10.
To gain a better understanding of the influence of ascites on DC activation, we explored the relative importance of ascites-derived versus autocrine IL-10 in our experimental system. Our aim was to deplete this protein from ascites before addition of the fluid to cell cultures, allowing us to discriminate between the distinct influences of autocrine versus paracrine IL-10. The establishment of a suitable protocol proved challenging, but we eventually succeeded and present here a method for selective depletion of proteins from ascites. In addition to IL-10, we expanded the application to another immunosuppressive factor, namely PGE2 (Kalinski, 2012), which we had also identified as a candidate protein in malignant ascites that may partially impair DC activation (unpublished data).
2. Material and methods
2.1. Patient specimens
The collection of ascites for this study was approved by the Guy's Research Ethics Committee (REC Number: 09/H0804/45). Informed consent was obtained from all participants. Ascites from patients suffering from advanced stage (FIGO stage III or IV) high grade serous ovarian carcinoma was collected by paracentesis (St Thomas' Hospital, London, UK). Upon receipt, ascites samples were centrifuged for 10 min at 270g, allowing the separation of the fluid into a cellular fraction and non-cellular supernatant. Only the non-cellular supernatant was used in this study. The material was stored in aliquots at − 80 °C. Upon thawing, before cytokine depletion and use in cell cultures, the non-cellular fluid was passed through a syringe-driven sterile filter of 0.2 μm pore size (Appleton Woods, Birmingham, UK).
2.2. Neutralizing antibodies
IL-10 neutralizing antibody (clone 25,209; RRID: AB_358066) and the corresponding mouse IgG2b isotype control antibody (clone 20,116; RRID: AB_357344) were from R&D Systems (Abingdon, UK). Neutralizing αPGE2 antibody (clone 2B5; mouse IgG1; RRID: AB_327973) was from Cayman Europe (Tallinn, Estonia) and the corresponding mouse IgG1 isotype control antibody (clone 11,711; RRID: AB_357346) was purchased from R&D Systems.
2.3. Depletion of IL-10 using protein G-coated agarose beads
IL-10 neutralizing antibody or relevant mouse IgG2b isotype control antibody were added to sterile filtered ascites at concentrations of 5 μg/ml or 20 μg/ml and the samples were incubated for 30 min at 4 °C. After addition of Protein G agarose beads (Thermo Scientific, Cramlington, UK) (100 μl beads per 1 ml of ascites) the samples were incubated for 30 min at room temperature (RT). With a binding capacity of 2 mg of antibody per 100 μl the amount of Protein G-coupled agarose beads exceeded the amount of used antibody by 400 × (5 μg/ml) and 100 × (20 μg/ml) fold, respectively. Samples were then centrifuged at 420g for 5 min, and the supernatant was carefully harvested. Centrifugation at identical speed and duration was repeated to ensure complete removal of residual agarose beads.
2.4. Depletion of IL-10 using protein G coated magnetic beads
For immobilization of αIL-10 antibody or relevant mouse IgG2b isotype control antibody on PureProteome Protein G Magnetic Beads (Millipore, Livingston, UK), the manufacturer's protocol was followed. Briefly, 50 μl Protein G Magnetic Beads were incubated with 10 μg/ml αIL-10 neutralizing antibody or respective mouse IgG2b isotype control antibody for 10 min at RT with continuous mixing. Beads were then washed in 500 μl PBS containing 0.1% Tween 20 with the help of the PureProteome magnetic stand (Millipore). Subsequently, 200 μg chromatographically purified mouse IgG (Invitrogen, Paisley, UK) was added to each sample in order to saturate vacant IgG-binding sites on Protein G Magnetic Beads. Incubation for 10 min at RT with continuous mixing was followed by 3 × washing in 500 μl PBS containing 0.1% Tween 20. Antibody-coated Protein G Magnetic Beads were added to 1 ml sterile filtered ascites and incubated for 1 h or overnight at 4 °C with continuous mixing. After incubation, magnetic beads were removed by placing the microcentrifuge tubes into the magnetic stand and transfer of the bead-free ascites into a fresh microcentrifuge tube. This procedure was repeated two more times to ensure complete removal of magnetic beads.
2.5. Depletion of IL-10 or PGE2 using NHS coated magnetic beads
For conjugation of αIL-10 antibody, αPGE2 antibody or relevant isotype control antibodies (mouse IgG2b or IgG1) to PureProteome N-Hydroxysuccinimide (NHS) FlexiBind Magnetic Beads (Millipore), the manufacturer's protocol was followed. To ensure successful coupling of antibody to the PureProteome NHS Magnetic Beads, the antibodies must be present at a concentration of 2 mg/ml or above. The stock concentration of both isotype control antibodies as well as the αIL-10 antibody was 500 μg/ml and a further concentration was therefore required to reach 2 mg/ml. For this, a defined amount of antibody (100 μg) in solution was pipetted into an Amicon® Ultra-0.5 30 K Device (Millipore), and centrifuged at 14,000 × g for 7 min. The obtained concentrated antibody solution was recovered from the device and the volume was adjusted to 50 μl with PBS, resulting in a final antibody concentration of 2 mg/ml. The αPGE2 antibody was supplied at a concentration of 2 mg/ml and no further concentration was therefore required.
For the coupling of antibodies to PureProteome NHS Magnetic Beads, 100 μl bead slurry was added to a microcentrifuge tube (Costar® Sigma-Aldrich), and beads were washed 1 × with ice-cold equilibration buffer (1 mM HCl; Millipore) with the help of PureProteome Magnetic Stand (Millipore). 50 μl of respective antibody solution (2 mg/ml) was added immediately, followed by incubation for 2 h at RT. Subsequently, beads were washed 5 × in the magnetic stand with quench buffer (100 mM Tris-HCl, 150 mM NaCl, pH 8.0; Millipore) followed by an incubation of 2 h at RT in quench buffer. The beads were subsequently washed 4 × with wash/coupling buffer (PBS, pH 7.4; Millipore) and finally resuspended in 100 μl of PBS. Antibody-coated beads were stored at 4 °C until used. For the depletion, the αIL-10 or αPGE2 antibody-coated beads were incubated with ascites from ovarian carcinoma patients (20 μl antibody-conjugated bead slurry per 1 ml ascites) overnight at 4 °C with continuous mixing. To control for the specificity of the depletion, isotype control mouse IgG coated beads were incubated with ascites samples for mock depletion. After overnight incubation, beads were removed from ascites samples using the PureProteome Magnetic Stand. Ascites depleted of IL-10, PGE2 and mock-depleted samples were stored at − 80 °C until their use in activation assays.
2.6. Detection of IL-10 or PGE2 in depleted ascites samples
To verify successful depletion of IL-10 or PGE2 from ascites, levels of IL-10 were measured by Flow Cytomix® human IL-10 Simplex Kit (eBioscience, Hatfield, UK). The manufacturer's protocol for Simplex Kits was followed using dilution buffers, reagents and a 96-well filter plate provided in the Flow Cytomix Basic Kit (eBioscience). Samples were transferred into FACS tubes for acquisition on the FACS Canto II (BD), and data was analyzed and quantified using Flow Cytomix® Analysis Software (eBioscience).
For measurement of PGE2, High Sensitivity PGE2 Parameter Assay kit (Arbor Assays, Ann Arbor, MI, U.S.A.) was used. The manufacturer's protocol was followed, using a pre-coated 96-well plate, dilution buffers and reagents provided in the kit. Optical density (OD) was measured on an ELISA microplate reader (Biorad, Hemel Hempstead, UK) at 450 nm and all samples were evaluated with reference to the standard curve for PGE2 quantification.
2.7. Detection of mouse IgG1 and mouse IgG2b in depleted ascites samples
For detection of mouse IgG1 or mouse IgG2b in ascites after depletion, Immuno MAXISORP ELISA 96-well plates (Nunc, Roskilde, Denmark) were coated with purified anti-mouse IgG1 antibody (rat IgG clone RMG1–1; BioLegend; RRID: AB_315061) or purified anti-mouse IgG2b antibody (rat IgG clone RMG2b-1; BioLegend; RRID: AB_315065) diluted in PBS (for both anti-mouse IgG1 and IgG2b 2 μg/ml coating antibody concentration was used). Plates were incubated overnight at 4 °C, and subsequently washed twice with wash buffer (PBS containing 0.05% Tween 20) before non-specific binding sites were blocked by incubating all wells with PBS containing 10% heat-inactivated FCS; Sigma Aldrich) for 2 h at RT. After washing twice, standard (mouse IgG1 clone 11,711 or mouse IgG2b clone 20,116; both R&D Systems) and samples were added to triplicate wells at appropriate dilutions in PBS. The anticipated IgG concentrations in ascites were difficult to estimate, and therefore a wide range of sample dilutions (undiluted, 1:10, 1:100, 1:1000, 1:10,000) was used in all ELISA experiments to ensure that the values fell within the sensitivity range of the assay. After overnight incubation at 4 °C, plates were washed 4 × with wash buffer and biotinylated anti-mouse IgG detection antibody (goat Ig clone Poly4053; BioLegend; RRID: AB_315006) was added to all wells (0.5 μg/ml final antibody concentration) and allowed to incubate for 1 h at RT. After 4 × washing, ExtrAvidin®-Alkaline Phosphatase (Sigma-Aldrich) was added to all wells (1:5000 dilution in PBS) and plates were incubated for 1 h at RT. Plates were washed 4 ×, followed by application of SIGMAFAST pNitrophenyl phosphate (PNPP) and buffer Tablets (Sigma-Aldrich) dissolved in deionised water. The OD was measured on an ELISA microplate reader (Biorad) at 405 nm and all samples were evaluated with reference to the standard curve for protein quantification. The ELISA had a sensitivity of 0.1 ng/ml for both mouse IgG2b and mouse IgG1.
2.8. Detection of human IgG in ascites samples
For detection of human IgG in ascites samples before and after depletion, Human IgG total Ready-SET-Go!® kit (Affymetrix eBioscience, Hatfield, UK) was used. The manufacturer's protocol was followed, using Immuno MAXISORP ELISA 96-well plates together with antibodies, dilution buffers and reagents provided in the kit. The OD was measured on an ELISA microplate reader at 450 nm and all samples were evaluated with reference to the standard curve for IgG quantification.
2.9. Activation assay of monocyte-derived DC with LPS and ascites
The collection of peripheral blood for this study was approved by the Guy's Research Ethics Committee (REC Number: 09/H0804/45). Informed consent was obtained from all participants who gave blood by venipuncture. Peripheral blood from healthy volunteers was obtained by venipuncture and collected in heparinized vacutainer tubes (BD). Peripheral blood mononuclear cells (PBMC) were obtained from whole blood by Ficoll (Lymphocyte Separation Medium 1077; PAA Laboratories, Yeovil Somerset, UK) density gradient centrifugation. CD14+ monocytes were isolated from PBMC by negative selection using the EasySep® Human Monocyte Enrichment Kit (StemCell Technologies, Grenoble, France) following the manufacturer's instructions. Enriched CD14+ monocytes were seeded on 6-well-plates at a density of 6-9 × 106 cells/well in 3 ml complete RPMI 1640 medium supplemented with 10% FCS, 2 mM glutamine, 100 U/ml penicillin, 100 mg/ml streptomycin and 50 μM 2-Mercaptoethanol. Granulocyte-macrophage colony stimulating factor (GM-CSF) and IL-4 (both R&D Systems) were added at a concentration of 20 ng/ml each. Cells were cultured at 37 °C in a 5% CO2 atmosphere for 7 days. GM-CSF and IL-4 were replenished on day 2 and day 5 along with 500 μl fresh complete RPMI 1640 medium per well. Cells were harvested on day 7 of culture. Subsequently, monocyte-derived DC were seeded on 96-well tissue culture plates at a density of 1 × 105 cells/well in complete RPMI 1640 medium, with a final well volume of 200 μl. Triplicate wells were used for all samples. The reagents were added to the cells in the following order: First the neutralizing or control antibody was added, followed by the ascites and then the LPS. TLR4 agonist LPS was used at 1 μg/ml, and IL-10 depleted or undepleted, sterile filtered ascites was added to respective wells to reach 10% final cell culture concentration. αIL-10 neutralizing antibody or mouse IgG2b isotype control antibody were added at a concentration of 5 μg/ml where indicated. Cells were incubated for 18 h overnight at 37 °C in a 5% CO2 atmosphere. Supernatants were harvested and analyzed for IL-6 levels by ELISA.
2.10. Detection of IL-6 levels in cell culture supernatants
Immuno MAXISORP ELISA 96-well plates were coated with 4 μg/ml αIL-6 capture antibody (rat IgG1 clone MQ2-13A5; BD; RRID: AB_398568) diluted in 0.1 M Na2HPO4, pH 9.0 and incubated overnight at 4 °C. Plates were washed 2 × with PBS containing 0.05% Tween 20 and non-specific binding sites were blocked by incubating all wells with blocking buffer (PBS containing 2.5% FCS and 0.02% NaN3) for 2 h at RT. After washing twice, IL-6 standard (BD) and samples were added to triplicate wells at appropriate dilutions in blocking buffer. Samples without LPS were measured neat. Samples stimulated with LPS were diluted 1:50 in order to ensure that the values fell within the sensitivity range of the assay. After overnight incubation at 4 °C, plates were washed 4 × and αIL-6 detection antibody (rat IgG2a clone MQ2-39C3; BD; RRID: AB_395470) diluted in blocking buffer was added to all wells at a concentration of 2.5 μg/ml and allowed to incubate for 1 h at RT. After four washes, ExtrAvidin®-Alkaline Phosphatase was added to all wells and plates were incubated for 1 h at RT. Again plates were washed 4 × followed by application of SIGMAFAST PNPP and buffer Tablets dissolved in deionised water. OD was measured on an ELISA microplate reader and all samples were evaluated with reference to the standard curve for IL-6 quantification.
2.11. Statistical analysis
Data were analyzed using PRISM Graph Software (La Jolla, California, U.S.A.). Appropriate statistical tests were applied for different experiments, taking into consideration the composition of each experiment, relevant experimental cohorts and scientific questions. Differences were considered significant at a p-value < 0.05.
3. Results
3.1. Depletion of IL-10 from ovarian carcinoma associated ascites using αIL-10 antibody and protein G-coupled agarose beads
The initial approach we took to deplete IL-10 from ascites comprised addition of IL-10 specific neutralizing antibody to ascites, enabling IL-10 in ascites to bind to this antibody, forming antibody-antigen complexes. Subsequently, we intended to remove such complexes by centrifugation after addition of agarose beads coated with Protein G, a streptococcal cell wall protein with a high affinity to the Fc and Fab regions of immunoglobulins (Grubb et al., 1982, Kraulis et al., 1996). The affinity of Protein G varies considerably for antibodies of different isotypes and host species. Its very high affinity for mouse IgG2b antibody, the isotype of the αIL-10 neutralizing antibody, made Protein G a suitable candidate for this depletion.
In our initial experiment, we used two different concentrations of αIL-10 neutralizing antibody. Two ovarian carcinoma ascites samples were incubated with 5 μg/ml αIL-10 antibody, while for four ascites samples, the concentration of 20 μg/ml αIL-10 antibody was chosen. To determine whether IL-10 was successfully removed from all samples, the depletion was verified by measurement of IL-10 in the depleted samples. Indeed, IL-10 was not detectable in the samples depleted with the antibody concentration of 20 μg/ml (data not shown). While this could be interpreted as successful IL-10 depletion from ascites, we could not exclude the possibility that complexes of IL-10 and αIL-10 antibody were still present and that IL-10 had, in fact, not been depleted but was simply neutralized and therefore not detectable. Even more importantly, we had to exclude the possibility that unbound αIL-10 antibody with the capacity to neutralize autocrine DC-derived IL-10 had remained in the ascites samples undergoing the depletion procedure. We therefore determined whether αIL-10 antibody had been completely removed by Protein G agarose beads by measuring the concentration of mouse IgG2b antibody in the depleted ascites samples (Fig. 1A). The two samples depleted with a concentration of 5 μg/ml αIL-10 antibody showed levels of approximately 4 μg/ml mouse IgG2b, indicating that 80% of the αIL-10 antibody remained in the supernatant after centrifugation. Among the four samples depleted using 20 μg/ml, the IgG2b concentrations ranged from 13 to 17 μg/ml. The residual αIL-10 antibody posed a considerable problem, as upon addition of the depleted ascites to cell cultures, residual unbound IL-10-specific antibody could potentially neutralize autocrine IL-10.
Fig. 1.
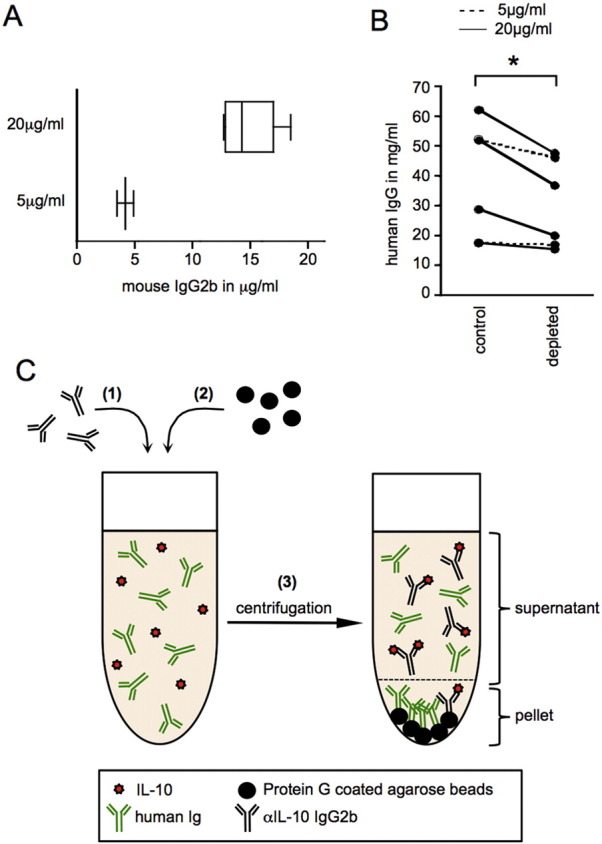
Depletion of IL-10 from ascites using αIL-10 neutralizing antibody and Protein G-coated agarose beads.
(A) Levels of mouse IgG2b αIL-10 neutralizing antibody in ascites samples were measured after depletion of IL-10 using Protein G-coated agarose beads. IL-10 depletion was performed with 5 μg/ml neutralizing antibody (lower panel) in two (n = 2) and with 20 μg/ml neutralizing antibody (top panel) in four (n = 4) ascites samples. The mouse IgG2b antibody concentration was determined by sandwich ELISA. (B) Human IgG levels in ascites were measured by sandwich ELISA before (control) and after depletion with Protein G-coated agarose beads (depleted). The two experiments where 5 μg/ml neutralizing antibody was used are shown as dashed lines (n = 2), while in the remaining experiments (n = 4) shown as solid lines, a concentration of 20 μg/ml neutralizing antibody was used. Wilcoxon matched pairs test, two-tailed. * = p < 0.05 (C) Schematic depiction of the pitfalls encountered with this experimental approach. (1) Neutralizing αIL-10 mouse IgG2b antibody is added to ovarian carcinoma associated ascites, which contains a high concentration of human IgG. Upon incubation, the neutralizing αIL-10 antibody binds IL-10 present in ascites. (2) Protein G-coated agarose beads are added to the sample. Human IgG competes with IL-10-specific mouse IgG2b antibody for Protein G-binding sites. (3) Centrifugation creates a pellet of agarose beads with predominantly bound human immunoglobulins, leaving IL-10-specific antibody in form of immune complexes with IL-10 or in form of free antibody not bound to its antigen behind in the ascites.
Given that ascites is abundant with soluble proteins including immunoglobulins (Yildirim et al., 2002), we hypothesized that the Protein G-coated agarose beads not only reacted with the IL-10-specific mouse antibody, but also bound to human immunoglobulins present in ascites. Competition between the human immunoglobulins and the αIL10 antibody for binding by protein G-coupled agarose beads could explain why the depletion of the latter was inefficient. To explore whether this was the case, we measured the concentration of human IgG in ascites before depletion and thereafter (Fig. 1B). In all six ascites samples, we detected reduced human IgG levels after depletion, indicating that human IgG was being partially depleted with the Protein G-coated agarose beads used in this experiment. The levels of IgG in different ascites samples differed considerably, and it appears that in samples with higher IgG content, the reduction in IgG levels after depletion was particularly dramatic. These findings support the notion that direct competition with human IgG in ascites can interfere with the depletion of particular ascites-associated factors using neutralizing antibodies in combination with protein G-coupled beads, as schematically illustrated in Fig. 1C.
To summarize, the specific depletion of IL-10 from ovarian carcinoma ascites using Protein G-coated agarose beads for removal of complexes of IL-10 and αIL-10 antibody was not successful, and a different protocol for IL-10 depletion had to be identified.
3.2. Depletion of IL-10 from ascites using magnetic protein G beads pre-coated with αIL-10 specific neutralizing antibody
Despite the problems we encountered with the use of Protein G coated agarose beads as explained above, Protein G remained an agent worth considering for our purposes because of its very high affinity to immunoglobulins and its frequent use in antibody purification and immobilization protocols. To avoid the direct competition for binding sites between neutralizing antibody and ascites-intrinsic proteins, in our next approach we pre-coated magnetic Protein G beads with αIL-10 antibody before addition of such complexes to ascites samples. We hoped that competition between the neutralizing IL-10-specific antibody and the human immunoglobulins in ascites would be reduced if the former had been bound to magnetic Protein G-coated beads prior to addition to ascites samples.
The binding capacity of the used magnetic beads coated with Protein G was very high (175 μg IgG per 50 μl according to the supplier), and saturating the beads using αIL-10 neutralizing antibody was not feasible due to the high costs. However, we were reluctant to use partially saturated Protein G beads, since our previous protocol has shown that Protein G binds human IgG from ascites, leading to its partial depletion and resulting in an altered composition of ascites samples. To avoid this, after coating beads with αIL-10 specific antibody, we saturated the beads using chromatographically purified mouse IgG before using the pre-coated beads for depletion of IL-10 from ascites.
In our first depletion attempt with this method, we incubated three ascites samples with the pre-coated beads overnight with continuous mixing. After removal of bead complexes by magnetic separation, we measured IgG2b levels in the depleted samples. The amount of αIL-10 antibody used to pre-coat the magnetic beads had been 10 μg. Disappointingly, the detected levels of mouse IgG2b in these samples reached levels of 10 μg or beyond (Fig. 2A), indicating that during incubation, αIL-10 antibody and/or chromatographically purified mouse IgG antibody had dissociated from the beads. We suspected that this may have been due to the long overnight incubation time, and hence repeated the experiment with one ascites sample, reducing the incubation time to 1 h. However, even upon reduction of the incubation time, the level of mouse IgG2b detected in the depleted ascites sample was approaching 10 μg (Fig. 2A). To establish whether mouse IgG2b had been displaced as a result of competition with human IgG, we determined the concentration of human IgG in ascites samples before and after depletion (Fig. 2B). While in three samples we noted a slight decrease in IgG levels, this was not as prominent as seen previously with Protein G-coated agarose beads. Since the exact interactions between the different sources of antibodies and the magnetic Protein G-coupled beads during incubation and magnetic separation are unknown, it remains unclear to what extent αIL-10 antibody bound to Protein G-coupled beads is replaced by human IgG (Fig. 2C). Independent of the alteration of the ascites composition with regards to human IgG levels with this experimental protocol, the residual mouse IgG2b antibody in depleted samples still posed a considerable problem. We therefore concluded that despite its high affinity to mouse IgG, Protein G is not suitable for antibody-mediated depletion of specific cytokines from ascites. Collectively, these findings prompted us to explore alternative methods for IL-10 depletion, with specific focus on improving the stability of the binding of αIL-10 antibody to magnetic particles.
Fig. 2.
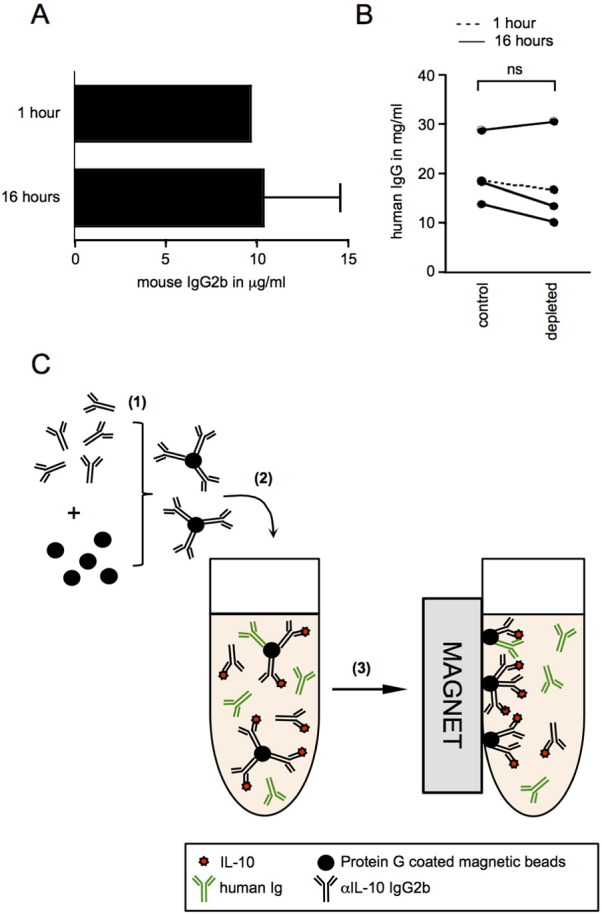
Depletion of IL-10 from ascites using Protein G magnetic beads coated with αIL-10 neutralizing antibody.
(A) Levels of mouse IgG2b αIL-10 neutralizing antibody in ascites samples were measured after depletion of IL-10 using Protein G coated magnetic beads. The depletion was performed with 1 h (n = 1) and 16 h (n = 3) incubation periods for αIL-10 antibody-coated beads in ascites. The mouse IgG2b concentration was quantified by sandwich ELISA. (B) Human IgG levels in ascites were measured by sandwich ELISA before (control) and after depletion with Protein G-coated magnetic beads (depleted). Values before and after 16 h incubation of ascites with antibody-coated Protein G beads are connected by a solid line, while the dashed line indicates human IgG values in ascites before and after an incubation of 1 h. Wilcoxon matched pairs test, two-tailed; ns = not significant. (C) Schematic illustration of the drawbacks of this experimental method. (1) Protein G-coated magnetic beads are incubated with mouse IgG2b αIL-10 neutralizing antibody. The antibody binds to Protein G beads with high affinity. (2) The beads pre-coated with αIL-10-specific antibody are added to ascites to allow binding of IL-10. Partial displacement of mouse IgG2b αIL-10 neutralizing antibody and/or mouse IgG2b control antibody is observed. (3) After removal of Protein G magnetic beads and bound immunoglobulins with the help of a magnet, αIL-10 antibody either with or without bound IL-10 as well as the isotype control antibody are left behind in the ascites.
3.3. Successful depletion of IL-10 from ascites using neutralizing antibodies covalently bound to NHS ester coated magnetic beads
Commercially available magnetic beads coated with NHS ester groups (Millipore) have the ability to covalently bind free amine residues and can be used to permanently immobilize a range of proteins including antibodies. Since the bonds between the neutralizing antibody and the beads are covalent, competition with ascites-associated antibodies should not pose a problem and displacement of the neutralizing antibody should be negligible (Fig. 3A). We, therefore, hypothesized that incubation of such antibody-coupled magnetic beads generated with αIL-10 neutralizing antibody supports the removal of IL-10 from ascites by magnetic separation without affecting the human IgG levels in the samples.
Fig. 3.
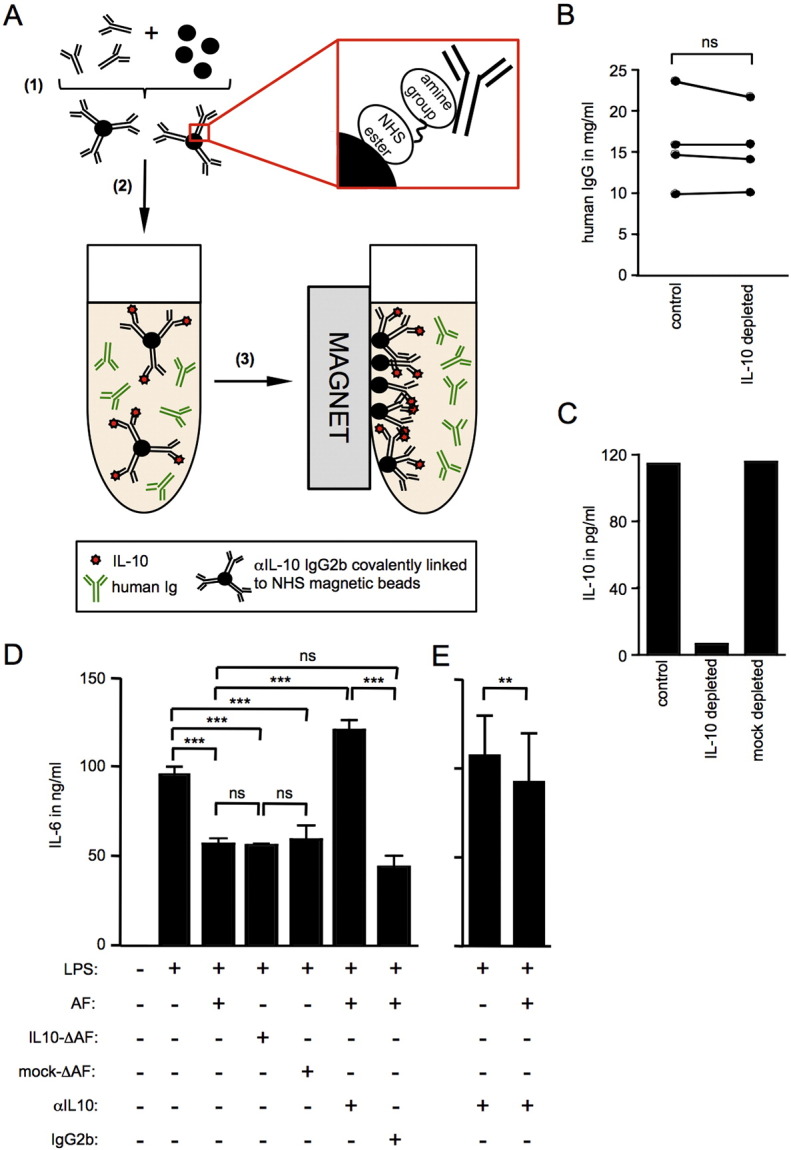
Successful depletion of IL-10 from ovarian carcinoma associated ascites using NHS magnetic beads for investigation of paracrine effects.
Schematic display of IL-10 depletion with αIL-10 antibody covalently bound to magnetic beads. (1) Magnetic NHS ester beads are biochemically linked to mouse IgG2b αIL-10 neutralizing antibody or isotype control antibody. The bonds between the NHS ester group on the beads and free amines of the antibody are covalent, permanently immobilizing the antibody on the bead surface. (2) Antibody-coated beads are added to ascites and incubated overnight to allow specific binding of IL-10 to its neutralizing antibody. (3) A magnet is used to remove complexes of magnetic beads, αIL-10 neutralizing antibody and IL-10. (B) Human IgG levels in ascites were measured by sandwich ELISA before (control) and after depletion αIL-10 antibody bound to magnetic NHS beads (IL-10 depleted). n = 4; Wilcoxon matched pairs test, two-tailed; ns = not significant. (C) IL-10 levels in depleted versus non-depleted control or mock-depleted ascites sample were measured by FlowCytomix bead ELISA (n = 1). The IL-10 level in the depleted sample (6.41 pg/ml) falls below assay sensitivity (27 pg/ml). (D) Monocyte-derived DC were stimulated with 1 μg/ml LPS in the presence or absence of 10% ascites from ovarian carcinoma patients which had previously been depleted of IL-10 (IL10-ΔAF) or which had undergone mock depletion (mock-ΔAF). Alternatively, DC cultures were treated with LPS and untreated ascites fluid (AF) in the presence of αIL-10 neutralizing antibody (5 μg/ml); n = 3 (3 independent experiments; monocyte-derived DC from 1 healthy donor, cultured with three ascites samples from three individual ovarian carcinoma patients). IL-6 levels were measured in cell culture supernatants by sandwich ELISA. One-way ANOVA (Friedman test with Bonferroni post-test); ***p < 0.001. (E) As in D; n = 6 (monocyte-derived DC from 3 healthy donors, cultured with two ascites samples from two individual ovarian carcinoma patients). Wilcoxon matched pairs test, two-tailed; **p < 0.01.
We applied this method to deplete IL-10 from four ovarian carcinoma associated ascites samples. In order to confirm that the αIL-10 neutralizing antibody is not displaced from the beads during the depletion process, we measured mouse IgG2b antibody levels in IL-10- and mock-depleted ascites. The mouse IgG2b specific ELISA of the depleted samples showed residual αIL-10 antibody levels of 0.5–1.3 ng/ml (Table 1). The manufacturer's antibody titration guidelines suggest that concentrations of this antibody of 10 ng/ml or above are necessary to achieve neutralization of 0.1 ng/ml IL-10. In our experimental system, the levels of autocrine IL-10 produced by monocyte-derived DC after TLR-mediated activation reached levels of 1 ng/ml or higher (data not shown), which would require the addition of at least 100 ng/ml of αIL-10 antibody for successful neutralization. Further, when using ascites in cell culture, it was typically not used neat but was added to cell culture media at a final concentration of 10%, leading to a further 10-fold dilution of residual αIL-10 antibody. We were therefore confident that this amount of neutralizing antibody would not interfere with autocrine IL-10 production in our cell cultures and was therefore negligible. Furthermore, we determined the concentration of human IgG in ascites before and after depletion (Fig. 3B). Our results show that human IgG was not depleted from ascites by this protocol.
Table 1.
Covalent binding of αIL-10 neutralizing antibody to NHS coated magnetic beads leads to minimal residual αIL-10 antibody in depleted ascites samples.
| Samples | αIL-10 antibody | IgG2b isotype |
|---|---|---|
| #1 | 1.3 ng/ml | 2.0 ng/ml |
| #2 | 0.8 ng/ml | 2.3 ng/ml |
| #3 | 0.5 ng/ml | 2.5 ng/ml |
| #4 | 0.6 ng/ml | 2.5 ng/ml |
Levels of mouse IgG2b antibody were measured in four individual ascites samples after incubation with NHS magnetic beads bound to αIL-10 or isotype control antibody. The mouse IgG2b concentration was quantified by sandwich ELISA.
The successful depletion of IL-10 was further verified by IL-10 specific bead ELISA (Fig. 3C and Table 2). IL-10 levels of 2.9–6.41 pg/ml were detected in the depleted samples, which was considerably below assay sensitivity of 27 pg/ml. Importantly, mock-depleted control samples treated with beads coupled to mouse IgG2b isotype control antibody contained IL-10 at levels identical to undepleted ascites (Fig. 3C and Table 2).
Table 2.
IL-10 depletion using αIL-10 antibody bound to NHS magnetic beads leads to removal of IL-10 from ascites.
| Samples | IL-10 before depletion | IL-10 after depletion | IL-10 after mock depletion |
|---|---|---|---|
| #1 | 114.0 pg/ml | 6.4 pg/ml | 115.2 pg/ml |
| #2 | 48.0 pg/ml | 4.4 pg/ml | 49.6 pg/ml |
| #3 | 37.9 pg/ml | 2.9 pg/ml | 36.8 pg/ml |
| #4 | 47.8 pg/ml | 5.2 pg/ml | 48.2 pg/ml |
Levels of IL-10 were determined in four ascites samples before and after IL-10 depletion. IL-10 dropped considerably below assay sensitivity (27 pg/ml) in all samples depleted with αIL-10 antibody, while levels remained unchanged in mock depleted samples. The IL-10 concentration was quantified by Flow Cytomix bead ELISA.
In summary, removal of IL-10 from ascites with the use of NHS-coated magnetic beads proved successful.
3.4. Depletion of IL-10 from ascites allows for clear distinction between in vitro effects of paracrine ascites-derived versus autocrine IL-10
To identify the contribution of paracrine versus autocrine IL-10 in our experimental system, we conducted an assay using monocyte-derived dendritic cells (DC) activated by TLR4 agonist LPS in presence or absence of ovarian carcinoma-associated ascites depleted of IL-10. TLR4-mediated activation of monocyte-derived DC in presence of ascites is partially impaired (Fig. 3D). Neutralization of IL-10 by addition of a IL-10 specific antibody indeed showed dramatic impact on monocyte-derived DC activation levels, restoring LPS-induced IL-6 production beyond the levels observed upon LPS-mediated activation in the absence of ascites (Fig. 3D). However, since IL-10 neutralization in the presence of ascites affects autocrine DC-derived and paracrine ascites-associated IL-10 alike, these data cannot be used to clarify their respective influence on overall activation levels. Interestingly, levels of IL-6 production in the presence of IL-10 neutralizing antibody are higher for samples activated in the absence of ascites than in the presence of ascites (Fig. 3E) indicating the presence of other ascites-associated factors that reduce IL-6 induction. In order to investigate the specific role of paracrine ascites-associated IL-10, ascites samples that had been depleted of IL-10 were employed. Depletion of IL-10 prior to addition of ascites to cell cultures leaves autocrine IL-10 unaffected while eliminating the effect of ascites-derived IL-10. Interestingly and somewhat unexpectedly, addition of IL-10 depleted ascites did not restore nor even alter IL-6 production in response to LPS suggesting that paracrine ascites-derived IL-10 does not directly contribute to the suppression of DC activation by ovarian carcinoma-associated ascites (Fig. 3D). This finding further supports the interpretation that other immunosuppressive factors in ovarian carcinoma-associated ascites impair DC activation.
3.5. Depletion of PGE2 from ascites using neutralizing antibodies covalently bound to NHS ester coated magnetic beads
We wanted to test whether other immunosuppressive factors can be successfully depleted using antibody covalently bound to NHS ester coated beads and decided to use this protocol for depletion of ascites-associated PGE2. For this, we coated NHS beads with PGE2-specific antibody or mouse IgG1 isotype control antibody and followed the procedure as described for IL-10 for depletion of two ovarian carcinoma ascites samples. As with the IL-10 depletion, levels of PGE2 specific neutralizing antibody in the depleted samples were in the range of 1–1.3 ng/ml and therefore negligible (Table 3), confirming that detachment of the covalently bound antibody during incubation with ascites is minimal. In addition, PGE2 removal from depleted samples was verified by PGE2-specific ELISA (Fig. 4). After one round of depletion, we saw a considerable decrease in PGE2 levels in both depleted samples. However, PGE2 levels had decreased from 158.7 pg/ml to 23.5 pg/ml in one sample and from 597.5 pg/ml to 240.5 pg/ml in the other sample representing an 85% and 60% reduction in PGE2 levels, respectively. It appears that the αPGE2 antibody was used at suboptimal levels reaching saturation and was, therefore, unable to mediate complete PGE2 depletion. We repeated the depletion procedure, adding fresh αPGE2-coated NHS beads to both samples for one (sample #1) or two (sample #2) more depletion rounds. After these additional incubation steps, PGE2 was undetectable in both samples (Fig. 4). As expected, PGE2 levels in the ascites samples remained unaffected by mock depletion with mouse IgG1 isotype control antibody-coupled beads.
Table 3.
Covalent binding of αPGE2 neutralizing antibody to NHS magnetic beads leads to minimal residual αPGE2 antibody in depleted ascites samples.
| Samples | αPGE2 antibody | IgG1 isotype |
|---|---|---|
| #1 | 1.3 ng/ml | 0.4 ng/ml |
| #2 | 1.3 ng/ml | 0.2 ng/ml |
Levels of mouse IgG1 αPGE2 neutralizing antibody and respective isotype control antibody were measured in ascites samples after depletion of PGE2 using NHS magnetic beads. The mouse IgG1 concentration was quantified by sandwich ELISA.
Fig. 4.
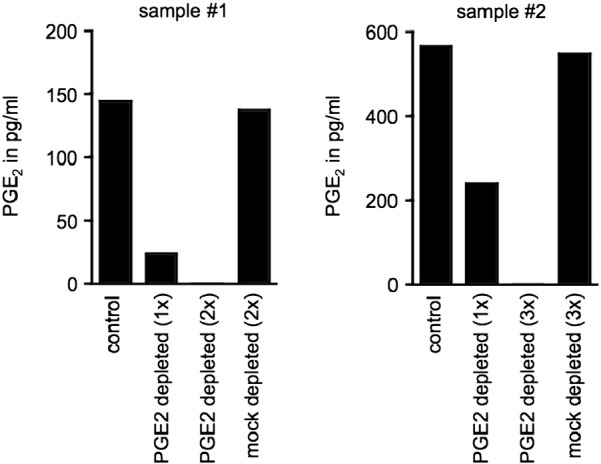
Depletion of PGE2 from ovarian carcinoma associated ascites using NHS magnetic beads.
PGE2 levels in mock-depleted control versus PGE2-depleted ascites samples after depletion using NHS magnetic beads coated with αPGE2 antibody. Each graph represents one individual ascites sample (n = 2). Two (sample #1) or three (sample #2) rounds of depletion were performed to ensure complete removal of PGE2 from ascites. PGE2 levels were measured by sandwich ELISA.
In summary, we have shown that NHS-coated magnetic beads can be covalently coupled to antibodies, and that such antibody-coupled beads can be used for specific and complete removal of selected proteins such as IL-10 and PGE2 from ovarian carcinoma-associated ascites.
4. Discussion
Ovarian carcinoma associated ascites is abundant with proteins, and their characterization is of notable interest in the field of cancer immunology and immunotherapy (Kipps et al., 2013). > 200 proteins can be identified in the cell-free fraction of ascites (Gortzak-Uzan et al., 2008), and the myriad of soluble components makes selective depletion of a particular protein challenging. To identify a suitable protocol for successful removal of immunosuppressive factors such as IL-10 and PGE2 from the fluid, it was imperative to take the composition of ascites into careful consideration.
In our initial depletion attempts for removal of αIL-10 antibody, we relied on Protein G. While its high affinity for murine IgG rendered Protein G an excellent candidate for binding and depletion of the neutralizing αIL-10 mouse IgG2b antibodies, its high affinity to human IgG and the abundance of the latter in ascites posed considerable obstacles (Bjorck and Kronvall, 1984). Direct competition of human IgG with αIL-10 antibody for Protein G binding sites not only resulted in residual αIL-10 antibody in depleted samples, but also altered ascites composition through partial removal of human IgG by Protein G coupled beads. Although we have not tested whether ascites-derived human IgG influences DC in our assays, we wanted to avoid altering any parameters other than IL-10 or PGE2 levels in ascites, and ensure the depletion was restricted to these proteins. Surprisingly, the level of depletion of human IgG from the ascites samples was reduced to non-significant levels upon pre-incubation of the neutralizing αIL-10 antibody with the protein G coupled magnetic beads as compared to adding beads and neutralizing antibody to the samples without pre-incubation, while the dislocation of neutralizing αIL-10 and/or isotype control mouse IgG2b antibody was still immense. It is, therefore, unclear whether the dislocation of antibody from the beads took place due to competition with the human IgG present in the ascites or whether antibody was simply mechanically sheared off during the incubation. Unexpectedly, the reduction in the pre-incubation time from 16 h to 1 h did not noticeably alter the amount of dislocated mouse IgG2b antibody, so we decided to abandon this approach.
Since the binding of antibody to beads based on the interaction of protein G with the antibody Fc region was not robust enough for our purposes, we decided to pursue a strategy based on covalent linkage of the antibody to the beads. Upon covalent binding of free amine groups on the antibody to magnetic beads harbouring reactive NHS ester groups, we finally achieved highly specific removal of IL-10 and PGE2. The procedure ensures complete depletion of the desired factors while leaving human IgG levels in depleted ascites unaffected. The established protocol is likely to be applicable for the selective removal of a multitude of other proteins from ascites, for which specific antibodies are available. We are confident that this method is further transferrable to depletion of tumour conditioned media, serum, or any other biological fluid. In this context, it is important to note that one round of depletion was sufficient to remove IL-10 but not PGE2 from the ascites samples in our study. How many rounds are required for complete removal of a targeted factor will depend on the absolute levels of the factor in the sample, the affinity of the antibody to its ligand and the efficacy of coupling the antibody to the NHS magnetic beads and will have to be determined for each antibody and sample type individually. In order to ensure complete depletion of the targeted factor in all samples, it is imperative to determine the levels of the targeted factor before and after depletion for all samples.
Our data clearly illustrate the vital importance of discrimination between the contributions of autocrine versus extrinsic factors. The down-modulation of DC activation by autocrine TLR-induced IL-10 has been described and has prompted us to clarify whether paracrine ascites-associated IL-10 contributed to the suppression in DC activation observed in our experimental system (Moore et al., 2001, Chang et al., 2007). In our activation assay, use of neutralizing antibodies alone deceptively implicates IL-10 as the driving factor of ascites-induced suppression, and it is only through the method of depletion that these observations could be further elucidated, showing that, in fact, after complete removal of IL-10 from ascites, suppression of LPS-mediated DC activation persists. This implies that paracrine ascites-associated IL-10 is redundant in our experimental system and that factors other than IL-10 within ascites play an important suppressive role. We have followed up on this observation and have investigated the mechanism of immunosuppression that impairs DC activation in the presence of ovarian carcinoma in a more extensive research study (manuscript in preparation).
We demonstrate here the relevance of distinguishing the differential influences of tumour-derived versus autocrine factors on DC and their activation with TLR agonists. These observations can be extrapolated and should be considered in future studies using a wide variety of cell subsets and culture conditions. Selective depletion of proteins can contribute to the elucidation of immunoregulatory or -stimulatory cytokine networks, which constitutes an important step towards a thorough and profound understanding of the complex interactions within the tumour environment and the establishment of more elaborate immunotherapeutic concepts.
Acknowledgements
This work was funded in part by a Cancer Research UK Career Development Award (A5593) to SD, which also supported EB in form of a Cancer Research UK PhD Studentship (A11089).
References
- Bjorck L., Kronvall G. Purification and some properties of streptococcal protein G, a novel IgG-binding reagent. J. Immunol. 1984;133:969–974. [PubMed] [Google Scholar]
- Boonstra A., Rajsbaum R., Holman M., Marques R., Asselin-Paturel C., Pereira J.P., Bates E.E., Akira S., Vieira P., Liu Y.J., Trinchieri G., O'Garra A. Macrophages and myeloid dendritic cells, but not plasmacytoid dendritic cells, produce IL-10 in response to MyD88- and TRIF-dependent TLR signals, and TLR-independent signals. J. Immunol. 2006;177:7551–7558. doi: 10.4049/jimmunol.177.11.7551. [DOI] [PubMed] [Google Scholar]
- Chang W.L., Baumgarth N., Eberhardt M.K., Lee C.Y., Baron C.A., Gregg J.P., Barry P.A. Exposure of myeloid dendritic cells to exogenous or endogenous IL-10 during maturation determines their longevity. J. Immunol. 2007;178:7794–7804. doi: 10.4049/jimmunol.178.12.7794. [DOI] [PubMed] [Google Scholar]
- Chen L.L., Ye F., Lu W.G., Yu Y., Chen H.Z., Xie X. Evaluation of immune inhibitory cytokine profiles in epithelial ovarian carcinoma. J. Obstet. Gynaecol. Res. 2009;35:212–218. doi: 10.1111/j.1447-0756.2008.00935.x. [DOI] [PubMed] [Google Scholar]
- Giuntoli R.L., 2nd, Webb T.J., Zoso A., Rogers O., Diaz-Montes T.P., Bristow R.E., Oelke M. Ovarian cancer-associated ascites demonstrates altered immune environment: implications for antitumor immunity. Anticancer Res. 2009;29:2875–2884. [PubMed] [Google Scholar]
- Gortzak-Uzan L., Ignatchenko A., Evangelou A.I., Agochiya M., Brown K.A., St Onge P., Kireeva I., Schmitt-Ulms G., Brown T.J., Murphy J., Rosen B., Shaw P., Jurisica I., Kislinger T. A proteome resource of ovarian cancer ascites: integrated proteomic and bioinformatic analyses to identify putative biomarkers. J. Proteome Res. 2008;7:339–351. doi: 10.1021/pr0703223. [DOI] [PubMed] [Google Scholar]
- Grubb A., Grubb R., Christensen P., Schalen C. Isolation and some properties of an IgG Fc-binding protein from group A streptococci type 15. Int. Arch. Allergy Appl. Immunol. 1982;67:369–376. doi: 10.1159/000233049. [DOI] [PubMed] [Google Scholar]
- Jarrossay D., Napolitani G., Colonna M., Sallusto F., Lanzavecchia A. Specialization and complementarity in microbial molecule recognition by human myeloid and plasmacytoid dendritic cells. Eur. J. Immunol. 2001;31:3388–3393. doi: 10.1002/1521-4141(200111)31:11<3388::aid-immu3388>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Kalinski P. Regulation of immune responses by prostaglandin E2. J. Immunol. 2012;188:21–28. doi: 10.4049/jimmunol.1101029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipps E., Tan D.S., Kaye S.B. Meeting the challenge of ascites in ovarian cancer: new avenues for therapy and research. Nat. Rev. Cancer. 2013;13:273–282. doi: 10.1038/nrc3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis P.J., Jonasson P., Nygren P.A., Uhlen M., Jendeberg L., Nilsson B., Kordel J. The serum albumin-binding domain of streptococcal protein G is a three-helical bundle: a heteronuclear NMR study. FEBS Lett. 1996;378:190–194. doi: 10.1016/0014-5793(95)01452-7. [DOI] [PubMed] [Google Scholar]
- Loercher A.E., Nash M.A., Kavanagh J.J., Platsoucas C.D., Freedman R.S. Identification of an IL-10-producing HLA-DR-negative monocyte subset in the malignant ascites of patients with ovarian carcinoma that inhibits cytokine protein expression and proliferation of autologous T cells. J. Immunol. 1999;163:6251–6260. [PubMed] [Google Scholar]
- Matte I., Lane D., Laplante C., Rancourt C., Piche A. Profiling of cytokines in human epithelial ovarian cancer ascites. Am. J. Cancer Res. 2012;2:566–580. [PMC free article] [PubMed] [Google Scholar]
- Melichar B., Savary C., Kudelka A.P., Verschraegen C., Kavanagh J.J., Edwards C.L., Platsoucas C.D., Freedman R.S. Lineage-negative human leukocyte antigen-DR + cells with the phenotype of undifferentiated dendritic cells in patients with carcinoma of the abdomen and pelvis. Clin. Cancer Res. 1998;4:799–809. [PubMed] [Google Scholar]
- Moore K.W., de Waal Malefyt R., Coffman R.L., O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- Mustea A., Konsgen D., Braicu E.I., Pirvulescu C., Sun P., Sofroni D., Lichtenegger W., Sehouli J. Expression of IL-10 in patients with ovarian carcinoma. Anticancer Res. 2006;26:1715–1718. [PubMed] [Google Scholar]
- Mustea A., Braicu E.I., Koensgen D., Yuan S., Sun P.M., Stamatian F., Lichtenegger W., Chen F.C., Chekerov R., Sehouli J. Monitoring of IL-10 in the serum of patients with advanced ovarian cancer: results from a prospective pilot-study. Cytokine. 2009;45:8–11. doi: 10.1016/j.cyto.2008.10.019. [DOI] [PubMed] [Google Scholar]
- Nowak M., Glowacka E., Szpakowski M., Szyllo K., Malinowski A., Kulig A., Tchorzewski H., Wilczynski J. Proinflammatory and immunosuppressive serum, ascites and cyst fluid cytokines in patients with early and advanced ovarian cancer and benign ovarian tumors. Neuro Endocrinol. Lett. 2010;31:375–383. [PubMed] [Google Scholar]
- Punnonen R., Teisala K., Kuoppala T., Bennett B., Punnonen J. Cytokine production profiles in the peritoneal fluids of patients with malignant or benign gynecologic tumors. Cancer. 1998;83:788–796. doi: 10.1002/(sici)1097-0142(19980815)83:4<788::aid-cncr24>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Santin A.D., Bellone S., Ravaggi A., Roman J., Smith C.V., Pecorelli S., Cannon M.J., Parham G.P. Increased levels of interleukin-10 and transforming growth factor-beta in the plasma and ascitic fluid of patients with advanced ovarian cancer. BJOG. 2001;108:804–808. doi: 10.1111/j.1471-0528.2001.00206.x. [DOI] [PubMed] [Google Scholar]
- Yigit R., Massuger L.F., Figdor C.G., Torensma R. Ovarian cancer creates a suppressive microenvironment to escape immune elimination. Gynecol. Oncol. 2010;117:366–372. doi: 10.1016/j.ygyno.2010.01.019. [DOI] [PubMed] [Google Scholar]
- Yigit R., Figdor C.G., Zusterzeel P.L., Pots J.M., Torensma R., Massuger L.F. Cytokine analysis as a tool to understand tumour-host interaction in ovarian cancer. Eur. J. Cancer. 2011;47:1883–1889. doi: 10.1016/j.ejca.2011.03.026. [DOI] [PubMed] [Google Scholar]
- Yildirim B., Sezgin N., Sari R., Sevinc A., Hilmioglu F. Complement and immunoglobulin levels in serum and ascitic fluid of patients with spontaneous bacterial peritonitis, malignant ascites, and tuberculous peritonitis. South. Med. J. 2002;95:1158–1162. [PubMed] [Google Scholar]
- Zhou J., Ye F., Chen H., Lv W., Gan N. The expression of interleukin-10 in patients with primary ovarian epithelial carcinoma and in ovarian carcinoma cell lines. J. Int. Med. Res. 2007;35:290–300. doi: 10.1177/147323000703500302. [DOI] [PubMed] [Google Scholar]