

Abstract
The development of functional peripheral ganglia requires a balance of specification of both neuronal and glial components. In the developing dorsal root ganglia (DRGs), these components form from partially-restricted bipotent neuroglial precursors derived from the neural crest. Work in mouse and chick has identified several factors, including Delta/Notch signaling, required for specification of a balance of these components. We have previously shown in zebrafish that the Sry-related HMG domain transcription factor, Sox10, plays an unexpected, but crucial, role in sensory neuron fate specification in vivo. In the same study we described a novel Sox10 mutant allele, sox10baz1, in which sensory neuron numbers are elevated above those of wild-types. Here we investigate the origin of this neurogenic phenotype. We demonstrate that the supernumerary neurons are sensory neurons, and that enteric and sympathetic neurons are almost absent just as in classical sox10 null alleles; peripheral glial development is also severely abrogated in a manner similar to other sox10 mutant alleles. Examination of proliferation and apoptosis in the developing DRG reveals very low levels of both processes in wild-type and sox10baz1, excluding changes in the balance of these as an explanation for the overproduction of sensory neurons. Using chemical inhibition of Delta-Notch-Notch signaling we demonstrate that in embryonic zebrafish, as in mouse and chick, lateral inhibition during the phase of trunk DRG development is required to achieve a balance between glial and neuronal numbers. Importantly, however, we show that this mechanism is insufficient to explain quantitative aspects of the baz1 phenotype. The Sox10(baz1) protein shows a single amino acid substitution in the DNA binding HMG domain; structural analysis indicates that this change is likely to result in reduced flexibility in the HMG domain, consistent with sequence-specific modification of Sox10 binding to DNA. Unlike other Sox10 mutant proteins, Sox10(baz1) retains an ability to drive neurogenin1 transcription. We show that overexpression of neurogenin1 is sufficient to produce supernumerary DRG sensory neurons in a wild-type background, and can rescue the sensory neuron phenotype of sox10 morphants in a manner closely resembling the baz1 phenotype. We conclude that an imbalance of neuronal and glial fate specification results from the Sox10(baz1) protein’s unique ability to drive sensory neuron specification whilst failing to drive glial development. The sox10baz1 phenotype reveals for the first time that a Notch-dependent lateral inhibition mechanism is not sufficient to fully explain the balance of neurons and glia in the developing DRGs, and that a second Sox10-dependent mechanism is necessary. Sox10 is thus a key transcription factor in achieving the balance of sensory neuronal and glial fates.
Introduction
The neural crest is a fascinating cell-type due to the diversity of cell-types derived from it, including diverse peripheral neurons, all peripheral glia, multiple pigment and skeletal cell-types, and various adult stem cells, including those for adult pigment cells[1]. One key question in development is how cells of different types are produced in the correct numbers, and the neural crest has been an excellent model system for addressing this issue [2]. Originating at the boundary of the neural plate and the non-neural ectoderm, neural crest cells delaminate from the dorsal neural tube, before undergoing extensive migration throughout the body [3, 4]. Among the derivatives of those migrating on the medial migration pathway between the spinal cord and the somites are the dorsal root ganglia (DRGs), consisting of both peripheral neurons and satellite glia, together with the Schwann cells that cover the peripheral nerve axons.
Work in both mouse and zebrafish has shown that the Sry-related HMG box 10 (Sox10) gene is crucial for the specification of all non-skeletogenic fates from the neural crest, which are absent or strongly reduced in numbers in strong loss-of-function mutants[5–13]. In each case, SOX10 drives expression of lineage-specific transcription factors that control the differentiation of the individual cell-types; in the Sox10 mutants, transcription of these factors is severely reduced[14]. This process is best characterised in the melanocyte, where SOX10 drives expression of microphthalmia-related transcription factor (Mitf; mitfa in zebrafish), which encodes a basic Helix-Loop-Helix Leucine Zipper transcription factor and is a master regulator for melanocyte development[10, 11, 15–19]. For all glial cells, the lineage-specific transcription factors include SOX10 itself and Pax3[14, 20–23]. In the case of the DRG neurons, fate specification depends upon transcriptional activation of neurogenin1 (neurog1) and neurog2, which encode bHLH transcription factors[24–29], likely acting with other factors[30]. The role for Sox10 in activating neurog1 has been somewhat controversial. In mouse, careful studies of the DRG phenotype of the loss-of-function mutants focussed on the glial phenotype; the DRG sensory neuron phenotype was interpreted as a secondary consequence of the failure of glial differentiation[20, 31]. In contrast, our studies in zebrafish showed a clear role for Sox10 in regulating early neurog1 expression, and thus fate specification, which was strongly reduced in strong loss-of-function mutants [13].
The related question of how the balance of sensory neuron and satellite glial cell numbers is achieved has also received attention. With Sox10 required for both fates, it seemed that other factors must be key. Sensory neuron specification, at least in mouse (but not zebrafish[32]), depends upon Wnt signaling driving transcriptional activation of Neurog1/2[33, 34]. Likewise, glial fate specification is stimulated by Neuregulin signaling[35, 36]. Neuregulin signaling mediated by ErbB3b/ErbB2 is also required for formation of DRGs in zebrafish, perhaps due to defects in medial pathway migration of neural crest cells[37, 38]. Signaling for sensory neuron specification, but not for other functions of ErbB3b/ErbB2 signaling including glial development, are mediated through a complex formed around the scaffold protein Sorbs3[39]. Sonic hedgehog signaling also plays a role in determining sensory neuron number [40]. A key role is played by Delta-NotchDelta-Notch signaling, via lateral inhibition [41]. In mouse neural crest stem cells, stimulation of Delta-Notch signaling drives specification of glial fate[42]. Studies in chick have shown that nascent sensory neurons express Delta1 while non-neuronal cells in the ganglia express high levels of Notch1 [43]. As a consequence, in cells adjacent to those expressing Delta1 Neurog1 expression is inhibited, neuronal differentiation is delayed and glial fate specification proceeds [25, 43]. Consistent with this, transgenic suppression of Notch signaling in the neural crest of mouse also results in a neurogenic phenotype[44, 45]. In zebrafish, where the initial DRGs are very small, each consisting of only 2–5 neurons and around 6–10 support cells in 5 days post fertilisation (dpf) larvae, substantial growth of the DRGs occurs during larval development[46]. Recently, expression of notch1a, deltaA and deltaD has been noted in non-neuronal cells in DRGs[47]. Furthermore Delta-Notch signaling has been shown to contribute to the balanced production of neurons and glia during larval growth of the DRGs[47].
We identified the zebrafish sox10baz1 (hereafter referred to as baz1) mutant during a sox10 allele screen [13]. Our initial observations showed that it combined features typical of other sox10 mutant alleles, such as absence of normal melanocytes and of peripheral glial myelin basic protein (mbp) expression, and with a strong effect on xanthophores and iridophores that resembles, but is not quite as severe as in, a null allele (e.g. sox10m618, hereafter m618). Thus it appeared to be a strong hypomorph for the former cell-types, but a weaker hypomorph for the latter. More remarkably, certain neurons of the trunk and tail were more abundant than in wild-type siblings (a hypermorphic phenotype), in striking contrast to the dramatic reduction of these cells in all other sox10 mutant alleles [13]. Sequencing identified the causal lesion as a single nucleotide substitution resulting in a V117M change in the HMG domain of the protein. Here, we provide a comprehensive characterisation of this unique sox10 mutant phenotype. Our data demonstrate that the supernumerary neurons are DRG sensory neurons, that peripheral (as well as oligodendrocyte) glial fates are highly reduced, and that the neurons form precociously in a classic neurogenic phenotype. We show that this DRG sensory neurogenic phenotype can be partially reproduced by chemical manipulation of Delta-Notch signaling in early neural crest development. Consistent with this, we show expression of delta genes in nascent DRGs and in vivo activation of Notch signaling in non-neuronal cells in these early DRGs. Thus, Delta-Notch signaling plays a role in generating the balance of derivatives in the early DRGs. However, we show that changes in Notch signaling alone cannot explain the quantitative aspects of the baz1 phenotype. In silico modelling suggests that the V117M mutation is likely to change the flexibility of the DNA-binding HMG domain, consistent with the fate-specific differences in the severity of the Sox10(baz1) phenotype. Importantly, we show that Sox10(baz1), like Sox10(Wild-Type, WT), but in contrast to classic loss-of-function Sox10(m618), can readily activate the neurog1 promoter in zebrafish embryos. Consistent with this, ectopic expression of neurog1 in a sox10 loss-of-function context rescues the sensory neuronal phenotype. We propose that the baz1 DRG phenotype results from severe disruption of the process of glial specification, whilst remarkably sparing sensory neuron specification; we suggest that a crucial aspect of Sox10-dependent glial specification is a Notch-independent repression of neurog1 expression that acts alongside Delta-Notch signaling in these nascent sensory neurons to prevent supernumerary neuronal differentiation. The baz1 mutant DRG phenotype indicates that Sox10 is important not just for specification of DRG cell-fates, but also for the balance in the production of both neurons and glia.
Results
Supernumerary neurons in Sox10baz1 mutants express sensory, but not sympathetic or enteric markers, and form precociously
In our initial description of the sox10baz1 phenotype (hereafter referred to as baz1), we showed that at 5 days post-fertilisation (dpf) embryos homozygous for this allele display an approximately 2-fold increase in Elav1/Hu-expressing neurons relative to WT siblings at locations where dorsal root ganglia (DRG) develop, and that this phenotype could be detected as early as 36 hours post-fertilisation (hpf) by monitoring the expression of the proneural gene neurog1[13]. To address the mechanisms underlying this surprising phenotype, we first sought to confirm that the supernumerary neurons are indeed DRG sensory neurons. Like neurog1, expression of neuronal differentiation1 (neurod1) labels the sensory neuron lineage [24, 48]. In baz1 mutant embryos, neurod1 expression was seen in a substantially increased number of cells on the medial pathway, in a pattern that strikingly resembled that of Elav1/Hu (Fig 1; [13]). Counts demonstrated that neurod1+ cells were increased around 3-fold compared with WT siblings (Fig 1C). Furthermore, neurod1 expression was seen precociously, with expression in mutants extending further posteriorly (i.e. to developmentally younger regions of the embryo) at both 36 and 45 hpf (Fig 1A and 1B, right panels). Similarly, supernumerary cells in baz1 mutants were clearly islet1-positive (S1 Fig), consistent with them being specified as sensory neurons.
Fig 1. Precocious and supernumerary sensory neuron specification in sox10baz1 mutants.

A,B) neurod1 expression is seen in more cells (close-ups in left panels; arrowheads indicate a subset of neurod1+ cells) and extending more posteriorly (right panels; arrowhead marks posteriormost neurod1+ DRG) in baz1 mutants compared with WT siblings at both 36 and 45 hpf. C) Counts of neurod1+ cells on one side of embryo at 36 and 45 hpf embryos (N = 11 for all conditions except 36 hpf baz1, where N = 13). baz1 mutants significantly different to WT siblings (Student’s t test; ***, p<0.0001. In this and all subsequent images, embryos are shown in lateral view with dorsal to the top and anterior to the left, unless otherwise stated. Scale bar, 100 μm.
In contrast to sensory neuron markers, markers of other NCC-derived peripheral neurons were absent both at their endogenous site of expression and along the medial pathway (Fig 2). We had previously shown that Elav1/Hu+-enteric neurons were lacking in baz1 mutant embryos [13]. Here, we complement this observation by showing that phox2b-expressing enteric progenitors were also missing from the developing gut of baz1 mutants at 72 hpf, and no sign of ectopic phox2b expression was detected on the medial pathway (Fig 2A and 2B). Likewise, immunofluorescent detection of the sympathetic neuron marker Tyrosine Hydroxylase (TH) was strongly reduced in baz1 mutants at 7 dpf with no expression on the medial pathway (Fig 2C–2N, quantified in R); unexpectedly, embryos occasionally showed ectopic TH-expressing neurons associated with the Posterior Lateral Line nerve (PLLn; Fig 2L–2N). These data strongly suggest that the supernumerary neurons detected on the medial pathway of baz1 mutant embryos are restricted to the sensory lineage.
Fig 2. Enteric precursors are absent in baz1 mutants and supernumerary neurons in trunk and tail do not express markers of non-sensory neuronal types.

A,B) phox2b expression at 72 hpf in wild-types (A) is restricted to the enteric neuron progenitors, but is absent from baz1 embryos (B). Tyrosine hydroxylase (TH) immunofluorescence reveals differentiated sympathetic neurons at 7 dpf in WT (C-E, I-K), whereas these are much reduced in baz1 mutants (F-H, L-M). White arrows indicate a subset of TH+ neurons. Inset in J represents enlargement of boxed area to show autonomic neuron chain more clearly. Occasional baz1 mutants show anti-TH immunofluorescence in neurons (*) associated with the PLLn (O-Q). DIC (C,F,I,L,O) and immunofluorescent TH images (D,G,J,M,P) are merged in panels E,H,K,N,Q respectively. sb, swim bladder; vs, ventral stripe. R) Quantitation (mean ± s.d.) of anti-TH positive sympathetic neurons in WT (N = 16) and baz1 mutants (N = 16). * indicates significant difference (two-tailed t test, p<0.001) Scale bar, 50 μm(C).
To explore further the temporal aspects of the formation of these supernumerary sensory neurons we compared the distribution of Elav1/Hu antigen with Tg(sox10(4.9):GFP)ba2, in which GFP initially labels all neural crest cells, including neural progenitors on the medial pathway, but later is maintained only in glial cells (Fig 3; [13]). Using GFP as a short-term lineage marker for neural crest-derived DRG progenitors, we noticed that in WT embryos Elav1/Hu immunofluorescence was seen in only a few GFP+ cells in the anterior trunk by 36 hpf, and not until 48 hpf were substantial numbers detected posteriorly (Fig 3A, 3D and 3G; quantified in panels M-O). At these stages many Elav1/Hu-;GFP+ progenitors were seen on the medial pathway. In sox10m618 mutants (hereafter referred to as m618), reduced numbers of cells express Elav1/Hu, and most cells remain Elav1/Hu-;GFP+ (Fig 3C, 3F and 3I). In contrast, baz1 mutants show substantial numbers of Elav1/Hu+;GFP+ double positive cells in both the anterior and posterior trunk from 36 hpf onwards, and by 48 hpf have approximately three times as many as stage-matched WT siblings, although we note that not all GFP+ cells express Elav1/Hu (Fig 3B, 3E, 3H and 3M–3O). By 5 dpf, the GFP signal is often strongly reduced, especially in sox10 mutants, due to a failure of the GFP to persist in unspecified precursors; nevertheless, DRG neurons are still distinctive due to their combination of spatial localisation and Elav1/Hu immunofluorescence. Elevated numbers of neurons remain, but there are now fewer than at earlier stages (Fig 3J–3L and 3P).
Fig 3. Medial pathway neural precursors undergo precocious and supernumerary differentiation into neurons in baz1 mutants.

Confocal images of developing trunk DRGs of WT (A, D, G, J), baz1 (B, E, H, K) and m618 mutants (C, F, I, L) showing Elav1/Hu (red) and sox10:GFP (green) at each of 36 (A-C), 42 (D-F), 48 hpf (G-I) and 5 dpf (J-L). Arrowheads indicate subset of Elav1/Hu+ DRG sensory neurons. M-P) Counts (mean±s.d.) of trunk (Tr) and tail (Ta) and total (TOT) Elav1+ cells in DRGs of baz1 (yellow) and m618 (blue) mutants and their respective WT siblings. Significantly elevated numbers of neurons are indicated (two-tailed Student’s t test; **, p<0.01; ***, p<0.001). Note in panels J-L) that variable prominence of Elav1/Hu detection in spinal cord is an artefact of antibody penetration into CNS at this late stage. Scale bar, 50 μm.
To confirm these observations, we performed time-lapse studies of neuronal specification in baz1 mutant embryos and their WT siblings. We generated fish carrying the Tg(-4.9sox10:GFP)ba2 and Tg(-8.4neurog1:nRFP)sb3 transgenes and bred them onto the baz1 mutant background [13, 49]. In these fish, early neural crest cells (NCCs), including the nascent DRG, are labelled with GFP and neuronal specification is indicated by the initiation of RFP expression in a subset of these cells (S1 and S2 videos; S2 Fig); time-lapse focused on the posterior trunk, and typically began at around 36 hpf and continued for up to 25 hours. In WT fish, RFP expression was not normally detected in NCCs at the beginning of the analysis, but isolated sensory neurons were specified during the next day of development. In contrast, baz1 mutants regularly showed RFP expression in sensory neurons from the beginning of the time-lapse and multiple NCCs in each ganglion begin expressing RFP over the course of the analysis.
Finally, we addressed whether a change in the balance of proliferation or apoptosis in the DRG could account for the increased numbers of sensory neurons in baz1 mutant embryos. Apoptosis rates were assessed by TUNEL in which DRG cells were identified by GFP expression from the Tg(-4.9sox10:GFP)ba2 transgene at 36 and 48 hpf in baz1 mutants and their WT siblings. Although TUNEL+ cells were detected in embryos at all stages, apoptosis rates in the DRGs were negligible in either genetic context (Table 1). Likewise, proliferation rates in DRG were determined using phospho-Histone H3 (pH3) immunofluorescence in baz1 mutants and their WT siblings at 36, 42 and 48 hpf; DRG cells were identified by GFP fluorescence from the Tg(-4.9sox10:GFP)ba2 transgene (36 hpf) or anti-Elav1/Hu immunofluorescence (42 and 48 hpf). In all cases, although pH3+ cells were detected widely in these embryos, proliferation rates in the DRGs were also negligible (Table 2).
Table 1. Apoptosis in the DRGs is negligible in baz1 mutants and their WT siblings.
| WT | baz1 | |||||
|---|---|---|---|---|---|---|
| Stage (hpf) | No. Embryos | Total No. DRGs assessed | Total no. GFP+;TUNEL+ DRG cells | No. Embryos | Total No. DRGs assessed | Total no. GFP+;TUNEL+ DRG cells |
| 36 | 15 | 195 | 1 | 10 | 130 | 3 |
| 48 | 10 | 250 | 0 | 8 | 200 | 0 |
Table shows number of embryos scored, number of DRGs assessed per embryo and total number of TUNEL+ DRG cells in all embryos scored.
Table 2. Proliferation in the DRGs is negligible in baz1 mutants and their WT siblings.
| WT | baz1 | |||||||
|---|---|---|---|---|---|---|---|---|
| Stage (hpf) | No. Embryos | Total No. DRGs assessed | Total no. GFP+;pH3+ DRG cells | Total no. Elav1/Hu+;pH3+ DRG cells | No. Embryos | Total No. DRGs assessed | Total no. GFP+;pH3+ DRG cells | Total no. Elav1/Hu+;pH3+ DRG cells |
| 36 | 22 | 286 | 6 | nd | 8 | 104 | 1 | nd |
| 42 | 16 | 320 | nd | 2 | 3 | 60 | nd | 0 |
| 48 | 15 | 375 | nd | 3 | 5 | 125 | nd | 2 |
Table shows number of embryos scored, number of DRGs assessed per embryo and total number of pH3+ DRG cells in all embryos scored. nd, not determined
Taken together, these data suggest that the supernumerary neurons detected in the DRG of baz1 mutant embryos at 48 hpf are specified sensory neurons, whereas in WT siblings at the same stage many NCCs appear to remain in a progenitor state. We also conclude that the balance of proliferation and apoptosis in the DRG is unlikely to contribute significantly to the phenotype observed in baz1 mutants.
In contrast to the unique sensory neuron phenotype, baz1 mutants show a severe defect in glial fate specification and differentiation that is comparable to, but slightly weaker than, that of other sox10 alleles we have studied ([50]; S3 Fig). Thus, sox10 expression associated with the trunk medial pathway in baz1 mutants showed a similar pattern to WT, and was more prominent than in m618 mutants (compare S3 Fig panel C with panels G and K, white asterisks); interestingly, the developmentally younger DRGs in the tail more closely resembled those in m618 (compare S3 Fig panels D with H and L, white asterisks). Not withstanding the mutant nature of the proteins encoded by these transcripts, the fact that sox10 transcripts are detectable enables us to use them as a marker of sox10-expressing cell states, including neural progenitor cells (early) and differentiating glia (later). We interpret these data to suggest that sox10 expression is initially normal, but then fails to be maintained in putative glia in the sox10 mutant alleles, and that this loss is more pronounced in m618. We also saw a very strong reduction in the number of foxd3-expressing Schwann cell precursors at 48 hpf on both the spinal nerves (S3 Fig panels M,P) and the PLLn (S3 Fig panels N,Q), with remaining cells on the PLLn concentrated near the base of the nerve (S3 Fig panel Q). Absence of peripheral glia on the PLLn results in supernumerary production of lateral line neuromasts [51]; using isl1 as a marker for neuromasts [52], we saw that baz1 mutants also showed precocious differentiation of neuromasts (S3 Fig panels O,R), confirming the absence of PLLn Schwann cells. In summary, our analysis shows a consistent picture of reduced glial cell development, with much reduced numbers of peripheral glial cells and no sign of Schwann cell differentiation.
In addition, oligodendrocyte differentiation is impaired in baz1 mutants. Sox10 expression also marks oligodendrocyte precursors and differentiating oligodendrocytes, and Sox10 is required for oligodendrocyte differentiation. We asked whether baz1 mutants showed defects in oligodendrocyte development. Using sox10 expression in the ventral CNS as a marker of oligodendrocyte precursors, and dispersed expression in scattered cells throughout the CNS as a marker of oligodendrocytes, we saw no change in oligodendrocyte precursors and specification in baz1 mutants (S4 Fig panels A-F). In contrast, mbp expression in differentiating oligodendrocytes is reduced, indicating that oligodendrocyte differentiation is impaired in baz1 mutants (S4 Fig panels G,H).
Supernumerary neurons and Notch signaling
The sensory neuron phenotype we describe in baz1 mutants shows the hallmarks of a neurogenic phenotype, with supernumerary and precocious neuronal differentiation at the expense of glial cell-types (Fig 3, S2 Fig; [13]). Classically, such phenotypes result from disruption of Notch signaling. To explore a potential role for Notch signaling in DRG development, we first asked if the expression of Notch pathway components could be detected in the nascent DRGs. For this, we explored the patterns of expression of jagged and delta ligands at stages from 24 to 38 hpf, focusing on the posterior trunk region; overlapping expression with neural crest cells in the nascent DRG was identified by their co-expression of sox10, foxd3 or neurog1. As expected, the expression of jagged genes was generally detected at low levels. While we confirmed the previously published patterns of expression, for instance of jag1b and jag2 in pronephric duct, notochord and spinal cord, we did not see pronounced expression of jag1a, jag1b or jag2 in nascent DRGs (data not shown), consistent with the findings of Zecchin et al [53]; over-developing the in situ hybridisations highlighted a low level expression of jag1a expression in cells adjacent to, but not overlapping with, the nascent DRGs (data not shown).
In contrast, we found clear expression of delta ligands in the nascent DRGs, identified by neurog1 expression. We examined all four delta genes, but found that only two (deltaA and deltaD), showed overlapping or adjacent expression with neurog1-positive DRG cells, consistent with recently published data [47]. At 30 hpf, deltaA and neurog1 showed strong colocalisation in individual cells of the nascent DRG (Fig 4A–4C and data not shown). At later stages (shown at 38 hpf in Fig 4D–4F), deltaA and neurog1 remain expressed in the nascent DRGs, but only weakly, preventing unequivocal assessment of co-localisation at the single cell level. Similarly, deltaD expression showed co-localisation with neurog1 in individual cells of the nascent DRGs at these same stages (Fig 4G–4I and data not shown). We confirmed these observations at 30 hpf for the deltaA and deltaD genes using colocalisation with the alternative DRG marker, neurod1 (S5 Fig). We conclude that Delta-Notch signaling, mediated through one or both of DeltaA or DeltaD, might contribute to fate specification within zebrafish DRG.
Fig 4. deltaA and deltaD gene expression overlaps with neurog1 in the nascent DRGs.

A-C) deltaA expression (red) clearly overlaps with neurog1 (green) in the nascent DRG (arrows) at 30 hpf. D-F) At 38 hpf, deltaA expression is clearly seen in the DRGs, but weaker signals make it difficult to discern if expression is in the same cells as express neurog1 or simply in other cells of the ganglia. G-I) deltaD expression (red) clearly overlaps with neurog1 (green) in the nascent DRG (arrows) at 38 hpf. All main panels are confocal images of fluorescent dual-color in situ hybridisations in lateral view, with insets showing y-z planes (left) and x-z planes (above) for each. Insets in the bottom right of panels C, F and I show enlargements of the double-labeled cells indicated by the arrows. nc, notochord; sc, spinal cord.
We then asked whether DRG cells were responding to these ligands by activating the Notch pathway. For this, we used the Notch reporter line, Tg(12xNRE:eGFP), bearing twelve Notch Responsive Elements (NRE) upstream of the coding sequence of eGFP [54]. DRG cells were identified using a Tg(sox10(7.2):mRFP) reporter line [55]. As previously described for another Notch reporter line, strong eGFP expression is detected in all blood vessels [56], but we also see weaker signals overlapping the DRG (arrows and asterisk in Fig 5A–5G). While co-expression of eGFP and RFP was best seen at 48 hpf, overlap between the Notch-reporter and the DRG marker was detected as early as 36 hpf (Fig 5A–5G; data not shown). We conclude that Delta-Notch signaling is active within neural crest cells of the nascent DRG in zebrafish embryos starting at a stage before 36 hpf. These data are fully consistent with the delta expression patterns reported above and with the idea that DRG neuronal numbers are regulated, at least in part, by this pathway.
Fig 5. Notch signaling is active in nascent DRGs.

Neural crest cells in the DRGs of 48 hpf embryos (arrows) were readily identified by expression of mRFP reporter in sox10:mRFP fish (A). These overlapped with eGFP expression from the 12xNRE:eGFP (Notch signaling) reporter (B), as shown in superimposed image (C and D). Close-ups of individual DRGs labelled with asterisk are shown in panels E and F. Panel G shows DRGs (white arrows, yellow signals) as single plane acquisitions, in lateral view (top left), transversal view (top right), dorsal view (bottom left) and mini global view (bottom right). Note that in all panels eGFP is also strongly visible in blood vessels, as expected for the Notch reporter. nc, notochord; sc, spinal cord.
Finally, we asked whether loss of Notch signaling phenocopies the baz1 DRG phenotype. Classical zebrafish mutants affecting Delta-Notch signaling show early defects in neural crest specification [27], precluding examination of later roles in DRG formation. Notch signaling depends upon γ-secretase-mediated cleavage of Notch receptor that can be blocked with DAPT, a γ -secretase inhibitor that has been widely used to study Notch function in zebrafish [57]. We, thus, asked whether DAPT treatment could modulate the number of DRG sensory neurons. We found that a 30–72 hpf treatment window (i.e. extending throughout the initial phase of sensory neuron specification) resulted in a significant increase in DRG sensory neurons in both trunk and tail, although the degree of increase was not as striking as in the baz1 mutants (Fig 6A–6H, quantified in 6I; compare Figs 1 and 3). As reported before, treated embryos showed a bending of the antero-posterior axis (Fig 6C, compare Fig 6A), but are otherwise morphologically grossly normal. To verify the specificity of this effect, we used a second γ -secretase inhibitor, Compound E [58], which gave a quantitatively comparable increase in DRG sensory neuron numbers (Fig 6J). We conclude that DRG neuron number in zebrafish, as in mouse and chick, is regulated by Notch signaling, a conclusion also reached in a recent, independent study [47].
Fig 6. Notch inhibition phenocopies the sox10baz1 DRG phenotype.

A-H) Posterior trunks of WT embryos treated with 100 μM DAPT from 30–72 hpf (C,D,G,H) and controls exposed to DMSO carrier alone (A,B,E,F). E-F show enlargements of A-D. Treated embryos show dorsally-curved body axis (C). Immunofluorescence for Elav1/Hu revealed increased numbers of DRG sensory neurons (arrowhead). I,J) Quantitation of DRG sensory neurons in 5 dpf embryos after treatment with 100 μM DAPT (I, orange) or 100 μM Compound E (J, orange) from 30–72 hpf; compared with control DMSO-treated embryos (yellow in I and J), sensory neuron numbers are significantly increased (***) when Notch signaling is inhibited. While trunk refers to the entire somitic region from posterior to the otic vesicle to the anus, tail refers to the post-anal body; posterior trunk refers to the 7 most posterior somatic segments dorsal to the yolk sac extension. N = 13 (Control, I), 13 (DAPT, I), 16 (Control DMSO, J) and 15 (Comp. E, J). Significance of elevated numbers of neurons was evaluated by two-tailed Student’s t test (**, p<0.01; ***, p<0.001).
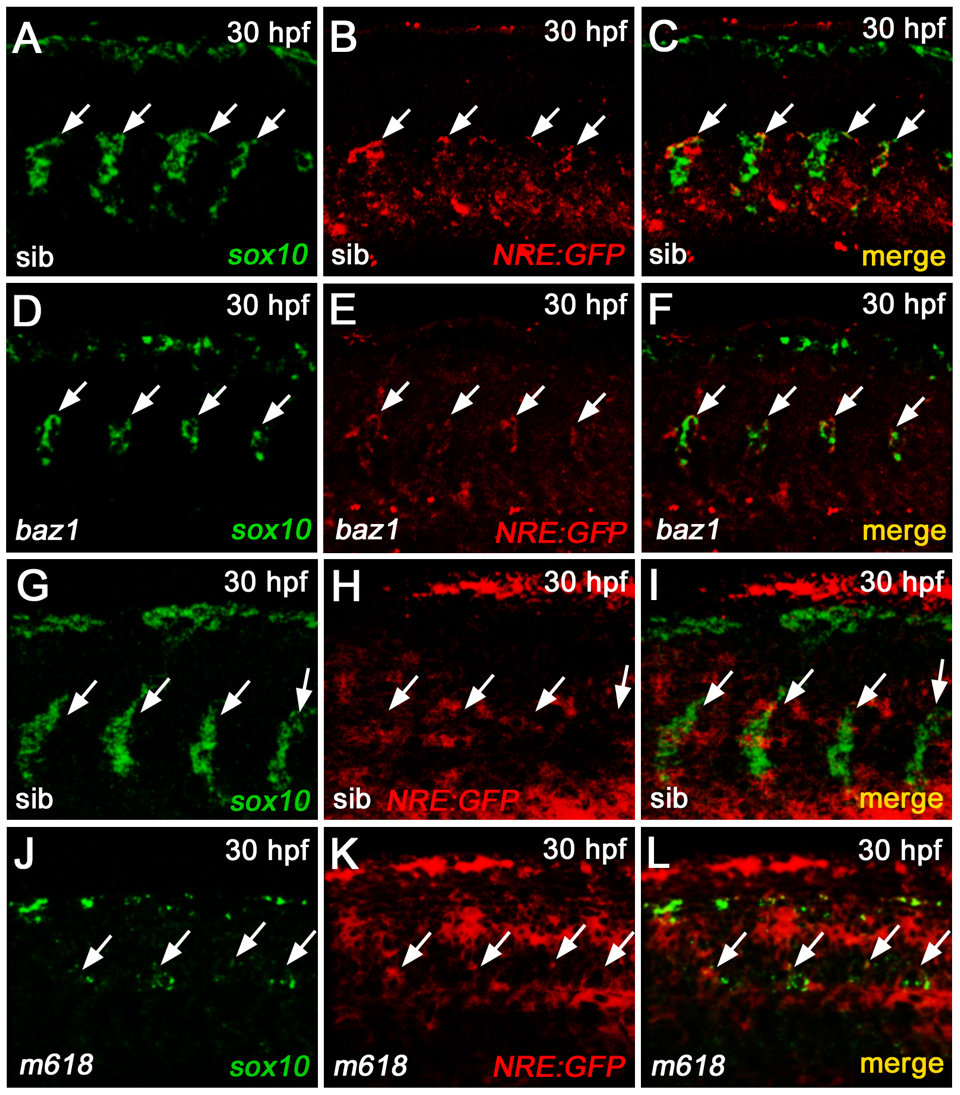
Implicit in the above analysis is the assumption that Notch signaling is affected in, and thus likely contributes to, the baz1 mutant phenotype, whereas in m618 mutants, Notch signaling was likely to remain intact. However, it is conceivable that Notch signaling might be unaffected in both mutant contexts, and that differences in another mechanism are solely responsible for the distinct sensory neuron phenotypes of m618 and baz1 mutants. To test our assumption, we evaluated Notch signaling in the DRGs of m618 and baz1 mutants, by crossing the Tg(12xNRE:eGFP) reporter onto the respective sox10 mutant backgrounds. Assessing DRG cells at 48 hpf, while we consistently observed Notch reporter activity in m618 mutants (Fig 7B), DRG signals were considerably weakened in baz1 mutants (Fig 7E). Quantification of the fluorescence intensity from the DRGs confirmed that Notch signaling in baz1 mutants was reduced by around 50% (Fig 7F), whereas in m618 mutants levels were not significantly changed compared with WT siblings (Fig 7C). Reduction of Notch signaling in baz1 and normal activation in m618 mutants was also observed using an additional Notch responsive line expressing a nuclear-localized red protein Tg(EPV.Tp1-Mmu.Hbb:NLS-mCherry)[59], thus confirming our previous results with an independently generated Notch reporter allele (data not shown). In all cases fluorescence from the dorsal aorta and intersomitic vessels was comparable between mutants and their respective WT siblings. We saw consistent reduction in Notch reporter expression in the neural tube of baz1 mutants compared with WT siblings, whereas in m618 mutants Notch signaling appeared, if anything, to be elevated compared to siblings (Fig 7B, 7C, 7E and 7F; S6 Fig). It is likely that the elevated levels in m618 mutants may at least in part be an artefact of the lack of melanin (which will partially quench the fluorescent signal). However, the significantly lowered expression in baz1 mutants is unexpected, but conceivably results from differences in oligodendrocyte progenitor development in these two different mutant alleles; this will require further investigation. Meanwhile, in the context of DRG development, we conclude that, as predicted, Notch signaling remains active in the DRGs of m618 mutants, but is significantly reduced in baz1 mutants. Analysis of gfp transcriptional expression in these lines, using sox10 to label medial pathway neural crest cells in the nascent DRGs confirms this interpretation (S7 Fig).
Fig 7. Quantification of Notch reporter differences in m618 and baz1 mutants at 48 hpf.

A-F) confocal acquisitions of WT control (A,D) and mutant (B, m618; E, baz1) trunk regions, followed by Notch reporter (NRE:EGFP) fluorescence analysis (C,F). A slight increase of reporter signal is detected in the neural tube (nt) and dorsal root ganglia (drg, arrowhead) of m618 mutants (B) compared to controls (A), while a decrease of signal is detected in the same regions of baz1 mutants (compare E with D). Relative fluorescence intensity (RFI) in aorta (a) and intersomitic vessels (v) appears unmodified in all conditions. n = 6 measurements per condition. n.s. = not significant; ** = p<0.01; *** = p<0.001.
Sox10(baz1) protein retains transcriptional activity at the neurog1 locus
The baz1 DRG phenotype is consistent with a decrease of Notch signaling. However, the quantitative difference in the increase in neuronal number in baz1 compared with the maximum effect of chemical inhibition of Notch signaling suggested this is, at best, only a partial explanation. The baz1 mutation results in a single amino acid substitution (V117M) in the DNA binding HMG domain [13]. Molecular dynamics simulation studies confirmed the expectation that the Sox10(baz1) substitution results in relatively subtle changes in the protein, most strikingly an apparent reduction in the flexibility of the HMG domain (S8 Fig). Whilst these observations will require detailed experimental confirmation, they are consistent with the hypothesis that Sox10(baz1) may show differential affinity for the regulatory sequences of target genes, retaining transcriptional activity at some target genes whilst losing activity at others.
We have previously shown that null alleles of sox10 display a substantial reduction in DRG sensory neurons that correlates with reduced expression of neurog1 [13]; Neurog1 is the obligate proneural gene for DRG sensory neuron specification in zebrafish [29]. We have also shown that Sox10(WT) but not Sox10(m618) drives ectopic neurog1 transcription when mis-expressed [13]. Given the generally strong loss-of-function nature of the baz1 phenotype, the increase in neurog1 expressing cells on the medial pathway in baz1 embryos is particularly surprising. Since it seemed unlikely that this increase simply reflected a reduction in Notch activity, we asked whether Sox10(baz1) retains the ability to drive neurog1 expression. To test this, we took advantage of a previously reported Tg(-8.4neurog1:GFP)sb1 transgenic line that largely replicates the endogenous neurog1 expression pattern in neuronal precursors, including the DRG sensory neurons [49]. Consistent with our published results, GFP transcription from the transgene was not driven by mis-expression of Sox10(m618) (Fig 8B). In contrast, Sox10(baz1) induced GFP transcription in a manner similar to Sox10(WT) (Fig 8A and 8C), indicating that the baz1 mutation does not affect Sox10 transcriptional activity at the neurog1 locus. These results suggest that the increase in DRG sensory neurons seen in baz1 mutants is not solely due to a reduction in Notch signaling.
Fig 8. Sox10(baz1) retains transcriptional activity at neurog1.
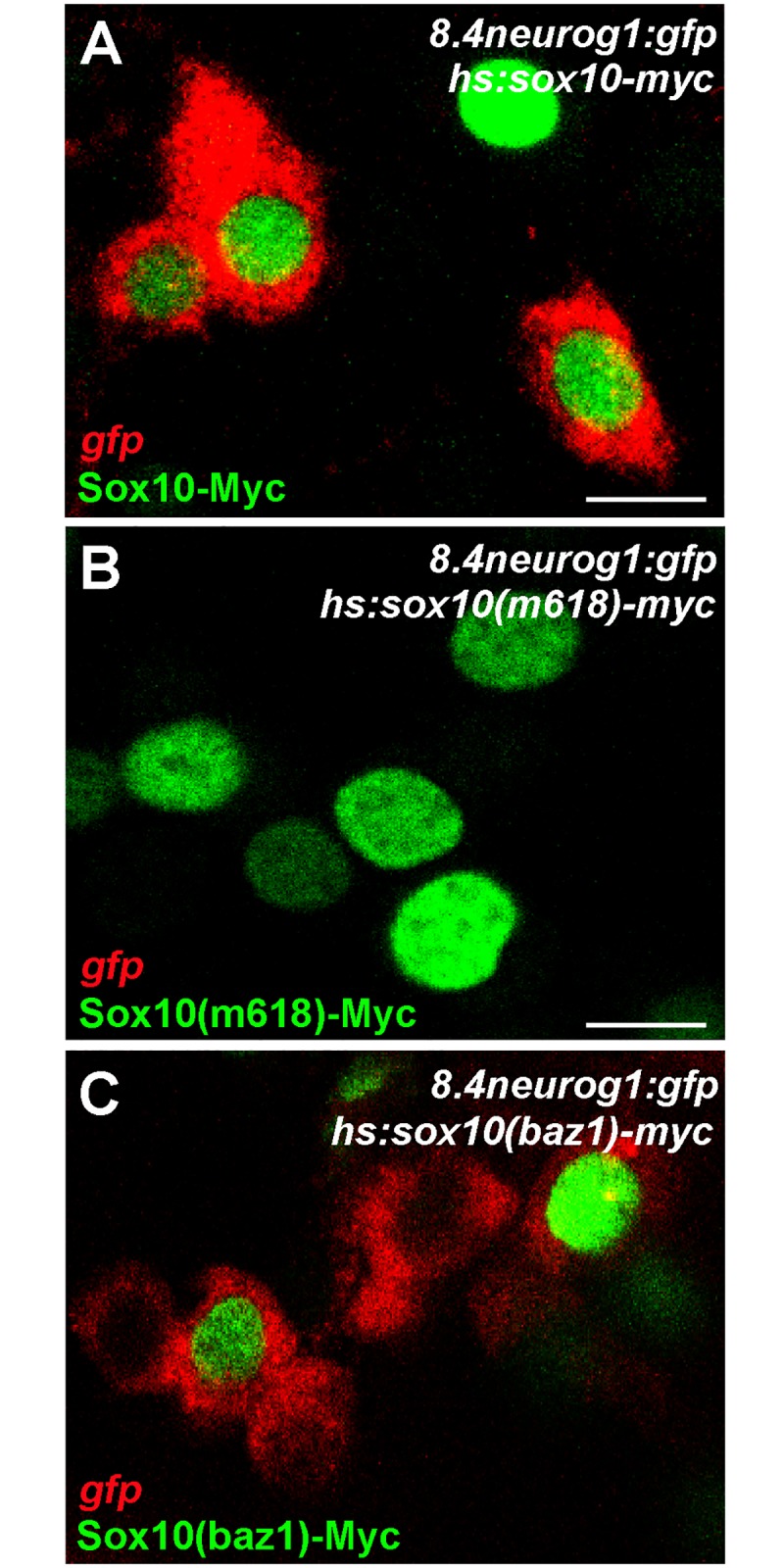
Tg(8.4neurog1:GFP) embryos were injected with expression plasmids encoding variants of Sox10. Embryos were heat-shocked at 7 hpf and fixed at 10 hpf. Expression of the Sox10 variant proteins was detected by immunofluorescent detection of their Myc tags (green) and activation of the neurog1 reporter gene is seen by In situ hybridisation against the gfp mRNA (red). Note how both WT and baz1 proteins activate Tg(8.4neurog1:GFP) reporter expression (A C), whereas m618 does not (B). Apparent non-autonomous activation of gfp after expression of Sox10(baz1) but not Sox10(WT) nor Sox10(m618) was reproducibly seen, but always restricted to cells adjacent to those expressing Sox10(baz1); the mechanism underlying this remains unclear but could be related to differential stability of the various Sox10 proteins.
Neurog1 rescues sensory neurons in sox10 morphants
Our data clearly indicated both that neurog1 is precociously and ectopically expressed in nascent baz1 DRG and that Sox10(baz1) protein retains the capacity to contribute to this expression ([13] and this report). To address whether an increase in neurog1 expression is sufficient to promote DRG neuron specification as in baz1 mutants, we mis-expressed Neurog1 using the Tg(hsp70l:neurog1)ups1 line [60]. When Tg(hsp70l:neurog1)ups1 embryos were heat shocked in a WT background a robust increase in DRG sensory neuron cell numbers is detected (Fig 8B and 8C’, quantified in A). We next asked if mis-expression in the absence of Sox10 activity rescues DRG sensory neuron specification. For these experiments we used a morpholino to phenocopy the sox10 mutant phenotype. This morpholino has previously been shown to phenocopy the pigment cell phenotypes of the sox10m618 allele [61], but its effect on sensory neurons had not been assessed. As expected, injection of the morpholino into WT embryos resulted in a strong decrease in sensory neuron numbers compared with controls (Fig 9D and 9D’, quantified in A). Activation of Tg(hsp70l:neurog1)ups1 transgene in sox10 morphant embryos, however, resulted in dramatic rescue of DRG neuron cell numbers (Fig 9D and 9E’, quantified in A); the rescued DRG neurons were frequently arranged as highly expanded DRG, qualitatively phenocopying the baz1 phenotype. We conclude that in the baz1 mutants, Sox10(baz1)-dependent activation of neurog1 expression functions in conjunction with reduced Notch signaling to drive the specification of supernumerary DRG neurons.
Fig 9. Global mis-expression of Neurog1 is sufficient to phenocopy the baz1 mutant DRG phenotype.

(A) Quantification of mean (±s.d.) total numbers of Elav1/Hu+ DRG neurons in tail (blue), trunk (green) or total (red). The number of embryos analysed was 6 (WT control), 6 (hs:ngn1), 8 (MO sox10) and 7 (MO sox10, hs:ngn1). Significance of differences as determined by two-tailed Student’s t test comparisons is indicated by * (p<0.05), ** (p<0.001) and *** (p<0.0005). Control (B-C’) or sox10-morpholino injected (D-E’) embryos were left uninduced (B,B’,D,D’) or subjected to heat-shock (C,C’,E,E’) and subsequently processed for Elav1/Hu immunofluorescence at 5 dpf; embryos were heat-shocked at 28hpf for 1 hour. Panels show lateral views of trunk (B,C,D,E) and close-ups of representative DRGs (B’,C’,D’,E’).
Discussion
In this study, we have comprehensively extended the description of the neural phenotypes of the baz1 mutants, showing clearly that the glial, as well as enteric and sympathetic neuron phenotypes are similar to other strong loss-of-function sox10 alleles. Interestingly, the baz1 sensory neuron phenotype is restricted to the DRGs and does not affect the placode-derived neurons of the cranial ganglia (data not shown). We provide evidence arguing against any changes in the balance of DRG cell proliferation/apoptosis underlying the baz1 phenotype. Instead, using molecular markers and timelapse studies with double transgenic reporter lines, we note the strikingly precocious and supernumerary nature of that DRG sensory neuron phenotype. These features are typical of defects in lateral inhibition processes, driven by Notch signaling. Previous work in mouse and chick had shown that Delta-Notch signaling controls neuronal versus glial fate choice in the DRGs[42, 43], leading us to test the hypothesis that this mechanism was conserved in zebrafish DRGs. We used two Notch signaling inhibitors (DAPT and compound E) in a 30–72 hpf time-window, combined with transgenic reporter studies to show that Notch signaling is active in the DRGs at these developmental stages. Our expression studies of all known Notch ligands clearly implicate DeltaA and/or DeltaD, and not the other Deltas nor the Jaggeds, as the relevant signals. Although the jagged genes’ expression patterns do not overlap with neurog1, sox10 or foxd3, making a direct role in DRG development unlikely, the wide expression of jag1a in neural cells close to cells expressing these markers, allows the possibility that Jag1a might have some role in DRG development. Indeed, expression of Jagged1 has been documented in mouse DRGs, at the E10.5 developmental stage [62]. However, our studies of the effects of Jag1a knockdown using previously characterised morpholinos [63] resulted in significant increases in isl1+ Rohon-Beard neurons but showed no effects on numbers of either isl1+ motor neurons nor sox10+ neural crest cells in the DRG domain (NT, unpub. data). These data indicate that Notch signaling in the early stages of DRG development depends predominantly on DeltaA and DeltaD. McGraw et al. [47] also used DAPT treatment, independently supported by a transgenic approach to inhibit Notch signaling, to assess the requirement for Notch signaling in regulating neuron cell number in larval DRGs. Their treatment windows partially overlapped those used in our study, extending from 2 to 5 dpf, and achieved changes in DRG neuron number quantitatively comparable to those observed in our Notch inhibitor studies described here. McGraw and colleagues demonstrated the expression of deltaA and deltaD in cells adjacent to differentiating DRG sensory neurons and suggested that Delta-Notch signaling in DRG progenitors controls neuron cell number. Here, we confirm and extend their observations using i) co-expression of in situ markers neurog1 and neurod1 with delta gene expression to show that early neuronal progenitors are expressing delta genes, but importantly also ii) by directly demonstrating that activation of Notch signaling is detectable in vivo in the early DRGs.
Our study and others’ [47, 64] have left open the possibility of non-autonomous effects of the Notch signaling, but the demonstration here of activated Notch signaling in DRGs, coupled with our and the Raible groups [47] demonstration of expression of Delta-Notch signaling components in these cells, provides strong support for the idea of an autonomous function in the DRGs. Furthermore, recent in vivo studies in mouse using a Wnt1-Cre transgene to target the neural crest showed an autonomous role for Notch signaling in mouse DRGs[44, 45], so it seems that Delta-Notch signaling in the nascent DRG, and then continuing through larval development as neurons are added in the zebrafish [47], is a conserved mechanism crucial for the balanced production of neurons and glia in vivo. Importantly, consistent with this proposal, our direct assessment of Notch signaling levels in the sox10 mutant DRGs clearly shows that Notch signaling is active in m618 mutants at levels comparable to WT siblings, but that there is substantial reduction in autonomous signaling activity in baz1 mutants.
It might seem that a natural interpretation of our baz1 data is that in this sox10 mutant allele DRG development occurs in the absence of effective Delta-Notch signaling, perhaps because expression of one or more signaling components is Sox10-dependent and thus compromised in the mutant DRGs i.e. the DRG phenotype is simply a neurogenic phenotype. This conclusion would be consistent with observations from mouse, showing that Notch-1 expression in nascent DRGs is much reduced in Sox10Dom homozygotes[20]. But we note that that same study showed that Sox10Dom mutants have a reduced number of DRG sensory neurons compared to WT siblings, and this is consistent with the typical loss-of-function sox10 phenotype seen in zebrafish [10, 13, 50]. Furthermore, in zebrafish, we show directly here that while Notch signaling is reduced in the developing DRGs of baz1 mutants, it is apparently intact in the more typical m618 mutant allele. Thus, the molecular explanation for the differential sensory neuron phenotypes is that Sox10 regulates expression of neurog1 [13], and this is missing in these typical sox10 mutants, whereas in baz1 mutants neurog1 activation by Sox10 is retained and Notch signaling is impaired. Finally, we note that quantitatively the effects on DRG sensory neuron numbers of disrupting Delta-Notch signaling using two different drugs (DAPT and Compound E) or a transgenic approach (c. 25–30% increase; [47] and this study) are consistently distinct from that of the baz1 mutant (c. 250% increase; [13] and this study). This suggests that the explanation for the baz1 phenotype must be more than simply loss of Delta-Notch signaling.
In this context, the surprisingly nuanced effects on neural crest development in the unique baz1 mutant is revealing. These mutants combine a strong loss-of-function phenotype for pigment cells and glia with a gain of function-type DRG sensory neuron phenotype. Our sox10 in situ data indicate that a key role for Sox10 in maintenance of its own expression in glial cells is impaired; this is an important observation since Sox10 is a key transcription factor driving glial cell differentiation[20, 31, 65–68]. Thus glial development is largely prevented, consistent with the precocious differentiation of secondary neuromasts shown here. Crucially, however, Sox10-mediated transcriptional activation of neurog1 is substantially normal in DRG sensory neurons. The subtle substitution (V117M) of hydrophobic residues in the Sox10(baz1) mutant DNA-binding HMG domain would seem compatible with sequence-specific alterations in DNA binding properties, a view supported by our initial in silico modelling approaches. These simulations suggested that the baz1 mutation might result in modifications to the flexibility of the HMG domain structure, but it will require more detailed molecular dynamic and experimental structural analyses to explore the impact of the substitution in detail. For now, we predict that expression of key targets in glial (e.g. sox10) and melanocyte (e.g. mitfa) development fails as in other loss-of-function alleles, whereas a select subset of targets (e.g. neurog1) remain activated at approximately normal levels.
How do these effects combine to generate a neurogenic sensory neuron phenotype in the DRGs? Studies in mouse Sox10 mutants have nicely demonstrated that extracellular signals influencing neural crest cell fate decisions are differentially interpreted depending upon the cellular context and levels of Sox10 protein (and thus, presumably, Sox10 activity)[67]. We propose a model that combines our data with the current understanding of the cellular origin of DRG neurons so as to explain this unique phenotype (Fig 10). An elegant study by the Raible group established that zebrafish DRGs are derived from bipotent sensory neuroglial precursors that express low level neurog1[29]. They showed that in normal development a subset of these progenitors maintain and upregulate neurog1 and thus become sensory neurons, whilst the remaining cells downregulate neurog1 expression and become glial cells; furthermore, in neurog1 mutants, all progenitors adopt a glial fate. We note the neurogenic phenotype and recalling its established mechanism, we assume that Sox10 dependent alterations of Delta-Notch signaling are significant. In a WT situation (normal Sox10), Sox10 promotes upregulation of neurog1 in some precursors as they adopt a neuronal fate. Positive feedback between Neurog1 and Delta expression in the nascent sensory neurons results in enhanced Delta-Notch signaling and, together with Sox10-dependent maintenance of sox10 expression, this promotes glial fate choice in Notch-responsive cells, achieving a balance of cell-types. In m618 mutants, Sox10-dependent neurog1 and sox10 expression are both impaired, resulting in reduced numbers of sensory neurons and a complete absence of glial differentiation; hence, many DRG progenitors fail to become specified and remain in a progenitor state. Our data shows that Sox10(m618) protein is incapable of activating neurog1 expression, so the ‘escaper’ neurons are clearly not Sox10-dependent; indeed, in previous work we showed that production of the remaining sensory neurons depends upon Sox9b activity [13]. The number of sensory neurons formed in the m618 mutant DRGs is restricted to a WT number, likely due to the intact Notch signaling that we show here. In contrast, in baz1 mutants, Sox10(baz1) retains the ability to activate neurog1 expression so that sensory neuron specification occurs normally. But in addition, Delta-Notch signaling fails to be activated and Sox10-dependent activation of neurog1 in neighbouring progenitors in the nascent DRG fails to be repressed. As the neuronal fate is not correctly repressed in putative “glial precursors”, this results in a neurogenic phenotype. As described so far, this would be expected to result in a DRG phenotype equivalent to that generated by inhibition of Notch signaling i.e. a relatively modest increase in DRG neuron numbers. So, how do we explain the elevated numbers of sensory neurons in the baz1 DRGs? We deduce that Sox10 must have a role in repressing neuronal development in glial progenitors that is independent of Delta-Notch signaling. As noted, in baz1 mutants, Sox10-dependent positive feedback on sox10 expression is reduced too, so that glial fates fail to be specified. The baz1 phenotype demonstrates that one important aspect of this glial fate specification process is a further negative feedback on neuronal development, one that is independent of Delta-Notch signaling but which reinforces Notch signaling-dependent repression of neurog1 expression (Factor X in Fig 10). Although the molecular details of this mechanism remain to be discovered, the baz1 phenotype demonstrates that in its absence persistent neurog1 expression will drive the majority of DRG progenitors to adopt a sensory neuronal fate, consistent with our neurog1 overexpression data which in both wild-type and sox10 loss-of-function situations promotes formation of DRGs with strongly elevated sensory neuron numbers. Our study of the effects of over-expressing the different Sox10 proteins (Fig 8) shows unexpected Sox10-;Neurog1+ cells adjacent to the expected Sox1+;Neurog1+ cells where Sox10(baz1) was overexpressed. These might be explained by destabilisation of the Sox10(baz1) protein or, more unexpectedly, by an apparent short-range, non-autonomous effect of Sox10(baz1). The former suggestion would be consistent with the general loss-of-function type phenotype displayed by this mutant; in this context, it might be that the ability to maintain activation of neurog1 would be exceptional.
Fig 10. Model of fate specification in DRG development.

A) Integrating our data from this study with previous work we suggest how the initial DRG consisting of bipotent DRG progenitors (black rectangles) become specified (second row) and the resultant cell-fates adopted (bottom row). We outline the process in WT (neuron (N, cyan) and glial fates (G, magenta); first column), baz1 (neuron only; second column), m618 (failure to undergo fate specification; third column) and neurog1 mutants (all adopt glia; fourth column). The model summarises at each stage the relative levels of inputs (large font is high activity, small font is weak activity) from each of Sox10, Neurog1, Notch signaling and a Sox10-dependent Factor X, proposed to provide a repressive input on neurog1 expression. Colour coding of text indicates function of each input (i.e. Neurog1 drives sensory neuron, Notch signaling and Factor X drive glial specification), but Sox10 (which has independent roles in specification of both fates) is shown in both colours where WT, and in blue in baz1 and in black in m618 to reflect the neural fate specification activity retained in these mutants. In the case of Sox10 input, note that also font size in neighbouring cells refers to activity with respect to fate being adopted by each cell, since baz1 mutant reveals that these roles can be separated. The underlying gene regulatory network is outlined in panel B). Asterisk in m618 cells denotes that sensory neuron specification in m618 mutants occurs in the absence of Sox10 activity, as a result of Sox9b-mediated transcription of neurog1[13]. See text for further details. We note that the data from Fig 8 indicates the possibility that Sox10(baz1) may have unique non-autonomous effects on neurog1 expression, which, if confirmed, would require modification of the model proposed.
Our data showing neurog1 transgene activation by Sox10 proteins (Fig 9) strongly support previous data indicating a direct role for Sox10 in DRG sensory neuron specification via activation of neurog1 expression[13]. Our transgene data indicate that crucial regulatory elements lie in the region 8.4 kb upstream of the transcriptional start site; consistent with this, we note the presence of multiple Sox10 binding sites in this interval (RM and PB, unpub. data). Further work to characterise the specific binding sites and their regulation will be necessary to fully understand sensory neuron specification. To the best of our knowledge it remains unclear if this mechanism is conserved in mouse [13, 20, 31], although we find conserved Sox10 binding sites upstream of neurog1 and neurog2 in mouse.
Together, our data indicates that in the zebrafish DRGs, Sox10 has a crucial role in establishing the ratios of neuronal and glial cell-types, through roles in fate specification of both sensory neurons and glia (via transcriptional activation of neurog1 expression in nascent neurons and sox10 maintenance in prospective glial cells, respectively), but also by a mechanism of neuronal fate repression in prospective glial cells that is independent of Notch signaling. A detailed dissection of the molecular mechanisms underlying this intriguing and unexpected role will depend upon extensive characterisation of the structure of the gene regulatory network underlying glial fate specification and differentiation.
Materials and methods
Fish husbandry and zebrafish strains
All animals were handled in accordance with the relevant national and international guidelines. Animals in the Kelsh lab are housed in a facility certified by the Home Office, and the work was approved by the University of Bath Animal Welfare and Ethical Review Body and performed under Home Office Project Licenses PPL30/2415 and PPL30/2937. Animals in the Blader lab were housed in a facility certified by the French Ministry of Agriculture: approval ID B-31-555-10. The work in the Blader lab is monitored by a local ethics committee (FRBT C2EA-01). All animals in the Tiso lab were handled under the Italian Ministry of Health authorization n. 407/2015 (UniPD Ethical Committee Reference: OPBA n. 50/2014). Embryos were collected from natural matings and staged (hours post fertilization (hpf) at 28.5°C) according to Kimmel et al. [69]. They were incubated at 28.5°C in 100ml petri dishes containing embryo medium (0.137M NaCl, 5.4mM KCl, 0.25mM Na2HPO4, 0.44mM KH2PO4, 1.3mM CaCl2, 1.0mM MgSO4, 4.2mM NaHCO3). Strains used were as described previously: sox10m618 and sox10baz1 [10, 13]; Tg(neurog1(-8.4):nRFP)sb3, Tg(neurog1(-8.4):EGFP)sb1Tg and Tg(neurog1(-3.1):EGFP)sb2Tg [49]; Tg(hsp70l:neurog1)ups1 [60], Tg(-4.9sox10:eGFP)ba2 [13], Tg(sox10(7.2):mRFP) [55]; Tg(−2.4 kb neurod1:GFP)ia50 [70]; Tg(12xNRE:eGFP) and Tg(12xNRE:mCherry) [54]. Homozygous mutant embryos were readily identified by pigmentation phenotype.
Whole-mount in situ hybridisation, antibody staining and TUNEL analysis, imaging and statistical analysis
These were performed as previously described [13], with mutants and siblings in same tube and >20 embryos processed per condition and representative embryos selected for imaging. Experiments were performed at least in duplicate. Fluorescent dual color In situ hybridization, based on FastRed/FastBlue double staining, was performed as described in Lauter et al. [71]. In situ probes were as follows: deltaA, deltaB, and deltaD [72], deltaC [73], foxd3[74], jgd1a, jgd1b, jgd2 [53], mbp[75], neurog1[28], neurod[28], notch1a [76], notch1b [77], notch2 [78], notch3 [79], sox10 [10], and egfp (Tol2 Kit vector). Antibody staining was largely performed as described in [40]. Prior to primary antibody incubation, embryos were permeabilized with 5μl/ml of proteinase K for 30 mins at 37°C, then rinsed with 5% goat serum at RT for 5 mins and washed 3x1 hour with distilled water at RT. Antibodies used were Hu/Elav1 (1:700 mAb 16A11)[80], Tyrosine Hydroxylase (1:500, Immunostar, Cat. # 22941) and Alexa Fluor 488 goat-mouse IgG (1:750, Molecular Probes, Cat. # A11001). For post mortem imaging, embryos were cleared and mounted in glycerol/PBS buffer. For in vivo imaging of transgenic lines, embryos were anesthetized in tricaine (0.016%) and mounted in 1% low melting agarose/PBS buffer. Embryos were analysed and imaged using either an LSM510 confocal microscope (Carl Zeiss Imaging) or an Eclipse E800 (Nikon) microscope using appropriate filters and a SPOT digital camera (Diagnostic Instruments) or an AxioImager M2 with Apotome 2 (Zeiss), or using a spectral confocal microscope (Leica SP5), able to selectively acquire near-red fluorescence from FastRed and far-red emission from FastBlue. Image analysis and 3D processing were performed using either ZEN 2009 (Carl Zeiss Imaging) or Volocity 6.0 software (Perkin Elmer). Images were minimally processed using Photoshop (Adobe, Creative Suite 6) to adjust levels, contrast and brightness. Fluorescence quantification was obtained with the Measurements tool of the Volocity 6.0 software, by fluorescent dot counting performed on maximum projection images of selected anatomical regions (ROI dimension: 10 μm x 10 μm x 50 μm; 6 measurements/condition). Counts of DRG cells were performed on 1 side of an embryo only. Statistical analysis was performed using the Prism Statistical Package (GraphPad, San Diego, CA).
Timelapse studies
For timelapse analysis of DRG development embryos were mounted in 0.6% low melting point agarose in glass-bottomed dishes after dechorionation and anaesthesis in 0.004% tricaine. Imaging was performed using either LSM510META or LSM5 Live confocal microscopes (Carl Zeiss Imaging), with image processing using either ZEN 2009 (Carl Zeiss Imaging) or Imaris 7.1 software (Bitplane A6, Zurich, Switzerland). Embryos were staged by position of PLL primordium [69] at start of experiment. Up to 4 embryos were imaged simultaneously, with wild-type (WT) and baz1 mutants mounted alongside each other. Images were taken at 30 min intervals for up to c. 25 h.
Notch inhibitor studies
Notch inhibitor studies were performed on dechorionated embryos (dishes were coated in 2% agarose to prevent adhesion of embryos to dish during treatment. DAPT-treatment was performed according to Geling et al (2002) [57], using 100 μM DAPT in 2% DMSO in embryo medium. Compound E[58] was used in a similar way[81], with embryos treated with 100μM (diluted into 2% DMSO) working solution. In all cases, dishes were wrapped in foil during treatment to exclude light.
Neurog1 regulation assays
PCR fragments were inserted into a previously described vector to generate Myc-tagged fusion proteins for Sox10(WT), Sox10(m618) and Sox10(baz1) under the control of the hsp70l promoter [60]. These plasmids were injected into embryos from the Tg(neurog1(-8.4):EGFP)sb1 line. Embryos were subsequently heat-shocked for 1 hour starting at 7 hpf, and fixed at 9.5 hpf. Finally, in situ hybridisations coupled with Myc immunodetections were performed to identify cells expressing the fusion proteins (anti-Myc “9E10”; [82]) and to address whether expression of the reporter transgene (gfp mRNA) had been induced in these cells. Three independent injection/induction experiments were performed for each Sox10 variant; at least 40 embryos were injected with each construct. While embryos injected with hs:sox10(m618) never resulted in ectopic activation of the neurog1(-8.4):EGFP transgene despite all embryos displaying cells expressing Myc-tagged protein after heatshock, both hs:sox10(WT) and hs:sox10(baz1) injection/induction resulted in robust ectopic gfp expression. In the case of hs:sox10(WT) injection/induction, ectopic expression of gfp was restricted to Myc-positive cells whereas gfp+/Myc-negative cells were often seen adjacent to gfp+/Myc+ cells when injection/induction was performed with the hs:sox10(baz1) transgene.
Molecular modeling
3D prediction
HMG box from human SOX-9 bound to the DNA sequence 5'-CTCTTTGAGAAG-3' has been used as template structure (pdb: 4EUW). Given that wild type and baz1 mutant proteins share 98% sequence identity with the template, backbone and conserved amino-acids have been preserved. Non-conserved amino acids side chains have been reconstructed using the side-chain placement tool SCWRL 4.0 [83].
MD simulation
All simulation steps were carried over using the AMBER03 force field [84] within the GROMACS 5.04 (http://www.gromacs.org/) software package [85] for both wild type and baz1 mutant structural models. The simulation was run in a cubic box with a 1.0 nm edge length and the system was solvated with Simple Point Charge water (SPC), a generic equilibrated 3-point solvent model. In order to neutralize the systems, 15 sodium ions were added to replace 15 water molecules. Electrostatic interactions were calculated by the Particle Mesh Ewald (PME) method [86] and All bond lengths were constrained by the LINCS algorithm [87]. Energy minimization was performed and the systems were subjected to a steepest descent energy minimization until reaching a tolerance force of no greater than 1000 kJ mol-1 nm-1. Equilibration of the solvent and ions around the protein-DNA complex was conducted in two phases. The system was slowly heated up from 0 K to 300 K under an NVT ensemble (constant Number of particles, Volume, and Temperature) for 100-ps. After temperature equilibration, pressure stabilization was carried out under an NPT ensemble for 100-ps, wherein the Number of particles, Pressure, and Temperature are all constant and the Parrinello-Rahman barostat was used [88]. The integration time step was 2 fs for both NVT and NPT. Complete MD simulation was run for 100000000 steps (200 ns) at a constant temperature of 300 K and a constant pressure of 1 atm. The trajectories were obtained using “gmx trjconv” GROMACS tool, corrected for periodicity, and analyzed by using “gmx rms” GROMACS utility in order to obtain the Root-Mean-Square Deviation (RMSD). Principal Components Analysis (PCA) has been carried out on the final trajectories by building the covariance matrix of the atomic fluctuations using the “gmx covar”. The set of eigenvectors and eigenvalues was obtained from the diagonalisation of the matrix using “gmx anaeig” tool. The first three principal components representing the largest-amplitude collective motions have been considered.
Supporting information
A, B) Expression of isl1 at 48 hpf in WT (A) and baz1 (B) is seen in DRGs (*), but is especially prominent in the baz1 mutants where supernumerary cells are often more ventrally positioned and thus more prominent.
(TIF)
Extracted frames from S1 video (WT, left) and S2 video (baz1, right) of 4 segments of trunk of Tg(-4.9sox10:eGFP)ba2; Tg(neurog1(-8.4)):nRFP). Arrows indicate subset of DRG neurons (nRFP+). Time since timelapse initiated indicated in Hrs;Mins.
(JPG)
{kind=link}
A-L) sox10 expression at 48 hpf. Columns show sox10-expressing cells in head (A, E, I), lateral pathway of trunk (B, F, J), medial pathway of trunk (C, G, K) and tail (D, H, L) respectively of 48 hpf WT (A-D), baz1 (E-H) and m618 (I-L) mutant embryos. Glia of cranial ganglia and Schwann cell precursors on cranial and PLL nerve (white arrowheads) are prominent in WT, but highly reduced or nearly absent in baz1 and m618 mutants respectively. Cells of DRGs (white asterisk) form a prominent segmentally reiterated pattern on the medial pathway of trunk and tail of WT, but are reduced in sox10 mutants; note that in tail, where cells are developmentally younger, the DRG patterns are more similar. Xanthoblasts (black asterisk) are prominent on lateral pathway of WTs, but reduced and absent in baz1 and m618 mutants respectively. M,N,P,Q) foxd3 expression at 48 hpf. Expression in Schwann cell precursors associated with spinal nerves forms segmentally reiterated pattern readily seen in WT (M), but numbers of cells are much reduced in baz1 (P). foxd3-expressing Schwann cell precursors (white arrowhead) on PLLn are prominent in WT (N), but highly reduced in number in baz1 at 48 hpf (Q), where they are usually seen only anteriorly. O,R) Supernumerary neuromasts in baz1 mutants. Whole mount In situ hybridisation with isl1 probe in 48 hpf WT (O) and baz1 mutant (R). Note supernumerary neuromasts (black arrowheads) in baz1 mutant. Photos all from single, typical individual of each genotype, after PTU treatment.
(TIF)
A,B) Oligodendrocyte precursors at 48 hpf in trunk spinal cord express sox10 and are indistinguishable in number and distribution in WT (A) and baz1 mutant (B). C,D) sox10 expression in dispersing oligodendrocyte of hindbrain; note that numbers and dorsoventral distribution strongly resemble wild-type siblings. E,F) sox10 expression in oligodendrocyte progenitors in the ventralmost spinal cord are unaffected at 72 hpf; region of somites 7–11 is shown. G,H) Oligodendrocyte differentiation is abnormal as shown by strongly decreased mbp expression in hindbrain of 72 hpf embryo.
(TIF)
A-C) deltaA expression (red) is faint, but overlaps with neurod1 (green) in the nascent DRG (arrows) at 30 hpf. D-F) At 30 hpf, deltaD expression (red) overlaps with neurod1 (green) in the nascent DRG (arrows). G-I) deltaA expression (red) clearly overlaps with deltaD (green) in the nascent DRG (arrows) at 30 hpf. All main panels are confocal images of fluorescent dual-color In situ hybridisations in lateral view, with insets showing y-z planes (left) and x-z planes (above) for each. nc, notochord; sc, spinal cord.
(TIF)
A-D) bright field (A,C) and fluorescent (B,D) views of control (ctrl) and mutant (m618, baz1) embryos in Notch reporter (NRE:EGFP) transgenic background. m618 embryos and their controls do not show dramatic differences in Notch reporter activation, while baz1 mutants exhibit decreased fluorescent signals compared to controls. All panels display 36 hpf embryos in lateral view, anterior to the left. Scale bar: 1 mm.
(JPG)
{kind=link}
A-F) fluorescent WISH analysis in baz1 and WT sib shows a decrease of sox10 and Notch reporter transcription in the DRG regions (arrows) of the mutant (D-F), compared to the control (A-C). G-L) fluorescent WISH analysis in m618 and WT sib shows a decrease of sox10 and persistency of Notch reporter transcription in the DRG regions (arrows) of the mutant (J-L), compared to the control (G-I). All panels display embryonic trunk regions at 30 hpf, in lateral view with anterior to the left.
(JPG)
{kind=link}
The results of the principal component analysis of the first three largest-amplitude collective motions are reported. A,B) Energy minimized models of wild type (A) and Sox10(baz1)(B) mutant respectively. Both minimum and maximum extremes of the fluctuations are shown as white ribbons and the average structure is shown as black ribbon. Valine and methionine are highlighted in sticks. C-F) Root mean square deviation (RMSD; y-axis) of each structure of the MD trajectory with the corresponding energy minimized structure from 200 ns MD simulation (x-axis time in ns). Panels show the backbone (C,D) and DNA fluctuations (E,F) of wild type (C,E) and Sox10(baz1)(D,F).
(TIF)
Arrow indicates typical DRG sensory neuron.
(AVI)
Arrows indicates subset of DRG sensory neurons.
(AVI)
Acknowledgments
We are grateful to the Biotechnological and Biological Sciences Research Council (http://www.bbsrc.ac.uk/), grant BB/D000440/1, RNK) and University of Bath (http://www.bath.ac.uk/) for funding this work, as well as to the Uehara Memorial Foundation (http://www.ueharazaidan.or.jp/) and a Wellcome Trust VIP award (http://www.wellcome.ac.uk/; MN), Royal Society International Collaboration Grant JP100027 (https://royalsociety.org/) to RNK and CONACyT (http://www.conacyt.mx/) grant 329640/384511 to KCS. NT is supported by the EU ZF-HEALTH grant (CT-2010-242048) (www.zf-health.org), by the French Muscular Dystrophy Association (AFM Telethon project POLYGON, 18572) (www.afm-telethon.com), by the Ministry of Health grant RF-2010-2309484 (www.salute.gov.it) and by the University of Padova projects OPTOZEN (CPDA128151) and TIGRE (CPDA148582) (http://www.unipd.it). GB is recipient of a post-doc fellowship “Assegno di Ricerca Junior” (CPDR124317/12) from the University of Padova (http://www.unipd.it). ST is supported by PRAT grant CPDA138081/13 from University of Padova (http://www.unipd.it). Work by RM and PB was funded by the Fondation ARC (http://www.fondation-arc.org; grant SFI20101201699).
We are grateful to Donna Zhang and Maria Daskalaki for technical assistance, to Richard Spire and Marc Shedden for fish care, and to Jean van den Elsen for advice on structural modelling. We gratefully acknowledge Silvia Munoz-Decalzo, Chris Dooley, Alessandro Mongera and Virgilio Failla for technical advice on timelapse imaging and processing.
Data Availability
All original numerical datasets are provided in the University of Bath Research Data Archive at https://doi.org/10.15125/BATH-00164.
Funding Statement
We are grateful to the Biotechnological and Biological Sciences Research Council (http://www.bbsrc.ac.uk/), grant BB/D000440/1, RNK) and University of Bath (http://www.bath.ac.uk/) for funding this work, as well as to the Uehara Memorial Foundation (http://www.ueharazaidan.or.jp/) and a Wellcome Trust VIP award (http://www.wellcome.ac.uk/; MN), Royal Society International Collaboration Grant JP100027 (https://royalsociety.org/) to RNK and CONACyT (http://www.conacyt.mx/) grant 329640/384511 to KCS. NT is supported by the EU ZF-HEALTH grant (CT-2010-242048) (www.zf-health.org), by the French Muscular Dystrophy Association (AFM Telethon project POLYGON, 18572) (www.afm-telethon.com), by the Ministry of Health grant RF-2010-2309484 (www.salute.gov.it) and by the University of Padova projects OPTOZEN (CPDA128151) and TIGRE (CPDA148582) (http://www.unipd.it). GB is recipient of a post-doc fellowship “Assegno di Ricerca Junior” (CPDR124317/12) from the University of Padova (http://www.unipd.it). ST is supported by PRAT grant CPDA138081/13 from University of Padova (http://www.unipd.it). Work by RM and PB was funded by the Fondation ARC (http://www.fondation-arc.org; grant SFI20101201699). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Le Douarin NM, Kalcheim C. The Neural Crest. 2nd ed Cambridge: Cambridge University Press; 1999. [Google Scholar]
- 2.Nitzan E, Krispin S, Pfaltzgraff ER, Klar A, Labosky PA, Kalcheim C. A dynamic code of dorsal neural tube genes regulates the segregation between neurogenic and melanogenic neural crest cells. Development. 2013;140(11):2269–79. Epub 2013/04/26. 10.1242/dev.093294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mayor R, Theveneau E. The neural crest. Development. 2013;140(11):2247–51. Epub 2013/05/16. 10.1242/dev.091751 [DOI] [PubMed] [Google Scholar]
- 4.Milet C, Monsoro-Burq AH. Neural crest induction at the neural plate border in vertebrates. Dev Biol. 2012;366(1):22–33. Epub 2012/02/07. 10.1016/j.ydbio.2012.01.013 [DOI] [PubMed] [Google Scholar]
- 5.Herbarth B, Pingault V, Bondurand N, Kuhlbrodt K, Hermans-Borgmeyer I, Puliti A, et al. Mutation of the Sry-related Sox10 gene in Dominant megacolon, a mouse model for human Hirschsprung disease. Proc Natl Acad Sci U S A. 1998;95(9):5161–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuhlbrodt K, Herbarth B, Sock E, Hermans-Borgmeyer I, Wegner M. Sox10, a novel transcriptional modulator in glial cells. J Neurosci. 1998;18(1):237–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pingault V, Bondurand N, Kuhlbrodt K, Goerich DE, Prehu MO, Puliti A, et al. SOX10 mutations in patients with Waardenburg-Hirschsprung disease. Nat Genet. 1998;18(2):171–3. 10.1038/ng0298-171 [DOI] [PubMed] [Google Scholar]
- 8.Southard-Smith EM, Kos L, Pavan WJ. Sox10 mutation disrupts neural crest development in DOM Hirschsprung mouse model. 1061–4036. 1998;18(1):60–4. [DOI] [PubMed] [Google Scholar]
- 9.Bondurand N, Pingault V, Goerich DE, Lemort N, Sock E, Caignec CL, et al. Interaction among SOX10, PAX3 and MITF, three genes altered in Waardenburg syndrome. Hum Mol Genet. 2000;9(13):1907–17. [DOI] [PubMed] [Google Scholar]
- 10.Dutton KA, Pauliny A, Lopes SS, Elworthy S, Carney TJ, Rauch J, et al. Zebrafish colourless encodes sox10 and specifies non-ectomesenchymal neural crest fates. Development. 2001;128(21):4113–25. [DOI] [PubMed] [Google Scholar]
- 11.Elworthy S, Lister JA, Carney TJ, Raible DW, Kelsh RN. Transcriptional regulation of mitfa accounts for the sox10 requirement in zebrafish melanophore development. Development. 2003;130(12):2809–18. [DOI] [PubMed] [Google Scholar]
- 12.Mollaaghababa R, Pavan WJ. The importance of having your SOX on: role of SOX10 in the development of neural crest-derived melanocytes and glia. Oncogene. 2003;22(20):3024–34. 10.1038/sj.onc.1206442 [DOI] [PubMed] [Google Scholar]
- 13.Carney TJ, Dutton KA, Greenhill E, Delfino-Machin M, Dufourcq P, Blader P, et al. A direct role for Sox10 in specification of neural crest-derived sensory neurons. Development. 2006;133(23):4619–30. 10.1242/dev.02668 [DOI] [PubMed] [Google Scholar]
- 14.Kelsh RN. Sorting out Sox10 functions in neural crest development. Bioessays. 2006;28(8):788–98. 10.1002/bies.20445 [DOI] [PubMed] [Google Scholar]
- 15.Lee M, Goodall J, Verastegui C, Ballotti R, Goding CR. Direct regulation of the microphthalmia promoter by Sox10 links Waardenburg-Shah syndrome (WS4)-associated hypopigmentation and deafness to WS2. J Biol Chem. 2000;275:37978–83. 10.1074/jbc.M003816200 [DOI] [PubMed] [Google Scholar]
- 16.Potterf SB, Furumura M, Dunn KJ, Arnheiter H, Pavan WJ. Transcription factor hierarchy in Waardenburg syndrome: regulation of MITF expression by SOX10 and PAX3. Hum Genet. 2000;107(1):1–6. [DOI] [PubMed] [Google Scholar]
- 17.Verastegui C, Bille K, Ortonne JP, Ballotti R. Regulation of the microphthalmia-associated transcription factor gene by the Waardenburg syndrome type 4 gene, SOX10. J Biol Chem. 2000;275(40):30757–60. 10.1074/jbc.C000445200 [DOI] [PubMed] [Google Scholar]
- 18.Aoki Y, Saint-Germain N, Gyda M, Magner-Fink E, Lee YH, Credidio C, et al. Sox10 regulates the development of neural crest-derived melanocytes in Xenopus. Dev Biol. 2003;259(1):19–33. [DOI] [PubMed] [Google Scholar]
- 19.Hou L, Arnheiter H, Pavan WJ. Interspecies difference in the regulation of melanocyte development by SOX10 and MITF. Proc Natl Acad Sci U S A. 2006;103(24):9081–5. 10.1073/pnas.0603114103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Britsch S, Goerich DE, Riethmacher D, Peirano RI, Rossner M, Nave KA, et al. The transcription factor Sox10 is a key regulator of peripheral glial development. Genes Dev. 2001;15(1):66–78. 10.1101/gad.186601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wegner M, Stolt CC. From stem cells to neurons and glia: a Soxist's view of neural development. Trends Neurosci. 2005;28(11):583–8. Epub 2005/09/06. 10.1016/j.tins.2005.08.008 [DOI] [PubMed] [Google Scholar]
- 22.Prasad MK, Reed X, Gorkin DU, Cronin JC, McAdow AR, Chain K, et al. SOX10 directly modulates ERBB3 transcription via an intronic neural crest enhancer. BMC Dev Biol. 2011;11:40 Epub 2011/06/16. 10.1186/1471-213X-11-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jacob C, Lotscher P, Engler S, Baggiolini A, Varum Tavares S, Brugger V, et al. HDAC1 and HDAC2 control the specification of neural crest cells into peripheral glia. J Neurosci. 2014;34(17):6112–22. Epub 2014/04/25. 10.1523/JNEUROSCI.5212-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greenwood AL, Turner EE, Anderson DJ. Identification of dividing, determined sensory neuron precursors in the mammalian neural crest. Development. 1999;126(16):3545–59. [DOI] [PubMed] [Google Scholar]
- 25.Perez SE, Rebelo S, Anderson DJ. Early specification of sensory neuron fate revealed by expression and function of neurogenins in the chick embryo. Development. 1999;126(8):1715–28. [DOI] [PubMed] [Google Scholar]
- 26.Ma Q, Anderson DJ, Fritzsch B. Neurogenin 1 null mutant ears develop fewer, morphologically normal hair cells in smaller sensory epithelia devoid of innervation. J Assoc Res Otolaryngol. 2000;1(2):129–43. 10.1007/s101620010017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cornell RA, Eisen JS. Delta/Notch signaling promotes formation of zebrafish neural crest by repressing Neurogenin 1 function. Development. 2002;129(11):2639–48. Epub 2002/05/17. [DOI] [PubMed] [Google Scholar]
- 28.Blader P, Fischer N, Gradwohl G, Guillemont F, Strahle U. The activity of neurogenin1 is controlled by local cues in the zebrafish embryo. Development. 1997;124(22):4557–69. [DOI] [PubMed] [Google Scholar]
- 29.McGraw HF, Nechiporuk A, Raible DW. Zebrafish dorsal root ganglia neural precursor cells adopt a glial fate in the absence of neurogenin1. J Neurosci. 2008;28(47):12558–69. Epub 2008/11/21. 10.1523/JNEUROSCI.2079-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pavan WJ, Raible DW. Specification of neural crest into sensory neuron and melanocyte lineages. Dev Biol. 2012;366(1):55–63. Epub 2012/04/03. 10.1016/j.ydbio.2012.02.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sonnenberg-Riethmacher E, Miehe M, Stolt CC, Goerich DE, Wegner M, Riethmacher D. Development and degeneration of dorsal root ganglia in the absence of the HMG-domain transcription factor Sox10. Mech Dev. 2001;109(2):253–65. [DOI] [PubMed] [Google Scholar]
- 32.Dorsky RI, Moon RT, Raible DW. Control of neural crest cell fate by the Wnt signalling pathway. Nature. 1998;396(6709):370–3. Epub 1998/12/09. 10.1038/24620 [DOI] [PubMed] [Google Scholar]
- 33.Lee HY, Kleber M, Hari L, Brault V, Suter U, Taketo MM, et al. Instructive role of Wnt/beta-catenin in sensory fate specification in neural crest stem cells. Science. 2004;303(5660):1020–3. 10.1126/science.1091611 [DOI] [PubMed] [Google Scholar]
- 34.Hari L, Brault V, Kleber M, Lee HY, Ille F, Leimeroth R, et al. Lineage-specific requirements of beta-catenin in neural crest development. J Cell Biol. 2002;159(5):867–80. 10.1083/jcb.200209039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shah NM, Anderson DJ. Integration of multiple instructive cues by neural crest stem cells reveals cell-intrinsic biases in relative growth factor responsiveness. 0027–8424. 1997;94(21):11369–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shah NM, Marchionni MA, Isaacs I, Stroobant P, Anderson DJ. Glial growth-factor restricts mammalian neural crest stem-cells to a glial fate. Cell. 1994;77(3):349–60. [DOI] [PubMed] [Google Scholar]
- 37.Honjo Y, Kniss J, Eisen JS. Neuregulin-mediated ErbB3 signaling is required for formation of zebrafish dorsal root ganglion neurons. Development. 2008;135(15):2615–25. Epub 2008/07/05. 10.1242/dev.022178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dooley CM, Mongera A, Walderich B, Nusslein-Volhard C. On the embryonic origin of adult melanophores: the role of ErbB and Kit signalling in establishing melanophore stem cells in zebrafish. Development. 2013;140(5):1003–13. Epub 2013/02/01. 10.1242/dev.087007 [DOI] [PubMed] [Google Scholar]
- 39.Malmquist SJ, Abramsson A, McGraw HF, Linbo TH, Raible DW. Modulation of dorsal root ganglion development by ErbB signaling and the scaffold protein Sorbs3. Development. 2013;140(19):3986–96. Epub 2013/09/06. 10.1242/dev.084640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ungos JM, Karlstrom RO, Raible DW. Hedgehog signaling is directly required for the development of zebrafish dorsal root ganglia neurons. Development. 2003;130(22):5351–62. 10.1242/dev.00722 [DOI] [PubMed] [Google Scholar]
- 41.Cornell RA, Eisen JS. Notch in the pathway: the roles of Notch signaling in neural crest development. Semin Cell Dev Biol. 2005;16(6):663–72. Epub 2005/08/02. 10.1016/j.semcdb.2005.06.009 [DOI] [PubMed] [Google Scholar]
- 42.Morrison SJ, Perez SE, Qiao Z, Verdi JM, Hicks C, Weinmaster G, et al. Transient Notch activation initiates an irreversible switch from neurogenesis to gliogenesis by neural crest stem cells. Cell. 2000;101(5):499–510. [DOI] [PubMed] [Google Scholar]
- 43.Wakamatsu Y, Maynard TM, Weston JA. Fate determination of neural crest cells by NOTCH-mediated lateral inhibition and asymmetrical cell division during gangliogenesis. Development. 2000;127(13):2811–21. Epub 2000/06/14. [DOI] [PubMed] [Google Scholar]
- 44.Hu ZL, Shi M, Huang Y, Zheng MH, Pei Z, Chen JY, et al. The role of the transcription factor Rbpj in the development of dorsal root ganglia. Neural Dev. 2011;6:14 Epub 2011/04/23. 10.1186/1749-8104-6-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mead TJ, Yutzey KE. Notch pathway regulation of neural crest cell development in vivo. Dev Dyn. 2012;241(2):376–89. Epub 2012/01/26. 10.1002/dvdy.23717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.An M, Luo R, Henion PD. Differentiation and maturation of zebrafish dorsal root and sympathetic ganglion neurons. J Comp Neurol. 2002;446(3):267–75. [DOI] [PubMed] [Google Scholar]
- 47.McGraw HF, Snelson CD, Prendergast A, Suli A, Raible DW. Postembryonic neuronal addition in zebrafish dorsal root ganglia is regulated by Notch signaling. Neural Dev. 2012;7:23 Epub 2012/06/29. 10.1186/1749-8104-7-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee JE, Hollenberg SM, Snider L, Turner DL, Lipnick N, Weintraub H. Conversion of Xenopus ectoderm into neurons by NeuroD, a basic helix-loop-helix protein. Science. 1995;268:836–44. [DOI] [PubMed] [Google Scholar]
- 49.Blader P, Plessy C, Strahle U. Multiple regulatory elements with spatially and temporally distinct activities control neurogenin1 expression in primary neurons of the zebrafish embryo. Mech Dev. 2003;120(2):211–8. [DOI] [PubMed] [Google Scholar]
- 50.Kelsh RN, Eisen JS. The zebrafish colourless gene regulates development of non-ectomesenchymal neural crest derivatives. Development. 2000;127:515–25. [DOI] [PubMed] [Google Scholar]
- 51.Lopez-Schier H, Hudspeth AJ. Supernumerary neuromasts in the posterior lateral line of zebrafish lacking peripheral glia. Proc Natl Acad Sci U S A. 2005;102(5):1496–501. Epub 2005/01/29. 10.1073/pnas.0409361102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Higashijima S, Hotta Y, Okamoto H. Visualization of cranial motor neurons in live transgenic zebrafish expressing green fluorescent protein under the control of the islet-1 promoter/enhancer. 0270–6474. 2000;20:206–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zecchin E, Conigliaro A, Tiso N, Argenton F, Bortolussi M. Expression analysis of jagged genes in zebrafish embryos. Dev Dyn. 2005;233(2):638–45. Epub 2005/04/15. 10.1002/dvdy.20366 [DOI] [PubMed] [Google Scholar]
- 54.Moro E, Vettori A, Porazzi P, Schiavone M, Rampazzo E, Casari A, et al. Generation and application of signaling pathway reporter lines in zebrafish. Mol Genet Genomics. 2013;288(5–6):231–42. Epub 2013/05/16. 10.1007/s00438-013-0750-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kucenas S, Takada N, Park HC, Woodruff E, Broadie K, Appel B. CNS-derived glia ensheath peripheral nerves and mediate motor root development. Nat Neurosci. 2008;11(2):143–51. Epub 2008/01/08. 10.1038/nn2025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Parsons MJ, Pisharath H, Yusuff S, Moore JC, Siekmann AF, Lawson N, et al. Notch-responsive cells initiate the secondary transition in larval zebrafish pancreas. Mech Dev. 2009;126(10):898–912. 10.1016/j.mod.2009.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Geling A, Steiner H, Willem M, Bally-Cuif L, Haass C. A gamma-secretase inhibitor blocks Notch signaling in vivo and causes a severe neurogenic phenotype in zebrafish. EMBO Rep. 2002;3(7):688–94. 10.1093/embo-reports/kvf124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Seiffert D, Bradley JD, Rominger CM, Rominger DH, Yang F, Meredith JE Jr., et al. Presenilin-1 and -2 are molecular targets for gamma-secretase inhibitors. J Biol Chem. 2000;275(44):34086–91. 10.1074/jbc.M005430200 [DOI] [PubMed] [Google Scholar]
- 59.Schiavone M, Rampazzo E, Casari A, Battilana G, Persano L, Moro E, et al. Zebrafish reporter lines reveal in vivo signaling pathway activities involved in pancreatic cancer. Dis Model Mech. 2014;7(7):883–94. Epub 2014/06/01. 10.1242/dmm.014969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Madelaine R, Blader P. A cluster of non-redundant Ngn1 binding sites is required for regulation of deltaA expression in zebrafish. Dev Biol. 2011;350(1):198–207. Epub 2010/11/03. 10.1016/j.ydbio.2010.10.025 [DOI] [PubMed] [Google Scholar]
- 61.Dutton K, Dutton JR, Pauliny A, Kelsh RN. A morpholino phenocopy of the colourless mutant. Genesis. 2001;30(3):188–9. [DOI] [PubMed] [Google Scholar]
- 62.Mitsiadis TA, Henrique D, Thesleff I, Lendahl U. Mouse Serrate-1 (Jagged-1): expression in the developing tooth is regulated by epithelial-mesenchymal interactions and fibroblast growth factor-4. Development. 1997;124(8):1473–83. Epub 1997/04/01. [DOI] [PubMed] [Google Scholar]
- 63.Zecchin E, Filippi A, Biemar F, Tiso N, Pauls S, Ellertsdottir E, et al. Distinct delta and jagged genes control sequential segregation of pancreatic cell types from precursor pools in zebrafish. Dev Biol. 2007;301(1):192–204. Epub 2006/10/25. 10.1016/j.ydbio.2006.09.041 [DOI] [PubMed] [Google Scholar]
- 64.Gray M, Moens CB, Amacher SL, Eisen JS, Beattie CE. Zebrafish deadly seven functions in neurogenesis. Dev Biol. 2001;237(2):306–23. Epub 2001/09/07. 10.1006/dbio.2001.0381 [DOI] [PubMed] [Google Scholar]
- 65.Schlierf B, Werner T, Glaser G, Wegner M. Expression of connexin47 in oligodendrocytes is regulated by the Sox10 transcription factor. J Mol Biol. 2006;361(1):11–21. Epub 2006/07/11. 10.1016/j.jmb.2006.05.072 [DOI] [PubMed] [Google Scholar]
- 66.Ito Y, Wiese S, Funk N, Chittka A, Rossoll W, Bommel H, et al. Sox10 regulates ciliary neurotrophic factor gene expression in Schwann cells. Proc Natl Acad Sci U S A. 2006;103(20):7871–6. Epub 2006/05/11. 10.1073/pnas.0602332103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Paratore C, Goerich DE, Suter U, Wegner M, Sommer L. Survival and glial fate acquisition of neural crest cells are regulated by an interplay between the transcription factor Sox10 and extrinsic combinatorial signaling. Development. 2001;128(20):3949–61. Epub 2001/10/20. [DOI] [PubMed] [Google Scholar]
- 68.Peirano RI, Goerich DE, Riethmacher D, Wegner M. Protein zero gene expression is regulated by the glial transcription factor Sox10. 0270–7306. 2000;20(9):3198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of Embryonic Development of the Zebrafish. Dev Dynamics. 1995;203:253–310. [DOI] [PubMed] [Google Scholar]
- 70.Casari A, Schiavone M, Facchinello N, Vettori A, Meyer D, Tiso N, et al. A Smad3 transgenic reporter reveals TGF-beta control of zebrafish spinal cord development. Dev Biol. 2014;396(1):81–93. Epub 2014/10/07. 10.1016/j.ydbio.2014.09.025 [DOI] [PubMed] [Google Scholar]
- 71.Lauter G, Soll I, Hauptmann G. Two-color fluorescent in situ hybridization in the embryonic zebrafish brain using differential detection systems. BMC Dev Biol. 2011;11:43 Epub 2011/07/06. 10.1186/1471-213X-11-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Haddon C, Smithers L, Schneider-Maunory S, Coche T, Henrique D, Lewis J. Multiple delta genes and lateral inhibition in zebrafish primary neurogenesis. Development. 1998;125(3):359–70. [DOI] [PubMed] [Google Scholar]
- 73.Smithers L, Haddon C, Jiang Y-J, Lewis J. Sequence and embryonic expression of deltaC in the zebrafish. Mechanisms of Development. 2000;90:119–23. [DOI] [PubMed] [Google Scholar]
- 74.Odenthal J, Nusslein-Volhard C. fork head domain genes in zebrafish. Dev Genes Evol. 1998;208(5):245–58. [DOI] [PubMed] [Google Scholar]
- 75.Brosamle C, Halpern ME. Characterization of myelination in the developing zebrafish. Glia. 2002;39(1):47–57. 10.1002/glia.10088 [DOI] [PubMed] [Google Scholar]
- 76.Jiang YJ, Brand M, Heisenberg CP, Beuchle D, Furutani-Seiki M, Kelsh RN, et al. Mutations affecting neurogenesis and brain morphology in the zebrafish, Danio rerio. Development. 1996;123:205–16. Epub 1996/12/01. [DOI] [PubMed] [Google Scholar]
- 77.Pasini A, Jiang YJ, Wilkinson DG. Two zebrafish Notch-dependent hairy/Enhancer-of-split-related genes, her6 and her4, are required to maintain the coordination of cyclic gene expression in the presomitic mesoderm. Development. 2004;131(7):1529–41. Epub 2004/03/17. 10.1242/dev.01031 [DOI] [PubMed] [Google Scholar]
- 78.Thisse B, Pfumio S, Fürthauer M, B L, Heyer V, Degrave A, et al. Expression of the zebrafish genome during embryogenesis. ZFIN online publication. 2001.
- 79.Itoh M, Chitnis AB. Expression of proneural and neurogenic genes in the zebrafish lateral line primordium correlates with selection of hair cell fate in neuromasts. Mechanisms of Development. 2001;102:263–6. [DOI] [PubMed] [Google Scholar]
- 80.Marusich MF, Furneaux HM, Henion PD, Weston JA. Hu neuronal proteins are expressed in proliferating neurogenic cells. J Neurobiol. 1994;25(2):143–55. 10.1002/neu.480250206 [DOI] [PubMed] [Google Scholar]
- 81.Bernardos RL, Lentz SI, Wolfe MS, Raymond PA. Notch-Delta signaling is required for spatial patterning and Muller glia differentiation in the zebrafish retina. Dev Biol. 2005;278(2):381–95. 10.1016/j.ydbio.2004.11.018 [DOI] [PubMed] [Google Scholar]
- 82.Evan GI, Lewis GK, Ramsay G, Bishop JM. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol Cell Biol. 1985;5(12):3610–6. Epub 1985/12/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Krivov GG, Shapovalov MV, Dunbrack RL Jr. Improved prediction of protein side-chain conformations with SCWRL4. Proteins. 2009;77(4):778–95. Epub 2009/07/16. 10.1002/prot.22488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Duan Y, Wu C, Chowdhury S, Lee MC, Xiong GM, Zhang W, et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem. 2003;24(16):1999–2012. 10.1002/jcc.10349 [DOI] [PubMed] [Google Scholar]
- 85.Pronk S, Pall S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29(7):845–54. 10.1093/bioinformatics/btt055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A Smooth Particle Mesh Ewald Method. J Chem Phys. 1995;103(19):8577–93. [Google Scholar]
- 87.Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: A linear constraint solver for molecular simulations. J Comput Chem. 1997;18(12):1463–72. [Google Scholar]
- 88.Parrinello M, Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys. 1981;52:7182–90. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A, B) Expression of isl1 at 48 hpf in WT (A) and baz1 (B) is seen in DRGs (*), but is especially prominent in the baz1 mutants where supernumerary cells are often more ventrally positioned and thus more prominent.
(TIF)
Extracted frames from S1 video (WT, left) and S2 video (baz1, right) of 4 segments of trunk of Tg(-4.9sox10:eGFP)ba2; Tg(neurog1(-8.4)):nRFP). Arrows indicate subset of DRG neurons (nRFP+). Time since timelapse initiated indicated in Hrs;Mins.
(JPG)
A-L) sox10 expression at 48 hpf. Columns show sox10-expressing cells in head (A, E, I), lateral pathway of trunk (B, F, J), medial pathway of trunk (C, G, K) and tail (D, H, L) respectively of 48 hpf WT (A-D), baz1 (E-H) and m618 (I-L) mutant embryos. Glia of cranial ganglia and Schwann cell precursors on cranial and PLL nerve (white arrowheads) are prominent in WT, but highly reduced or nearly absent in baz1 and m618 mutants respectively. Cells of DRGs (white asterisk) form a prominent segmentally reiterated pattern on the medial pathway of trunk and tail of WT, but are reduced in sox10 mutants; note that in tail, where cells are developmentally younger, the DRG patterns are more similar. Xanthoblasts (black asterisk) are prominent on lateral pathway of WTs, but reduced and absent in baz1 and m618 mutants respectively. M,N,P,Q) foxd3 expression at 48 hpf. Expression in Schwann cell precursors associated with spinal nerves forms segmentally reiterated pattern readily seen in WT (M), but numbers of cells are much reduced in baz1 (P). foxd3-expressing Schwann cell precursors (white arrowhead) on PLLn are prominent in WT (N), but highly reduced in number in baz1 at 48 hpf (Q), where they are usually seen only anteriorly. O,R) Supernumerary neuromasts in baz1 mutants. Whole mount In situ hybridisation with isl1 probe in 48 hpf WT (O) and baz1 mutant (R). Note supernumerary neuromasts (black arrowheads) in baz1 mutant. Photos all from single, typical individual of each genotype, after PTU treatment.
(TIF)
A,B) Oligodendrocyte precursors at 48 hpf in trunk spinal cord express sox10 and are indistinguishable in number and distribution in WT (A) and baz1 mutant (B). C,D) sox10 expression in dispersing oligodendrocyte of hindbrain; note that numbers and dorsoventral distribution strongly resemble wild-type siblings. E,F) sox10 expression in oligodendrocyte progenitors in the ventralmost spinal cord are unaffected at 72 hpf; region of somites 7–11 is shown. G,H) Oligodendrocyte differentiation is abnormal as shown by strongly decreased mbp expression in hindbrain of 72 hpf embryo.
(TIF)
A-C) deltaA expression (red) is faint, but overlaps with neurod1 (green) in the nascent DRG (arrows) at 30 hpf. D-F) At 30 hpf, deltaD expression (red) overlaps with neurod1 (green) in the nascent DRG (arrows). G-I) deltaA expression (red) clearly overlaps with deltaD (green) in the nascent DRG (arrows) at 30 hpf. All main panels are confocal images of fluorescent dual-color In situ hybridisations in lateral view, with insets showing y-z planes (left) and x-z planes (above) for each. nc, notochord; sc, spinal cord.
(TIF)
A-D) bright field (A,C) and fluorescent (B,D) views of control (ctrl) and mutant (m618, baz1) embryos in Notch reporter (NRE:EGFP) transgenic background. m618 embryos and their controls do not show dramatic differences in Notch reporter activation, while baz1 mutants exhibit decreased fluorescent signals compared to controls. All panels display 36 hpf embryos in lateral view, anterior to the left. Scale bar: 1 mm.
(JPG)
A-F) fluorescent WISH analysis in baz1 and WT sib shows a decrease of sox10 and Notch reporter transcription in the DRG regions (arrows) of the mutant (D-F), compared to the control (A-C). G-L) fluorescent WISH analysis in m618 and WT sib shows a decrease of sox10 and persistency of Notch reporter transcription in the DRG regions (arrows) of the mutant (J-L), compared to the control (G-I). All panels display embryonic trunk regions at 30 hpf, in lateral view with anterior to the left.
(JPG)
The results of the principal component analysis of the first three largest-amplitude collective motions are reported. A,B) Energy minimized models of wild type (A) and Sox10(baz1)(B) mutant respectively. Both minimum and maximum extremes of the fluctuations are shown as white ribbons and the average structure is shown as black ribbon. Valine and methionine are highlighted in sticks. C-F) Root mean square deviation (RMSD; y-axis) of each structure of the MD trajectory with the corresponding energy minimized structure from 200 ns MD simulation (x-axis time in ns). Panels show the backbone (C,D) and DNA fluctuations (E,F) of wild type (C,E) and Sox10(baz1)(D,F).
(TIF)
Arrow indicates typical DRG sensory neuron.
(AVI)
Arrows indicates subset of DRG sensory neurons.
(AVI)
Data Availability Statement
All original numerical datasets are provided in the University of Bath Research Data Archive at https://doi.org/10.15125/BATH-00164.