

Abstract
Soybean lipoxygenase-1 (SLO-1) catalyzes the C-H abstraction from the reactive carbon (C-11) in linoleic acid as the first and rate-determining step in the formation of alkylhydroperoxides. While previous labeling strategies have focused on deuterium labeling to ascertain the primary and secondary kinetic isotope effects for this reaction, there is an emerging interest and need for selectively enriched 13C isotopologues. In this report, we present synthetic strategies for site-specific 13C labeled linoleic acid substrates. We take advantage of a Corey-Fuchs formyl to terminal 13C-labeled alkyne conversion, using 13CBr4 as the labeling source, to reduce the number of steps from a previous fatty acid 13C synthetic labeling approach. The labeled linoleic acid substrates are useful as nuclear tunneling markers and for extracting active site geometries of the enzyme-substrate complex in lipoxygenase.
Keywords: Secondary isotope effects, hydrogen tunneling, lipoxygenase, linoleic acid
Graphical Abstract

Lipoxygenases, widely represented in animals, plants, fungi and prokaryotes, have gained much attention in the field due to observations of enormous, non-classical primary deuterium kinetic isotope effects (KIEs) in the range of 20–120 for the native enzymes.1 These enzymes catalyze the C-H abstraction from long chain polyunsaturated fatty acids in the process of preparing diverse (per)oxidized metabolites, important for growth, development and pathogenic defense in plants and various cell signaling and inflammatory response pathways in animals.2 Importantly, soybean lipoxygenase-1 (SLO-1) has emerged as one of the paradigmatic enzymes in hydrogen tunneling, because the rate-determining hydrogen atom abstraction in the native form is associated with a very large primary KIEs at room temperature (ca. 80), in a nearly temperature independent manner (ΔEa = 0.9 kcal/mol).3 As shown in Figure 1, for the SLO-1 reaction, a mononuclear, nonheme ferric hydroxide accepts the transferred electron and proton, respectively, generating a ferrous water cofactor and a pentadienyl radical. Molecular oxygen is then inserted into the backbone, selectively at position 13 for wild-type SLO-1.1a, 4 A second round of proton-coupled electron transfer (PCET) regenerates the active ferric-hydroxide cofactor and yields the product, 13S-hydroperoxyoctadecadienoic acid. Also shown in Figure 1 are two amino acid sidechains, Leu546 and Leu754, which position the reactive carbon in proximity to the ferric-hydroxide reactive center and are integral for efficient hydrogen wave function overlap in SLO-1.5
Figure 1.

Mechanism of linoleic acid peroxidation by soybean lipoxygenase-1. The first and final steps of the reaction are associated with proton-coupled electron transfer (PCET) processes. The important, conserved aliphatic residues (Leu546 and Leu754) that position the reactive carbon (C-11 of linoleic acid) against the active site ferric hydroxide cofactor in SLO-1 are modeled for reference.
Inflated, non-classical primary deuterium isotope effects (kH/kD > 7), together with deviations in Swain-Schaad relationships (especially for secondary D/T KIEs) and temperature independent KIEs, are considered as kinetic signatures for full tunneling in enzymatic reactions occurring at room temperature.6 In the case of lipoxygenases, many synthetic strategies have been reported for site-specific deuterium labeled substrates, but there has also been an emerging interest in 13C labels for these compounds. While the tunneling properties have been ascribed to the transferred hydrogen, recent evidence in SLO-1 reaction points towards the tunneling of heavy atoms in the backbone of the substrate.7 In that study, the 13C KIEs were measured from natural abundance 13C using an NMR approach designed for small molecules by Singleton.8 To overcome the relatively large errors associated with these measurements for enzymatic reactions, a high precision 1H-detected 13C heteronuclear single quantum coherence (HSQC) 2 dimensional (2D) NMR approach has been previously reported for the determination of such 13C KIEs. 9 This technique requires the site-specific labeling of the carbon atom of interest and a carbon atom, adjacent to the target carbon atom so that they are spin-coupled, as a reporter.10 While 1 dimensional (1D) 13C NMR techniques are typically easier and faster and can be utilized to test these competitive isotope effects,10 the HSQC approach9 may offer enhanced signal-to-noise, which is particularly advantageous in the lipoxygenase reaction that is limited to low (micromolar) substrate concentrations. In addition, site-specific 13C labeling of arachidonic acid has previously served to assign the magnetic resonance spectral contributions of the delocalized radicals, generated upon reaction with prostaglandin H synthase.11 While arachidonic acid is the preferred substrate of mammalian lipoxygenases2a and exhibits rate constants and deuterium KIE for the reaction with SLO-1 that is comparable to linoleic acid, the latter is considered the physiological substrate for the plant enzyme.12 This manuscript describes synthetic strategies for generating site-specific 13C enriched linoleic acids. These labeled substrates can be applied to evaluate the extent of tunneling in the substrate backbone from 13C KIEs by 1D 13C or 2D HSQC NMR techniques, to assign magnetic resonance signatures of the substrate radicals in the lipoxygenase reaction, and/or to resolve the elusive donor-acceptor distances in various lipoxygenase enzymes using magnetic resonance techniques sensitive to electron-nuclear couplings.13
Our first target is the isotopologue with 13C enriched at the reactive carbon, C-11, of linoleic acid (11-[13C]-1), which, as shown in Scheme 1, was synthesized in two parts with an unlabeled fragment, 2-(dec-9-yn-1-yloxy)tetrahydro-2H-pyran (3) and a 13C labeled fragment, 1-bromo-2-octyne (1-[13C]-4). As shown for example in Scheme 1, the two fragments 3 and 4 were joined via a Cu(I)-assisted Grignard coupling reaction. A similar strategy was previously used in our laboratory with 1,1-[2H2]-4 and 9-decynoic acid that produced low yields of the desired, stable intermediate in the synthesis of monodeuterated or dideuterated linoleic acids.14 To overcome these previous pitfalls, we started with commercially available 9-decyn-1-ol (2), which we protected through the overnight reaction of 3,4-dihydro-2H-pyran (DHP) to generate 3. In this manner, only 1 eq. of the ethylmagnesium bromide (EtMgBr) is required to deprotonate the terminal alkyne, 3. This strategy produced reasonable yields (ca. 70 %) of the desired 11-[13C]-2-(octadeca-9,12-diyn-1-yloxy)tetrahydro-2H-pyran, 11-[13C]-5. In the course of arachidonic acid synthesis for prostaglandin H synthase substrates, Peng et al. followed a similar approach that led to high yields (86%).11 One caveat is that a fraction of the starting material 3 is virtually inseparable from product 11-[13C]-5 with silica gel purifications. We maintained the contaminating starting material from the coupling step to the end, because it did not interfere with the final steps. In our final purification step, linoleic acid could be separated cleanly and easily from this impurity using a reverse phase C18 column.
Scheme 1.

Synthesis of 11-[13C] -1. The asterisks denote the position of the 13C label.
After the coupling that generates 11-[13C]-3, the linoleic acid substrate could be completed (Scheme 1) by deprotection of 11-[13C]-5, liberating the corresponding alcohol, 11-[13C]-6, followed by oxidation to the acid, 11-[13C]-7, with Jones reagent and hydrogenation to linoleic acid, 11-[13C]-1. The hydrogenation reaction was performed with standard Lindlar catalyst and H2. Further, our hydrogenation reactions were carried out in presence of quinolones which enhances selectivity for 9Z, 12Z alkene production.15 HPLC analyses of the purified final products indicated only linoleic acid (C18:2 Δ9,12) with no detectable amounts of oleic (C18:1 Δ9) or stearic (C18:0) acids, consistent with no over-reduction. 1H NMR spectra were consistent with cis,cis-9,12 isomer. An alternative approach, described by our group previously,16 catecholborane can also be used to achieve reduction selectivity.15
The 13C labeled 1-bromo-2-octyne fragment, 1-[13C]-4, was synthesized as described in Scheme 2. First, 2-octyn-1-ol (10) was generated from the deprotonation of the commercially available 1-heptyne 8 with EtMgBr that was subsequently reacted with 13C-paraformaldehyde ([13C]-9). This route has been described previously for the synthesis of 1-[2H]-10 and 1,1-[2H2]-10.14, 17 Bromination of 1-[13C]-10 compounds to the corresponding desired 1-[13C]-4 was carried out cleanly with Ph3P/Br2.
Scheme 2.
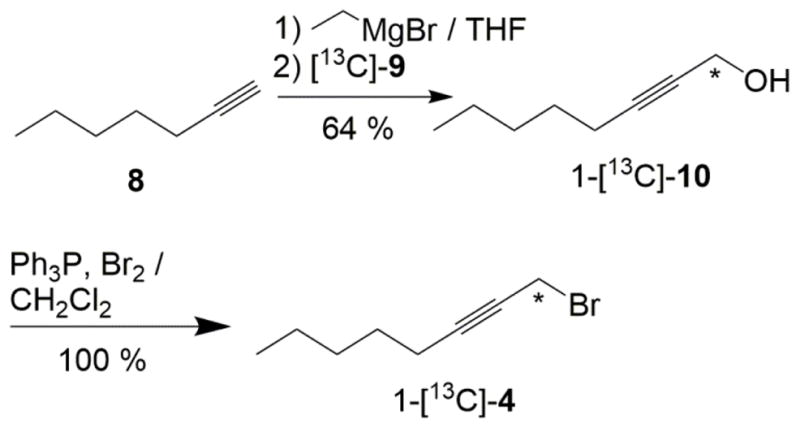
Synthesis of fragment 1-[13C]-4 using 13C-paraformaldehyde, [13C]-9. The asterisks denote the position of the 13C label.
As depicted in Scheme 3, the linoleic acids, 10-[13C]-11,11-[2H2]-1 and 10,11-[13C2]-11,11-[2H2]-1 were also synthesized in two parts, with a common 13C labeled C-1 to C-10 fragment (2). The label (12C or 13C) at reactive carbon in the final product, 1, was controlled by the nature of the carbon isotope in the 1-bromo-2-octyne (3) fragment. In addition, deuterium was also included at the reactive carbon (C-11) for both linoleic acids in Scheme 3. Our lab has previously shown that for the WT SLO-1 enzyme chemistry is partially rate limiting with the protium substrate under certain conditions, but becomes fully rate limiting with deuterated substrate.1a Therefore, dideuterated substrates would be required to isolate the intrinsic KIE of the reactive carbon from NMR competitive measurements.
Scheme 3.

General strategy outline for 2H, 13C isotope labeling of linoleic acid substrates. The asterisks denote the position of the 13C label. For details about the synthetic steps, refer to Scheme 1.
The protection of the 1,9-nonanediol (11) resulted in a mixture of starting material 11, desired single protected 9-((tetrahydro-2H-pyran-2-yl)oxy)nonan-1-ol (12) and fully protected species. Because the starting material 11 was inexpensive and the desired compound 12 was easily separable with silica gel purification, we could prepare this compound on a relatively large scale. As illustrated in Scheme 4, oxidation of the alcohol 12 to the corresponding 9-((tetrahydro-2H-pyran-2-yl)oxy)nonan-1-ol (13) was carried out with pyridinium chlorochromate (PCC) and sodium acetate to maintain the THP protecting group. The highlight within this synthesis is the preparation of 2-(10-[13C]-dec-9-yn-1-yloxy)tetrahydro-2H-pyran (10-[13C]-3). This step was accomplished by a formyl-to-ethynyl conversion, first described by Corey and Fuchs,18 using the aldehyde 13 and triphenylphosine-carbon tetrabromide-13C. The inclusion of 1 eq. of anhydrous triethylamine19 during the addition of the aldehyde 13 to the stirring triphenylphosphine-carbon tetrabromide solution in CH2Cl2 was essential to produce the desired 2-((10-[13C]-10,10-dibromodec-9-en-1-yl)oxy)tetrahydro-2H-pyran (10-[13C]-14). Due to the cost of the 13C isotopologue, we reduced the starting equivalents of 13C-carbontetrabromide from the recommended 2 eq.18–19 to 1 eq. in this report. Even with only 1 eq. of 13CBr4 (and 2 eq. Ph3P), we were able to cleanly produce the desired 10-[13C]-14 in very satisfactory yields (ca. 90%).
Scheme 4.

Synthesis of common fragment 10-[13C]-3.
It has been suggested that alkyne lithium salts couple with appropriate electrophiles in a two-step, one pot synthesis.19 It could be envisioned that addition of 2 eq. of a strong base such as lithium diisopropylamide (LDA) would sequentially transform 10-[13C]-14 to a lithium alkyne salt. Subsequent addition of 4 would, in principle, act as a proper electrophile and undergo carbon-carbon bond formation, thus generating the desired 9,12-diyne. However, attempts to directly couple the lithium alkyne salt, generated in situ, with natural abundance 1-bromo-2-octyne (4) were not successful and upon work up gave only the alkyne 10-[13C]-3. We therefore reacted the dibromoalkene, 10-[13C]-14, with excess LDA at −70°C to ensure conversion to the lithium alkyne salt, which was warmed to room temperature and quenched to isolate the desired 2-(10-[13C]-dec-9-yn-1-yloxy)tetrahydro-2H-pyran (10-[13C]-3) in good yield (90%; Scheme 4).
For the synthesis of 1,1-[2H2]-10 and 1-[13C]-1,1-[2H2]-10, the 2H and/or 13C isotopologues were introduced onto 1-heptyne 8 using paraformaldehyde-d2 and paraformaldehyde-13C-d2, respectively, forming the corresponding labeled 2-octyn-1-ol (1,1-[2H2]-10 and 1-[13C]-1,1-[2H2]-10). The route is comparable to that described for the synthesis of 1-[13C]-10 in Scheme 2. Paraformaldehyde allows us to add not only 13C at the reactive carbon, but also deuterium. The compound 1,1-[2H2]-10 has also been prepared previously via reduction of the commercially available methyl 2-octynoate with LiAlD4.16 Reduction of 2-octynoate with LiAlD4 produces slightly better yields of the 1,1-[2H2]-2-octyn-1-ol, but requires either additional purification or reaction steps to remove unwanted side products. For the synthesis of the deuterium and 13C labeled 2-octyn-1-ol, the most direct route is through the use of paraformaldehyde-d2-13C, as outlined here. As illustrated in Scheme 2, bromination of these 2-octyn-1-ol (10) labels generated the respective labeled 1-bromo-2-octyne (4). The coupling and final steps for these labeled compounds are identical to those described above in Scheme 1 for 11-[13C]-1 and gave similar yields and purities for final products.
Unimolecular rate (kcat) and Michaelis (KM) constants were measured from the reaction of the linoleic acids synthesized here with WT SLO-1 at 30°C (see Table 1). Michaelis-Menton saturation curves were plotted from measuring the rate of product formation, monitored spectrophotometrically from the absorbance at 234 nm,20 as a function of substrate concentration (2–80 μM). The kinetic parameters of 10-[13C]-11,11-[2H2]-1 and 10,11-[13C2]-11,11-[2H2]-1 are compared to the commercially available perdeuterated substrate, extracted from alagal cells.21 It is important to note that the kcat for the perdeuterated substrate (kcat = 5.7 s−1) is taken from a recent report22 and the apparent slight elevation of this value in reference to the kcat magnitudes for the deuterated, carbon-13 labeled substrates, reported here (Table 1), is well within the typical variation from different enzyme preparations. Importantly, the KM values for all substrates are identical. These data are validations to the synthesized compounds, 10-[13C]-11,11-[2H2]-1 and 10,11-[13C2]-11,11-[2H2]-1, as bona fide lipoxygenase substrates.
Table 1.
Kinetic analysis of the 2H, 13C labeled substrates with WT SLO-1.a
| Linoleic acid | kcat (s−1) | KM (μM) |
|---|---|---|
| [2H31]-1b | 5.7 (0.1) | 7.8 (0.5) |
| 11,11-[2H2]-1c | 4.5 (0.2) | 7.7 (1.3) |
| 10-[13C]-11,11-[2H2]-1d | 4.7 (0.2) | 7.8 (1.0) |
| 10,11-13C2-11,11-[2H2]-1d | 4.5 (0.2) | 7.6 (1.0) |
Reactions were conducted with 3.5 nM WT SLO, prepared as previously described,5 at 30°C in 0.1 M borate, pH 9.0 buffer. Substrates were prepared at 1 mM working stocks in 0.1 M sodium borate (pH 9.0) buffer. Prior to use, the concentration of the effective substrate was confirmed enzymatically. Each kinetic data set was conducted with 7 substrate concentrations. For each substrate analyzed, the resulting kinetic parameters are averages from 3 individual data sets.
Substrate is commercially available from Cambridge Isotopes. The first order rate (kcat) is from ref 22
Synthesized from our lab previously; kcat is consistent with previous report20.
Synthesized here.
While there are several reports that describe routes to synthesize lipoxygenase substrates (linoleic and arachidonic acids) that are enriched with deuterons at various positions along the backbone (see for examples, refs 11, 14, 16–17, 23), this report describes the strategy to label backbone carbons site-selectively in linoleic acid. An alternative approach has been reported for the site-specific 13C labeling of arachidonic acids for substrates of PGHS and lipoxygenase. 11 In that report, Peng et al. used potassium 13C-cyanide to add the 13C label, primarily due to its comparatively low cost. Here, we chose to use the Corey-Fuchs aldehyde-alkyne conversion with 13C-carbon tetrabromide as one source for the 13C label, because this approach, although using a slightly more expensive starting material, led to a shorter route (11 steps vs 17 steps in ref 11) for the final product.
Supplementary Material
Strategies for site-specific 13C isotope labeling of fatty acids
Cu(I)-mediated cross coupling reactions
Application of Corey-Fuchs aldehyde-alkyne conversion for selective 13C insertion
Acknowledgments
Financial support was provided by the National Institutes of Health GM118117-01 to J.P.K.; A.R.O was supported by NIH postdoctoral fellowship F32 GM113432. The authors thank Prof. Carolyn Bertozzi for the use of the Biotage HPLC. The authors also thank Drs. Shenshen Hu, Eric Koehn, John Latham, Jianyu Zhang, and Gaëlle Deshayes for helpful discussions.
Footnotes
Supporting Information available. Experimental procedures and NMR characterization of synthetic compounds.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) Glickman MH, Klinman JP. Nature of rate-limiting steps in the soybean lipoxygenase-1 reaction. Biochemistry. 1995;34:14077–14092. doi: 10.1021/bi00043a013. [DOI] [PubMed] [Google Scholar]; (b) Su C, Sahlin M, Oliw EH. Kinetics of manganese lipoxygenase with a catalytic mononuclear redox center. J Biol Chem. 2000;275:18830–18835. doi: 10.1074/jbc.M001408200. [DOI] [PubMed] [Google Scholar]; (c) Segraves EN, Holman TR. Kinetic investigations of the rate-limiting step in human 12- and 15-lipoxygenase. Biochemistry. 2003;42:5236–5243. doi: 10.1021/bi0273462. [DOI] [PubMed] [Google Scholar]; (d) Carr CAM, Klinman JP. Hydrogen tunneling in a prokaryotic lipoxygenase. Biochemistry. 2014;53:2212–2214. doi: 10.1021/bi500070q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Brash AR. Lipoxygenases: occurrence, functions, catalysis, and acquisition of substrate. J Biol Chem. 1999;274:23679–23682. doi: 10.1074/jbc.274.34.23679. [DOI] [PubMed] [Google Scholar]; (b) Haeggstrom JZ, Funk CD. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem Rev. 2011;111:5866–5898. doi: 10.1021/cr200246d. [DOI] [PubMed] [Google Scholar]
- 3.Klinman JP. Importance of protein dynamics during enzymatic C-H bond cleavage catalysis. Biochemistry. 2013;52:2068–2077. doi: 10.1021/bi301504m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Collazo L, Klinman JP. Control of the position of oxygen delivery in soybean lipoxygenase-1 by amino acid side chains within a gas migration channel. J Biol Chem. 2016;291:9052–9059. doi: 10.1074/jbc.M115.709154. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Coffa G, Schneider C, Brash A. A comprehensive model of positional and stereo control in lipoxygenases. Biochem Biophys Res Commun. 2005;338:87–92. doi: 10.1016/j.bbrc.2005.07.185. [DOI] [PubMed] [Google Scholar]
- 5.Hu S, Sharma SC, Scouras AD, Soudackov AV, Carr CAM, Hammes-Schiffer S, Alber T, Klinman JP. Extremely elevated room-temperature kinetic isotope effects quantify the critical role of barrier width in enzymatic C-H activation. J Am Chem Soc. 2014;136:8157–8160. doi: 10.1021/ja502726s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Knapp MJ, Klinman JP. Environmentally coupled hydrogen tunneling. Eur J Biochem. 2002;269:3113–3121. doi: 10.1046/j.1432-1033.2002.03022.x. [DOI] [PubMed] [Google Scholar]; (b) Klinman JP. Linking protein structure and dynamics to catalysis: the role of hydrogen tunneling. Phil Trans R Soc B. 2006;361:1323–1331. doi: 10.1098/rstb.2006.1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meyer MP, Klinman JP. Investigating inner-sphere reorganization via secondary kinetic isotope effects in the C-H cleavage reaction catalyzed by soybean lipoxygenase: tunneling in the substrate backbone as well as the transferred hydrogen. J Am Chem Soc. 2011;133:430–439. doi: 10.1021/ja1050742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singleton DA, Thomas AA. High-precision simultaneous determination of multiple small kinetic isotope effects at natural abundance. J Am Chem Soc. 1995;117:9357–9358. [Google Scholar]
- 9.Manning KA, Sathyamoorthy B, Eletsky A, Szyperski T, Murkin AS. Highly precise measurement of kinetic isotope effects using 1H-detected 2D [13C,1H]-HSQC NMR spectroscopy. J Am Chem Soc. 2012;134:20589–20592. doi: 10.1021/ja310353c. [DOI] [PubMed] [Google Scholar]
- 10.Chan J, Lewis AR, Gilbert M, Karwski M-F, Bennet AJ. A direct NMR method for the measurement of competitive kinetic isotope effects. Nature Chem Biol. 2010;6:405–407. doi: 10.1038/nchembio.352. [DOI] [PubMed] [Google Scholar]
- 11.Peng S, McGinley CM, van der Donk WA. Synthesis of site-specifically labeled arachidonic acids as mechanistic probes for prostaglandin H synthase. Org Lett. 2004;6:349–352. doi: 10.1021/ol0361711. [DOI] [PubMed] [Google Scholar]
- 12.Jacquot C, Peng S, van der Donk WA. Kinetic isotope effects in the oxidation of arachidonic acid by soybean lipoxygenase-1. Bioorg Med Chem Lett. 2008;18:5959–5962. doi: 10.1016/j.bmcl.2008.08.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Yang TC, Wolfe MD, Neibergall MB, Mekmouche Y, Lipscomb JD, Hoffman BM. Substrate binding to the NO-ferro-naphthalene 1,2-dioxygenase studied by high-resolution Q-band pulsed 2H-ENDOR spectroscopy. J Am Chem Soc. 2003;125:7056–7066. doi: 10.1021/ja0214126. [DOI] [PubMed] [Google Scholar]; (b) Martinie RJ, Livada J, Chang W, Green MT, Krebs C, Bollinger JM, Jr, Silakov A. Experimental correlation of substrate position with reaction outcome in the aliphatic halogenase, SyrB2. J Am Chem Soc. 2015;137:6912–6919. doi: 10.1021/jacs.5b03370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rickert K, Klinman JP. Nature of hydrogen transfer in soybean lipoxygenase 1: separation of primary and secondary isotope effects. Biochemistry. 1999;38:12218–12228. doi: 10.1021/bi990834y. [DOI] [PubMed] [Google Scholar]
- 15.Oger C, Balas L, Durand T, Galano J-M. Are alkyne reductions chemo-, regio-, and stereoselective enough to provide pure (Z)-olefins in polyfunctionalized bioactive molecules? Chem Rev. 2013;113:1313–1350. doi: 10.1021/cr3001753. [DOI] [PubMed] [Google Scholar]
- 16.Meyer MP, Klinman JP. Synthesis of linoleic acids combinatorially labeled at the vinylic positions as substrates for lipoxygenases. Tetrahedron Lett. 2008;49:3600–3603. doi: 10.1016/j.tetlet.2008.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jacquot C, Wecksler AT, McGinley CM, Segraves EN, Holman TR, van der Donk WA. Isotope sensitve branching and kinetic isotope effects in the reaction of deuterated arachidonic acids with human 12- and 15-lipoxygenases. Biochemistry. 2008;47:7295–7303. doi: 10.1021/bi800308q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Corey EJ, Fuchs PL. A synthetic method for formyl --> ethynl conversion. Tetrahedron Lett. 1972;13:3769–3772. [Google Scholar]
- 19.Grandjean D, Pale P, Chuche J. An improved procedure for aldehyde-to-alkyne homologation via 1,1-dibromoalkenes; synthesis of 1-bromoalkynes. Tetrahedron Lett. 1994;35:3529–3530. [Google Scholar]
- 20.Knapp MJ, Rickert K, Klinman JP. Temperature-dependent isotope effects in soybean lipoxygenase-1: Correlaing hydrogen tunneling with protein dynamics. J Am Chem Soc. 2002;124:3865–3874. doi: 10.1021/ja012205t. [DOI] [PubMed] [Google Scholar]
- 21.Sharma SC, Klinman JP. Kinetic detection of orthogonal protein and chemical coordinates in enzyme catalysis: double mutants of soybean lipoxygenase. Biochemistry. 2015;54:5447–5456. doi: 10.1021/acs.biochem.5b00374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Offenbacher AR, Hu S, Poss EM, Carr CAM, Scouras AD, Iavarone AT, Palla A, Alber T, Fraser JS, Klinman JP. Identification of a surface protein loop as the source of thermal activation during C-H cleavage by tunneling. 2016 [Google Scholar]
- 23.Peng S, Okeley NM, Tsai AL, Wu G, Kulmacz RJ, van der Donk WA. Synthesis of isotopically labeled arachidonic acids to probe reaction mechanism of prostaglandin H synthase. J Am Chem Soc. 2002;124:10785–10796. doi: 10.1021/ja026880u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.