

Abstract
Advanced paternal age is linked to complications in pregnancy and genetic diseases in offspring. Aging results in excess reactive oxygen species (ROS) and DNA damage in spermatozoa; this damage can be transmitted to progeny with detrimental consequences. Although there is a loss of antioxidants with aging, the impact on aging male germ cells of the complete absence of either catalase (CAT) or superoxide dismutase 1 (SOD1) has not been investigated. We used CAT-null (Cat−/−) and SOD1-null (Sod−/−) mice to determine whether loss of these antioxidants increases germ cell susceptibility to redox dysfunction with aging. Aging reduced fertility and the numbers of Sertoli and germ cells in all mice. Aged Sod−/− mice displayed an increased loss of fertility compared to aged wild-type mice. Treatment with the pro-oxidant SIN-10 increased ROS in spermatocytes of aged wild-type and Sod−/− mice, while aged Cat−/− mice were able to neutralize this ROS. The antioxidant peroxiredoxin 1 (PRDX1) increased with age in wild-type and Cat−/− mice but was consistently low in young and aged Sod−/− mice. DNA damage and repair markers (γ-H2AX and 53BP1) were reduced with aging and lower in young Sod−/− and Cat−/− mice. Colocalization of γ-H2AX and 53BP1 suggested active repair in young wild-type mice but reduced in young Cat−/− and in Sod−/− mice and with age. Oxidative DNA damage (8-oxodG) increased in young Sod−/− mice and with age in all mice. These studies show that aged Sod−/− mice display severe redox dysfunction, while wild-type and Cat−/− mice have compensatory mechanisms to partially alleviate oxidative stress and reduce age-related DNA damage in spermatozoa. Thus, SOD1 but not CAT is critical to the maintenance of germ cell quality with aging.
Keywords: aging, oxidative stress, Sertoli cells, spermatogenesis
INTRODUCTION
Spermatogenesis is a continuous process that allows males to produce sperm from the onset of puberty and well into old age. Although spermatozoa are continuously produced with advanced paternal age (APA), there is a growing body of evidence indicating that APA is associated with negative impact on the quality of male germ cells [1]. Epidemiological studies indicate that APA may be a factor that is as critical as maternal age in determining pregnancy outcomes and offspring health. The effects of APA on pregnancy include increased time to pregnancy [2], preimplantation loss [3], and miscarriage [4]. The children of older fathers show increased risk for congenital birth defects and diseases such as achondroplasia, schizophrenia [5], and autism [6, 7].
The free radical theory of aging [8, 9] suggests that oxidative stress due to excessive free radicals or reactive oxygen species (ROS) is responsible for aging [10, 11]. ROS are the natural by-products of cellular metabolism and are involved in various cellular functions [12, 13]. If not maintained within normal physiological levels, ROS can damage cellular macromolecules, inducing stress signaling and, at high levels, cell death [14]. Thus, cellular antioxidant defenses [15] that neutralize ROS are essential, particularly for the protection of rapidly dividing male germ cells. The antioxidant superoxide dismutases (SODs) break down superoxide (O2−) radicals into hydrogen peroxide (H2O2), which can then be further neutralized by either catalase (CAT) or peroxiredoxins (PRDXs) to water. While CAT is localized to the peroxisomes and responds to high levels of H2O2, the PRDXs are widely distributed in various subcellular organelles [16, 17] and neutralize low levels of H2O2, hydroxyl (OH−) and peroxynitrite (ONOO−) radicals [18, 19].
Aging results in the loss of antioxidant activity and elevated levels of ROS [20, 21]. That this imbalance between ROS and antioxidants causes oxidative stress is well documented in the male reproductive tract [22, 23] and in the spermatozoa of aging rodents [21]. Despite the presence of complex antioxidant pathways with redundant antioxidant enzymes, germ-line knockouts (KOs), such as thioredoxins 2 and 3, which are part of the thioredoxin-glutoredoxin system, display increased susceptibility in spermatozoa to age-related oxidative stress [24]. Gene expression studies using isolated germ cells from aging rodents have highlighted that the antioxidants SOD1 and CAT play a critical role in the response to oxidative stress during aging [25, 26]. Mice lacking SOD1 (Sod−/−) show increased spermatogenic cell susceptibility to heat stress [27] and have spermatozoa with impaired fertilizing ability [28]; to the best of our knowledge, there are no studies examining the germ cells of CAT-null (Cat−/−) mice. The effects of aging on the quality of male germ cells in Sod−/− and Cat−/− mice have yet to be investigated. In addition to SOD1 and CAT, reduced levels of antioxidant PRDX1 have been linked to pathologies caused by increased oxidative stress, such as testicular torsion and varicocele [29], as well as male infertility [30, 31]. Furthermore, deficiencies in PRDX1 are associated with accelerated aging [32, 33]. The role of PRDX1in the aging testis remains unclear.
In this study, we investigated the effects of the loss of SOD1 and CAT in both young and aged Sod−/− and Cat−/− mice on testicular germ cells. Furthermore, to assess response to oxidative stress, mice were exposed to an acute oxidative insult by the pro-oxidant molsidomine (SIN-10) followed by measurement of ROS levels in live spermatocytes. We hypothesize that germ cells from aged mice lacking SOD1 will display increased ROS and greater susceptibility to DNA damage, while aged mice lacking CAT compensatory antioxidant will show a milder phenotype.
MATERIALS AND METHODS
Animals
The mice used for this study were bred in-house. C57BL6 strain wild-type (WT) and Sod−/− (B6.129S7-Sod1tm1Leb/DnJ) breeders were purchased from the Jackson Laboratory. Cat−/− global KO breeders [34] were acquired from Dr. Eugene Chen at the University of Michigan. Sod−/− mice have exons 1 and 2 replaced with a PGK-hprt expression cassette [35], and Cat−/− mice have part of intron 4 and exon 5 replaced by a neocassette [34]. All mice were on C57BL/6 background and aged 3 mo (young) and 18 mo (aged). Mice were housed at the McIntyre Animal Resources Centre at McGill University, maintained under controlled temperature (22°C) and lighting (12L:12D), and provided with food and water ad libitum. Genotypes were determined by polymerase chain reaction (PCR) analysis of DNA extracted from tail biopsies using Qiagen DNeasy Tissue Kits (Qiagen). Sod−/− and Cat−/− genotyping was done using PCR protocols provided by the Jackson Laboratory and Dr. Eugene Chen, respectively. All animal studies were conducted in accordance with the principles and procedures outlined in the Guide to the Care and Use of Experimental Animals prepared by the Canadian Council on Animal Care (McGill Animal Care Committee protocol #4687).
Fertility Studies
To assess the fertility of male mice, we bred young WT female mice with either 3-mo (young) or 12-mo (aged) WT, Sod−/−, or Cat−/− males. The numbers of pups per litter that were sired from these breeding pairs were counted for each mouse group. The results were presented as number of pups per litter sired by 3-mo and 12-mo males of each genotype. The number of litters examined per experimental group ranges from 11 to 14. While all female breeders were WT and aged between 3 and 6 mo, male breeders were chosen from each genotype (WT, Sod−/−, or Cat−/−) and aged 12 mo, which is typically the oldest age of retired breeders [36].
Animal Treatments
Oxidative stress was induced with administration of molsidomine (SIN-10) (Sigma Aldrich Canada). Mice aged 3 or 18 mo were given a single intra-peritoneal injection of 30 mg/kg of body weight 2 h prior to being euthanized [37]. SIN-10 treatment time was chosen based on previous studies that indicated that serum levels of the SIN-10 metabolite SIN-1 peaked at 2 h [38], and experimental trials in which the treatment time of 2 h was found to induce a peak increase in ROS in pachytene spermatocytes from young WT animals (Supplemental Figure S1A; Supplemental Data are available online at www.biolreprod.org). The SIN-10 dose was based on information in the literature [37, 38] and preliminary dose-response trials using aged animals to measure ROS response to the oxidative insult (Supplemental Figure S1B). In control animals, SIN-10 was replaced with saline injections. After each treatment, mice were euthanized using carbon dioxide followed by cervical dislocation. Tissues were removed, weighed, flash frozen, and stored at −80°C for later analyses. Five to seven animals were used for each of the four groups of animals (young and old serving as either control or SIN-10 treated) for both the Sod−/− and the Cat−/−studies.
Spermatocyte ROS Measurements
Mice were euthanized 2 h after SIN-10 or saline (control) injections, and their testes were removed. Preparation of testicular germ cell suspensions was adapted from a previously described method [39]. Briefly, the testes were decapsulated, and the parenchyma was minced using a scalpel until in a semiliquid state and mixed with an equal volume of phenol red-free media (Sigma Aldrich Canada); the cell suspension was strained through a 56-μm nylon mesh. The cells were then diluted and counted using a hemocytometer, and 150 000 cells were aliquoted per tube from the mixed testicular cells.
The mixed testicular cells were then incubated for 30 min at 32°C with Hoechst (2,5′-bi-1H-benzimidazole, 2′-[4-ethoxyphenyl]-5-[4-methyl-1-piperazinyl]) (ThermoFisher) and probes to measure ROS that included CellROX DeepRed Reagent and peroxynitrite sensor aminophenyl fluorescein (Invitrogen). After incubation, the cells were washed in media and transferred to a 96-well Cell Carrier (PerkinElmer) plate with an optically clear bottom. The plate was immediately scanned using the Operetta (PerkinElmer) high-content imaging system, and images were then analyzed using Columbus software (version 2.5.0; PerkinElmer) with a previously validated algorithm designed to select for spermatocytes [40]. The overall ROS and peroxynitrite intensities in spermatocytes were measured per well per cell using this software. Information for each probe used is listed in Supplemental Table S1.
Sperm Counts
Testicular and epididymal sperm counts were done as previously described [41]. Briefly, a frozen testis or epididymis from each mouse was weighed and homogenized (Polytron PTA7, setting 5; Brinkman Instruments) for two 15-sec periods separated by a 30-sec interval in 2 ml of 0.9% NaCl (Millipore), 0.1% thimerosal (Sigma), and 0.5% Triton X-100 (BDH). Sperm heads were counted in aliquots of diluted homogenate using a hemocytometer. Sperm counts from the testis and epididymis are shown as the number of sperm × 106 per tissue.
Testis Fixation
Young and aged mice were euthanized from each group (WT, Sod−/−, and Cat−/−); their testes were removed and immersion fixed in modified Davidson fixative [42] for 2 h, then the testes were sliced in half and further fixed for 6–8 h. After fixation, the testes were transferred to 70% ethanol, further dehydrated, and then embedded in paraffin.
Histology
Several samples from different testes were sectioned (4 μm) and placed on a single slide; each had sections from different mice of the same group, with the addition of one WT control animal on each slide. The slides were deparaffinized (Histoclear; Diamed Inc.) and loaded in 30-slide batches into the Tecan GenePaint (Tecan) solvent delivery robotic system, programmed to rehydrate the tissues by passing them through a graded ethanol series, and then stained with Periodic Acid Schiff (PAS) (Sigma) according to the manufacturer's protocol. The slides were mounted with Permount (Fisher Scientific) and coverslips and left to dry before observation with a light microscope. Tubule diameters and epithelial heights were measured using ImageJ (NIH) for 200 tubules per animal, with five to six animals per experimental group. Using similar methods, a fixed epididymis from reach mouse was sectioned (4 μm) and stained using Toluidine Blue (Sigma) following the manufacturer's protocol.
Immunofluorescence
The Tecan GenePaint robotic system was used to do automated immunofluorescence labeling of batches of slides. Before loading the slides, they were deparaffinized with Histoclear, rehydrated through a graded ethanol series, washed in PBS for 5 min, and finally boiled in sodium citrate buffer (10 mM sodium citrate, 0.05% Tween 20, pH 6.0) for 30 min. The slides were left to cool to room temperature for approximately 20 min and then rinsed in PBS and loaded into the Tecan GenePaint robotic system, which was programmed to add blocking reagents (10% normal goat serum) for 1 h, primary antibodies for 1 h, secondary antibodies for 45 min, and 4′,6′-diamidino-2-phenylindole (DAPI) combined with lectin PNA from Arachis hypogaea (peanut) Alexa Fluor 647 conjugate (ThermoFisher) (used as a staging tool) for 10 min. Finally, slides were mounted with ProLong Gold antifade mountant (ThermoFisher), left to dry at room temperature for 24 h, and then stored at 4°C before imaging. Information for each antibody used is listed in Supplemental Table S1.
Oxidative damage-induced DNA lesions, assessed using 8-oxo-2′-deoxyguanosine (8-oxodG) as a marker, was detected in sperm heads by immunofluorescence, as previously described [25]. Briefly, frozen slides with sperm smears were placed in PBS for 5 min and blocked in goat serum for 30 min. Slides were then incubated in primary antibody specific for 8-oxodG (1:10) overnight at 48°C (controls incubated in serum alone). Slides were washed in PBS and incubated in goat-anti-mouse conjugated to Alexafluor 488 (ThermoFisher) for 30 min, washed in PBS, and mounted with DAPI. Using a Zeiss LSM 510 Axiovert confocal microscope to visualize sperm, a minimum of 100 spermatozoa per slide per sample were counted as either positive or negative for 8-oxodG. Positive control samples were pretreated for 30 min with 5% H2O2 (Sigma).
Measurement of Immunofluorescence Intensity
Sequential images were captured (resolution 2488 × 2488) using a multiphoton Leica TCS SP8 MP microscope with 20× and 63× objectives. Quantification of positive cells per tubule was assessed using Imaris version 3.8 software (Bitplane). Briefly, tubules were manually selected and cells identified by Imaris using DAPI nuclear stain; intensity data for each cell for each channel were exported. Data from control slides with no primary antibody and slides with blocking peptides were used to set baseline thresholds and the intensity range for each marker.
Statistical Analyses
All statistical analyses were done using two-way ANOVA followed by Bonferroni tests or one-way ANOVA with Newman-Keuls tests with GraphPad Prism 4.0; significance was set at P < 0.05.
RESULTS
The Effects of Aging on Body and Tissue Weights, Sperm Counts, and Fertility
WT mice at 3 mo had significantly higher body weights than KO mice of the same age. Mice were littermates, indicating that the body weight was reduced in response to the antioxidant loss in Sod−/− and Cat−/− mice (Fig. 1A). Aging resulted in increased body weight for all animals. WT mice had the greatest increase in weight, while Cat−/− and Sod−/− mice had significantly lower body weights compared to WT (Fig. 1A). Fertility was unaffected at 3 mo among WT, Cat−/−, or Sod−/−mice. By 12 mo, the number of pups per litter was significantly reduced in all mice; however, the number of pups per litter in Sod−/− mice at this age was significantly lower than that in aged controls (Fig. 1B).
FIG. 1.

Reproductive organ weights and testis and epididymal sperm counts in aged wild-type (WT), Cat−/− and Sod−/− mice. Body weights increased with age in WT, Cat−/−, and Sod−/− mice (A), while Cat−/−, and Sod−/− mice remained consistently lower in weight than WT mice. The numbers of pups per litter decreased with aging (B) and were significantly lower in aged Sod−/− mice. Testis weights (C) were reduced in Cat−/− and Sod−/− compared to WT mice. Testis sperm counts (D) were lower in young Sod−/− mice and all aged mice. Epididymal weights were not significantly changed (E); sperm count in the epididymis (F) reflected the changes observed in the testis sperm counts. Mean values with SEM error bars; two-way ANOVA with Bonferroni test; *P < 0.05; **P < 0.01; ***P < 0.001, n = 10–14 (A, B, D, F), n = 5–8 (E, G), n = 4–5 (C, F).
Testis weight remained the same with age; Cat−/− and Sod−/− mice maintained lower testis weights compared to WT mice at both 3 and 18 mo (Fig. 1C). Testes sperm counts were significantly reduced in young Sod−/− mice; by 18 mo, all mice had significantly reduced testis sperm counts (Fig. 1D). There were no significant changes observed in epididymal weights with either aging or KO mice (Fig. 1E). Yet, similar to the testis sperm counts, the epididymal sperm counts were significantly reduced in young Sod−/− mice compared to WT; epididymal sperm counts were significantly reduced in all mice with aging (Fig. 1F). Since tissue weights were only minimally affected, expressing sperm counts per milligram of tissue, as opposed to per tissue, resulted in the same significant differences (data not shown).
Epididymal histology revealed lumina filled with sperm in the caput and cauda of young WT mice; in aged WT mice, some sloughed-off cells and sperm were visible in the epididymal lumina. Similar observations were made in the caput and cauda epididymides of young and aged Cat−/− mice (Supplemental Figure S2). In contrast, at 3 mo, Sod−/− mice displayed sloughed-off cells in all segments of the epididymis, with collections of cells in the lumina; by 18 mo, the epididymal epithelia of the different segments were visibly altered with accumulation of a few sloughed-off cells and fluids in the epididymal lumina (Supplemental Figure S2).
Accelerated Loss of Germ Cells and Sertoli Cells with Aging in Antioxidant KO Mice
Testes histology (Fig. 2, A–F) indicated loss of germ cells in the seminiferous tubules of all mice with aging (Fig. 2, B, D, and F). Seminiferous tubule diameters and epithelium heights were significantly reduced with aging in WT mice (Fig. 2, G and H). In the testes of young Cat−/− mice, some seminiferous tubule lumina were filled with sloughed-off cells (Fig. 2C). Compared to WT mice, young Cat−/− mice had significantly smaller seminiferous tubule diameters and epithelial heights; diameters were maintained in aged Cat−/− animals, while epithelial heights were significantly reduced at 18 mo (Fig. 2H). Young Sod−/− mice had significantly reduced tubule diameters compared to WT; these were further reduced with aging (Fig. 2G). The seminiferous tubule epithelial heights were significantly smaller in young Sod−/− mice when compared to WT and were unaltered with aging (Fig. 2H).
FIG. 2.

Histological analyses of young and aged Cat−/− and Sod−/− mouse testes. Testis histology in young and aged mice stained with PAS-hematoxylin (A–F). Seminiferous tubule diameters (G) were reduced in knockout (KO) mice and in all mice with aging. The seminiferous tubule epithelium height (H) was reduced in KO mice, with the most extreme loss in height in Sod−/− mice. Loss of tubule height was observed with aging in all mice. Mean values with SEM error bars; two-way ANOVA with Bonferroni test; *P < 0.05; **P < 0.01; ***P < 0.001, n = 6 (A–F); n = 8 (G, H). Bar = 100 μm.
Using ZBTB16 as a marker for spermatogonia and vimentin as a Sertoli cell marker (Fig. 3A), we found that the mean germ cell numbers per seminiferous tubule were significantly lower in Cat−/− and Sod−/− mice compared to WT at 3 mo; all mice displayed a loss in germ cell numbers with aging (Fig. 3B). This loss of germ cells in aged animals was significant at all stage groupings. Sertoli cell numbers were reduced only in young Sod−/− compared to WT mice; this reduction was significant in the later stages of spermatogenesis in stage groupings VIII–IX and X–XII. Aging resulted in the loss of Sertoli cells in all mice; interestingly, aged Cat−/− mice displayed significantly higher numbers of Sertoli cells compared to aged Sod−/− mice (Fig. 3C), indicative of possible compensatory protective mechanisms in Cat−/− mice. The numbers of spermatogonia among all groups were unaltered (Supplemental Figure S3). Serum levels of gonadotropins LH and FSH and serum testosterone were not significantly changed between WT, Cat−/−, and Sod−/−mice or with aging (Supplemental Figure S4).
FIG. 3.

Increased Sertoli cell loss in Sod−/− mice and with aging in all mice. Representative image with DAPI (nuclear stain), ZBTB16 (spermatogonial marker), and vimentin (Sertoli marker in the testis epithelium) (A). Imaris quantification of the number of germ cells per tubule (B) and the number of Sertoli cells per tubule (C) are also shown sorted by stage groupings. Mean values with error bars (±SEM); two-way ANOVA with Bonferroni test; *P < 0.05; **P < 0.01; ***P < 0.001; n = 4. Bar = 20 μm.
Spermatocyte ROS in SIN-10-Treated Mice
Mice were treated with the pro-oxidant SIN-10 (Fig. 4A), their germ cells were isolated, and the mean ROS intensities were quantified following spermatocyte selection. Young WT mice responded to the oxidative insult with no change in spermatocyte ROS versus saline-treated controls, suggesting effective removal of cellular ROS (Fig. 4B). However, spermatocytes from aged WT mice were not able to remove ROS following oxidative insult and displayed significantly increased ROS with SIN-10 treatment (Fig. 4B). In contrast, spermatocytes from both young and aged Cat−/− mice were able to respond to SIN-10-induced oxidative insult by neutralizing ROS efficiently (Fig. 4C). In Sod−/− mice, the ROS levels of spermatocytes showed similar responses to SIN-10 as WT mice; spermatocytes from young mice were able to neutralize SIN-10-induced ROS, but in the aged mice they were not able to neutralize this ROS (Fig. 4D). Based on MitoSOX measurements, spermatocyte O2− levels were not significantly changed among the groups (data not shown), while ONOO− measurements, detected using aminophenyl fluorescein, displayed identical results to those seen with overall cellular ROS (Supplemental Figure S5).
FIG. 4.
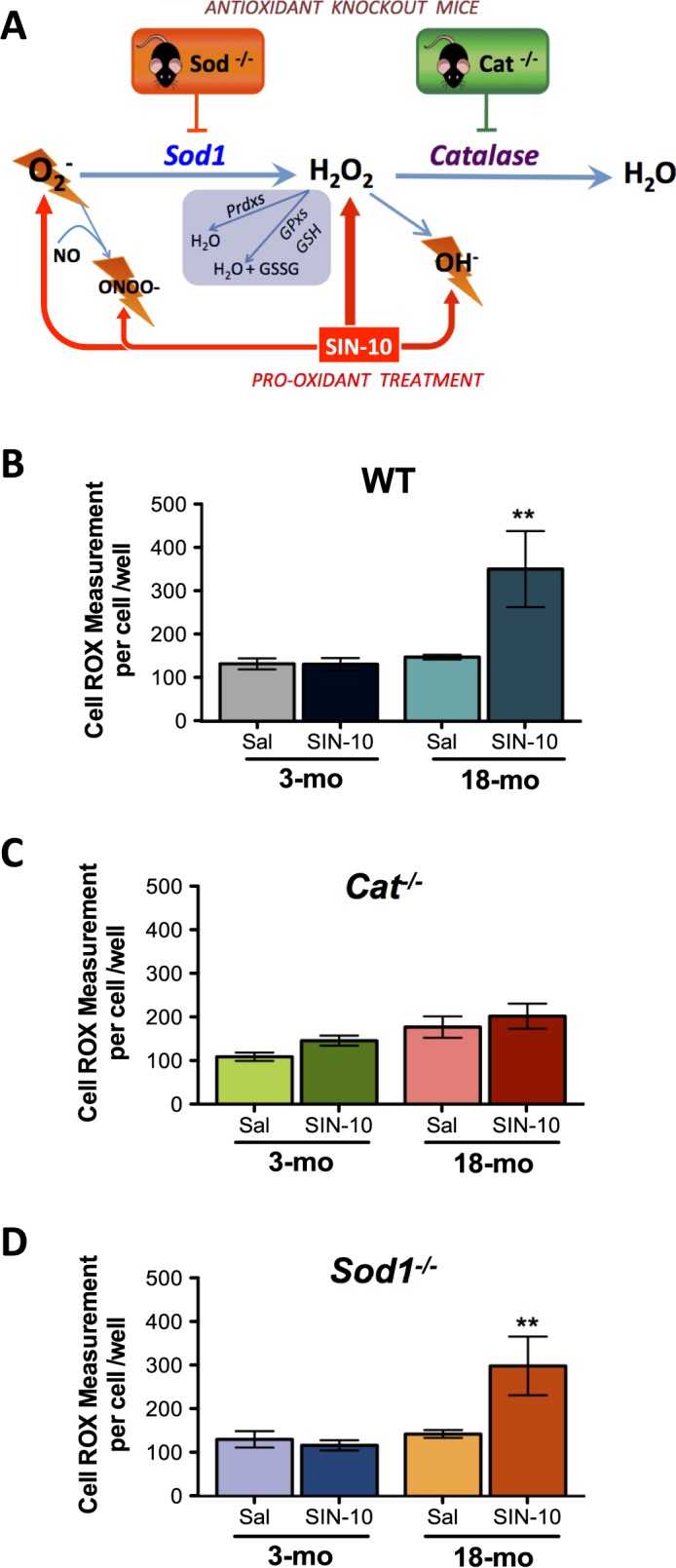
Redox dysfunction with aging in WT and Sod−/− spermatocytes and efficient management of oxidative stress in spermatocytes from Cat−/− mice following SIN-10 induced oxidative insult. Schematic of SIN-10 induced ROS generation (A). Overall ROS, measured using CellROX, showed efficient removal of ROS in all young animals, WT (B), Cat−/− (C), and Sod−/− (D). While aged Cat−/− mice responded to SIN-10 with removal of ROS, aged WT and Sod−/− did not efficiently remove ROS following SIN-10 treatment (D). Mean values shown with error bars (±SEM); one-way ANOVA with Newman-Keuls; **P < 0.01, n = 4–5.
PRDX1 Localized to Germ Cell Cytoplasm and the Acrosome
Live cell ROS measurements indicated that ONOO− radicals were generated in the spermatocytes following SIN-10 treatment in aged WT and Sod−/− (Supplemental Figure S5), so we decided to further investigate the antioxidant involved in peroxynitrite neutralization, PRDX1 [33]. In addition, PRDX1 has been associated with aging and oxidative stress-linked male infertility [29]. Interestingly, previous studies localized PRDX1 specifically to the Leydig cells in the mouse testis [43]. Similar to these studies, we localized PRDX1 to the Leydig cells (Supplemental Figure S6); however, unlike these previous studies, PRDX1 was also detected in the cytosol of spermatocytes and developing spermatids, specifically within the developing acrosome (Fig. 5A, orange arrow heads indicate PRDX1 in the acrosome). Furthermore, the localization of PRDX1 revealed that there was a progressive increase in PRDX1 immunoreactivity in germ cells as they developed through the different stages of spermatogenesis (Fig. 5A).
FIG. 5.

Immunolocalization of PRDX1 to the developing acrosome and inhibition of the increase in PRDX1 with aging in Sod−/− mice. PRDX1 was localized in the seminiferous tubules to the cytoplasm of spermatocytes (portions of the images showing this are magnified in insets) and within the acrosome of spermatids (A) (see insets). Imaris quantified percentage of PRDX1-positive cells per tubule showed increased PRDX1 with age in WT and Cat−/− mice; however, Sod−/− mice did not display this increase in PRDX1 with age (B). Mean values shown with error bars (±SEM); two-way ANOVA with Bonferroni test; *P < 0.05; **P < 0.01; ***P < 0.001; n = 5. Bar = 20 μm.
Up-Regulation of PRDX1 with Aging Occurs in All Mice, Except Sod−/− Mice
The percentage of PRDX1-positive cells per seminiferous tubule section was quantified in the testes of each experimental group. The results showed that PRDX1-positive cells increased with age in WT and Cat−/− mice, with the most significant increase seen during stages VIII–IX (Fig. 5B). There was no significant difference between WT and Cat−/− mice at 3 mo, but by 18 mo, WT mice had significantly more PRDX1-positive cells than Cat−/− mice (Fig. 5B), particularly during spermatogenic stages VIII–IX. Young Sod−/− mice displayed significantly fewer PRDX1-positive cells per tubule compared to young WT mice; this trend was most evident during the later stage groupings 8–9 and 10–12. Unlike the increases observed in WT and Cat−/− mice, in Sod−/− mice there were no significant changes in the percentage of PRDX1-positive cells per seminiferous tubule with aging (Fig. 5B). No significant changes were observed between young and aged Sod−/− in PRDX1 at any stage groupings. Overall, compared to both aged WT and Cat−/− mice, the percentages of PRDX1-positive cells per tubule were significantly lower in the aged Sod−/− mice (Fig. 5B).
Increased DNA Double-Strand Breaks and Reduced Repair in Aged Mice
DNA double-strand breaks (DBSs) lead to the modification of histone H2AX; phosphorylation of this histone forms γ-H2AX, which then recruits DNA damage repair proteins, such as p53 binding protein 1 (53BP1). γ-H2AX is normally present within germ cells going through meiosis in the testis [44]. The percentage of cells positive for γ-H2AX per tubule in young WT mice is indicated in Figure 6A. No significant differences were observed in γ-H2AX-positive cells in young WT, Cat−/−, or Sod−/− mice. However, during stages I–IV, young Cat−/− displayed a significantly higher percentage of γ-H2AX-positive cells (Fig. 6A). Aging caused the percentage of γ-H2AX-positive cells to decrease in WT mice at all stages. In contrast, aging resulted in an increased percentage of γ-H2AX-positive cells in both Cat−/− or Sod−/− mice compared to WT control mice; this was most pronounced during stages V–VII, suggesting persistent DNA damage in Cat−/− and Sod−/− mice with advanced age (Fig. 6A). The DNA damage repair response occurs with the accumulation of 53BP1 at the site of γ-H2AX foci. The percentage of 53BP1-positive cells was highest in young WT mice; this was evident throughout all stages of spermatogenesis (Fig. 6B). Overall 53BP1 was reduced significantly in both young Cat−/− and Sod−/− mice; this was significant in stages I–IV, V–VII, and VIII–IX. In addition, aging resulted in a decrease in 53BP1-positive cells in all mice compared to young WT mice (Fig. 6B). Nevertheless, in aged Cat−/− and Sod−/− mice, significantly higher numbers of 53BP1-positive cells were evident in the later stages VIII–IX and X–XII.
FIG. 6.
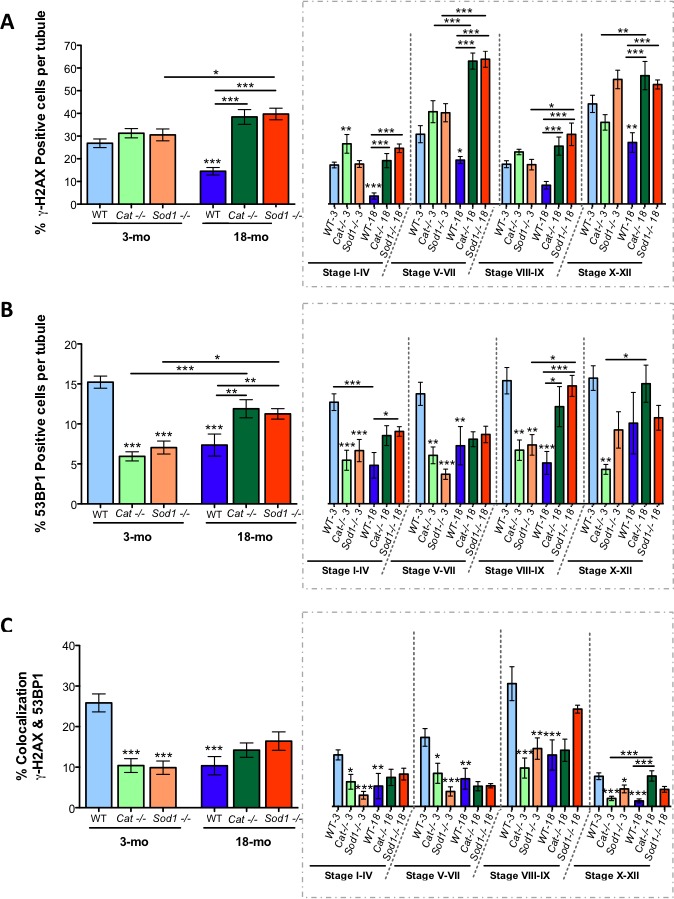
Reduced DNA damage repair in seminiferous tubules of young Cat−/− and Sod−/− mice and in all aged mice. The percentage of γ-H2AX-positive cells per seminiferous tubule increased with age in knockout (KO) mice (A). The percentage of 53BP1-positive cells per tubule was reduced in young KO mice and increased in aged KO mice, while the opposite was seen in aged WT mice (B). Colocalization of γ-H2AX- and 53BP1-positive cells showed decreased DNA damage repair in young KO mice and in all aged mice (C). Mean values shown with error bars (±SEM); two-way ANOVA with Bonferroni test; *P < 0.05; **P < 0.01; ***P < 0.001; n = 5.
The percentage of cells with colocalization of γ-H2AX and 53BP1, an indication of DNA damage repair, showed that repair was decreased in young Cat−/− and Sod−/− mice, which had fewer γ-H2AX- and 53BP1-positive cells (Fig. 6C). In addition, stage-specific identification of DNA damage repair was clear, with stages VIII–IX being the highest for colocalization, while stages X–XII displayed the lowest colocalization. Aging led to fewer cells with colocalized γ-H2AX and 53BP1 in all mice, across all spermatogenic stages, suggesting that WT, Cat−/−, and Sod−/− may have less DSB repair occurring in aged control and KO mice.
Increased Oxidative Stress-Induced DNA damage in the Spermatozoa of Sod−/− Mice Regardless of Age
Immunofluorescence was used to determine the extent of 8-oxodG, an ROS-induced DNA damage lesion, in spermatozoa (Fig. 7A). Positive control samples pretreated with H2O2 had 100% 8-oxodG-positive spermatozoa. The lowest numbers of 8-oxodG-positive spermatozoa were found in young WT mice. The percentage of 8-oxodG-positive spermatozoa increased in WT mice as a result of aging (Fig. 7B). 8-OxodG-labeled spermatozoa were significantly elevated in young Cat−/− compared to young WT mice; however, the proportion of these 8-oxodG lesions in Cat−/− mice remained unaltered with aging. The Sod−/− mice displayed the highest percentage of 8-oxodG-positive spermatozoa (approximately 80%) in both young and aged groups, showing the highest amount of ROS-induced damage regardless of age (Fig. 7B).
FIG. 7.

Elevated oxidative stress-induced DNA damage in young and aged Sod−/− mice. Sample immunofluorescence images of sperm heads, with 8-oxodG (green) counterstained with DAPI (blue); inset shows negative control (A). The quantified percentage of 8-oxodG-positive sperm heads showed an increase in 8-oxodG with aging in WT and an increased 8-oxodG in Cat−/− mice that remained at the same level with aging. The highest amounts of 8-oxodG were observed in Sod−/− mice in both the young and the aged groups. Mean values shown with error bars (±SEM); two-way ANOVA with Bonferroni multiple comparison test; *P < 0.05; ***P < 0.001; n = 5. Bar = 2.5 μm.
DISCUSSION
Aging is associated with the loss of antioxidants and increased ROS; previous studies have indicated that germ cells are sensitive to aging-associated oxidative stress [21, 25, 26]. To determine the consequences of losing key antioxidants, either CAT or SOD1, on maturing male germ cells, we bred and aged Cat−/− and Sod−/− mice and examined the effects of aging on their developing testicular germ cells.
The original studies that had generated these null mutant (Cat−/− and Sod−/−) mice found them to be viable and phenotypically normal despite the lack of these major antioxidant enzymes [34, 35]. Later studies examining the lack of SOD1 revealed increased vulnerability to ROS and also displayed a 30% reduction in lifespan [45–47]; these are highly significant findings in the context of aging studies. Similar to recent studies [48, 49], our examination of the overall health of these animals revealed that the loss of either antioxidant, CAT or SOD1, resulted in mice weighing less than their WT littermates at both 3 and 18 mo of age. These results were not surprising considering that CAT imparts various metabolic health benefits and SOD1 is a major cytoplasmic superoxide scavenger with minimal compensatory mechanisms [50–52]; thus, the global and chronic loss of these antioxidants could contribute to the overall decrease in the weights of these mice compared to WT mice throughout their lives.
Although it is natural to hypothesize that the loss of an antioxidant will result in accelerated aging, the results of systemic oxidative stress are difficult to predict. The effects of compensatory mechanisms and chronic stress, which are observed with aging, make predicting the effects of antioxidant null mutations particularly difficult. Previous studies found both Cat−/− and Sod−/− mice to be fertile [34, 35]. However, many null mutation mouse studies have reported male mice to be fertile and overlooked the possible effects on sperm counts and testicular histology. Mice can have very large reductions in their sperm counts [53, 54] and reduced Sertoli cell numbers [54, 55] and yet be fertile. Our data on breeding indicate that despite a clear reduction in the number of sperm in young Sod−/− mice, these males show no difference in fertility (number of pups per litter). Sperm counts in all mice declined with aging. In addition, aging reduced fertility in all groups of mice, with significantly fewer pups per litter sired by 12-mo-old males. The 12-mo-old Sod−/− mice displayed the most severe reduction in pup numbers.
Testis weights were consistently lower in KO mice compared to WT mice, while epididymal weights were unaltered. The histology of aged testes displayed the typical presentations associated with aging, including loss of germ cells and shrunken seminiferous tubules [56]. Further analyses revealed that, in addition to the loss of germ cells, Sertoli cell numbers were reduced in young Sod−/− mice and all aged mice.
Epididymal histology revealed that the most severely affected epididymides were in aged Sod−/− mice; all segments of the epididymides showed degenerated epithelia and cellular debris in the lumina. The epididymides from aged Sod−/− mice also had few or no visible spermatozoa (Supplemental Figure S2), providing corroborating evidence of the significant decline in spermatozoa counts and supporting the reduced fertility observed in Sod−/− mice. SOD1 is expressed in the epithelium of the epididymis from the caput to the cauda [57], accompanied by an extracellular secreted SOD (SOD3) [58] as well as several other antioxidants, including GPX1, 3, 4, and 5 and GSH [59]. Aging decreases antioxidants GPX4 and 5 in the epididymis [57]; the lack of SOD1 in Sod−/− mice could further exacerbate the redox dysfunction occurring with aging. Future investigations will address changes in other antioxidants and delve into the mechanisms contributing to the advanced Sod−/− epididymal degeneration that was observed with aging.
The spermatocytes of aged WT and Sod−/− mice were least able to cope with elevated ROS after mice were administered SIN-10 as an oxidative insult. As seen with previous studies on the Cat−/− mice [34, 60], the lifelong absence of CAT could lead to the up-regulation of compensatory antioxidant systems. The protective effects observed in the Cat−/− Sertoli cell number with aging could be attributed to compensatory antioxidants, such as PRDX1. These compensatory mechanisms are demonstrated in the spermatocytes of aged Cat−/− mice, which are able to detoxify SIN-10-induced ROS, suggesting that similar mechanisms may be protecting Sertoli cell numbers with aging in these animals.
Currently, there are no data to suggest that compensation could be occurring in Sod−/− mice. One particular antioxidant that is affected by both aging and oxidative stress and is linked to male infertility is PRDX1 [29]. As previously reported, PRDX1 was localized to Leydig cells [42]; however, we also found it localized to the cytoplasm of spermatocytes and developing acrosome of spermatids. We found that PRDX1 was altered with aging and reduced in the germ cells of null mutant mice. More specifically, aging resulted in an increase in PRDX1 in the germ cells of WT and Cat−/− mice, confirming the hypothesized genetic robustness in the Cat−/− mice model [61, 62]. In Sod−/− mice, PRDX1 was consistently at lower levels than WT and was not increased with aging. This lower PRDX1 in Sod−/− mice points toward redox dysfunction as the primary cause of increased vulnerability in the Sod−/− mice. More specifically, the low levels of PRDX1 in the germ cells of aging Sod−/− mice suggest that compensatory removal of radicals was disrupted and that the normal expression of antioxidant was altered. Furthermore, the ability of high levels of ROS to hyperoxidize antioxidants, in particular PRDXs [29, 63], contributes to the hypothesis that antioxidants may become catalytically inactive during aging [29, 63, 64]. While antioxidant enzymatic activity needs to be examined in the aging testis of these mice, the present findings indicate that the loss of SOD1 may result in increased redox dysfunction in the germ cells of aging mice.
Although previous studies have confirmed glutathione peroxidase 1 (GPX1) activity to be unaltered between WT and Cat−/− mice [33] and in Sod−/− mice [26], it remains possible that the compensatory mechanisms observed between Cat−/− may be due to the thioredoxin-glutoredoxin system [24]. Further studies are required to clarify this point and to determine the effects of aging on the thioredoxin-glutoredoxin antioxidants in these KO mice.
Erythrocytes are constantly under oxidative stress; however, the absence of CAT and GPX1 does not lead to hemolytic anemia due to damage and premature death of erythrocytes [65, 66]. The only antioxidant KO that did display this vulnerability was the PRDX1 KO [67], leading our investigation toward PRDX1. The altered PRDX1 levels with aging and in the KO animals suggests that although PRDXs are not considered to be primary enzymatic scavengers, they can act in compensatory ROS removal with aging.
Next, we decided to examine DNA damage and repair markers in the seminiferous tubules of these animals. The DNA DSB marker γ-H2AX increased with aging in animals with null mutations, indicating the presence of elevated levels of DSBs in the germ cells of both aged Cat−/− and Sod−/− mice. Normally, in response to this damage, elevated DNA damage-repair protein 53BP1 accumulates at the site of DSBs. 53BP1 accumulation at the site of γ-H2AX leads to downstream signaling for DNA damage repair, with NHEJ repair promoted by 53BP1. We speculate that DNA damage repair mechanisms could be compromised in the young KO animals, which display fewer 53BP1-positive cells. Our results indicate that with aging, additional oxidative stress induces increased γ-H2AX in these animals and results in increased 53BP1 accumulation to promote signaling for DNA damage repair. Moreover, the colocalization of γ-H2AX and 53BP1 revealed active repair in young WT mice but reduced DNA repair in all other mice. In support of these findings, recent studies with models of controlled DNA DSBs suggest that DNA damage itself can contribute to deregulation of genes during aging [68, 69].
Finally, our examination of 8-oxodG, a DNA lesion that is directly induced by oxidative stress, corroborated previous observations that 8-oxodG significantly increased in spermatozoa with aging [60]. In addition, these results suggested that the greatest oxidative damage occurred in the spermatozoa of both young and aged Sod−/− mice. In addition, this damage was not repaired in Sod−/− mice during spermatogenesis. While damage was detected in young Cat−/− mice, this damage was not further increased by aging, suggesting that while loss of CAT increased DNA damage in spermatozoa of young animals, aging did not further contribute to this oxidative stress-induced damage. These findings reinforce the importance of maintaining SOD1 to prevent irreversible oxidative stress induced damage [11, 45], leading us to postulate that in the absence of SOD1, ONOO− could be the species of radicals inflicting excessive damage in Sod−/− spermatozoa.
In conclusion, the present study displays the complexity of the antioxidant systems and their differential effects on the quality of aging male germ cells. The loss of CAT resulted in loss in germ cells but no loss in Sertoli cells in young mice. Furthermore, spermatocytes from aged Cat−/− mice displayed an enhanced ability to detoxify ROS following SIN-10 treatment. Aged Cat−/− mice had significantly increased levels of PRDX1, indicating that the up-regulation of this antioxidant may have increased ROS removal capabilities. However, the levels of DNA damage in the spermatozoa from young animals were still high in Cat−/− mice. The loss of SOD1 was markedly detrimental when compared to CAT loss. The negative consequences of SOD1 loss, in young and aged mice, were evident through reduced sperm counts; abnormal epididymal histology, significant germ cell and Sertoli cell loss, and increased spermatozoa DNA damage were observed. However, the effects on fertility were manifested only with aging. Finally, the inability of spermatocytes from Sod−/− mice to remove ROS following SIN-10 demonstrated the loss of redox capabilities, while the failure of Sod−/− mice to up-regulate PRDX1 with aging identified redox dysfunction.
In conclusion, the results of these studies may have clinical implications in relation to sperm selection for artificial reproductive technologies. The present study suggests that SOD1-mimicking compounds may represent a critical component of potential antioxidant supplements for infertile/subfertile men and aspiring aging fathers who have spermatozoa with high levels of oxidative damage.
ACKOWLEDGMENT
The authors wish to thank Dr. Eugene Chen (University of Michigan) for providing the catalase KO mice and Dr. Cristian O'Flaherty (McGill University) for the gift of anti-PRDX antibodies. Thanks also to Trang Luu for her technical assistance, Yaned Gaitan for her assistance with Tecan GenePaint facilities, and Dr. Wolfgang Reintsch for his help with confocal imaging and Imaris analysis. Serum hormones were assayed by Ligand Core at the University of Virginia.
Footnotes
These studies were funded by grants MOP-89767 and TE1-138298 from CIHR. B.R. is a James McGill Professor.
REFERENCES
- Lawson G, Fletcher R. Delayed fatherhood. J Fam Plan Reprod Health Care. 2014;40:283–288. doi: 10.1136/jfprhc-2013-100866. [DOI] [PubMed] [Google Scholar]
- Hassan MA, Killick SR. Effect of male age on fertility: evidence for the decline in male fertility with increasing age. Fertil Steril. 2003;79(suppl 3):1520–1527. doi: 10.1016/s0015-0282(03)00366-2. [DOI] [PubMed] [Google Scholar]
- Serre V, Robaire B. Paternal age affects fertility and progeny outcome in the Brown Norway rat. Fertil Steril. 1998;70:625–631. doi: 10.1016/s0015-0282(98)00259-3. [DOI] [PubMed] [Google Scholar]
- Kleinhaus K, Perrin M, Friedlander Y, Paltiel O, Malaspina D, Harlap S. Paternal age and spontaneous abortion. Obstet Gynecol. 2006;108:369–377. doi: 10.1097/01.AOG.0000224606.26514.3a. [DOI] [PubMed] [Google Scholar]
- Martin AK, Robinson G, Reutens D, Mowry B. Clinical and parental age characteristics of rare copy number variant burden in patients with schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2015;168B:374–382. doi: 10.1002/ajmg.b.32321. [DOI] [PubMed] [Google Scholar]
- Lampi KM, Hinkka-Yli-Salomäki S, Lehti V, Helenius H, Gissler M, Brown AS, Sourander A. Parental age and risk of autism spectrum disorders in a Finnish national birth cohort. J Autism Dev Disord 43; 2013:2526–2535. doi: 10.1007/s10803-013-1801-3. [DOI] [PubMed] [Google Scholar]
- Sandin S, Lichtenstein P, Kuja-Halkola R, Larsson H, Hultman CM, Reichenberg A. The familial risk of autism. J Am Med Assoc. 2014;311:1770–1777. doi: 10.1001/jama.2014.4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman D. Free radical theory of aging. Triangle. 1973;12:153–158. [PubMed] [Google Scholar]
- Harman D. The free radical theory of aging. Antioxid Redox Signal. 2003;5:557–561. doi: 10.1089/152308603770310202. [DOI] [PubMed] [Google Scholar]
- Desai N, Sabanegh E, Jr, Kim T, Agarwal A. Free radical theory of aging: implications in male infertility. Urology. 2010;75:14–19. doi: 10.1016/j.urology.2009.05.025. [DOI] [PubMed] [Google Scholar]
- Aitken RJ. A free radical theory of male infertility Reprod Fertil Dev 1994. 6 19 23; discussion 23-24 [DOI] [PubMed] [Google Scholar]
- Finkel T. Signal transduction by reactive oxygen species in non-phagocytic cells. J Leukoc Biol. 1999;65:337–340. doi: 10.1002/jlb.65.3.337. [DOI] [PubMed] [Google Scholar]
- O'Flaherty CM, Beorlegui NB, Beconi MT. Reactive oxygen species requirements for bovine sperm capacitation and acrosome reaction. Theriogenology. 1999;52:289–301. doi: 10.1016/S0093-691X(99)00129-6. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JM. Free radicals, lipid peroxidation, and cell damage. Lancet. 1984;2:1095. doi: 10.1016/s0140-6736(84)91530-7. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Antioxidant defence mechanisms: from the beginning to the end (of the beginning) Free Radic Res. 1999;31:261–272. doi: 10.1080/10715769900300841. [DOI] [PubMed] [Google Scholar]
- Seo MS, Kang SW, Kim K, Baines IC, Lee TH, Rhee SG. Identification of a new type of mammalian peroxiredoxin that forms an intramolecular disulfide as a reaction intermediate. J Biol Chem. 2000;275:20346–20354. doi: 10.1074/jbc.M001943200. [DOI] [PubMed] [Google Scholar]
- Wood ZA, Schroder E. Robin Harris J, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2003;28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- Rhee SG, Woo HA, Kil IS, Bae SH. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J Biol Chem. 2012;287:4403–4410. doi: 10.1074/jbc.R111.283432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann CA, Cao J, Manevich Y. Peroxiredoxin 1 and its role in cell signaling. Cell Cycle. 2009;8:4072–4078. doi: 10.4161/cc.8.24.10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershon H, Gershon D. Detection of inactive enzyme molecules in ageing organisms. Nature. 1970;227:1214–1217. doi: 10.1038/2271214a0. [DOI] [PubMed] [Google Scholar]
- Weir CP, Robaire B. Spermatozoa have decreased antioxidant enzymatic capacity and increased reactive oxygen species production during aging in the Brown Norway rat. J Androl. 2007;28:229–240. doi: 10.2164/jandrol.106.001362. [DOI] [PubMed] [Google Scholar]
- Mueller A, Hermo L, Robaire B. The effects of aging on the expression of glutathione S-transferases in the testis and epididymis of the Brown Norway rat. J Androl. 1998;19:450–465. [PubMed] [Google Scholar]
- Jervis KM, Robaire B. The effects of long-term vitamin E treatment on gene expression and oxidative stress damage in the aging Brown Norway rat epididymis. Biol Reprod. 2004;71:1088–1095. doi: 10.1095/biolreprod.104.028886. [DOI] [PubMed] [Google Scholar]
- Smith TB, Baker MA, Connaughton HS, Habenicht U, Aitken RJ. Functional deletion of Txndc2 and Txndc3 increases the susceptibility of spermatozoa to age-related oxidative stress. Free Radic Biol Med. 2013;65:872–881. doi: 10.1016/j.freeradbiomed.2013.05.021. [DOI] [PubMed] [Google Scholar]
- Paul C, Nagano M, Robaire B. Aging results in differential regulation of DNA repair pathways in pachytene spermatocytes in the Brown Norway rat. Biol Reprod. 2011;85:1269–1278. doi: 10.1095/biolreprod.111.094219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaratnam J, Paul C, Robaire B. Male rat germ cells display age-dependent and cell-specific susceptibility in response to oxidative stress challenges. Biol Reprod. 2015;93:72. doi: 10.1095/biolreprod.115.131318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii T, Matsuki S, Iuchi Y, Okada F, Toyosaki S, Tomita Y, Ikeda Y, Fujii J. Accelerated impairment of spermatogenic cells in SOD1-knockout mice under heat stress. Free Radic Res. 2005;39:697–705. doi: 10.1080/10715760500130517. [DOI] [PubMed] [Google Scholar]
- Tsunoda S, Kawano N, Miyado K, Kimura N, Fujii J. Impaired fertilizing ability of superoxide dismutase 1-deficient mouse sperm during in vitro fertilization. Biol Reprod. 2012;87:121. doi: 10.1095/biolreprod.112.102129. [DOI] [PubMed] [Google Scholar]
- Gong S, San Gabriel MC, Zini A, Chan P, O'Flaherty C. Low amounts and high thiol oxidation of peroxiredoxins in spermatozoa from infertile men. J Androl. 2012;33:1342–1351. doi: 10.2164/jandrol.111.016162. [DOI] [PubMed] [Google Scholar]
- Liu Y, O'Flaherty C. Oxidative stress alters thiol redox status of peroxiredoxin 1 and 6 and impairs rat sperm quality. Asian J Androl. doi: 10.4103/1008-682X.170863. (in press). Published online ahead of print 29 January 2016; DOI: 10.4103/1008-682X.170863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Liu N, Zeng H. Peroxiredoxin I in sperm and reactive oxygen species in seminal plasma in patients with idiopathic asthenozoospermia. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2014;39:842–848. doi: 10.3969/j.issn.1672-7347.2014.08.015. [DOI] [PubMed] [Google Scholar]
- Chu KL, Lew QJ, Rajasegaran V, Kung JT, Zheng L, Yang Q, Shaw R, Cheong N, Liou YC, Chao SH. Regulation of PRDX1 peroxidase activity by Pin1. Cell Cycle. 2013;12:944–952. doi: 10.4161/cc.23916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann CA, Krause DS, Carman CV, Das S, Dubey DP, Abraham JL, Bronson RT, Fujiwara Y, Orkin SH, Van Etten RA. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature. 2003;424:561–565. doi: 10.1038/nature01819. [DOI] [PubMed] [Google Scholar]
- Ho YS, Xiong Y, Ma W, Spector A, Ho DS. Mice lacking catalase develop normally but show differential sensitivity to oxidant tissue injury. J Biol Chem. 2004;279:32804–32812. doi: 10.1074/jbc.M404800200. [DOI] [PubMed] [Google Scholar]
- Matzuk MM, Dionne L, Guo Q, Kumar TR, Lebovitz RM. Ovarian function in superoxide dismutase 1 and 2 knockout mice. Endocrinology. 1998;139:4008–4011. doi: 10.1210/endo.139.9.6289. [DOI] [PubMed] [Google Scholar]
- Fox JG, Barthold S, Davisson M, Newcomer CE, Quimby FW, Smith A. The Mouse in Biomedical Research: Diseases New York: Academic Press, 2006. [Google Scholar]
- Meinertz T, Brandstatter A, Trenk D, Jahnchen E, Ostrowski J, Gartner W. Relationship between pharmacokinetics and pharmacodynamics of molsidomine and its metabolites in humans. Am Heart J. 1985;109:644–648. doi: 10.1016/0002-8703(85)90671-4. [DOI] [PubMed] [Google Scholar]
- Rosenkranz B, Winkelmann BR, Parnham MJ. Clinical pharmacokinetics of molsidomine. Clin Pharmacokinet. 1996;30:372–384. doi: 10.2165/00003088-199630050-00004. [DOI] [PubMed] [Google Scholar]
- Shepherd RW, Millette CF, DeWolf WC. Enrichment of primary pachytene spermatocytes from the human testis. Gamete Res. 1981;4:487–498. [Google Scholar]
- Selvaratnam J. Aging male germ cells: responses to oxidative stress and the effects of altered antioxidant status. Montreal, Canada: McGill University; 2016. Thesis. [Google Scholar]
- Robaire B, Ewing LL, Zirkin BR, Irby DC. Steroid delta4-5alpha-reductase and 3alpha-hydroxysteroid dehydrogenase in the rat epididymis. Endocrinology. 1977;101:1379–1390. doi: 10.1210/endo-101-5-1379. [DOI] [PubMed] [Google Scholar]
- Latendresse JR, Warbrittion AR, Jonassen H, Creasy DM. Fixation of testes and eyes using a modified Davidson's fluid: comparison with Bouin's fluid and conventional Davidson's fluid. Toxicol Pathol. 2002;30:524–533. doi: 10.1080/01926230290105721. [DOI] [PubMed] [Google Scholar]
- Lee K, Park JS, Kim YJ. Soo Lee YS, Sook Hwang TS, Kim DJ, Park EM, Park YM. Differential expression of Prx I and II in mouse testis and their up-regulation by radiation. Biochem Biophys Res Commun. 2002;296:337–342. doi: 10.1016/s0006-291x(02)00801-x. [DOI] [PubMed] [Google Scholar]
- Hamer G, Roepers-Gajadien HL, van Duyn-Goedhart A, Gademan IS, Kal HB, van Buul PP, de Rooij DG. DNA double-strand breaks and gamma-H2AX signaling in the testis. Biol Reprod. 2003;68:628–634. doi: 10.1095/biolreprod.102.008672. [DOI] [PubMed] [Google Scholar]
- Elchuri S, Oberley TD, Qi W, Eisenstein RS. Jackson Roberts L, Van Remmen H, Epstein CJ, Huang TT. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene. 2005;24:367–380. doi: 10.1038/sj.onc.1208207. [DOI] [PubMed] [Google Scholar]
- Flood DG, Reaume AG, Gruner JA, Hoffman EK, Hirsch JD, Lin YG, Dorfman KS, Scott RW. Hindlimb motor neurons require Cu/Zn superoxide dismutase for maintenance of neuromuscular junctions. Am J Pathol. 1999;155:663–672. doi: 10.1016/S0002-9440(10)65162-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller FL, Song W, Liu Y, Chaudhuri A, Pieke-Dahl S, Strong R, Huang TT, Epstein CJ, Roberts LJ, II, Csete M, Faulkner JA, Van Remmen H. Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radic Biol Med. 2006;40:1993–2004. doi: 10.1016/j.freeradbiomed.2006.01.036. [DOI] [PubMed] [Google Scholar]
- Noda Y, Ota K, Shirasawa T, Shimizu T. Copper/zinc superoxide dismutase insufficiency impairs progesterone secretion and fertility in female mice. Biol Reprod. 2012;86:16. doi: 10.1095/biolreprod.111.092999. [DOI] [PubMed] [Google Scholar]
- Sentman ML, Granstrom M, Jakobson H, Reaume A, Basu S, Marklund SL. Phenotypes of mice lacking extracellular superoxide dismutase and copper- and zinc-containing superoxide dismutase. J Biol Chem. 2006;281:6904–6909. doi: 10.1074/jbc.M510764200. [DOI] [PubMed] [Google Scholar]
- Lee HY, Choi CS, Birkenfeld AL, Alves TC, Jornayvaz FR, Jurczak MJ, Zhang D, Woo DK, Shadel GS, Ladiges W, Rabinovitch PS, Santos JH, et al. Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell Metab. 2010;12:668–674. doi: 10.1016/j.cmet.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- Umanskaya A, Santulli G, Xie W, Andersson DC, Reiken SR, Marks AR. Genetically enhancing mitochondrial antioxidant activity improves muscle function in aging. Proc Natl Acad Sci U S A. 2014;111:15250–15255. doi: 10.1073/pnas.1412754111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson DF, Fowler KJ, Earle E, Saffery R, Kalitsis P, Trowell H, Hill J, Wreford NG, de Kretser DM, Cancilla MR, Howman E, Hii L, et al. Centromere protein B null mice are mitotically and meiotically normal but have lower body and testis weights. J Cell Biol. 1998;141:309–319. doi: 10.1083/jcb.141.2.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar TR, Varani S, Wreford NG, Telfer NM, de Kretser DM, Matzuk MM. Male reproductive phenotypes in double mutant mice lacking both FSHβ and activin receptor IIA. Endocrinology. 2001;142:3512–3518. doi: 10.1210/endo.142.8.8336. [DOI] [PubMed] [Google Scholar]
- Kumar TR, Wang Y, Lu N, Matzuk MM. Follicle stimulating hormone is required for ovarian follicle maturation but not male fertility. Nat Genet. 1997;15:201–204. doi: 10.1038/ng0297-201. [DOI] [PubMed] [Google Scholar]
- Takano H, Abe K. Age-related histologic changes in the adult mouse testis. Archi Histol Jpn. 1987;50:533–544. doi: 10.1679/aohc.50.533. [DOI] [PubMed] [Google Scholar]
- Jervis KM, Robaire B. Dynamic changes in gene expression along the rat epididymis. Biol Reprod. 2001;65:696–703. doi: 10.1095/biolreprod65.3.696. [DOI] [PubMed] [Google Scholar]
- Perry AC, Jones R, Isolation Hall L. and characterization of a rat cDNA clone encoding a secreted superoxide dismutase reveals the epididymis to be a major site of its expression. Biochem J. 1993;293(pt 1):21–25. doi: 10.1042/bj2930021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenzi A, Gandini L, Picardo M, Tramer F, Sandri G, Panfili E. Lipoperoxidation damage of spermatozoa polyunsaturated fatty acids (PUFA): scavenger mechanisms and possible scavenger therapies. Front Biosci. 2000;5:1–15. doi: 10.2741/lenzi. [DOI] [PubMed] [Google Scholar]
- Johnson RM, Ho YS, Yu DY, Kuypers FA, Ravindranath Y, Goyette GW. The effects of disruption of genes for peroxiredoxin-2, glutathione peroxidase-1, and catalase on erythrocyte oxidative metabolism. Free Radic Biol Med. 2010;48:519–525. doi: 10.1016/j.freeradbiomed.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitano H. Biological robustness. Nat Rev Genet. 2004;5:826–837. doi: 10.1038/nrg1471. [DOI] [PubMed] [Google Scholar]
- Gu Z, Steinmetz LM, Gu X, Scharfe C, Davis RW, Li WH. Role of duplicate genes in genetic robustness against null mutations. Nature. 2003;421:63–66. doi: 10.1038/nature01198. [DOI] [PubMed] [Google Scholar]
- O'Flaherty C, de Souza AR. Hydrogen peroxide modifies human sperm peroxiredoxins in a dose-dependent manner. Biol Reprod. 2011;84:238–247. doi: 10.1095/biolreprod.110.085712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molin M, Yang J, Hanzen S, Toledano MB, Labarre J, Nystrom T. Life span extension and H(2)O(2) resistance elicited by caloric restriction require the peroxiredoxin Tsa1 in Saccharomyces cerevisiae. Mol Cell. 2011;43:823–833. doi: 10.1016/j.molcel.2011.07.027. [DOI] [PubMed] [Google Scholar]
- Ho YS, Magnenat JL, Bronson RT, Cao J, Gargano M, Sugawara M, Funk CD. Mice deficient in cellular glutathione peroxidase develop normally and show no increased sensitivity to hyperoxia. J Biol Chem. 1997;272:16644–16651. doi: 10.1074/jbc.272.26.16644. [DOI] [PubMed] [Google Scholar]
- Ho YS, Xiong Y, Ma W, Spector A, Ho DS. Mice lacking catalase develop normally but show differential sensitivity to oxidant tissue injury. J Biol Chem. 2004;279:32804–32812. doi: 10.1074/jbc.M404800200. [DOI] [PubMed] [Google Scholar]
- Neumann CA, Krause DS, Carman CV, Das S, Dubey DP, Abraham JL, Bronson RT, Fujiwara Y, Orkin SH, Van Etten RA. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature. 2003;424:561–565. doi: 10.1038/nature01819. [DOI] [PubMed] [Google Scholar]
- Kim J, Sturgill D, Tran AD, Sinclair DA, Oberdoerffer P. Controlled DNA double-strand break induction in mice reveals post-damage transcriptome stability Nucleic Acids Res 2016. 20; 44 e64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RR, Milholland B, de Bruin A, Curran S, Laberge RM, van Steeg H, Campisi J, Maslov AY, Vijg J. Controlled induction of DNA double-strand breaks in the mouse liver induces features of tissue ageing. Nat Commun. 2015;6:6790. doi: 10.1038/ncomms7790. [DOI] [PMC free article] [PubMed] [Google Scholar]