

Abstract
Accumulating evidence suggests that altered cellular metabolism is systemic in pulmonary hypertension (PH) and central to disease pathogenesis. However, bioenergetic changes in PH patients and their association with disease severity remain unclear. Here, we hypothesize that alteration in bioenergetic function is present in platelets from PH patients and correlates with clinical parameters of PH. Platelets isolated from controls and PH patients (n = 28) were subjected to extracellular flux analysis to determine oxygen consumption and glycolytic rates. Platelets from PH patients showed greater glycolytic rates than controls. Surprisingly, this was accompanied by significant increases in the maximal capacity for oxygen consumption, leading to enhanced respiratory reserve capacity in PH platelets. This increased platelet reserve capacity correlated with mean pulmonary artery pressure, pulmonary vascular resistance, and right ventricular stroke work index in PH patients and was abolished by the inhibition of fatty acid oxidation (FAO). Consistent with a shift to FAO, PH platelets showed augmented enzymatic activity of carnitine palmitoyltransferase-1 and electron transport chain complex II. These data extend the observation of a metabolic alteration in PH from the pulmonary vascular axis to the hematologic compartment and suggest that measurement of platelet bioenergetics is potentially useful in assessment of disease progression and severity.
Platelets pulmonary hypertension patients show increased mitochondrial reserve capacity due to enhanced fatty acid oxidation, and this parameter correlates with clinical hemodynamic parameters of disease.
Introduction
Pulmonary hypertension (PH) is a progressive vasculopathic disease characterized by elevated pulmonary artery pressure (PAP) and pulmonary vascular resistance (PVR). Pathogenesis of the disease involves vasoconstriction and vascular remodeling, which lead to vascular obstruction, right ventricular (RV) remodeling, and ultimately heart failure (1–4).
On a cellular level, altered metabolism, propagated by mitochondrial dysfunction, has emerged as a significant contributor to PH pathogenesis (5, 6). For example, endothelial and pulmonary artery smooth muscle cells from animal models and patients with PH exhibit a metabolic switch favoring glycolysis even in the presence of oxygen, and this is accompanied by altered mitochondrial electron transport chain (ETC) activities (7–9). This switch to aerobic glycolysis, reminiscent of malignant cells, is thought to at least partially underlie the apoptosis resistance and hyperproliferative cellular phenotype that contribute to vascular remodeling (10, 11). Notably, mitochondrial and metabolic aberrations are not confined to the pulmonary vasculature. Cardiac tissue and cells from animal models of PH have shown altered mitochondrial ETC enzyme expression, as well as changes in substrate utilization (12–17), and increased expression of glycolytic enzymes has also been reported in the skeletal muscle of PH patients (18). Consistent with a central role for this glycolytic shift in PH pathogenesis, pharmacologic therapy that suppresses glycolysis while enhancing oxidative phosphorylation attenuates PH progression (10, 15). Nonetheless, it remains unclear whether mitochondrial changes are present outside the pulmonary vascular system and how these changes correlate with clinical parameters of PH. Prior studies have utilized positron emission tomography (PET) imaging or muscle biopsies of select PH patients to demonstrate that metabolic changes are linked to hemodynamic alterations in humans (8, 18–20). However, PET imaging is limited in scope, and the small volume of muscle obtained from a biopsy is not sufficient to assess all aspects of mitochondrial function. Thus, a comprehensive assessment of bioenergetics has not been performed, and mitochondrial function is not routinely measured in PH patients.
Platelets are anucleate cytoplasmic fragments of megakaryocytes containing functional mitochondria. They represent a noninvasive source of human mitochondria for accurate assessment of bioenergetics and have been utilized to assess systemic metabolic changes in several diseases (21–24). Additionally, platelets are recognized to play a potential role in the pathogenesis of PH through propagation of intravascular thrombosis, excess aggregation, and platelet-endothelial cell interactions via secretion of platelet-derived molecules (25–27).
Here, we hypothesize that circulating platelets from PH patients exhibit altered bioenergetics, which correlate with clinical parameters of disease severity. We demonstrate that platelets from PH patients show a shift to glycolysis that is accompanied by an increase in respiratory reserve capacity due to enhanced fatty acid oxidation (FAO). We show that this dysfunction correlates with clinical parameters of hemodynamic and cardiac function, and we discuss these results in the context of the role of mitochondria in the pathogenesis of PH, as well as the potential to utilize platelets as a marker of bioenergetic dysfunction in PH.
Results
Platelets from PH patients show increased glycolysis and maximal respiratory capacity.
We first compared the bioenergetics of platelets isolated from healthy control subjects to those from subjects with WHO Group 1 PH (n = 28/group; See Table 1 for demographic and clinical parameters). The glycolytic rate of platelets from both groups was calculated by measurement of extracellular acidification rate (ECAR), which could be inhibited by 2-deoxy-D-glucose (2-DG) (Figure 1A). Similar to other cell types in PH (8, 15), glycolytic rate in platelets from PH patients was significantly increased compared with controls (5.04 ± 0.50 mpH/min vs. 2.99 ± 0.31 mpH/min; P < 0.01; Figure 1B).
Table 1. Subject demographics and clinical characteristics.

Figure 1. PH platelets have altered bioenergetics.

(A) Representative ECAR tracing in platelets from a control subject and a PH patient. (B) Basal glycolytic rate in control and PH subjects (n = 28/group). Data are mean ± SEM. (C) Representative platelet OCR profiles from 1 healthy control subject and a patient with PH. (D) Quantification of individual components of the platelet OCR profile in control and PH subjects (n = 28/group). Boxes show the 25%–75% percentile; whiskers show the minimum-maximum; lines represent the median; + represents the mean. (E) Mitochondrial superoxide production, measured by MitoSOX fluorescence, in control (n = 28) and PH (n = 19) platelets. Data are mean ± SEM. (F) Percent activated platelets, as measured by CD62 positivity, in control (n = 19) and PH (n = 16) subjects. Data are mean ± SEM. Unpaired 2-tailed t test was used to compare groups. *P < 0.01. Oligo, oligomycin; rote, rotenone.
Previous studies associate increased glycolysis with dysfunctional mitochondrial oxidative phosphorylation (8, 11, 28). Thus, we next measured platelet oxygen consumption rate (OCR). Surprisingly, platelets from PH patients showed an increase in absolute rates of basal respiration, proton leak, maximal uncoupled respiration, and nonmitochondrial OCR (Figure 1C). However, after correction for nonmitochondrial OCR, basal respiration, and proton leak did not differ significantly between groups (Figure 1D). In contrast, maximal uncoupled respiration was significantly greater in platelets from subjects with PH (227.2 ± 11.6 pmol O2/min; P < 0.0001) compared with controls (131.1 ± 10.2 pmol O2/min; Figure 1D). Physiologically, mitochondria respire at a submaximal level and maintain a reserve capacity that can be utilized if energy demand is increased (29). This reserve capacity, calculated as the difference between maximal and basal OCR, was significantly greater in platelets from PH patients (124.6 ± 11.1 pmol O2/min; P < 0.0001) compared with controls (42.1 ± 8.66 pmol O2/min; Figure 1D).
Notably, the alteration in bioenergetics observed in PH patients did not result in any change in mitochondrial oxidant production, measured by MitoSOX fluorescence (Figure 1E). Additionally, measurement of the platelet activation marker P-selectin (CD62) showed no change in platelet activation in PH patients (Figure 1F).
Importantly, the majority of the PH cohort were on pulmonary vasodilator therapy at the time of bioenergetic profile assessment. Because these medications potentially affect platelet function, we ascertained whether these medications altered platelet bioenergetics. Examination of platelet bioenergetic parameters stratified by PH medication showed no difference in respiration, glycolysis, or oxidant production, regardless of the use of phosphodiesterase-5 (PDE5) inhibitors, prostacyclin analogues (PC), or endothelin receptor antagonists (ERA; Table 2).
Table 2. PH platelet bioenergetics are not affected by pulmonary vasodilator therapies.

Enzymatic activities are altered in PH.
To determine the mechanism underlying increased glycolysis and enhanced maximal capacity in PH platelets, we next measured the expression and activity of key enzymatic regulators of glucose oxidation and oxidative phosphorylation. Consistent with increased glycolysis, PH platelets showed a slight trend to decreased pyruvate dehydrogenase (PDH) activity (Figure 2A), though no change in pyruvate dehydrogenase kinase (PDK) expression was observed (Figure 2, B and C).
Figure 2. Altered enzymatic activity in PH platelets.

(A) Enzymatic activity of PDH in platelets from control (n = 7) and PH (n = 9) subjects. Data are mean ± SEM. (B) Representative Western blots for PDK1 and integrin αIIβ expression in platelets from control and PH subjects. (C) Quantification of PDK1/integrin αIIβ protein expression in platelets from control (n = 17) and PH patients (n = 17). (D) Representative Western blots for complexes (Cx) I, II, IV, and V and integrin αIIβ expression in platelets from control and PH subjects. (E) Quantification of complex/integrin αIIβ protein expression in platelets from control (n = 8) and PH patients (n = 6). (F) Enzymatic activity (fold-change of control) of platelet mitochondrial Cx I, II, and IV and citrate synthase (CS) from control (n = 12) and PH (n = 17) subjects. Boxes show the 25%–75% percentile; whiskers show the minimum-maximum; lines represent the median; + represents the mean. Unpaired 2-tailed t test was used to compare groups. *P < 0.01.
Measurement of the protein expression of the ETC complexes showed that platelets from PH patients had a significant increase in complex II protein expression, with a trend to decreased complex I expression (Figure 2, D and E). Consistent with this change in expression, complex II enzymatic activity was increased (2.66- ± 0.38-fold change; P < 0.05), and complex I activity was significantly decreased (0.54- ± 0.12-fold change; P < 0.01; Figure 2F). However, no change was observed in the enzymatic activity of complex IV or that of the matrix enzyme citrate synthase (Figure 2F).
Reserve capacity is dependent on increased FAO.
Fatty acid oxidation can inhibit glucose oxidation (via the Randle Cycle; ref. 30), and complex II activity has recently been shown to be stimulated by FAO (31). Thus, we next tested whether the enhanced respiratory capacity observed in PH platelets was due to FAO. Basal and maximal platelet OCR was measured in the presence of etomoxir (200 μmol/), an inhibitor of CPT1, which catalyzes fatty acid transport into the mitochondrion. As expected, etomoxir significantly decreased basal and maximal OCR in both control and PH platelets. Basal OCR was decreased to a similar extent in both groups (Figure 3A). However, etomoxir treatment decreased maximal respiration to a greater extent in platelets from PH patients (ΔOCR = 86.6 ± 12.5 pmol O2/min) compared with controls (ΔOCR = 45.1 ± 9.28 pmol O2/min), depleting the respiratory reserve and restoring the maximal respiratory capacity of PH platelets to the level of control platelets (Figure 3B). Consistent with this observation, platelets from PH patients showed a significant increase in CPT1 enzyme activity versus controls (1.32- ± 0.10-fold change; P < 0.05; Figure 3C). Taken together, these data suggest that increased FAO underlies enhanced respiratory capacity in PH platelets.
Figure 3. Increased reserve capacity in PH platelets is dependent upon enhanced FAO.

(A and B) Basal and maximal OCR in platelets from control (n = 13) and PH subjects (n = 16) untreated and treated with etomoxir. Boxes show the 25%–75% percentile; whiskers show the minimum-maximum; lines represent the median; + represents the mean. Two-way ANOVA with Bonferroni adjustment for multiple comparisons was used to compare groups. (C) Enzymatic activity of platelet CPT1 in control (n = 11) and PH (n = 12) subjects. Data are mean ± SEM. Unpaired 2-tailed t test used to compare groups. *P < 0.01.
Reserve capacity correlates with clinical parameters.
To determine whether altered bioenergetics are associated with clinical markers of PH, we next performed linear regression analysis between patient hemodynamic parameters and the reserve respiratory capacity in PH platelets (Figure 4, A–C). Platelet reserve capacity correlated positively with mean pulmonary artery pressure (mPAP; R2 = 0.168, P = 0.047; Figure 4A) and PVR (R2 = 0.176, P = 0.042; Figure 4B). Given that RV function is a key predictor of survival (32–34), we also checked for an association of RV stroke work index (RV SWI) to bioenergetics to find that platelet reserve capacity showed a significant correlation with RV SWI (R2 = 0.271, P = 0.032; Figure 4C) in PH patients.
Figure 4. PH platelet reserve capacity correlates with hemodynamic parameters.
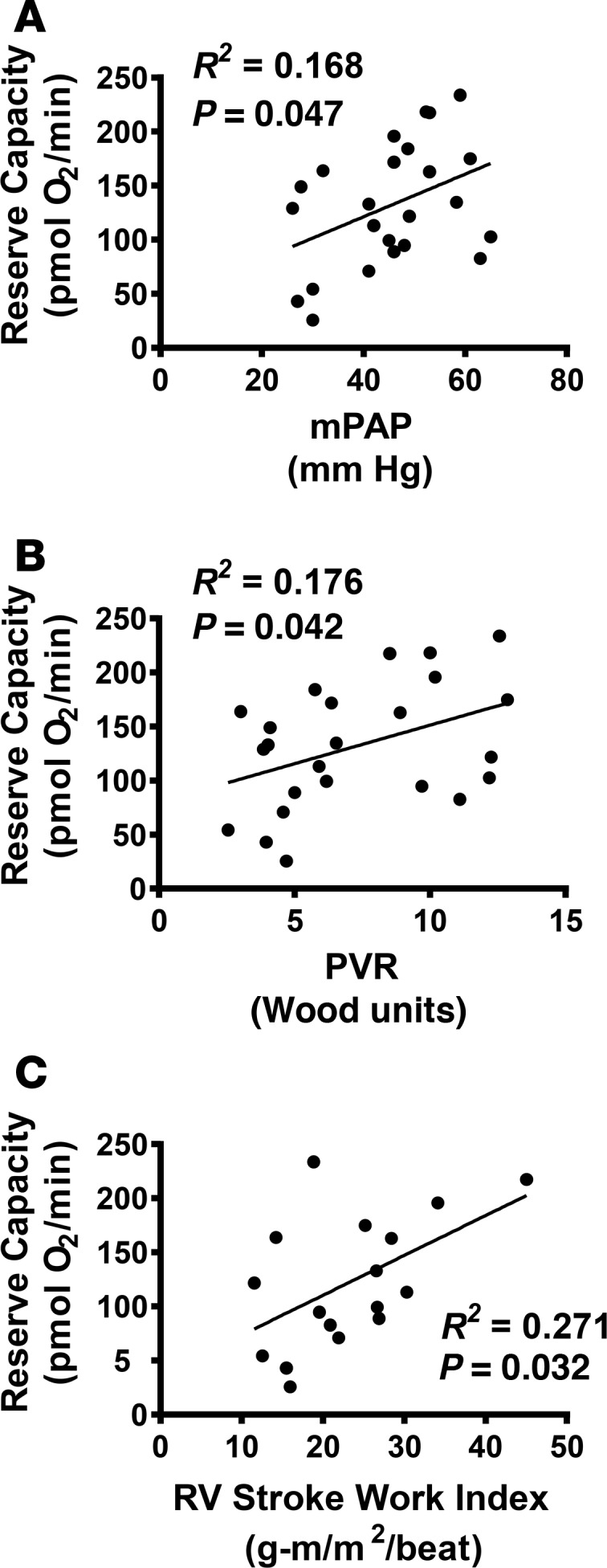
Linear regression of platelet respiratory reserve capacity with (A) mean pulmonary artery pressure (mPAP), (B) pulmonary vascular resistance (PVR), and (C) right ventricular stroke work index (RV SWI).
Discussion
In this study, we demonstrate that platelets from WHO Group 1 PH patients show altered bioenergetics compared with healthy controls. This alteration is characterized by increased glycolysis, which is accompanied by an augmented respiratory reserve capacity that is dependent on FAO. Notably, the increase in reserve capacity correlates positively with hemodynamic parameters, including mPAP, PVR, and RV SWI. These results are significant, as they confirm that PH patients have a platelet metabolic alteration that mirrors what has previously been observed in pulmonary vascular cells and cardiomyocytes. Further, the ability to measure these changes in platelets and their significant correlation with clinical parameters suggests that measurement of platelet bioenergetics could potentially be developed into a useful tool to assess clinical disease severity and progression.
Previous studies have suggested that oxidative phosphorylation in PH patients is depressed due to decreased mitochondrial protein expression or enzymatic damage (8, 11, 28). Contrary to these studies, our data demonstrate no change in basal respiration rate and significantly higher maximal respiratory capacity in platelets from PH patients. The reason for this difference between our data and prior studies may be due to differences in methodology or cell type. For example, Xu et al. demonstrated decreased respiration rates (measured by Clark-type oxygen electrode) in permeabilized endothelial cells with the supplementation of exogenous mitochondrial substrates (8), while the extracellular flux analysis employed in this study utilized intact cells and relied on the presence of endogenous substrate. Thus, it is possible that mitochondria from PH patients consume oxygen at a lower rate when other components of the cell are not considered but, in the intact cell, show no difference in respiratory rate. Further study is required to determine whether differences in OCRs between these two studies are due to methodological differences or whether the alterations we observe are specific to the platelet and not present in endothelial cells.
Cells generally work at a fraction of their maximal capacity and possess a large reserve capacity that enables upregulation of energy production under conditions of stress or excess energy demand (31, 35). Notably, the depletion of mitochondrial reserve capacity has been linked to increased cellular susceptibility to death in the presence of a number of stimuli (29, 36, 37). Thus, it is interesting to speculate that mechanisms to increase reserve capacity may underlie the apoptotic resistance observed in other cell types in PH (10, 11, 38). Our data suggest that substrate switching, particularly increased FAO, underlies this enhanced reserve capacity. These observations in platelets are consistent with prior studies linking FAO to increased reserve capacity in cardiomyocytes (7, 14). Our data also mirror prior studies in other tissues in PH demonstrating a decreased reliance on glucose oxidation (15) and increased FAO enzyme activity (13). The observed trend toward decreased PDH activity and increased CPT1 enzyme activity observed in the PH platelets provides an enzymatic basis for substrate switching. Collectively, these data suggest that substrate utility changes in PH are not limited to the pulmonary vasculature.
Our data show significant changes in the expression and activity of the ETC in PH platelets. We demonstrate a decrease in complex I protein expression and activity, which has been observed in other cell types derived from PH patients and animal models and is thought to underlie the glycolytic switch (11, 39). Notably, the greatest change was in the increased expression (1.5-fold) and activity (2.7-fold) of complex II, succinate dehydrogenase. Complex II is uniquely situated as part of the tricarboxylic acid (TCA) cycle, as well as an entry point of electrons into the ETC. Notably, complex II has recently been shown to act as a sensor for increased FAO, which upregulates its activity to facilitate increased reserve capacity in cardiomyocytes exposed to hypoxic stress (31). Interestingly, others have noted increased complex II activity in cardiomyocytes from animal models of PH, and this increase has been linked to exacerbated mitochondrial oxidant production and heart failure (17). Our study showed no significant difference in mitochondrial superoxide production in platelets from PH patients compared with healthy controls, suggesting potential cell type differences in mitochondrial redox signaling. Another explanation for the lack of increased mitochondrial oxidant production is that our cohort represents a group with compensated disease, as evidenced by normal cardiac output and index. Hence, a cohort of patients with more advanced or decompensated PH may reveal mitochondrial changes that are even more similar to those observed in animal models.
A recent study demonstrated that platelet activation induced by thrombin leads to increased platelet FAO and lipid mobilization through the actions of cytosolic phospholipase A2 (40). Although our data show upregulated FAO in PH platelets, we did not observe a significant increase in platelet activation markers. This is potentially due to the majority of our cohort being treated with PC, which have been demonstrated to directly inhibit platelet activation (41–43). Further study is required to define the lipid profile of platelets from PH patients and to determine whether (in the absence of medication) altered platelet bioenergetics are linked to changes in platelet function in PH. This is particularly important since platelet activation has historically been thought to play a prominent role in PH pathogenesis. In addition to mediating thrombosis, activated platelets store and secrete vasoactive and mitogenic mediators, including thrombospondin-1, thromboxane A2, and serotonin, that together cause vasoconstriction and propagate smooth muscle proliferation (reviewed in refs. 27, 44) — key events in the pathogenesis of PH.
In contrast to the potential effects of medication on platelet activation, our data suggest that the altered mitochondrial bioenergetics observed in PH platelets is not significantly affected by the major classes of PH therapeutics. However, this should be further examined in larger cohorts, as a potential limitation of our analysis was the small sample size of the subgroups on medication. Additionally, these data do not reveal whether the magnitude of bioenergetic change with disease progression is attenuated over the course of treatment. Studies evaluating these parameters over the course of disease treatment will be useful in determining whether changes in platelet bioenergetics can be utilized as markers to assess changes in systemic metabolic dysfunction in PH.
The finding that platelet respiratory reserve capacity correlates with mPAP and PVR presents a link between platelet mitochondrial function and disease severity. Notably, RV function predicts survival more accurately than PVR and is considered the key determinant of outcome in PH (32–34). The association of increased RV SWI, a measure of ventricular workload and contractility, with mitochondrial energetics in this cohort of well-compensated patients may suggest a role for enhanced mitochondrial capacity as an adaptive mechanism. However, it is interesting to speculate that reserve capacity may diminish as the RV decompensates. Further investigation is required to determine whether this association persists in patients with very advanced PH and heart failure. Notably, we did not assess whether platelet bioenergetic alterations correlated with brain natriuretic peptide (BNP) levels, an established marker of disease severity (45). Further, the majority of the current cohort were classified as WHO functional class 2 (moderate PH), thus we were underpowered to compare bioenergetics between functional classes. Additional investigation into the correlation of platelet reserve capacity and ECAR with other established markers of PH severity will be important to perform in larger future cohorts. Interestingly, associations have been demonstrated by utilizing [18F]fluoro-d-glucose (FDG) PET imaging of the heart in which increased glucose uptake correlated with hemodynamic parameters (19, 20). While FDG-PET imaging has been proposed as a technique for assessing PAH severity and right heart failure (19, 20), measurement of bioenergetics in a simple blood draw as demonstrated here may provide similar information more efficiently and be more inclusive of a broader population of patients.
Lastly, it is important to consider the present findings in the context of BM pathology, a well-documented characteristic of PH (46, 47). Several cell types from the myeloid lineage, including mesenchymal stem cells, fibrocytes, and hematopoietic stem cells, have been implicated in the pathogenesis of PH (47). For example, mast cells are known to be increased in vascular lesions of lungs explanted from PH patients (48, 49), and inhibition of mast cell degranulation in animal models of PH attenuates RV hypertrophy (50). Similarly, depletion of fibrocytes in a mouse model of PH inhibited pulmonary artery thickening (51). It is unclear whether these myeloid-derived cells are part of an adaptive response that leads to pathogenesis or whether they are dysfunctional. Our data show that platelets, which are formed in the BM and retain the mitochondria of their megakaryocyte precursors, have altered bioenergetics. This presents a mechanistic link between altered myeloid function and metabolic dysregulation of the pulmonary vasculature. While the status of cellular metabolism in other BM-derived cells in PH is currently unknown, this represents an interesting avenue of future study.
In summary, this study shows that patients with PH show aberrations in metabolism beyond the pulmonary artery and cardiac tissue, and that platelets from these patients can be used to assess mitochondrial changes. This metabolic alteration is characterized by an increased mitochondrial reserve capacity that is dependent on increased FAO and linked to an increase in complex II activity. Notably, these changes are not affected by common therapeutics, and they correlate with clinical hemodynamic parameters. The findings of this investigation warrant further study of platelet mitochondrial function in progression of PH and response to therapy, particularly in the context of emerging mitochondrial therapeutics.
Methods
Study population.
The PH group consisted of patients classified as WHO Group 1 based on ref. 52 presenting to the University of Pittsburgh Medical Center (UPMC) Heart and Vascular Institute Pulmonary Hypertension Clinic. The control group consisted of healthy age and sex-matched participants without known cardiopulmonary or hematologic disease. In the PH group, hemodynamic parameters (Table 1) were derived from right heart catheterization performed within 1 calendar year of bioenergetic assessment. WHO Functional Class and Etiology of PH was determined for 22 of the 28 subjects.
Platelet isolation.
Platelets were isolated by differential centrifugation and number quantified as previously described (23, 24). Briefly, venous blood was collected in citrate by standard venipuncture. Whole blood was centrifuged (150 g; 10 min) in the presence of PGI2 (1 μg/ml) to obtain platelet-rich plasma (PRP). Platelets were pelleted from PRP by centrifugation (1,500 g; 10 min), washed in erythrocyte lysis buffer containing PGI2, and then resuspended in modified Tyrode’s buffer (20 mmol/l HEPES, 128 mmol/l NaCl, 12 mmol/l bicarbonate, 0.4 mmol/l NaH2PO2, 5 mmol/l glucose, 1 mmol/l MgCl2, 2.8 mmol/L KCl, pH 7.4) (Sigma-Aldrich). CD41a expression measured by flow cytometry was used to confirm that platelets were >99% pure.
Measurement of platelet bioenergetics.
OCR and ECAR were measured in isolated platelets (50 × 106/well) by extracellular flux analysis (XF24, Seahorse Biosciences) as previously described (23). After measurement of basal OCR, OCR due to proton leak was determined by oligomycin A (2.5 μmol/l) treatment. Maximal uncoupled OCR was measured by the addition of the uncoupler carbonyl cyanide p-(trifluoro-methoxy) phenyl-hydrazone (FCCP; 0.7 μmol/l). Nonmitochondrial OCR (defined as the OCR of all cellular processes excluding mitochondrial respiration) was measured in the presence of rotenone (10 μmol/l). In a subset of samples, etomoxir (200 μmol/l) was added to quantify FAO-dependent OCR. Glycolytic rate was calculated by determining ECAR, which was sensitive to 2-DG (100 mmol/l) (Seahorse Bioscience).
Mitochondrial ROS generation.
Platelets were incubated with MitoSOX (Invitrogen; 5 μM, 10 minutes). Fluorescent intensity was measured kinetically at 510/580 nm.
Platelet activation.
Platelet activation was performed as previously described (23). Platelets were incubated with phycoerythrin-labeled (PE-labeled) mouse anti–human CD41a antibody (catalog 555467, BD Biosciences) and allophycocyanin-labeled (APC-labeled) mouse and anti–human CD62 antibody (catalog 550488, BD Biosciences; 30 min, 25oC) to measure surface P-selectin expression by flow cytometry (LSRFortessa with FASCDiva software; Becton Dickinson). Platelets were identified by their characteristic light scatter and CD41a antibody binding. Activated platelets are represented as percentage of 10,000 CD41a+ platelets exhibiting APC-CD62P fluorescence.
Mitochondrial enzyme expression.
Mitochondrial protein expression was measured by Western blot as previously described (23). Antibodies for complexes (Complex I MS112, Complex II MS204, Complex IV MS407, ATP synthase [Complex V] MS502) were purchased from MitoSciences, except integrin αIIβ antibody (catalog sc-166599, Santa Cruz Biotechnology Inc.). Antibody for PDK1 (catalog ab110335) was purchased from Abcam.
Carnitine palmitoyltransferase-1 (CPT1) and PDH activities.
CPT1 and PDH activities were determined in isolated platelets spectrophotometrically based on ref. 53 and utilizing a PDH activity kit (Pyruvate Dehydrogenase Enzyme Activity Microplate Assay Kit, catalog ab109902, Abcam), respectively. CPT1 activity of isolated platelets was determined by spectrophotometrically monitoring the generation of CoA-SH from 100 μM palmitoyl-CoA in the presence of 5 mM L-carnitine and 200 μM 5,5’-dinitro-bis-(2-nitrobenzoic acid) (DTNB) at an absorbance of 412 nm. PDH activity in platelets was quantified using a microplate assay kit according to manufacturer’s instructions (Abcam). Platelet proteins were solubilized by addition of detergent (1/10 volume) and then added to anti-PDH antibody–coated wells of a microplate to facilitate immunocapture of active PDH. PDH activity was then determined spectrophotometrically by monitoring the reduction of NAD+ to NADH, coupled to the reduction of a reporter dye at absorbance 450 nm.
ETC complex activity.
Enzymatic activity of complexes I, II, and IV and citrate synthase were performed as previously described (54).
Statistics.
Statistics were performed on Graphpad Prism 7.0 software. Data were compared by ANOVA or Student t test where appropriate. Correlations were determined by 2-tailed Pearson’s correlation and linear regression analysis with 95% CI. P < 0.05 was considered significant. Data are means ± SEM.
Study approval.
This study was approved by the IRB of the UPMC, and written informed consent was obtained from all subjects.
Author contributions
SS, MAS, and MTG conceived of and designed the study. MAS, PW, AW, and QLN evaluated the subjects and/or collected the clinical data. QLN and CC collected the experimental data. QLN analyzed the data. QLN, SS, MAS, and MTG wrote and/or edited the manuscript. All authors reviewed and approved the final version.
Acknowledgments
This work was supported by the Hemophilia Center of Western Pennsylvania (SS), NIH R01-GM113816-01 (SS) and AHA grant 16GRNT27740024 (SS), NIH 2P01HL103455-06 (MTG, SS, MAS)
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:JCI Insight. 2017;2(5):e91415. https://doi.org/10.1172/jci.insight.91415.
Contributor Information
Catherine Corey, Email: cgc9@pitt.edu.
Pamela White, Email: whitepm@upmc.edu.
Annie Watson, Email: watsonam3@upmc.edu.
Sruti Shiva, Email: sss43@pitt.edu.
References
- 1.Lai YC, Potoka KC, Champion HC, Mora AL, Gladwin MT. Pulmonary arterial hypertension: the clinical syndrome. Circ Res. 2014;115(1):115–130. doi: 10.1161/CIRCRESAHA.115.301146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tuder RM, Stacher E, Robinson J, Kumar R, Graham BB. Pathology of pulmonary hypertension. Clin Chest Med. 2013;34(4):639–650. doi: 10.1016/j.ccm.2013.08.009. [DOI] [PubMed] [Google Scholar]
- 3.Davies RJ, Morrell NW. Molecular mechanisms of pulmonary arterial hypertension: role of mutations in the bone morphogenetic protein type II receptor. Chest. 2008;134(6):1271–1277. doi: 10.1378/chest.08-1341. [DOI] [PubMed] [Google Scholar]
- 4.Bogaard HJ, Abe K, Vonk Noordegraaf A, Voelkel NF. The right ventricle under pressure: cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest. 2009;135(3):794–804. doi: 10.1378/chest.08-0492. [DOI] [PubMed] [Google Scholar]
- 5.Freund-Michel V, Khoyrattee N, Savineau JP, Muller B, Guibert C. Mitochondria: roles in pulmonary hypertension. Int J Biochem Cell Biol. 2014;55:93–97. doi: 10.1016/j.biocel.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 6.Paulin R, Michelakis ED. The metabolic theory of pulmonary arterial hypertension. Circ Res. 2014;115(1):148–164. doi: 10.1161/CIRCRESAHA.115.301130. [DOI] [PubMed] [Google Scholar]
- 7.Sutendra G, et al. Fatty acid oxidation and malonyl-CoA decarboxylase in the vascular remodeling of pulmonary hypertension. Sci Transl Med. 2010;2(44):44ra58. doi: 10.1126/scitranslmed.3001327. [DOI] [PubMed] [Google Scholar]
- 8.Xu W, et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci USA. 2007;104(4):1342–1347. doi: 10.1073/pnas.0605080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dromparis P, Sutendra G, Michelakis ED. The role of mitochondria in pulmonary vascular remodeling. J Mol Med. 2010;88(10):1003–1010. doi: 10.1007/s00109-010-0670-x. [DOI] [PubMed] [Google Scholar]
- 10.McMurtry MS, et al. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res. 2004;95(8):830–840. doi: 10.1161/01.RES.0000145360.16770.9f. [DOI] [PubMed] [Google Scholar]
- 11.Bonnet S, et al. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation. 2006;113(22):2630–2641. doi: 10.1161/CIRCULATIONAHA.105.609008. [DOI] [PubMed] [Google Scholar]
- 12.Gomez-Arroyo J, et al. Metabolic gene remodeling and mitochondrial dysfunction in failing right ventricular hypertrophy secondary to pulmonary arterial hypertension. Circ Heart Fail. 2013;6(1):136–144. doi: 10.1161/CIRCHEARTFAILURE.111.966127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Graham BB, et al. Severe pulmonary hypertension is associated with altered right ventricle metabolic substrate uptake. Am J Physiol Lung Cell Mol Physiol. 2015;309(5):L435–L440. doi: 10.1152/ajplung.00169.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fang YH, et al. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: exploiting Randle’s cycle. J Mol Med. 2012;90(1):31–43. doi: 10.1007/s00109-011-0804-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Piao L, et al. The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: resuscitating the hibernating right ventricle. J Mol Med. 2010;88(1):47–60. doi: 10.1007/s00109-009-0524-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sutendra G, et al. A metabolic remodeling in right ventricular hypertrophy is associated with decreased angiogenesis and a transition from a compensated to a decompensated state in pulmonary hypertension. J Mol Med. 2013;91(11):1315–1327. doi: 10.1007/s00109-013-1059-4. [DOI] [PubMed] [Google Scholar]
- 17.Redout EM, et al. Right-ventricular failure is associated with increased mitochondrial complex II activity and production of reactive oxygen species. Cardiovasc Res. 2007;75(4):770–781. doi: 10.1016/j.cardiores.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 18.Malenfant S, et al. Skeletal muscle proteomic signature and metabolic impairment in pulmonary hypertension. J Mol Med. 2015;93(5):573–584. doi: 10.1007/s00109-014-1244-0. [DOI] [PubMed] [Google Scholar]
- 19.Bokhari S, et al. PET imaging may provide a novel biomarker and understanding of right ventricular dysfunction in patients with idiopathic pulmonary arterial hypertension. Circ Cardiovasc Imaging. 2011;4(6):641–647. doi: 10.1161/CIRCIMAGING.110.963207. [DOI] [PubMed] [Google Scholar]
- 20.Ahmadi A, Ohira H, Mielniczuk LM. FDG PET imaging for identifying pulmonary hypertension and right heart failure. Curr Cardiol Rep. 2015;17(1):555. doi: 10.1007/s11886-014-0555-7. [DOI] [PubMed] [Google Scholar]
- 21.Avila C, et al. Platelet mitochondrial dysfunction is evident in type 2 diabetes in association with modifications of mitochondrial anti-oxidant stress proteins. Exp Clin Endocrinol Diabetes. 2012;120(4):248–251. doi: 10.1055/s-0031-1285833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sjövall F, Morota S, Hansson MJ, Friberg H, Gnaiger E, Elmér E. Temporal increase of platelet mitochondrial respiration is negatively associated with clinical outcome in patients with sepsis. Crit Care. 2010;14(6):R214. doi: 10.1186/cc9337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cardenes N, et al. Platelet bioenergetic screen in sickle cell patients reveals mitochondrial complex V inhibition, which contributes to platelet activation. Blood. 2014;123(18):2864–2872. doi: 10.1182/blood-2013-09-529420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu W, Cardenes N, Corey C, Erzurum SC, Shiva S. Platelets from Asthmatic Individuals Show Less Reliance on Glycolysis. PLoS ONE. 2015;10(7):e0132007. doi: 10.1371/journal.pone.0132007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson SR, Granton JT, Mehta S. Thrombotic arteriopathy and anticoagulation in pulmonary hypertension. Chest. 2006;130(2):545–552. doi: 10.1016/S0012-3692(15)51873-4. [DOI] [PubMed] [Google Scholar]
- 26.Crosswhite P, Sun Z. Molecular mechanisms of pulmonary arterial remodeling. Mol Med. 2014;20:191–201. doi: 10.2119/molmed.2013.00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zanjani KS. Platelets in pulmonary hypertension: a causative role or a simple association? Iran J Pediatr. 2012;22(2):145–157. [PMC free article] [PubMed] [Google Scholar]
- 28.Piao L, et al. FOXO1-mediated upregulation of pyruvate dehydrogenase kinase-4 (PDK4) decreases glucose oxidation and impairs right ventricular function in pulmonary hypertension: therapeutic benefits of dichloroacetate. J Mol Med. 2013;91(3):333–346. doi: 10.1007/s00109-012-0982-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hill BG, Dranka BP, Zou L, Chatham JC, Darley-Usmar VM. Importance of the bioenergetic reserve capacity in response to cardiomyocyte stress induced by 4-hydroxynonenal. Biochem J. 2009;424(1):99–107. doi: 10.1042/BJ20090934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Randle PJ. Regulatory interactions between lipids and carbohydrates: the glucose fatty acid cycle after 35 years. Diabetes Metab Rev. 1998;14(4):263–283. doi: 10.1002/(SICI)1099-0895(199812)14:4<263::AID-DMR233>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 31.Pfleger J, He M, Abdellatif M. Mitochondrial complex II is a source of the reserve respiratory capacity that is regulated by metabolic sensors and promotes cell survival. Cell Death Dis. 2015;6:e1835. doi: 10.1038/cddis.2015.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.D’Alonzo GE, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115(5):343–349. doi: 10.7326/0003-4819-115-5-343. [DOI] [PubMed] [Google Scholar]
- 33.Vonk Noordegraaf A, Galiè N. The role of the right ventricle in pulmonary arterial hypertension. Eur Respir Rev. 2011;20(122):243–253. doi: 10.1183/09059180.00006511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van de Veerdonk MC, et al. Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J Am Coll Cardiol. 2011;58(24):2511–2519. doi: 10.1016/j.jacc.2011.06.068. [DOI] [PubMed] [Google Scholar]
- 35.Nickens KP, Wikstrom JD, Shirihai OS, Patierno SR, Ceryak S. A bioenergetic profile of non-transformed fibroblasts uncovers a link between death-resistance and enhanced spare respiratory capacity. Mitochondrion. 2013;13(6):662–667. doi: 10.1016/j.mito.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yadava N, Nicholls DG. Spare respiratory capacity rather than oxidative stress regulates glutamate excitotoxicity after partial respiratory inhibition of mitochondrial complex I with rotenone. J Neurosci. 2007;27(27):7310–7317. doi: 10.1523/JNEUROSCI.0212-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sriskanthadevan S, et al. AML cells have low spare reserve capacity in their respiratory chain that renders them susceptible to oxidative metabolic stress. Blood. 2015;125(13):2120–2130. doi: 10.1182/blood-2014-08-594408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marsboom G, et al. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res. 2012;110(11):1484–1497. doi: 10.1161/CIRCRESAHA.111.263848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rafikov R, et al. Complex I dysfunction underlies the glycolytic switch in pulmonary hypertensive smooth muscle cells. Redox Biol. 2015;6:278–286. doi: 10.1016/j.redox.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Slatter DA, et al. Mapping the Human Platelet Lipidome Reveals Cytosolic Phospholipase A2 as a Regulator of Mitochondrial Bioenergetics during Activation. Cell Metab. 2016;23(5):930–944. doi: 10.1016/j.cmet.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mustard JF, Kinlough-Rathbone RL, Packham MA. Prostaglandins and platelets. Annu Rev Med. 1980;31:89–96. doi: 10.1146/annurev.me.31.020180.000513. [DOI] [PubMed] [Google Scholar]
- 42.Saniabadi AR, Lowe GD, Belch JJ, Barbenel JC, Forbes CD. Effect of prostacyclin (epoprostenol) on the aggregation of human platelets in whole blood in vitro. Haemostasis. 1984;14(6):487–494. doi: 10.1159/000215110. [DOI] [PubMed] [Google Scholar]
- 43.O’Connell C, et al. Comparative Safety and Tolerability of Prostacyclins in Pulmonary Hypertension. Drug Saf. 2016;39(4):287–294. doi: 10.1007/s40264-015-0365-x. [DOI] [PubMed] [Google Scholar]
- 44.Herve P, et al. Pathobiology of pulmonary hypertension. The role of platelets and thrombosis. Clin Chest Med. 2001;22(3):451–458. doi: 10.1016/S0272-5231(05)70283-5. [DOI] [PubMed] [Google Scholar]
- 45.Nagaya N, et al. Plasma brain natriuretic peptide as a prognostic indicator in patients with primary pulmonary hypertension. Circulation. 2000;102(8):865–870. doi: 10.1161/01.CIR.102.8.865. [DOI] [PubMed] [Google Scholar]
- 46.Nikolic I, Yu PB. The Role of Bone Marrow-derived Cells in Pulmonary Arterial Hypertension. What Lies Beneath? Am J Respir Crit Care Med. 2016;193(8):822–824. doi: 10.1164/rccm.201511-2293ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lanzola E, Farha S, Erzurum SC, Asosingh K. Bone marrow-derived vascular modulatory cells in pulmonary arterial hypertension. Pulm Circ. 2013;3(4):781–791. doi: 10.1086/674769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Savai R, et al. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186(9):897–908. doi: 10.1164/rccm.201202-0335OC. [DOI] [PubMed] [Google Scholar]
- 49.Montani D, et al. C-kit-positive cells accumulate in remodeled vessels of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2011;184(1):116–123. doi: 10.1164/rccm.201006-0905OC. [DOI] [PubMed] [Google Scholar]
- 50.Bartelds B, et al. Mast cell inhibition improves pulmonary vascular remodeling in pulmonary hypertension. Chest. 2012;141(3):651–660. doi: 10.1378/chest.11-0663. [DOI] [PubMed] [Google Scholar]
- 51.Frid MG, et al. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Pathol. 2006;168(2):659–669. doi: 10.2353/ajpath.2006.050599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Galiè N, Simonneau G. The Fifth World Symposium on Pulmonary Hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D1–D3. [DOI] [PubMed] [Google Scholar]
- 53.Linher-Melville K, Zantinge S, Sanli T, Gerstein H, Tsakiridis T, Singh G. Establishing a relationship between prolactin and altered fatty acid β-oxidation via carnitine palmitoyl transferase 1 in breast cancer cells. BMC Cancer. 2011;11:56. doi: 10.1186/1471-2407-11-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shiva S, et al. Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J Exp Med. 2007;204(9):2089–2102. doi: 10.1084/jem.20070198. [DOI] [PMC free article] [PubMed] [Google Scholar]