

Abstract
Quantification of stable isotope tracers has revealed the dynamic state of living tissues. A new form of imaging mass spectrometry quantifies isotope ratios in domains much smaller than a cubic micron, enabling measurement of cell turnover and metabolism with stable isotope tracers at the single-cell level with a methodology we refer to as multi-isotope imaging mass spectrometry. In a first-in-human study, we utilize stable isotope tracers of DNA synthesis and de novo lipogenesis to prospectively measure cell birth and adipocyte lipid turnover. In a study of healthy adults, we elucidate an age-dependent decline in new adipocyte generation and adipocyte lipid turnover. A linear regression model suggests that the aging effect could be mediated by a decline in insulin-like growth factor-1 (IGF-1). This study therefore establishes a method for measurement of cell turnover and metabolism in humans with subcellular resolution while implicating the growth hormone/IGF-1 axis in adipose tissue aging.
Utilizing a first-in-human application of multi-isotope imaging mass spectrometry, this study shows that new adipose tissue generation and lipid turnover declines with age.
Introduction
Rates of homeostatic cell turnover and cellular responses to tissue stress or injury have been actively debated for a range of organs. One such example is adipose tissue, for which estimated rates of homeostatic adipocyte turnover differ by over one order of magnitude (1–7). Landmark studies leveraging the global carbon-14 pulse that ensued in the 20th century after above-ground nuclear testing applied carbon-14 birth dating and population modeling to retrospectively infer the dynamics of human neurogenesis (8), cardiogenesis (9), adipogenesis (1), and adipocyte lipid turnover (10). The carbon-14 birth-dating methodology, however, cannot easily be applied to prospective human studies, because carbon-14 exposure is not a controllable variable and because cell birth or synthesis of new biomolecules is not directly measured. This provided rationale for translation of multi-isotope imaging mass spectrometry (MIMS) — an imaging method that enables multiplexed measurement of stable isotope tracers with subcellular resolution (11, 12).
The translation of MIMS to humans builds on extensive precedent for the safe utilization of stable isotopes in metabolic studies (13), including in vulnerable populations, such as pregnant women and critically ill infants (14). MIMS is made possible by an ion microscope (NanoSIMS) (15), which incorporates principles of secondary ion mass spectrometry (16, 17) and state-of-the-art ion optics. A sample surface is sputtered with a focused primary ion beam, triggering emission of atoms and molecular fragments, a fraction of which are ionized and transmitted to a mass spectrometer. Through improved spatial resolution (<50 nm) and mass resolution, more efficient secondary ion transmission, and the capability to accurately measure multiple ionic species in parallel, the NanoSIMS instrument facilitates the fundamental goal of MIMS: to quantitatively localize stable isotope tracers in subcellular domains within a reasonable amount of time (11). Here, we present a first-in-human MIMS study, in which leukocytosis is utilized as a model to develop the methodology, which in turn is applied to a proof-of-concept study of adipogenesis and adipocyte lipid turnover in subcutaneous adipose tissue (SAT).
Results
Measurement of human cell birth with stable isotope tracers and MIMS.
To establish a method for quantification of cell birth in humans, we adapted labeling protocols first developed for the measurement of cell division and fate in mice (12). We administered 15N-thymidine intravenously to 6 healthy adult volunteers for 48 hours (Table 1). Peripheral blood and SAT biopsies were collected after pulse and again after a 4-week chase, and the peripheral white blood cell fraction was utilized to validate the labeling protocol. Monocyte, granulocyte, and lymphocyte subpopulations were identified by their stereotypical appearance (Figure 1A). Labeled monocytes were evident in the peripheral blood at the conclusion of the 48-hour pulse (Figure 1B), consistent with monocyte dynamics in mice (18). In contrast, labeled lymphocytes were evident in circulation after a 1-month chase, consistent with a longer period of differentiation in the marrow before their release into the circulation. In the absence of more frequent sampling during label pulse and chase, these data cannot provide a complete picture of the dynamics of leukocyte birth and trafficking. These data do, however, establish the feasibility and reproducibility of detecting cell birth with MIMS after pulse-chase administration of stable isotope-tagged thymidine (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.90349DS1)
Table 1. Baseline characteristics of study participants.

Figure 1. Measurement of human cell birth with MIMS.

(A) 15N-thymidine–labeled leukocytes, collected after a 2-day intravenous labeling period or subsequent 4-week label-free chase. Quantitative mass images reveal subcellular details. High phosphate content in nucleic acid accounts for high signal in nuclei of 31P images. 32S images demonstrate intense signal in the granules contained in granulocytes. 15N/14N hue saturation intensity image shows label localized to the nuclei of cells born during label administration (arrows). The rainbow scale ranges from blue, set to natural ratio (0.37%, expressed as 0% above natural ratio), to red, where the ratio is above the natural ratio (200% = 3 times natural ratio). Scale bar: 5 μm. (B) Frequency of labeled leukocyte subsets after pulse or chase (n = 6 subjects). Data displayed as Tukey box-and-whisker plots. Mann-Whitney tests were used to assess significance. Note, the y axis ranges from 0%–20%. (C) Labeling schematic for protocol 2, in which subjects were administered intravenous 15N-thymidine continuously for 3 days concurrently with oral 2H-water. The expected effective labeling period for 2H-water is prolonged relative to 15N-thymidine due to a total body water half-life of ~1 week. (D) 15N-thymidine/2H-water doubly labeled cells (arrows). Scale bar: 5 μm. (E) Colocalization of label in D confirmed quantitatively by plotting pixel values, which demonstrate associations among 15N, 2H, and 31P signals, consistent with DNA labeling. Each plotted data point is extracted from a single pixel on the quantitative mass or ratio images. (F) Pixel analysis from 15N-thymidine–unlabeled cells in D demonstrates no positive correlation between the 2H and 31P signals. (G) Data from representative subject undergoing double-labeling protocol (n = 3). After pulse (left), 15N-thymidine–labeled monocytes demonstrate high 2H-water labeling relative to 15N-unlabeled cells. After chase (right), high 2H-labeling in 15N-thymidine–labeled lymphocytes relative to that observed in 15N-unlabeled cells. Rare 15N– outliers that are highly 2H-labeled likely represent cells born after cessation of 15N-thymidine labeling but within the effective 2H-water–labeling period shown schematically in C. Data displayed as Tukey box-and-whisker plots. Mann-Whitney tests were used to assess significance. b.g., background.
Practical considerations limit long-term intravenous administration of thymidine to detect rare birth events, which provided the rationale for the development of a second method, building on the precedent of long-term oral 2H-water administration as a tracer for DNA synthesis in bulk tissue samples (3). Although water is a nonspecific substrate for biosynthetic reactions, we reasoned that the rapid replication of cellular constituents, including DNA, during cell division would result in higher label incorporation relative to background incorporation due to homeostatic turnover of molecules in nondividing cells. Moreover, signal in highly stable constituents, such as DNA, should become more distinct during label-free chase with dilution of nonspecific background labeling. To establish the utility of 2H-water labeling, we recruited 3 healthy volunteers, to whom oral 2H-water was administered concurrently with intravenous 15N-thymidine (Figure 1C). Similar to the 2-day protocol, 15N-labeled cells at completion of pulse were primarily monocytes, whereas those labeled at the end of chase were primarily lymphocytes (Figure 1D). In both cell types, 15N+ cells were highly 2H labeled (Figure 1, D–G). In the lymphocyte population after chase, however, some 2H-labeled cells were 15N unlabeled, an observation that we attribute to the longer half-life of water in humans (19), relative to thymidine, resulting in prolongation of the effective labeling period with 2H-water relative to that achieved with a similar duration of 15N-thymidine administration. These data therefore support the utilization of either 15N-thymidine or 2H-water to track cell division, in vivo, in humans with MIMS.
MIMS demonstrates birth of new adipocytes in SAT.
To assess adipocyte birth, we analyzed SAT specimens collected from subjects administered intravenous 15N-thymidine for either 2 or 3 days (protocols 1 and 2), followed by a 1-month label-free chase. Through an approach previously applied to murine adipose tissue (6), we identified adipocyte nuclei by their close association to a defining lipid droplet. We analyzed adipose samples collected after a 1-month chase to account for the requisite time for progenitor differentiation, which occurs within approximately 2 weeks, based on labeling studies in mouse models. We detected five 15N-thymidine–labeled cells (Figure 2A, n = 5,033 total nuclei, n = 657 adipocyte nuclei); however, all of the labeled cells were nonadipocyte stromal-vascular cells. A calculation of the probability of obtaining this result for different hypothetical yearly adipocyte birth rates (Figure 2B) suggests that the true birth rate is less than approximately 75% per year (95% certainty).
Figure 2. Limited turnover of human SAT.
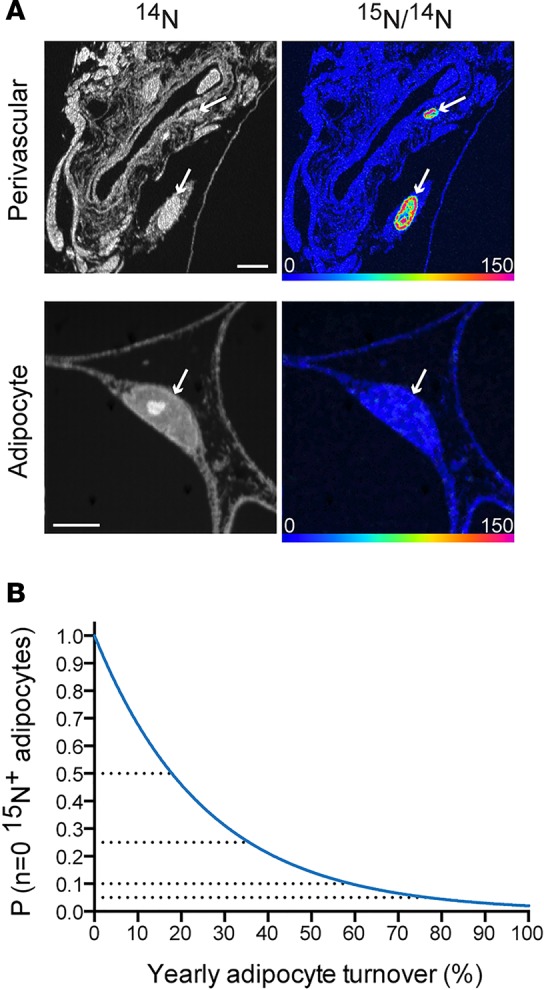
(A) MIMS analysis of SAT sections after 15N-thymidine pulse-chase revealed rare 15N-labeled stromal-vascular cells. Top row: 2 labeled cells (arrows) are found adjacent to a blood vessel. Bottom: unlabeled adipocyte nucleus (arrows). Scale bar: 5 μm. (B) The probability of observing no labeled adipocyte nuclei (n = 657 analyzed), given different yearly turnover rates (x axis). At a probability of 0.05, the yearly adipocyte turnover rate is 75%, suggesting that there is a 95% probability that the yearly adipocyte turnover rate is 75% or less.
In addition to the low rate of homeostatic adipocyte turnover, capturing rare labeling events in adipose tissue is further challenged by the large size of adipocytes relative to their nuclei, which results in a low frequency of adipocyte nuclei in any given imaging plane and large swaths of effective analytical dead space. We reasoned that an alternative approach to detecting rare adipocyte birth would be to increase the effective labeling frequency by extending the labeling duration. Therefore, we administered 2H-labeled water orally to 9 healthy adults for 4 weeks. The primary analytical endpoint was measurement of 2H-labeling in adipocyte nuclei after a 6-month label-free chase. At this time point, a small subset of adipocyte nuclei demonstrated markedly elevated 2H-labeling in a pattern similar to the chromatin, consistent with a mean frequency of 1.6% newly generated adipocytes (range = 0.7%–5.6%, Figure 3A).
Figure 3. Age-dependent decline in homeostatic adipogenesis in human SAT.

(A) 2H-labeled and unlabeled adipocyte nuclei after 4-week 2H-water–labeling pulse and 6-month chase. The 2H/1H HSI image (top right) shows label localized to the nucleus of a putative new adipocyte (arrows), in contrast to the unlabeled nucleus (bottom right, arrows). Scale ranges from background natural abundance to 100% (or 2 times) background. Scale bar: 5 μm. (B) Schematic depicting the hypothesized labeling of an adipocyte that matures during 2H-water labeling. 2H-labeling is expected to be high due to the developmental surge in lipid storage that occurs when a progenitor differentiates into a mature lipid-storing adipocyte. Prior analyses of the fate of 2H-water have established incorporation of 2H atoms by enzymatic reactions involved in pathways of intermediate metabolism, including gluconeogenesis, glycolysis, and the TCA cycle, thereby leading to the labeling of both the glycerol and fatty acid precursors of triglycerides. (C) 2H-high adipocyte lipid droplet (arrows). A small subset of such cells persisted after 6-month chase. (D) Correlation between the frequency of 2H-labeled adipocyte nuclei as shown in A and 2H-high adipocyte lipid droplets as shown in C at the 6-month chase time point (n = 9 subjects). (E) Negative correlation between projected adipocyte birth rate and subject age demonstrated for both measurements of new adipocyte formation (2H-labeled adipocyte nuclei and 2H-high adipocyte lipid droplets) (n = 9 subjects). (F) Positive correlation between projected yearly adipocyte birth rates and serum IGF-1 levels at 6-month chase time point (n = 9 subjects). (D–F) Data obtained by univariate regression.
The surge in lipid storage that occurs when progenitors differentiate into adipocytes provided a second avenue to identify new adipocytes because H atoms from water are introduced by several pathways of intermediate metabolism into glycerol and fatty acids, thereby resulting in the labeling of lipid with de novo lipogenesis (Figure 3B) (7, 20, 21). Indeed, at the conclusion of pulse labeling, we identified a subpopulation of adipocytes containing lipid 2H-label that was 3–7 SDs from the mean. We reasoned that such a population could arise due to one of two scenarios: either from de novo adipocyte differentiation or due to an intrinsically higher rate of lipid turnover. Adipocytes with higher lipid turnover, however, would also experience a more rapid rate of label decay during chase. Therefore, to distinguish these two possibilities, we analyzed the normalized distribution of adipocyte lipid droplets at the conclusion of 2H-water labeling and then again after 6 months of label-free chase. This analysis demonstrated a persistent subset of highly labeled adipocyte droplets after prolonged chase, consistent with their differentiation during 2H-water labeling (Figure 3C). Importantly, the frequency of putative new adipocytes based on high 2H-labeling positively correlated with the frequency of 2H-labeled adipocyte nuclei (Figure 3D), suggesting that the two approaches independently captured the frequency of new adipocyte formation. To project these frequencies into a yearly birth rate, we assumed that the effective labeling period conferred by 4 weeks of 2H-water administration would be extended by one half-life of total body water (~1 week) (19). This translated into a yearly adipocyte birth rate of approximately 11% (nuclei: median [interquartile range] = 11% [8, 60%]; lipid droplet: mean = 12% ± SD 8%).
The adipogenesis measurements described here are lower than rates derived from bulk measurement of 2H-water–labeled adipocytes isolated from subcutaneous adipose depots (0.16%–0.29% per day or 58%–106% per year) (7) and yet similar to the approximate 10% turnover derived from the 14C birth-dating model (1). One potential pitfall relevant to both methods is the reliance on purification and pooling of adipocytes from enzymatically digested adipose tissue. Indeed, the 2H-water–labeling study reported adipocyte purification efficacy of approximately 95% (7), whereas the 14C birth-dating study described optimization to ≥98% (1). Variable degrees of contamination by the more highly proliferative stromal-vascular fraction may account for previously reported differences in adipocyte birth. In taking advantage of the lateral imaging resolution of the MIMS instrument to distinguish adipocytes from adjacent interstitial structures, we propose that the present study is much less prone to such error. Moreover, because MIMS images are derived from the destructive sputtering and analysis of the surface atomic layers of the sample, they are representative of a small fraction of the standard 0.5-μm-thick sections, and therefore the depth resolution greatly exceeds the lateral resolution. This renders MIMS imaging much less prone to artifacts from compression of superimposed 3D objects into a 2D image. There is undoubtedly a low frequency of adipocytes that is excluded when the imaging plane captures the adipocyte nucleus en face, without sampling the lipid droplet. Such sampling errors should affect all adipocytes, whether labeled or unlabeled, and therefore are unlikely to represent a major source of error in the measurements of adipocyte birth.
MIMS demonstrates an age-dependent decline in human adipocyte birth.
Can this study also reveal insight into the mechanistic control of human adipogenesis? Given that aging is associated with declining organ function and regenerative capacity, we hypothesized that age might also be a critical factor in homeostatic adipogenesis. Indeed, regardless of whether we utilized measures based on nuclear or lipid droplet 2H-labeling, we observed an inverse association between age and adipocyte birth across the rather narrow age span of 23 to 38 years in this population, consistent with a decline in homeostatic adipogenesis with aging (Figure 3E; R = –0.72 to –0.80; P < 0.03). As gender may modulate adiposity, we considered whether the negative association between age and adipogenesis could be accounted for by gender. However, the aging effect remained significant, even after controlling for gender (P ≤ 0.04). Adipocyte development is at least partially under hormonal control, providing a rationale to investigate candidate hormonal regulators, selected based on prior evidence supporting a regulatory role in adipocyte development or body fat distribution (22–27). We measured serum levels of leptin, adiponectin, and insulin-like growth factor-1 (IGF-1), the latter of which is also known to decline with age (28). Although we were principally interested in plasma samples collected at the 6-month chase time point, the same time point at which adipogenesis was measured, we also report hormone levels in plasma samples collected from the study subjects at earlier time points (end of pulse and after 1-month chase). We found a significant positive association between IGF-1 (as measured at all 3 time points) and adipogenesis, as measured by nuclei at the 6-month chase time point (pulse [R = 0.83, P < 0.01], 1-month chase [R = 0.90, P < 0.001], and 6-month chase (R = 0.88, P < 0.01)], suggesting that IGF-1 may be a hormonal mediator of adipogenesis (Table 2). Similarly, there was a strong association between IGF-1 measured in the 6-month plasma samples and adipogenesis, as measured by lipid droplet 2H-labeling at 6 months (Figure 3F; R = 0.68, P = 0.04). When we controlled for IGF-1 in a multivariable linear regression model examining the relationship between age and adipogenesis, we found that the significant association between age and adipogenesis was weakened with low variance inflation factors, suggesting that the relationship between adipogenesis and age may in part be mediated by differences in IGF-1 levels (Table 3). In contrast, we did not find a strong association between adipogenesis and adiponectin or leptin. Together, these data suggest that the age-dependent decline in adipogenesis could potentially be partially determined by a similar age-dependent decline in IGF-1.
Table 2. Univariate associations between MIMS measurements of adipogenesis and lipid turnover as well as subject characteristics and hormone levels from protocol 3.

Table 3. Multivariable association between subject age and parameters of adipogenesis, controlling for IGF-1 levels in protocol 3.

MIMS demonstrates an age-dependent decline in adipocyte lipid turnover.
This study also offered the opportunity to interrogate the metabolic function of SAT through an analysis of the uptake and release of 2H-lipid label. Once label is taken up into the adipocyte lipid droplet due to de novo lipogenesis, the subsequent decay of that signal reflects lipid turnover. Indeed, an initial analysis of the labeling distribution of adipocyte data for all subjects at the end of a 1-month pulse and again after a 6-month chase demonstrated a downward shift in the labeling distribution, consistent with lipid turnover. The intersubject variability of lipid turnover in this relatively homogeneous cohort was remarkably broad (lipid half-life mean = 13.5 month ± SD 11.9 month; Figure 4). Although such variability is consistent with prior studies (7, 10), we observed a previously unappreciated association between subject age and lipid age, such that lipid turnover decreased with increasing age. This relationship held when controlling for gender (β = 2.5; P = 0.02) or BMI (β = 2.1, P < 0.04). Moreover, when we controlled for IGF-1 levels, we found that the relationship also remained significant, suggesting that the strong association between age and lipid turnover is at least partially independent of IGF-1, gender, or BMI. It may be that the current MIMS study revealed an aging effect that was not detected in the larger carbon-14 study, because the measurements were more direct. Alternatively, the differences may be attributable to differences in the study populations. Our study is smaller and therefore not powered to detect all determinants of adipogenesis or lipid turnover. In addition, the carbon-14 study merged cohorts with obesity and combined hyperlipidemia (10); whereas our smaller cohort included normal weight and slightly overweight individuals (BMI range 20–26). Given these caveats, a larger study specifically designed to test the aging hypothesis will be required to definitively determine the effect of age on the dynamics of adipocyte lipid turnover.
Figure 4. Age-dependent decline in lipid turnover in human subcutaneous adipocytes.

(A) Adipocyte lipid turnover assessed by the decay of 2H-lipid labeling over the course of a 6-month label-free chase. The “merged” analysis on the left includes pooled adipocyte measurements for all 9 subjects. On the right, the labeling distributions for two of the 9 subjects are shown, one with slow and one with fast lipid turnover. (B) Mean lipid age estimates are derived from the lipid half-life during label-free chase. The graph demonstrates a positive correlation between subject age and lipid age (n = 9 subjects). Data obtained by univariate regression.
Discussion
This first-in-human MIMS study establishes rates of adipogenesis and lipid turnover in the SAT of healthy adults, elucidating an age-dependent decline in both rates within a population of individuals with a narrow age range. These two indices reflect the function of two different cell types contained in adipose tissue. The mature adipocyte is defined by its capacity for storage and regulated release of lipids, whereas generation of new adipocytes is dependent on the self-renewal and differentiation of adipocyte progenitors. A decline in both of these seemingly disparate cellular functions, however, may in theory converge to reduce the plasticity of the subcutaneous depot. Therefore, these data raise the question of whether projection of the aging trend beyond the relatively young upper age limit of this cohort would contribute to the well-described loss of SAT volume and metabolically deleterious ectopic fat redistribution that occurs with advanced age (29, 30).
This study also implicates IGF-1 as a candidate determinant of subcutaneous adipogenesis, a concept consistent with the long-standing recognition that IGF-1 drives adipogenesis, in vitro (23), and more recent evidence that IGF-1 promotes adipocyte hyperplasia in mice (22). Given that impaired adipogenesis is also associated with insulin resistance (6, 31, 32), we speculate that a decline in IGF-1–mediated adipogenesis is one factor linking advanced age to insulin resistance. How might such a mechanism be reconciled with the reduction in insulin resistance reported in the contexts of growth hormone deficiency or resistance, in which levels of downstream IGF-1 are also reduced (33–35)? In contrast to the apparent metabolic advantage seen with severe, genetically mediated growth hormone resistance, levels of IGF-1 that fall in the low end of the physiologic range are associated with a higher risk of diabetes in aging adults (36). Moreover, IGF-1–associated genetic variants identified in GWAS studies are positively associated with insulin resistance (37). Therefore, we speculate that adipose tissue aging — and an associated impairment in adipogenesis — is a negative consequence of the age-dependent decline in IGF-1. This provides rationale for future investigations into whether the improvement in insulin sensitivity observed with IGF-1 therapy is at least partially mediated by stimulation of adipocyte progenitor differentiation (38, 39).
How does MIMS complement existing methods of studying human cell turnover and metabolism, such as the measurement of stable isotope tracers in bulk samples (3) or the 14C approach (8), whereby measurements of cell or molecular age are utilized to retrospectively infer turnover? We propose that MIMS presents a unique combination of capabilities, including (a) quantitative imaging at high magnification of individual cells in their natural microenvironment, (b) measurement of metabolic processes with a large array of commercially available or custom-synthesized stable isotope tracers, (c) multiplexed tracer measurements, and (d) perhaps most importantly, the possibility of performing prospective studies with control of the timing/duration of label exposure in order to allow assessment of interventions, ranging from environmental manipulations to novel therapies. The most important limitation of this methodology is perhaps that it requires tissue sampling, which, for most studies of internal tissues, will require recruitment of research subjects that are undergoing surgery on the organ of interest as part of their clinical care. Nonetheless, we propose this approach as a template of broad relevance to human studies of cell turnover, regeneration, and metabolic function in diverse tissues and cell types.
Methods
Human subjects
Eighteen subjects (Table 1) participated in one of three different stable isotope–labeling protocols described below. Healthy adult subjects were recruited through online advertisements. Inclusion criteria consisted of a BMI of between 20 and 27 kg/m2, no significant chronic medical disorders, and no current medications. Subjects were excluded if they had a history of keloid formation, active use of antiplatelet or antithrombotic medications, regular exercise more than 20 minutes per day, alcohol use in excess of 2 servings per day, drug use within the prior 6 months, active smoking within the prior 6 months, and/or if they had inadequate abdominal wall fat to allow safe biopsy. There were no specific dietary requirements for study participation, and subjects were not given specific instructions regarding diet. SAT biopsies were retrieved from the periumbilical region through the side wall of the umbilicus utilizing a Temno 14-gauge biopsy system (CareFusion) as described previously (40).
Stable isotope tracer protocols
Endotoxin-free 15N-thymidine and sterile 2H-water were obtained from Cambridge Isotope Laboratories Inc. Three labeling protocols were developed as described in more detail in Supplemental Figure 1 and as described below:
Protocol 1.
15N-thymidine was resuspended in 0.9% NaCl solution, filtered, and delivered intravenously as a 30-mg bolus, followed by a continuous 15 mg/h infusion for 48 hours (n = 6 subjects). At the conclusion of the 48-hour labeling period, peripheral blood and fat biopsies were collected. After completion of a 4-week label-free chase period, peripheral blood and fat biopsies were again collected.
Protocol 2.
15N-thymidine was resuspended in 0.9% NaCl solution, filtered, and delivered intravenously as a 30-mg bolus, followed by a continuous 15 mg/h infusion for 72 hours (n = 3 subjects). At the time the infusion was initiated, subjects consumed 400 ml sterile 2H-water. Subsequently, they ingested 60 ml sterile 2H-water daily for the remainder of the 72-hour labeling period. Fat biopsies and peripheral blood were collected at the conclusion of the labeling period and then again after a 4-week label-free chase.
Protocol 3.
Subjects presented to the Brigham and Women’s Hospital Clinical Research Center where they ingested 80 ml sterile 2H-water every 2 hours for 3 doses, for a total of 240 ml (n = 9 subjects). Subjects then consumed 60 ml sterile 2H-water daily for 4 weeks. Fat biopsies were performed at the conclusion of the labeling period and then again after 1 and 6 months of label-free chase. In the 3 months preceding the study, subjects were weight stable within 2 kg, as determined from either the medical record or self-report. Over the course of the 7-month protocol, weight stability was also maintained within 2 kg.
MIMS
Instrumental configuration.
MIMS was performed with modifications to previously described analytical methods, utilizing the prototype instrument and the NanoSIMS 50L (CAMECA) (6, 11, 12). The isotope ratio of 15N/14N (15N-thymidine labeling) was measured as previously described by quantifying the 12C15N–/12C14N– ratio (12). Analysis of 2H/1H (2H-water labeling) was derived by measuring 12C22H–/12C22H–, as previously described (41).
White blood cells.
Peripheral blood was collected into an EDTA-containing tube on ice. Samples were centrifuged for 10 minutes at 2,000 g. The buffy coat was collected and suspended in red blood cell lysis buffer (Invitrogen) on ice for 10 minutes. The suspension was then washed with PBS and fixed with 4% paraformaldehyde (PFA). Cells were pelleted, embedded in EPON or LR white resin, sectioned (0.5 μm), and mounted on silicon chips. Using mass images, and blind to the ratio images, white blood cells were categorized into 3 groups based on stereotypical cellular features, including whether granules were evident (granulocyte), the morphology of the nucleus (multi-lobulated=granulocyte, kidney bean shaped=monocyte, round=lymphocyte), and the nuclear-to-cytoplasmic ratio (scant cytoplasm=lymphocyte). Cells in which the imaging plane did not capture a sufficient cross section of the cell to make such a determination were excluded.
Adipose tissue.
Adipose tissue specimens were fixed with 4% PFA, processed, and analyzed with modifications to previously described methods. For analyses of adipose lipid droplets, samples were subjected to additional fixation with 2% osmium tetroxide (9 hours) followed by an overnight incubation with 1% osmium tetroxide and 1.5% potassium ferrocyanide prior to embedding in EPON or LR white resin. In the early phase of the study, samples were analyzed in automated chain analysis mode, covering large swaths of the section. Due to the low frequency of adipocyte nuclei in any given imaging plane, we assessed different strategies to increase yield, primarily through offline identification of regions of interest containing candidate nuclei, which allowed for targeted analysis of these preselected regions of interest. The first approach involved staining the sections with DAPI to identify all nuclei in the imaging plane. The second approach involved identification of candidate adipocyte nuclei using differential interference contrast microscopy (×50 objective). In this permissive approach to identification of adipocyte nuclei, the observer scanned the section and recorded regions of interest for all adipocyte nuclei. Images obtained with the NanoSIMS were analyzed in a customized plugin (OpenMIMS; https://github.com/BWHCNI/OpenMIMS) of ImageJ (NIH) and assigned an identity of adipocyte or stromal-vascular cell.
Hormonal analyses
Total adiponectin was measured in plasma by ELISA (R&D Systems) with an intra-assay coefficient of variation (CV) of 2.5%–4.7% and an interassay CV of 5.8%–6.9%. Total leptin was measured in plasma by ELISA (R&D Systems) with an intra-assay CV of 3.0%–3.3% and an interassay CV of 3.5%–5.4%. Total IGF-1 was measured in plasma by ELISA (R&D Systems) with an intra-assay CV of 3.5%–4.3% and an interassay CV of 7.5%–8.3%.
Statistics
To calculate how the observation of no 15N-thymidine–labeled nuclei (Figure 2) affects the probability of different yearly adipogenesis rates, we assumed homogeneity of the subjects and pooled the data, assuming that each measurement of an adipocyte nucleus was independent. The probability is expressed as P (Ravg yearly birth rate | no 15N+ nuclei) = [1 – R × (no. days labeling / 365)]total no. nuclei, where P represents probability, R represents the hypothetical yearly birth rate, and no 15N+ nuclei represents the observed absence of 15N-thymidine-labeled adipocyte nuclei.
Metrics of lipid turnover were determined using the lipid droplet–labeling distributions measured at the end of 2H-water labeling and again after 6 months of label-free chase. Standard half-life equations were utilized to determine lipid half-life and the related parameter of average lipid age: lipid t1/2 = chase duration / log2 (mean lipid label pulse/mean lipid label chase) and mean lipid age = | lipid t1/2 / ln(2), where ln represents natural logarithm function.
Statistical analyses were performed using JMP Prol 11.0 (SAS Institute Inc.) and Prism 6 (Graphpad). A 2-sided P value of less than 0.05 was considered significant. Means and SEM are reported unless the data were not normally distributed, in which case a median and interquartile range are reported. Univariate relationships were calculated using Pearson correlation coefficients or, if the data were not normally distributed, Spearman’s coefficients. Least-squares linear regression was used to control for variables in a multivariable analysis. As adipogenesis calculated by nuclear 2H-labeling was nonnormally distributed even after attempts at normalization, we performed a least-squares linear regression using the nonnormally distributed data (reported in Table 3) and confirmed these results by (a) performing a rank-based analysis by ranking the data from 1 to 9 and using these ranked data in the least-squares linear regression model and (b) removing the largest value for the outcome variable, which normalized the data, and repeating the least-squares linear regression. Importantly, the results reported in Table 3 remained consistent with these two confirmatory analyses.
Study approval
The study was approved by the Partners Institutional Review Board, run by the Partners Healthcare Human Research Committee, Boston, Massachusetts, USA. Written informed consent was obtained from all subjects before their participation in the study.
Author contributions
EDR, RTL, CPL, and MLS conceptualized the study; CG, CPL, and MLS developed methodology; PKF, HL, and MLS performed formal analysis; CG, SK, ML, JPZ, JM, and MLS performed experiments; HFSC, FH, and DL provided resources; PKF and MLS wrote the manuscript; MLS facilitated visualization; RTL and MLS supervised the study; MLS was the project administrator; and EDR, RTL, CPL, and MLS acquired funding.
Supplementary Material
Acknowledgments
We thank CAMECA and F. Hillion for providing usage of a NanoSIMS instrument, which was used for the development of analytical strategies utilized in this study. We thank T. Perlstein for assistance with the human protocols. This work was funded by resources from the clinical research center and by a pilot grant (to EDR, RTL, CPL, and MLS) both from the Harvard Catalyst (NCATS/NIH UL1 TR001102 to EDR, RTL, CPL, and MLS), the Harvard Stem Cell Institute (to MLS), and Brigham and Women’s Hospital Funds (to MLS).
Footnotes
Conflict of interest: H. Francois-Saint-Cyr, F. Horreard, and D. Larson are employed by CAMECA, which manufactures the NanoSIMS instrument.
Reference information:JCI Insight. 2017;2(5):e90349. https://doi.org/10.1172/jci.insight.90349.
Contributor Information
Christelle Guillermier, Email: CGUILLERMIER@PARTNERS.ORG.
Mingyue Lun, Email: mlun@partners.org.
Jessica Milian, Email: jesmilian@gmail.com.
Hang Lee, Email: hlee5@partners.org.
Hugues Francois-Saint-Cyr, Email: Hugues.FSC@ametek.com.
Francois Horreard, Email: Francois.Horreard@ametek.com.
David Larson, Email: david.larson@ametek.com.
References
- 1.Spalding KL, et al. Dynamics of fat cell turnover in humans. Nature. 2008;453(7196):783–787. doi: 10.1038/nature06902. [DOI] [PubMed] [Google Scholar]
- 2.Rigamonti A, Brennand K, Lau F, Cowan CA. Rapid cellular turnover in adipose tissue. PLoS One. 2011;6(3):e17637. doi: 10.1371/journal.pone.0017637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neese RA, et al. Measurement in vivo of proliferation rates of slow turnover cells by 2H2O labeling of the deoxyribose moiety of DNA. Proc Natl Acad Sci USA. 2002;99(24):15345–15350. doi: 10.1073/pnas.232551499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jo J, et al. Hypertrophy and/or hyperplasia: dynamics of adipose tissue growth. PLoS Comput Biol. 2009;5(3):e1000324. doi: 10.1371/journal.pcbi.1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang QA, Tao C, Gupta RK, Scherer PE. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat Med. 2013;19(10):1338–1344. doi: 10.1038/nm.3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim SM, et al. Loss of white adipose hyperplastic potential is associated with enhanced susceptibility to insulin resistance. Cell Metab. 2014;20(6):1049–1058. doi: 10.1016/j.cmet.2014.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strawford A, Antelo F, Christiansen M, Hellerstein MK. Adipose tissue triglyceride turnover, de novo lipogenesis, and cell proliferation in humans measured with 2H2O. Am J Physiol Endocrinol Metab. 2004;286(4):E577–E588. doi: 10.1152/ajpendo.00093.2003. [DOI] [PubMed] [Google Scholar]
- 8.Spalding KL, Bhardwaj RD, Buchholz BA, Druid H, Frisén J. Retrospective birth dating of cells in humans. Cell. 2005;122(1):133–143. doi: 10.1016/j.cell.2005.04.028. [DOI] [PubMed] [Google Scholar]
- 9.Bergmann O, et al. Dynamics of cell generation and turnover in the human heart. Cell. 2015;161(7):1566–1575. doi: 10.1016/j.cell.2015.05.026. [DOI] [PubMed] [Google Scholar]
- 10.Arner P, et al. Dynamics of human adipose lipid turnover in health and metabolic disease. Nature. 2011;478(7367):110–113. doi: 10.1038/nature10426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lechene C, et al. High-resolution quantitative imaging of mammalian and bacterial cells using stable isotope mass spectrometry. J Biol. 2006;5(6):20. doi: 10.1186/jbiol42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steinhauser ML, et al. Multi-isotope imaging mass spectrometry quantifies stem cell division and metabolism. Nature. 2012;481(7382):516–519. doi: 10.1038/nature10734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hellerstein MK. In vivo measurement of fluxes through metabolic pathways: the missing link in functional genomics and pharmaceutical research. Annu Rev Nutr. 2003;23:379–402. doi: 10.1146/annurev.nutr.23.011702.073045. [DOI] [PubMed] [Google Scholar]
- 14.Steinhauser ML, Lechene CP. Quantitative imaging of subcellular metabolism with stable isotopes and multi-isotope imaging mass spectrometry. Semin Cell Dev Biol. 2013;24(8-9):661–667. doi: 10.1016/j.semcdb.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Slodzian G, Daigne B, Girard F, Boust F, Hillion F. Scanning secondary ion analytical microscopy with parallel detection. Biol Cell. 1992;74(1):43–50. doi: 10.1016/0248-4900(92)90007-N. [DOI] [PubMed] [Google Scholar]
- 16.Castaing R, Slodzian GJ. Optique corpusculaire-premiers essais de microanalyse par emission ionique secondaire. Microscopie. 1962;1:395–399. [Google Scholar]
- 17.Andersen CA, Hinthorne JR. Ion microprobe mass analyzer. Science. 1972;175(4024):853–860. doi: 10.1126/science.175.4024.853. [DOI] [PubMed] [Google Scholar]
- 18.Yona S, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38(1):79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hevesy G, Hofer E. Elimination of water from the human body. Nature. 1934;134:879. doi: 10.1038/134879a0. [DOI] [Google Scholar]
- 20.Turner SM, et al. Measurement of TG synthesis and turnover in vivo by 2H2O incorporation into the glycerol moiety and application of MIDA. Am J Physiol Endocrinol Metab. 2003;285(4):E790–E803. doi: 10.1152/ajpendo.00402.2002. [DOI] [PubMed] [Google Scholar]
- 21.Diraison F, Pachiaudi C, Beylot M. In vivo measurement of plasma cholesterol and fatty acid synthesis with deuterated water: determination of the average number of deuterium atoms incorporated. Metab Clin Exp. 1996;45(7):817–821. doi: 10.1016/S0026-0495(96)90152-3. [DOI] [PubMed] [Google Scholar]
- 22.Chang HR, Kim HJ, Xu X, Ferrante AW. Macrophage and adipocyte IGF1 maintain adipose tissue homeostasis during metabolic stresses. Obesity (Silver Spring) 2016;24(1):172–183. doi: 10.1002/oby.21354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith PJ, Wise LS, Berkowitz R, Wan C, Rubin CS. Insulin-like growth factor-I is an essential regulator of the differentiation of 3T3-L1 adipocytes. J Biol Chem. 1988;263(19):9402–9408. [PubMed] [Google Scholar]
- 24.Kim JY, et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest. 2007;117(9):2621–2637. doi: 10.1172/JCI31021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Machinal-Quélin F, Dieudonné MN, Leneveu MC, Pecquery R, Giudicelli Y. Proadipogenic effect of leptin on rat preadipocytes in vitro: activation of MAPK and STAT3 signaling pathways. Am J Physiol, Cell Physiol. 2002;282(4):C853–C863. doi: 10.1152/ajpcell.00331.2001. [DOI] [PubMed] [Google Scholar]
- 26.Yue R, Zhou BO, Shimada IS, Zhao Z, Morrison SJ. Leptin receptor promotes adipogenesis and reduces osteogenesis by regulating mesenchymal stromal cells in adult bone marrow. Cell Stem Cell. 2016;18(6):782–796. doi: 10.1016/j.stem.2016.02.015. [DOI] [PubMed] [Google Scholar]
- 27.Fu Y, Luo N, Klein RL, Garvey WT. Adiponectin promotes adipocyte differentiation, insulin sensitivity, and lipid accumulation. J Lipid Res. 2005;46(7):1369–1379. doi: 10.1194/jlr.M400373-JLR200. [DOI] [PubMed] [Google Scholar]
- 28.Rudman D, Kutner MH, Rogers CM, Lubin MF, Fleming GA, Bain RP. Impaired growth hormone secretion in the adult population: relation to age and adiposity. J Clin Invest. 1981;67(5):1361–1369. doi: 10.1172/JCI110164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fox CS, et al. Abdominal visceral and subcutaneous adipose tissue compartments: association with metabolic risk factors in the Framingham Heart Study. Circulation. 2007;116(1):39–48. doi: 10.1161/CIRCULATIONAHA.106.675355. [DOI] [PubMed] [Google Scholar]
- 30.Borkan GA, Hults DE, Gerzof SG, Robbins AH, Silbert CK. Age changes in body composition revealed by computed tomography. J Gerontol. 1983;38(6):673–677. doi: 10.1093/geronj/38.6.673. [DOI] [PubMed] [Google Scholar]
- 31.Majithia AR, et al. Rare variants in PPARG with decreased activity in adipocyte differentiation are associated with increased risk of type 2 diabetes. Proc Natl Acad Sci USA. 2014;111(36):13127–13132. doi: 10.1073/pnas.1410428111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arner E, et al. Adipocyte turnover: relevance to human adipose tissue morphology. Diabetes. 2010;59(1):105–109. doi: 10.2337/db09-0942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yakar S, et al. Inhibition of growth hormone action improves insulin sensitivity in liver IGF-1-deficient mice. J Clin Invest. 2004;113(1):96–105. doi: 10.1172/JCI17763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guevara-Aguirre J, et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci Transl Med. 2011;3(70):70ra13. doi: 10.1126/scitranslmed.3001845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guevara-Aguirre J, et al. GH receptor deficiency in Ecuadorian adults is associated with obesity and enhanced insulin sensitivity. J Clin Endocrinol Metab. 2015;100(7):2589–2596. doi: 10.1210/jc.2015-1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sandhu MS, Heald AH, Gibson JM, Cruickshank JK, Dunger DB, Wareham NJ. Circulating concentrations of insulin-like growth factor-I and development of glucose intolerance: a prospective observational study. Lancet. 2002;359(9319):1740–1745. doi: 10.1016/S0140-6736(02)08655-5. [DOI] [PubMed] [Google Scholar]
- 37.Dupuis J, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010;42(2):105–116. doi: 10.1038/ng.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morrow LA, O’Brien MB, Moller DE, Flier JS, Moses AC. Recombinant human insulin-like growth factor-I therapy improves glycemic control and insulin action in the type A syndrome of severe insulin resistance. J Clin Endocrinol Metab. 1994;79(1):205–210. doi: 10.1210/jcem.79.1.8027228. [DOI] [PubMed] [Google Scholar]
- 39.Moses AC, Young SC, Morrow LA, O’Brien M, Clemmons DR. Recombinant human insulin-like growth factor I increases insulin sensitivity and improves glycemic control in type II diabetes. Diabetes. 1996;45(1):91–100. doi: 10.2337/diab.45.1.91. [DOI] [PubMed] [Google Scholar]
- 40.Fazeli PK, et al. FGF21 and the late adaptive response to starvation in humans. J Clin Invest. 2015;125(12):4601–4611. doi: 10.1172/JCI83349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guillermier C, Steinhauser ML, Lechene CP. Quasi-simultaneous acquisition of nine secondary ions with seven detectors on NanoSIMS50L: application to biological samples. Surf Interface Anal. 2014;46:150–153. doi: 10.1002/sia.5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.