

Abstract
Biliary atresia is an obstructive cholangiopathy of infancy that progresses to end-stage cirrhosis. Although the pathogenesis of the disease is not completely understood, previous reports link TNFα to apoptosis of the bile duct epithelium in the presence of IFNγ. Here, we investigate if TNFα signaling regulates pathogenic mechanisms of biliary atresia. First, we quantified the expression of TNFA and its receptors TNFR1 and TNFR2 in human livers and found an increased expression of the receptors at the time of diagnosis. In mechanistic experiments using a neonatal mouse model of rhesus rotavirus–induced (RRV-induced) biliary atresia, the expression of the ligand and both receptors increased 6- to 8-fold in hepatic DCs and NK lymphocytes above controls. The activation of tissue NK cells by RRV-primed DCs was independent of TNFα-TNFR signaling. Once activated, the expression of TNFα by NK cells induced lysis of 55% ± 2% of bile duct epithelial cells, which was completely prevented by blocking TNFα or TNFR2, but not TNFR1. More notably, antibody-mediated or genetic disruption of TNFα-TNFR2 signaling in vivo decreased apoptosis and epithelial injury; suppressed the infiltration of livers by T cells, DCs, and NK cells; prevented extrahepatic bile duct obstruction; and promoted long-term survival. These findings point to a key role for the TNFα/TNFR2 axis on pathogenesis of experimental biliary atresia and identify new therapeutic targets to suppress the disease phenotype.
TNFα and TNFR2, but not TNFR1, are key effectors of bile duct injury and the obstructive phenotype of experimental biliary atresia.
Introduction
Biliary atresia is the most common obstructive cholangiopathy of childhood. Although the etiology is largely undefined, patient- and animal-based experiments point to a key role of the innate and adaptive immune responses in the pathogenesis of bile duct injury and obstruction (1). Studies using an experimental model of rhesus rotavirus–induced (RRV-induced) biliary atresia in newborn BALB/c mice have shown that the mechanisms of bile duct injury begins with the activation of DCs and NK cells, which target the bile duct epithelium (2, 3). This is followed by a tissue-specific expansion of CD8+ T cells, macrophages, and HMGB1- and prominin-1–expressing cells (4–7) and the expression of proinflammatory molecules such as IL-8, IFNγ, perforin, granzymes, and others (8–11). Interestingly, the incubation of cholangiocytes with IFNγ in a cell culture system does not induce cell injury but, when incubated in the presence of both IFNγ and TNFα, results in the activation of caspases and cholangiocyte apoptosis (12).
TNFα is a potent proinflammatory cytokine involved in systemic inflammation with a range of biological activities encompassing both beneficial and host-damaging effects linked to tumor cell apoptosis, sepsis, and autoimmune diseases (13). TNFα is expressed as a 26-kDa precursor transmembrane protein (mTNFα) and a 17-kDa secreted form. While the soluble form mediates pleiotropic effects such as cell proliferation, cytokine synthesis, and expression of adhesion molecules, the expression of activation-induced mTNFα on NK lymphocytes and other cells mediates cytotoxicity by means of cell-cell contact and preferential binding to TNFα receptor types 1 and 2 (TNFR1 and TNFR2) (14). In addition to acting as a ligand to TNFRs, TNFα elicits outside-to-inside signals (reverse signaling) (15). In the liver, TNFα/TNFR signaling has been linked to several biological processes, including liver regeneration (16), regulation of CD8+ T cell cytotoxicity (17), and mechanisms of fulminant hepatitis (18) and fibrogenesis (19). Based on our previous report that TNFα promotes cholangiocyte apoptosis in the presence of IFNγ (12), we directly explored whether TNFα plays a regulatory role in the pathogenesis of bile duct injury in biliary atresia. In livers of infants with the disease, we found increased expression of TNFR1 and TNFR2 mRNA at the time of diagnosis. In mechanistic experiments using the rotavirus-mouse model of biliary atresia, we found a differential expression of TNFα and TNFR2 in hepatic DCs, NK cells, and cholangiocytes. In mice, the disruption of any element of the TNFα-TNFR signaling suppressed the biliary atresia phenotype, with a greater role for TNFR2 in the cytokine response, cholangiocyte survival/death, and anatomical disruption of bile flow.
Results
Expression of TNFα and its receptors in experimental and human biliary atresia.
Based on the previous report that TNFα induces apoptosis in cholangiocytes in the presence of IFNγ in vitro and in mice with experimental biliary atresia (12), we first quantified the mRNA expression of TNFR1 and TNFR2 in livers of infants at the time of diagnosis; for TNFα, the expression was determined in the liver (mRNA) and serum (protein). The hepatic expression of TNFR1 and TNFR2 increased above controls, while the expression for TNFα did not change in the liver or serum (TNFα, serum; Figure 1, A and B). By immunostaining, TNFR2 expression was prominent in intrahepatic bile ducts of patients with biliary atresia, while TNFR1 signals were primarily localized to cholangiocytes in livers from normal and disease controls (Figure 1C). To directly explore whether TNFα-TNFR signaling is involved in pathogenic mechanisms of the disease, we next quantified mRNA for Tnfa, Tnfr1 and Tnfr2 genes in early phase of injury of extrahepatic bile ducts (3 days after RRV), lumen obstruction (7 days), and atresia (14 days) of neonatal BALB/c mice injected i.p. with 1.5 × 106 fluorescent forming units (ffu) of RRV; normal saline injection served as controls (9). Their expression increased in extrahepatic bile ducts at all time points, with greater levels for Tnfa and Tnfr2 (Figure 1D).
Figure 1. Expression of TNFα and TNFRs in humans and mice with biliary atresia.

(A) Liver mRNA expression for TNFA, TNFR1, and TNFR2 from microarray analysis of livers from infants with biliary atresia (n = 64) and intrahepatic cholestasis (n = 14) at diagnosis normalized to age-matched normal controls. (B) Scatter plots of serum TNFα levels in infants with biliary atresia at diagnosis (n = 11) and healthy infants (n = 6). (C) Representative immunohistochemical panels identify TNFR1+ signals (arrowheads) in livers from normal and disease controls (α-1 antitrypsin deficient, A1AT) relative to the less intense signals for TNFR2 (arrows). In contrast, TNFR2 stains strongly in patients with biliary atresia. PV, portal vein. (D) mRNA expression levels of Tnfa, Tnfr1, and Tnfr2 in extrahepatic bile ducts from mice at 3, 7, and 14 days after injection of saline or RRV by real-time PCR. Expression was normalized against Hprt. n = 4–5 mice/group/time point. Values expressed as mean ± SD. *P < 0.05,**P < 0.01, ***P < 0.001, 2-tailed parametric unpaired t test with Welch’s correction.
To better define the expression at the cellular level, we focused on NK cells and DCs based on their roles in regulating the onset of epithelial injury in experimental models (2). Flow cytometric analysis showed that the number of hepatic NK (CD3–CD49b+) cells expressing intracellular TNFα increased from undetectable in control mice to low level of detection at 3 days after RRV (P < 0.05) and to 6.3-fold at 7 days (P < 0.0001; Figure 2A); similar patterns and fold changes were found in DCs (CD11c+) (Figure 2B). Interestingly, the levels of TNFR1 did not change in NK cells and increased only at 7 days in DCs, but TNFR2 increased in both NK cells and DCs at 3 and 7 days after RRV (Figure 2C). The combined increase in the expression of TNFα and its receptors pointed to a potential role in the regulation of mechanisms of bile duct epithelial injury, with the potential for a preferential use of TNFR2 by NK cells and DCs based on their differential pattern of cellular expression.
Figure 2. Hepatic NK and DCs express TNFα.

Flow cytometric dot plot analyses showing the percent of (A) NK cells or (B) CD11c+ DCs from saline or RRV-challenged mice expressing TNFα, with an increased expression of intracellular TNFα at the onset of epithelial injury (day 3) and duct obstruction (day 7). Graphs on right show the percent populations and mean fluorescence intensities (MFI) of TNFα on NK and DCs. (C) Total number of DCs and NK cells expressing TNFR1 or -R2 at 3 and 7 days after saline or RRV infection. n = 4–5 specimens/group/time point with pools of 2–3 livers/specimen. Values expressed as mean ± SD. *P < 0.03, **P < 0.01, ***P < 0.001, 2-tailed parametric unpaired t test with Welch’s correction.
TNFα-TNFR signals and activation of NK cells by DCs.
To directly determine whether TNFα and its receptors regulate the ability of DCs to activate NK cells in response to RRV, we performed coculture activation assays using DCs from livers 3 days after infection with RRV and NK cells from RRV-naive mice matched by age in the presence of antibodies to block TNFα, TNFR1, or TNFR2; isotype antibodies were used as controls. Using flow cytometry to quantify the expression of Nkg2d and CD69 as markers of NK cell activation, we found that the individual or combined blocking of TNFR1 and/or TNFR2 on NK cells or of TNFα on DCs did not interfere with the activation of NK cells, as evidenced by the increase in the expression of Nkg2d or CD69 relative to isotype IgG controls (Figure 3, A and B). To more thoroughly examine the degree of activation, we quantified the expression of cytokines and chemokines in culture supernatants and found that blocking TNFα, TNFR1, and/or R2 suppressed but did not abolish the levels of TNFα, IL6, IL17a, CXCL9, CXCL10, CXCL1, and CCL5 (Figure 3, C and D). These findings suggested that the biological impacts of blocking TNFα-TNFR signals are limited to a modest suppression of cytokine/chemokine response, without substantially impairing the ability of DCs to activate NK cells in response to RRV infection. This lack of requirement for TNFα-TNFR signaling in the activation of NK cells by DCs raised the possibility that TNFα-TNFR may be important for the subsequent recognition and lysis of cholangiocytes by NK cells (a later phase of pathogenesis of biliary injury in experimental atresia).
Figure 3. Activation markers and cytokine/chemokine expression following blockade of TNFα or TNFR1/2.

Flow cytometric quantification of Nkg2d and CD69 in naive NK cells after coculture with RRV-primed DC in the presence of antibodies against TNFR1 and/or TNFR2 (A) or TNFα (B). Data are shown as % of cells and mean fluorescence intensities (MFI). (C and D) The concentration of individual cytokines/chemokines in the conditioned media from coculture experiments in which anti-TNFR1/2 antibodies were incubated with NK cells (C) or anti-TNFα antibodies with DC (D). n = 6 wells/condition with NK and DCs from pools of 3–4 livers. Values expressed as mean ± SD. *P < 0.05, ***P < 0.001, 2-tailed unpaired t test.
Requirement of TNFα-TNFR signaling for cholangiocyte lysis.
Using flow cytometry, we quantified the expression of TNFα, TNFR1, and TNFR2 in a murine cholangiocyte cell line (mCL) with and without exposure to RRV. At baseline, TNFα was expressed in 21% ± 4% of mCL and increased to 36% ± 4% (P < 0.003) at 24 hours of incubation with RRV at a multiplicity of infection of 100 (Figure 4A). In the same experimental approach, the surface expression of TNFR2 also increased from a baseline of 42% ± 1% to 60% ± 4% of cells (P < 0.003), while TNFR1 expression tended to decrease to 14% ± 1% from an initial detection of 21% ± 8% (P = 0.26). To examine whether this expression pattern is important for NK cell–induced lysis of cholangiocytes, we cultured hepatic NK cells from RRV-infected mice with mCL. As reported previously, RRV-primed but not -naive NK cells induced significant cholangiocyte lysis at 5 and 24 hours of culture (Figure 4B). In this assay, analysis of the supernatant revealed increased concentration of TNFα in wells containing RRV-primed NK cells at both time points (Figure 4C).
Figure 4. Expression of TNFα and TNFR1/2 in cholangiocytes and NK-mediated cytotoxicity.
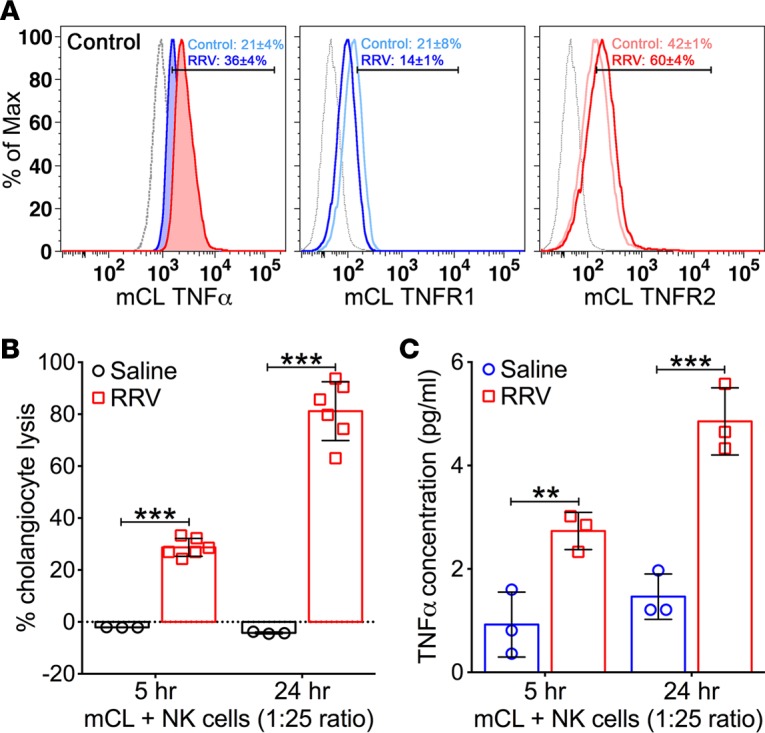
(A) Representative flow cytometry histograms show constitutive expression of TNFα, TNFR1, and TNFR2 by the cholangiocyte cell line (mCL) at baseline and after infection with RRV. Isotype stainings are shown as dotted gray lines; n = 6–8 replicates/staining. (B) LDH release assays show increased cholangiocyte lysis at 5 and 24 hours when mCL cells are cocultured with NK cells isolated from neonatal mice 7 days after injection of saline or RRV. n = 3–6 wells/time point. (C) Concentrations of TNFα in conditioned media from the cytotoxicity experiments with mCL and NK cells. Values are shown as mean ± SD. n = 3 wells/time point. **P < 0.01, ***P < 0.001, parametric 2-tailed t test with Welch’s correction.
To determine if cholangiocyte lysis was influenced by the expression of TNFα, we incubated naive and RRV-infected NK cells with anti-TNFα antibodies prior to coculture with cholangiocytes. After 5 hours, lysis of cholangiocytes was efficiently blocked by the antibodies at all target to effector cell ratios (Figure 5A). To precisely determine the contribution of the TNFα-TNFR axis, we modified the lysis assay to preincubate NK cells or cholangiocytes with antibodies to TNFR1 or TNFR2. The effect of blocking of TNFR1 on NK cells induced a statistically significant but modest decrease in cholangiocyte lysis, an effect that largely disappeared with increasing target to effector cell ratios. In contrast, blocking of both receptors completely suppressed cholangiocyte lysis, regardless of the cell ratio (Figure 5B). A greater impact for TNFR2 alone and the similar impact for blocking both TNFR1 and TNFR2 were shown when the antibodies were applied to cholangiocytes prior to coculture with RRV-primed NK cells (Figure 5C). Of note, inhibition of mCL lysis occurred despite the activation of NK cells, as evidenced by the high expression levels of Nkg2d or CD69 on NK cells at the conclusion of the coculture experiments (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.88747DS1). These data suggested that the expression of TNFα and its receptors regulate NK cell–mediated lysis of cholangiocytes in vitro, with greater contribution by TNFR2. They also raised the possibility that similar interactions may be important in vivo in the regulation of the tissue injury and clinical phenotype of experimental atresia.
Figure 5. Blocking antibodies to TNFα and TNFRs prevents cholangiocyte killing.
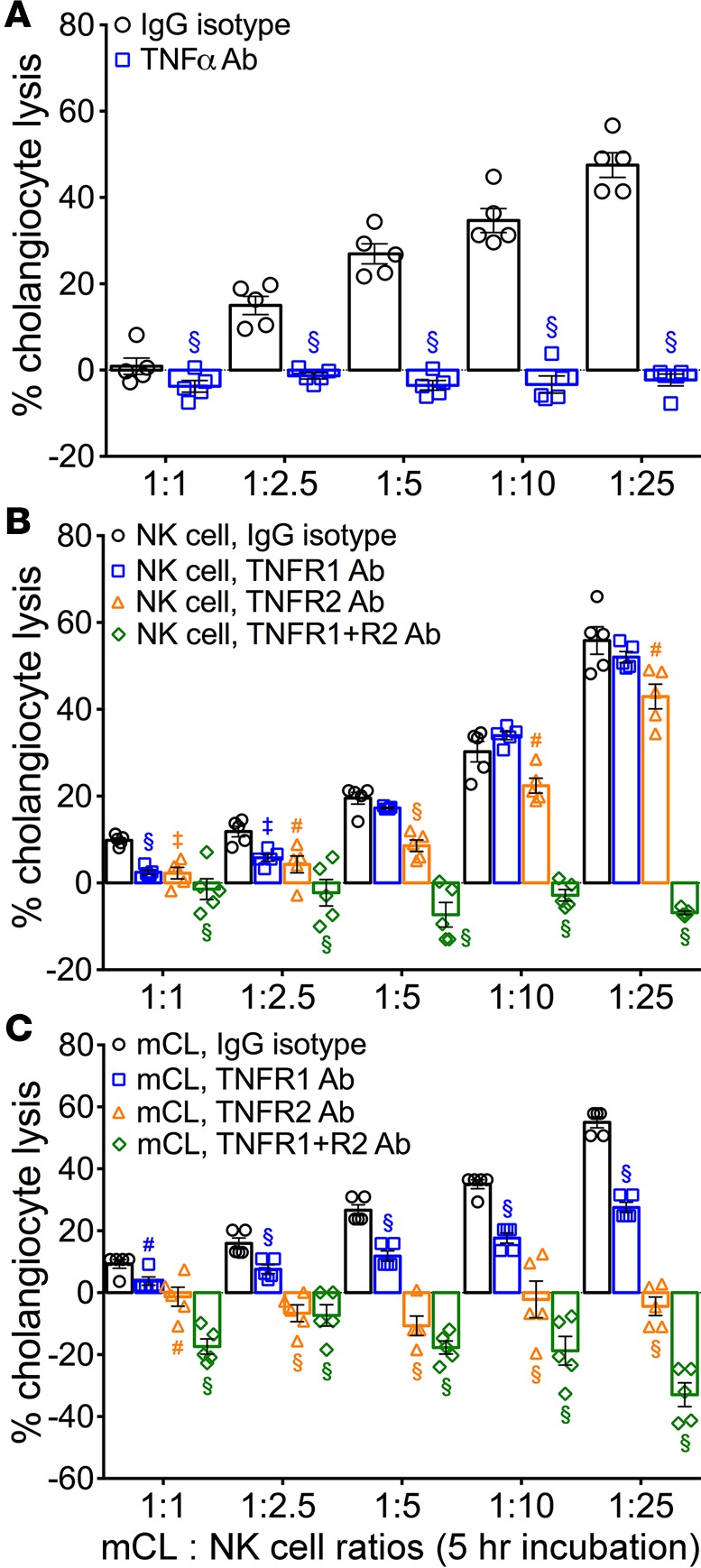
(A) LDH release by mCL cells after incubation with RRV-primed NK cells for 5 hours. Blocking antibodies to TNFα but not isotype lgG completely abolished cell lysis. B and C use the same experimental approach but use antibodies to TNFR1 and/or TNFR2. Results are representative of 2 independent experiments with NK cells obtained from a pool of 3–4 livers/sample. n = 4–6 wells/ratio/sample. Values depicted as mean ± SD, and levels of significance determined by 2-tailed parametric unpaired t test with Welch’s correction; #P < 0.05,‡P < 0.01, §P < 0.001.
Blocking of TNFα/TNFR prevents experimental biliary atresia.
To directly investigate the in vivo biological relevance of TNFα and TNFRs, we first examined whether interfering with TNFα function reduced the incidence of mucosal injury and biliary inflammation. Newborn mice were injected either with 0.9% saline or RRV within 24 hours of birth, and 1 day later were subjected to daily i.p. injections of 80 μg of rat anti-mouse TNFα antibodies (MP6-XT22) for 10 days; control mice received an equivalent amount of isotype rat IgG1 antibodies. Antibody injections were well tolerated, as evidenced by normal growth in saline-injected mice (receiving IgG isotype) beyond 2 weeks of age with normal liver and bile duct histology. RRV-infected control mice (which received isotype antibodies) developed persistent jaundice and showed 100% mortality by 18 days of life. In contrast, anti-TNFα antibodies prevented growth failure, recovered from jaundice, and improved long-term survival in more than 80% of mice (Figure 6A). Comprehensive histological analysis of livers showed improved liver histology (Figure 6B and Supplemental Figure 2), and extrahepatic bile ducts revealed an intact epithelium, minimal to no inflammation, and no lumen obstruction (Figure 6C), allowing mice to maintain bile flow (Table 1). Blocking TNFα significantly reduced the frequency of cells stained for activated caspase 3 in extrahepatic bile ducts (Figure 6D), the abundance of intrahepatic bile duct profiles (Figure 6E), and the tissue infiltration by effector CD4+/CD8+ T cells, DCs, and NK cells (Supplemental Figure 4A).
Figure 6. Anti-TNFα antibodies suppress the phenotype and bile duct injury.

(A) Daily treatment with anti-TNFα antibodies after RRV improved growth, recovery from jaundice, and survival of mice when compared with isotype and saline controls. n = 10–20 mice/group; ***P < 0.001 (lgG vs. TNFα Ab). (B and C) Representative H&E-stained sections of livers and extrahepatic bile ducts show suppression of liver inflammation and no bile duct obstruction in mice receiving anti-TNFα antibodies. Arrows denote intrahepatic bile ducts, and arrowheads show portal inflammation. (D) lmmunofluorescence staining for cytokeratin-19 (CK19, green) and activated caspase 3 (Casp3, red) shows dual-positive cells in bile ducts from lgG1- but not in TNFα Ab–treated mice. Arrowheads, CK19+Casp3+ cells; arrows, periductal Casp3 cells. (E) Representative immunostaining panels show decreased cholangiocyte profiles in livers of TNFα-blocked mice. Arrowheads, increased cholangiocyte profiles; arrows, normal ducts; asterisk, duct lumen; PV, portal vein. n = 4–6 mice/treatment group. Magnification of 400×; scale bar: 50 μm.
Table 1. Percentage of mice with lumen obstruction.

To investigate the relative contribution of TNFR1 and TNFR2 to TNFα-dependent signaling, we applied the same experimental approach to groups of mice that received 10 μg/mouse of blocking antibodies against TNFR1 (anti-CD120a) or TNFR2 (anti-CD120b). The improvement of cholestasis was more readily observed in RRV-infected mice receiving anti-TNFR2 antibodies (Figure 7A). More notably, the administration of anti-TNFR2 antibodies improved weight gain, prevented obstruction of extrahepatic bile ducts, and promoted long-term survival following RRV infection in a similar fashion (Figure 7A and Table 1). Examination of the livers and extrahepatic bile ducts (EHBDs) from mice surviving at day 31 showed normal parenchyma and portal tracts, and the extrahepatic bile duct had intact epithelium and a patent lumen (Figure 7B). Similar to findings seen following TNFα block in mice, treatment with anti-TNFR2 antibodies prevented activated caspase 3 staining (Figure 7C), the expansion of intrahepatic cholangiocyte profiles (Figure 7D), and decreased liver infiltration by T cells, DCs, and NK cells (Supplemental Figure 4B). To assess the effect of antibody blocking on downstream signals, we quantified the mRNA expression of Ask1 (for TNFR1) and Bmx (for TNFR2) and found both to be suppressed following the administration of respective antibodies (Supplemental Figure 5).
Figure 7. Improved duct injury, liver inflammation, and apoptosis after antibody blocking of TNFR1 or TNFR2.

(A) Daily treatment with antibodies to TNFR1 or TNFR2 after RRV improved growth, recovery from jaundice, and survival of mice. n = 10–20 mice/group; ***P < 0.001. (B) Representative H&E-stained sections of extrahepatic bile ducts and livers at day 31 after RRV infection show improvement in mice treated with antibodies to TNFR1 or TNFR2. Arrows denote normal intrahepatic bile ducts. (C) lmmunofluorescence staining for cytokeratin-19 (CK19, green) and activated caspase 3 (Casp31, red) shows dual-positive cells in bile ducts from TNFR1 but not TNFR2 Ab–treated mice. Arrowheads, CK19+Casp3+ cells; arrows, periductal Casp3 cells. (D) CK19 immunostaining in livers of TNFR1 and TNFR2 Ab–treated mice. Arrowheads, increased cholangiocyte profiles; block arrows, normal ducts. n = 4-6 mice/treatment group/day. PV, portal vein. Asterisks denote duct lumen. Magnification of 400×; scale bar: 50 μm.
Based on a previous report that the constitutional loss of TNFR1 in mice does not improve the experimental biliary atresia phenotype, we subjected Tnfr1 KO and Tnfr2 KO (Tnfr1–/–, Tnfr2–/–) mice to postnatal RRV infection and determined the clinical and tissue phenotypes in the first 3 weeks of life. Consistent with the previous report, Tnfr1–/– mice had poor growth, persistent jaundice, and poor long-term survival (Figure 8A), with obstruction of extrahepatic bile ducts that was indistinguishable from WT mice infected with RRV, while duct patency was preserved in saline-injected controls (Figure 8B). In contrast, analyses of Tnfr2–/– mice showed better growth, resolution of jaundice, and 50% survival, with mild cholangitis and patent extrahepatic bile ducts 14 days after RRV (Figure 8B). In the liver, portal inflammation was noticeably decreased in Tnfr2–/– mice, with preserved normal histology in saline-injected Tnfr1–/– and Tnfr2–/– mice (Supplemental Figure 3). Tnfr2–/– mice, but not Tnfr1–/–, prevented the accumulation of caspase 3–stained cells in bile ducts (Figure 8C), decreased bile duct profiles in portal tracts (Figure 8D), and reduced effector CD4+/CD8+ T cells, DCs, and NK cells (Supplemental Figure 4C). Altogether, these data showed that the constitutive loss of individual receptors enable KO mice to use TNFα-independent pathways to trigger tissue injury (albeit less efficiently in Tnfr2–/– mice), while the antibody blocking of TNFα or of its receptors in WT BALB/c mice more effectively block the development of key pathological features of experimental biliary atresia.
Figure 8. Genetic absence of TNFR2 but not TNFR1 suppresses the phenotype and bile duct injury.

(A) The clinical phenotypes of WT and Tnfr1–/– mice after RRV were similar, but Tnfr2–/– mice had resolution of jaundice over time and improved survival. n = 10–20 mice/group (WT vs. Tnfr1–/–), ***P < 0.001 (WT vs. Tnfr2–/–). (B) Representative H&E stain of longitudinal sections of extrahepatic bile ducts showing normal histology in saline-injected controls in both lines of KO mice, with obstructed duct lumen in Tnfr1–/– mice and patent duct in Tnfr2–/–mice 14 days after RRV. (C) Loss of Tnfr2 prevented caspase activation (Casp3, red) in epithelial (CK19, green) and submucosal cells. Arrowheads, CK19+Casp3+ cells; arrows, submucosal Casp3 cells. (D) Immunostaining for CK19 in livers from Tnfr1–/– and Tnfr2–/– mice. Arrowheads, increased cholangiocyte profiles; arrows, normal ducts. Asterisk denotes duct lumen. n = 4–6 mice/treatment group/day. Magnification of 400×; scale bar: 50 μm.
To identify potential molecules used by TNFR2 to regulate the tissue injury and obstructive phenotype, we utilized the analytical algorithm of the ToppGene Suite to select TNFR2 neighboring genes in a protein-protein interaction network. The network identified 29 proteins (Supplemental Figure 5A), of which 15 were encoded by genes with increased mRNA expression in extrahepatic bile duct using data from microarray-based genome-wide analysis at 3, 7, and 14 days after RRV infection (P < 0.05; Supplemental Figure 5B) (8). Among these genes, Stat1 and Irf1 had the highest fold changes in experimental mice above age-matched controls (Supplemental Figure 6) (20, 21). These data suggest a potential link between TNFR2 and Stat1/Irf1 circuits in controlling epithelial injury and duct obstruction.
Discussion
We found that integrity of the TNFα/TNFR2 circuit is necessary for the full phenotypic expression of experimental biliary atresia. Specifically, the disruption of individual elements of this circuit decreased the epithelial injury of extrahepatic bile ducts and largely prevented their obstruction, improved signs of cholestasis, and fostered long-term survival. Among the 3 molecules, antibody blocking of TNFα and TNFR2 had the greatest suppression of liver infiltration by T cells, DC, and NK cells; attenuated the lysis of cholangiocytes; and resulted in less tissue injury and better clinical improvement than the milder phenotype produced by blockade of TNFR1. Combining these findings with the increased expression of TNFR1 and TNFR2 in livers from infants at the time of diagnosis of biliary atresia strongly suggests that TNFα-mediated signaling plays key roles in pathogenesis of the disease.
The temporal requirement for the TNFα/TNFR2 signaling as a mechanism of epithelial injury in experimental biliary atresia occurs after NK cell activation. We reported previously that the activation of DCs is one of the initiating events triggered by RRV infection that activate NK cells to injure cholangiocytes (2). Consistent with this activation, we found an immediate increase in the expression of TNFα and both receptors in DCs within 3 days following RRV infection. However, blocking of TNFα and/or TNFR1/2 produced only a mild suppression of cytokine release, without interfering with the expression of activation markers by NK cells, suggesting that TNFα helps but is not required by DCs, which circumvent defective TNFα signals and are ultimately able to activate NK cells.
Following the activation of NK cells, TNFα signaling becomes more relevant in the injury of the duct epithelium. This is supported by the increased expression of TNFR2 in cholangiocytes and by the decrease in cholangiocyte apoptosis and lysis that results from blocking TNFα or TNFR2. In the liver, TNFα has been reported to promote tissue injury by the activation of proinflammatory circuits in several experimental models, including ischemia-reperfusion injury (22) and allograft rejection (23). The finding that it also triggers cell proliferation during liver regeneration points to different biological end-points depending on the type of injury or the preferential activation of individual receptors (24). Here, we showed that blocking TNFα/TNFR2 signals decrease the expansion of bile duct profiles in the liver, a hallmark feature of cholestatic injury. In NK cell–cholangiocyte lysis assays, cell death is more pronounced in the presence of TNFR2. One potential mechanism of cholangiocyte death is apoptosis, perhaps in synergy with IFNγ based on our previous studies showing that TNFα and IFNγ work together to promote cholangiocyte apoptosis in vitro (12). In agreement, blocking TNFR1 signals failed to reduce the epithelial injury seen in WT mice, which was significantly reduced by blocking of TNFα or TNFR2, promoting better long-term survival (above 80%). However, blocking of TNFR1 remains relevant to the full expression of the obstructive phenotype, as demonstrated by an improvement in long-term survival in about half of the mice well into adulthood. Collectively, these data suggest that effector mechanisms of experimental biliary atresia require an intact TNFα-TNFR2 signaling and identify the ligand and individual receptors (preferentially TNFR2) as potential molecular targets to decrease cell injury.
A previous report investigating the role of TNF-TNFR signaling in experimental atresia showed that mice genetically deficient in the receptor TNFR1 or treated with a TNFR1-Fc fusion protein Etanercept failed to lessen the severity of the disease (25). We found a similar lack of improvement in the obstructive phenotype in Tnfr1–/– mice; however, the findings may be explained by the different experimental approaches to inactivate TNF-TNFR1 signaling, with a notable difference in the delayed times of antibody administration used in the previous published study. In the current study, the early administration of anti-TNFR1 antibody improved symptoms and outcome in WT mice. Although we cannot discard the possibility that all or some of the beneficial effects of anti-TNFR1 antibody may result from nonspecific blocking of TNFR2, we favor a scenario in which the constitutive loss of TNFR1 enables the organism to use alternate molecular circuits to circumvent the inborn defect. In support of this scenario, the genetic loss of TNFR2 modestly improved the obstructive phenotype when compared with the more robust suppression of tissue injury and clinical outcome produced by anti-TNFR2 blocking antibodies.
The challenge in understanding the role of the immune system in pathogenesis of biliary atresia in humans is underscored by the lack of differences in the expression of TNFα, both in the serum and in the liver of children with biliary atresia. It is possible that its expression may have occurred at earlier phases of tissue injury, or that healthy infants may spontaneously have elevated levels of TNFα early in postnatal life as supported by the previous report of an increase in the first 40 days of life (26). In this context, our findings of increased expression of TNFR1 and TNFR2 mRNA in the livers, as well as increased immune-localization of TNFR2 to intrahepatic cholangiocytes at the time of diagnosis, point to a potential activation of this pathway at the tissue level, perhaps focusing the consequences of TNFα to the hepatobiliary system. A previous study reported that patients with lower bilirubin and AST levels requiring early transplantation after hepatoportoenterostomy correlated with TNFα levels, with an increase longitudinally from the time of diagnosis to 6 months after surgery (median value increase from 8–2,370 pg/ml) (27). Although these reports are interesting, we recognize the limited nature of these correlative studies and propose that a greater understanding of the strength of this association requires future studies that quantify the dynamic changes of TNFα or of its receptors over time and during different stages of the disease in affected children.
In summary, we found experimental evidence that TNFα and its receptor TNFR2 play a regulatory role in epithelial injury and bile duct obstruction in biliary atresia. Although the expression of the ligand and receptors is detected as early as the activation of DCs, the TNFα-TNFR2 signaling is more relevant to NK cell–mediated injury of the bile duct epithelium and amplification of the inflammatory response that ultimately obstructs the duct lumen and blocks bile flow. The combination of these experimental data with the increased expression of the TNFR1/2 in livers of affected children identify the ligand or individual receptors as potential therapeutic targets to suppress progression of liver disease. Whether these therapies apply to all or to a subgroup of patients requires a prospective assessment of the expression of the ligand (and/or its receptors) after corrective hepatoportoenterostomy, during progression of liver disease, and in the context of a clinical trial.
Methods
Supplemental Methods are available online with this article.
Mouse model of biliary atresia and blocking studies.
BALB/c mice were purchased from Charles River Laboratories and maintained in pathogen-free vivarium rooms equipped with a 12-hour dark-light cycle. Mice carrying the genetic inactivation of the Tnfr1 and Tnfr2 receptors on a C57BL/6 background were obtained from The Jackson Laboratory (stock numbers 003242 [Tnfr1] and 002620 [Tnfr2]) and sequentially back-crossed to BALB/c mice for >8 generations. Biliary atresia was induced in newborn BALB/c mice by i.p. inoculation of 1.5 × 106 ffu of RRV in the first 24 hours (9), with equivalent amount of 0.9% NaCl (saline) as control. For TNFα blocking studies, 80 μg of rat anti–mouse TNFα antibodies (clone MP6-XT2, Leinco Technologies Inc.) were injected daily after saline challenge or RRV challenge; control mice received equivalent rat IgG1 isotype antibodies. For TNFα receptor blocking, neonatal mice were challenged with RRV or saline solution followed by daily intraperitoneal injections of 10μg of either hamster anti-mouse CD120a (TNFR1) or CD120b (TNFR2) until day 7 of life; RRV and saline groups received an equivalent amount of Armenian hamster IgG antibodies. All groups of experimental mice were phenotyped daily as described previously (9). Mice were sacrificed at 3, 7, 14, or 31 days after saline or RRV injections with or without antibody treatment, and the gross appearance of individual organs was recorded. Livers were excised, and extrahepatic bile ducts and gallbladders were microdissected en bloc; the tissues were used either for mononuclear cell isolations or formalin fixation, or they frozen in liquid nitrogen per experimental design.
Gene and protein expression in human and murine tissues.
The liver gene expression for TNFA, TNFR1, and TNFR2 was extracted from genome-wide microarray experiments reported previously (8). In brief, RNA was isolated from liver biopsies at the time of diagnosis of biliary atresia or age-matched subjects with intrahepatic cholestasis (to serve as disease controls) and used to quantify mRNA expression with the GeneChip Human Gene 1.0 ST from Affymetrix; data are deposited in Gene Expression Omnibus (GEO) GSE46995 (8). For mice, the gene expression in extrahepatic bile ducts was quantified at different times points after RRV inoculation using specific primers and cycling conditions described in Supplemental Table 1, with normalization to endogenous Hprt as described previously (9). Cells undergoing apoptosis in extrahepatic bile ducts were detected by subjecting paraffin-embedded sections to sodium citrate antigen exposure, blocked with donkey serum and incubation with goat anti-cytokeratin 19 (clone M-17, Santa Cruz Biotechnology Inc.) and rabbit anti-activated caspase 3 (clone 9661, Cell Signaling Technology) antibodies. Specific signals were identified by fluorescent-conjugated Dylight-488/594 secondary antibodies. For protein quantification, the concentration of TNFα was measured using Luminex assays (Millipore) of serum specimens from 11 children with biliary atresia at the time of diagnosis (<4 months of age) and from 6 age-matched normal infants. To detect specific signals by immunostaining, formalin-fixed, paraffin-embedded sections of human wedge liver biopsies were subjected to citrate-based antigen retrieval, blocked with normal goat serum (Jackson ImmunoResearch) and incubated with anti-TNFR1 or TNFR2 antibodies followed by color development using DAB peroxidase substrate kit (Vector Laboratories).
Antibodies and reagents.
Neutralizing rat anti-mouse TNFα (clone MP6-XT22) and isotype rat IgG1 antibodies were obtained from BioLegend, while isotype Armenian hamster IgG antibodies were from BioXCell. Neutralizing anti-TNFR antibodies, hamster anti–mouse CD120a (TNFR1, clone 55R-170) and CD120b (TNFR2, clone TR75-54) were obtained from eBioscience. The following fluorescent antibodies were used to identify surface and intracellular signals by flow cytometry: PE-conjugated anti-TNFα (MP6-XT22), APC-conjugated anti-NK (CD49b; DX5), anti-CD8a (53-6.7), PerCP/Cy5.5-conjugated anti-CD3 (17A2), anti-CD11b (M1/70), anti-CD44 (IM7), anti-Nkp46 (29A1.4), and AF488-conjugated anti-CD11c (N418) (all from eBioscience); APC/Cy7-conjugated anti-CD45R/B220 (RA3-6B2), anti-CD69 (H1.2F3), anti-CD62L (MEL-14), PE-conjugated anti-CD120a (TNFR1; 55R-286), and anti-CD120b (TNFR2; TR75-89) (all from BioLegend); and PE-conjugated anti-NKG2D antibody (CD314; 191004) (from R&D systems). FITC-conjugated anti-pan cytokeratin (C-11; ab7753), unconjugated anti-CK19 (ab15463), anti-human TNFR1 (ab58436), and TNFR2 (ab117543) antibodies were from Abcam, and Dylight-488 and -594 conjugated secondary antibodies were obtained from Jackson ImmunoResearch. For Fc block, purified anti-FcγRII/III (CD16/CD32; 2.4G2) antibodies were obtained from BioXCell. Rabbit anti–activated caspase 3 (Asp175) (5A1E) mAb was from Cell Signaling Technology.
Liver cell isolation and flow cytometry.
To generate cell suspensions, livers from neonatal mice were perfused in situ through the portal vein with sterile PBS followed by dissociation using gentleMACS Tissue Dissociator (Miltenyi Biotec) according to the manufacturer’s instructions, followed by isolation of single-cell populations as previously described (2). For cellular phenotyping using surface markers, 1 × 105 to 1 × 106 cells were blocked with purified anti-FcRII/III (CD32/CD16) antibodies and then incubated with combinations of fluorochrome-conjugated antibodies in 200 μl PBS + 4% BSA at 4ºC for 45 minutes. Intracellular staining for cytokines was performed by first culturing freshly isolated hepatic mononuclear cell suspensions in complete media for 5 hours in the presence of 1 μl/1 × 106 cells of Brefeldin A solution (GolgiPlug; BD Biosciences) according to the manufacturer’s instructions, followed by labeling with fluorochrome-conjugated anti-cytokine antibodies using BD Cytofix/Cytoperm kit (BD Biosciences) for 45 minutes at 4ºC. Stained cells were analyzed using a FACSCanto III (BD Biosciences) flow cytometer collecting at least 100,000 events. Data were analyzed with FlowJo software version 9.7.6 (Tree Star Inc.) with forward/side scatter gating (to exclude cell debris) and secondary gatings relative to isotype controls to account for background fluorescence.
Cholangiocyte cytotoxicity assay.
Isolation of hepatic NK and coculture with cholangiocytes were done as described previously (3). Briefly, NK cells from livers of day 7–old saline and RRV challenged mice were purified using MACS columns and mouse CD49b (DX5) positive selection microbeads (Miltenyi Biotec). Enriched NK cells were washed twice with complete media (DMEM supplemented with 10% FCS, 1% L-glutamine and 0.5% antibiotics consisting of Penicillin-Streptomycin) and cocultured in increasing ratios with mCL for 5 hours. Cholangiocyte lysis determined by CytoTox 96 NonRadioactive Cytotoxicity Assay (Promega), which colorimetrically measures the release of lactate dehydrogenase (LDH). For experiments investigating the role of TNFα, anti-TNFα antibodies (MP6-XT22; 100 μg/ml) were added to the coculture medium. To determine the role of TNFR1 and/or TNFR2, NK cells or mCL were incubated with either 25 μg/ml of anti-TNFR1/R2 antibodies or the respective isotype antibodies for 1 hour at 4°C before using the cells in cytolytic assays. The percentage of cytotoxicity was calculated as 100 × ([experimental release] – [spontaneous release])/([maximal release] – [spontaneous release]). Spontaneous or maximal release was determined in presence of media alone or 1% Triton X-100 (Sigma-Aldrich), respectively. All assays were performed in a minimum of 6 wells/condition/time point.
DC-NK cocultures, protein quantification, and cell activation.
Cocultures of DCs and NK cells were performed as described previously (2). DCs and/or NK cells were preincubated with isotype or blocking antibodies to TNFα (MP6-XT22; 80 μg/ml) or TNFR1/TNFR2 (55R-170/TR75-54; 10 μg/ml) at 4˚C and washed twice with complete media before the cocultures; supernatants were collected at 24 hours after incubation to determine protein levels of TNFα, IL6, IL17A, CXCL9, CXCL10, CXCL1 (KC,) and CCL5 (RANTES) by Milliplex Assays (Millipore) using Luminex technology on the Bio-Plex (Bio-Rad) instrument. Cells were collected, washed with PBS + 4% BSA, labeled with fluorochrome-conjugated antibodies detecting NK cells, Nkg2d, and CD69 and analyzed on a FACSCanto III flow cytometer (BD Biosciences). All assays were done in a minimum of triplicate wells.
Statistics.
The numbers of mice or tissues used in each experiment are presented in the appropriate text or figure legends. In vitro experiments were performed at a minimum in triplicates for each assay/condition. Values are expressed as mean ± SD, and statistical significance was determined by 2-tailed Student’s unpaired t test with Welch’s correction (for growth charts) and Wilcoxon matched-pairs signed rank test (for symptoms of cholestasis) with a significance of P < 0.05. Survival curves were created by Kaplan-Meier analysis (Log-rank [Mantel-Cox] test) using the GraphPad Prism version 5.00 Software (GraphPad Software). Details of the statistical analyses for growth profiles, symptoms of cholestasis, and Kaplan-Meier survival charts are depicted in Supplemental Table 2.
Study approval.
Animal studies and experimental protocols were performed in strict accordance of the guidelines recommended by The Institutional Animal Care and Use Committee (IACUC; Protocol IACUC2013-0151) of Cincinnati Children’s Hospital Medical Center.
Author contributions
PS and JAB designed the research work. PS, TM, LY, and SG performed the animal experiments. PS, RM, and JAB analyzed the data. PS and JAB wrote and edited the manuscript. ZL performed bioinformatics analysis. All contributing authors assisted in revising the manuscript.
Supplementary Material
Acknowledgments
Supported by the NIH grants DK-64008 and DK-83781 to JAB, American Liver Foundation Liver Scholar Award to PS, and by the Integrative Morphology and the Gene and Protein Expression Cores of the Digestive Health Center (DK-78392). The studies were also supported by the support of the Junior Co-Operative Society of Cincinnati Children’s Hospital Medical Center. Serum and liver tissues were originally obtained as an ancillary study of the Childhood Liver Disease Research Network (DK-62497).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:JCI Insight. 2017;2(5):e88747. https://doi.org/10.1172/jci.insight.88747.
Contributor Information
Pranavkumar Shivakumar, Email: pranav.shivakumar@cchmc.org.
Tatsuki Mizuochi, Email: mizuochi_tatsuki@kurume-u.ac.jp.
Reena Mourya, Email: reena.mourya@cchmc.org.
Sridevi Gutta, Email: sridevi.gutta@cchmc.org.
Zhenhua Luo, Email: Zhenhua.Luo@cchmc.org.
References
- 1.Bessho K, Bezerra JA. Biliary atresia: will blocking inflammation tame the disease? Annu Rev Med. 2011;62:171–185. doi: 10.1146/annurev-med-042909-093734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saxena V, Shivakumar P, Sabla G, Mourya R, Chougnet C, Bezerra JA. Dendritic cells regulate natural killer cell activation and epithelial injury in experimental biliary atresia. Sci Transl Med. 2011;3(102):102ra94. doi: 10.1126/scitranslmed.3002069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shivakumar P, Sabla GE, Whitington P, Chougnet CA, Bezerra JA. Neonatal NK cells target the mouse duct epithelium via Nkg2d and drive tissue-specific injury in experimental biliary atresia. J Clin Invest. 2009;119(8):2281–2290. doi: 10.1172/JCI38879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shivakumar P, et al. Effector role of neonatal hepatic CD8+ lymphocytes in epithelial injury and autoimmunity in experimental biliary atresia. Gastroenterology. 2007;133(1):268–277. doi: 10.1053/j.gastro.2007.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mack CL, Tucker RM, Sokol RJ, Kotzin BL. Armed CD4+ Th1 effector cells and activated macrophages participate in bile duct injury in murine biliary atresia. Clin Immunol. 2005;115(2):200–209. doi: 10.1016/j.clim.2005.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qiu Y, et al. HMGB1-promoted and TLR2/4-dependent NK cell maturation and activation take part in rotavirus-induced murine biliary atresia. PLoS Pathog. 2014;10(3):e1004011. doi: 10.1371/journal.ppat.1004011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mavila N, et al. Expansion of prominin-1-expressing cells in association with fibrosis of biliary atresia. Hepatology. 2014;60(3):941–953. doi: 10.1002/hep.27203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bessho K, et al. Gene expression signature for biliary atresia and a role for interleukin-8 in pathogenesis of experimental disease. Hepatology. 2014;60(1):211–223. doi: 10.1002/hep.27045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shivakumar P, et al. Obstruction of extrahepatic bile ducts by lymphocytes is regulated by IFN-gamma in experimental biliary atresia. J Clin Invest. 2004;114(3):322–329. doi: 10.1172/JCI21153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shivakumar P, Mourya R, Bezerra JA. Perforin and granzymes work in synergy to mediate cholangiocyte injury in experimental biliary atresia. J Hepatol. 2014;60(2):370–376. doi: 10.1016/j.jhep.2013.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klemann C, et al. Interleukin 17, Produced by γδ T Cells, Contributes to Hepatic Inflammation in a Mouse Model of Biliary Atresia and Is Increased in Livers of Patients. Gastroenterology. 2016;150(1):229–241.e5. doi: 10.1053/j.gastro.2015.09.008. [DOI] [PubMed] [Google Scholar]
- 12.Erickson N, Mohanty SK, Shivakumar P, Sabla G, Chakraborty R, Bezerra JA. Temporal-spatial activation of apoptosis and epithelial injury in murine experimental biliary atresia. Hepatology. 2008;47(5):1567–1577. doi: 10.1002/hep.22229. [DOI] [PubMed] [Google Scholar]
- 13.Pfeffer K. Biological functions of tumor necrosis factor cytokines and their receptors. Cytokine Growth Factor Rev. 2003;14(3-4):185–191. doi: 10.1016/S1359-6101(03)00022-4. [DOI] [PubMed] [Google Scholar]
- 14.Holtmann MH, Schütz M, Galle PR, Neurath MF. Functional relevance of soluble TNF-alpha, transmembrane TNF-alpha and TNF-signal transduction in gastrointestinal diseases with special reference to inflammatory bowel diseases. Z Gastroenterol. 2002;40(8):587–600. doi: 10.1055/s-2002-33418. [DOI] [PubMed] [Google Scholar]
- 15.Juhász K, Buzás K, Duda E. Importance of reverse signaling of the TNF superfamily in immune regulation. Expert Rev Clin Immunol. 2013;9(4):335–348. doi: 10.1586/eci.13.14. [DOI] [PubMed] [Google Scholar]
- 16.Fausto N. Liver regeneration. J Hepatol. 2000;32(1 Suppl):19–31. doi: 10.1016/s0168-8278(00)80412-2. [DOI] [PubMed] [Google Scholar]
- 17.Zimmerer JM, et al. Cytotoxic effector function of CD4-independent, CD8(+) T cells is mediated by TNF-α/TNFR. Transplantation. 2012;94(11):1103–1110. doi: 10.1097/TP.0b013e318270f3c0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu H, Li H, Cao D, Wu Y, Chen Y. Tumor necrosis factor α (TNF-α) receptor-I is required for TNF-α-mediated fulminant virus hepatitis caused by murine hepatitis virus strain-3 infection. Immunol Lett. 2014;158(1-2):25–32. doi: 10.1016/j.imlet.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tarrats N, Moles A, Morales A, García-Ruiz C, Fernández-Checa JC, Marí M. Critical role of tumor necrosis factor receptor 1, but not 2, in hepatic stellate cell proliferation, extracellular matrix remodeling, and liver fibrogenesis. Hepatology. 2011;54(1):319–327. doi: 10.1002/hep.24388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Al-Lamki RS, et al. TNF receptors differentially signal and are differentially expressed and regulated in the human heart. Am J Transplant. 2009;9(12):2679–2696. doi: 10.1111/j.1600-6143.2009.02831.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Al-Lamki RS, et al. TNFR1- and TNFR2-mediated signaling pathways in human kidney are cell type-specific and differentially contribute to renal injury. FASEB J. 2005;19(12):1637–1645. doi: 10.1096/fj.05-3841com. [DOI] [PubMed] [Google Scholar]
- 22.Perry BC, Soltys D, Toledo AH, Toledo-Pereyra LH. Tumor necrosis factor-α in liver ischemia/reperfusion injury. J Invest Surg. 2011;24(4):178–188. doi: 10.3109/08941939.2011.568594. [DOI] [PubMed] [Google Scholar]
- 23.Adams DH, Afford SC. Effector mechanisms of nonsuppurative destructive cholangitis in graft-versus-host disease and allograft rejection. Semin Liver Dis. 2005;25(3):281–297. doi: 10.1055/s-2005-916320. [DOI] [PubMed] [Google Scholar]
- 24.Fujiyoshi M, Ozaki M. Molecular mechanisms of liver regeneration and protection for treatment of liver dysfunction and diseases. J Hepatobiliary Pancreat Sci. 2011;18(1):13–22. doi: 10.1007/s00534-010-0304-2. [DOI] [PubMed] [Google Scholar]
- 25.Tucker RM, Hendrickson RJ, Mukaida N, Gill RG, Mack CL. Progressive biliary destruction is independent of a functional tumor necrosis factor-alpha pathway in a rhesus rotavirus-induced murine model of biliary atresia. Viral Immunol. 2007;20(1):34–43. doi: 10.1089/vim.2006.0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sarandakou A, et al. Inflammatory cytokines in newborn infants. Mediators Inflamm. 1998;7(5):309–312. doi: 10.1080/09629359890811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Narayanaswamy B, Gonde C, Tredger JM, Hussain M, Vergani D, Davenport M. Serial circulating markers of inflammation in biliary atresia--evolution of the post-operative inflammatory process. Hepatology. 2007;46(1):180–187. doi: 10.1002/hep.21701. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.