

Abstract
This paper describes a procedure for transferring colloidal CdS and CdSe quantum dots (QDs) from organic solvents to water by exchanging their native hydrophobic ligands for phosphonopropionic acid (PPA) ligands, which bind to the QD surface through the phosphonate group. This method, which uses dimethylformamide as an intermediate transfer solvent, was developed in order to produce high-quality water soluble QDs with neither a sulfur-containing ligand nor a polymer encapsulation layer, both of which have disadvantages in applications of QDs to photocatalysis and biological imaging. CdS (CdSe) QDs were transferred to water with a 43% (48%) yield using PPA. The photoluminescence (PL) quantum yield for PPA-capped CdSe QDs is larger than that for QDs capped with the analogous sulfur-containing ligand, mercaptopropionic acid (MPA), by a factor of four at pH 7, and by up to a factor of 100 under basic conditions. The MPA ligands within MPA-capped QDs oxidize at Eox ~ +1.7 V vs. SCE, whereas cyclic voltammograms of PPA-capped QDs show no discerible oxidation peaks at applied potentials up to +2.5 V vs. SCE. The PPA-capped QDs are chemically and colloidally stable for at least five days in the dark, even in the presence of O2, and are stable when continuously illuminated for five days, when oxygen is excluded and a sacrificial reductant is present to capture photogenerated holes.
The unique properties of metal chalcogenide quantum dots (QDs) – namely, their spectrally narrow emission bands with with energies from the long-UV to the near-infrared, chemically tunable surfaces, large surface-area-to-volume ratios (~109 m-1), and high absorption cross-sections and photostability – make them promising as fluorescent bioimaging probes,1-8 as photosensitizers for redox catalysts, and as solo photocatalysts.9 Bioimaging and several types of catalysis, such as water splitting10-12 and the photosensitization of enzymes13-15, require the QDs to be transferred from organic solvents, in which they are typically prepared, to water via a ligand exchange. Several effective methods of phase transfer have been devised1, 16-21, but the most common have serious drawbacks for photocatalysis and imaging. The most popular method for water-solubilization of metal-chalcogenide QDs is the exchange of the native ligands for ligands with a hydrophilic tail and thiolate anchoring group, such as mercaptopropionic (or - undecanoic) acid9, 15, glutathione22, thiotic acid18, 23 and lipoic acid20, 24, or cysteine.25 For CdSe QDs, thiolate ligands in certain binding geometries introduce mid-gap localized trap states and accompanying picosecond-timescale decay pathways for the exciton, and thereby reduce the yield of both photoluminescence (PL), important for imaging, and extraction of holes, important for catalysis.8, 26-30 Growth of a shell of higher bandgap semiconductor in between the cadmium chalcogenide core and the ligand increases the brightness of the QD dramatically, and, in some cases, reduces the rate of hole trapping, but also increases the size of the QD, which sometimes reduces its access to intracellular compartments or makes it a less benign probe of biomolecular motion, and certainly inhibits hole extraction from QD photocatalysts and sensitizers. Additionally, the low oxidation potential of the thiolate group – +1.2 V vs. SCE for unbound mercaptopropionate and ~+1.7 V vs. SCE for Cd2+-bound mercaptopropionate acid, as shown below, for example – makes it susceptible to oxidation either by the photoexcited QD core or by dissolved O2. Disulfides have a relatively weak binding affinity for the metal ions on the QD surface than do thiolates,31 so this oxidation renders QDs coated in thiolates susceptible to aggregation and precipitation over time, and the disulfide ligands vulnerable to displacement by thiol-disulfide exchange in thiol-rich biological environments.32, 33 Finally, thiolate ligands can dramatically inhibit the catalytic activity of QD-sensitized metal-containing co-catalysts by binding to the metal.15, 34
An alternative method for water-solubilization of QDs is their encapsulation with polyethylene glycols16, 17, lipid micelles3, or amphiphilic polymers.35-37 Such an encapsulation scheme is very effective in stabilizing the QD and does not, in most cases, affect the exciton lifetime. The nanometers-thick ligand shell is problematic however because, in bio-imaging applications, it limits the rate of uptake of the QDs into cells, and, in catalytic applications, it slows diffusion of molecular substrates to and from the QD surface. Silica encapsulation has also been used for phase transfer, but results in the same inaccessibility of the QD core as does the polymer encapsulation.5, 38, 39 Optimization of colloidal QDs for two of their most promising applications – bioimaging and catalysis – therefore requires that these procedures be complemented by convenient, high-yielding procedures for organic-to-aqueous phase transfer through functionalization of the QD surface with sulfur-free, short-chain ligands.
We report here the phase transfer of CdSe and CdS QDs from organic solvent to water using phosphonopropionic acid (PPA) as a short-chain, sulfur-free water-solubilizing ligand. We utilize the high binding affinity of the phosphonate group of PPA to displace the native oleate ligands of CdS and CdSe QDs,40 and thereby transfer the QDs from hexanes to water with 43±9% (CdS) and 48±12% (CdSe) yield. The PPA-capped CdSe QDs have PL quantum yields (QYs) in water that are generally higher than QDs capped with the analogous thiol-containing ligand, mercaptopropionic acid (MPA): for example, at pH 11, the PL QY of the PPA-capped CdSe QDs was 1.6%, a factor of ~100 larger than that of the MPA-capped CdSe QDs. At pH values between 6 and 12, PPA-capped CdS and CdSe QDs are colloidally and chemically stable over five days when stored in water in the dark, even in the presence of oxygen. When stored in room light, exclusion of O2 and addition of a sacrificial reductant prevent corrosion of the QD surface by photogenerated holes. Importantly, the surface ligands of MPA-capped QDs oxidized at Eox ~ +1.7 V vs. SCE (Ip = -60 μA in acetonitrile), while the cyclic voltammagrams of PPA-capped QDs showed only small (capacitive and impurity) currents in the range of potentials scanned, up to +2.5 V vs. SCE.
RESULTS AND DISCUSSION
We synthesized oleate-capped CdS QDs using a method reported by Yu et al.41, 42 and oleate-capped CdSe QDs using a synthesis reported by Cao et al.43 Purification of the QDs was performed by alternate precipitation by acetone or methanol and resuspension in hexanes, see the Supporting Information for details. Both types of QDs are capped exclusively with oleate after purification. The CdS QDs we describe here have a first excitonic absorption at 410 nm (which corresponds to a radius of 1.9 nm) and our CdSe QDs have a first excitonic absorption at 533 nm (which corresponds to a radius of 1.4 nm).44
Figure 1A shows the five-step phase transfer of CdS or CdSe QDs from hexanes to water using PPA. We used 1H NMR to monitor the changes in the surface ligand composition throughout the procedure for the CdS QDs. Figure 1B shows the NMR spectrum of oleate capped CdS QDs after purification in benzene-d6, which we substitute for hexanes for NMR analysis because the signals of bound and free oleates are easily distinguished in this solvent. This spectrum contains a broad signal from bound oleate at 5.67 ppm and a sharp multiplet corresponding to free oleic acid/cadmium oleate at 5.49 ppm.45 The width of the bound oleate signal is due to the slow diffusion and the restricted rotational motion of molecules bound to nanoparticles.34, 40 Integration of the peak at 5.67 ppm, relative to a peak corresponding to the 18 protons of hexamethylcyclotrisiloxane at 0.13 ppm, shows that the average number of oleate ligands initially bound is 248 ± 11 per QD.
Figure 1.
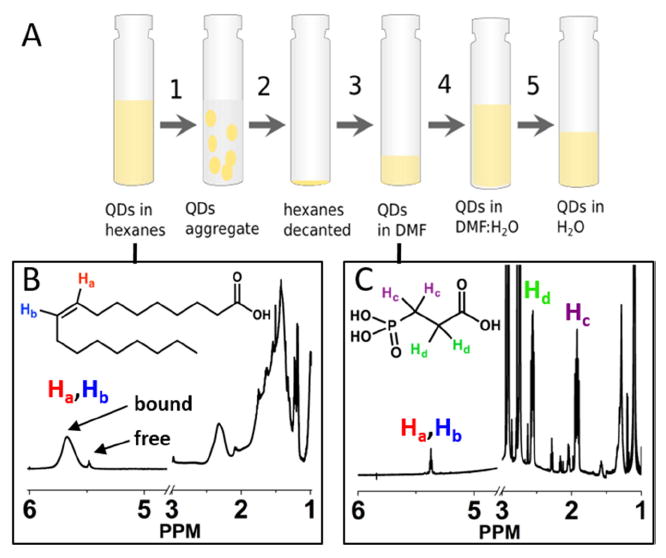
A) Scheme of the organic-to-aqueous ligand exchange procedure, in which the yellow shading represents the portion of the sample that contains the QDs: 1) add PPA to oleate-capped QDs in hexanes, 2) centrifuge and decant the supernatant, 3) redisperse QDs in DMF, 4) add 10 mM KOH solution in H2O, 5) wash with CHCl3 to remove DMF and evaporate the remaining chloroform with N2. B) 1H NMR spectrum of CdS QDs in C6D6 before addition of PPA. The spectrum contains a peak corresponding to the vinyl protons (“Ha” and “Hb”) of bound oleate at 5.67 ppm and a smaller peak corresponding to those protons on freely diffusing oleate at 5.49 ppm. All of the peaks between 1 ppm and 3 ppm correspond to the alkyl protons in oleate. C) 1H NMR spectrum of the PPA-capped QDs in DMF-d7. The two multiplets at 1.92 ppm (“Hc”) and 2.56 ppm (“Hd”) correspond to protons of free PPA. The spectrum also contains a small feature corresponding to residual free oleate (5.42 ppm, 15.7 ± 0.5 oleates per QD) and a small broad feature, not visible on this scale, corresponding to residual bound oleate (5.55 ppm, 2.5 ± 0.4 oleates per QD). The Supporting Information contains an NMR spectrum of the supernatant obtained after step 2.
In order to transfer oleate-capped QDs from hexanes to water, we first added 500 equivalents of PPA per QD as a 0.1 M solution in isopropanol to 3 mL of a 5 μM suspension of QDs in hexanes (Step 1 in Figure 1A). A few seconds after the addition of PPA, the QDs flocculated, due to displacement of oleate from the surface by the more polar PPA. Centrifugation of the suspension (Step 2) at 3500 rpm for 5 minutes left the hexanes layer completely colorless. We decanted the hexanes and resuspended the QDs (Step 3) in 2 mL N,N-dimethylformamide (DMF), or DMF-d7 for NMR analysis. We have found that direct addition of water to the QD pellet does not redisperse them, but addition of an intermediate solvent – DMF – enables their recovery. Formamide, ethylene glycol and propylene carbonate are also effective intermediate solvents; however, we used DMF because, unlike the other solvents mentioned, it could eventually be removed from an aqueous mixture by its extraction into chloroform (vide infra). Figure 1C shows the NMR spectrum of the PPA-capped CdS QDs in DMF-d7, where we observe a small amount of freely diffusing oleic acid (at 5.42 ppm) that was displaced by PPA, and freely diffusing PPA (at 2.56 ppm and 1.92 ppm) that did not adsorb to the QDs. We observe no signal from bound PPA, which is consistent with a previous report of highly broadened signals from protons on carbons alpha to the phosphonate group within phosphonate-bound ligands on CdSe QDs.40 Subtraction of the amount of free oleate present in the initial hexanes/benzene-d6 solution of QDs from the concentration of free oleate present after exchange steps 2 and 3 (in both the supernatant and the QD solution in DMF) yields 246±41 displaced oleate ligands per QD (see the Supporting Information, Figure S1). Subtraction of the total number of free PPA in the DMF-d7 and benzene-d6 phases from the amount of PPA added yields 269±31 PPA ligands bound per QD. We therefore conclude that each PPA ligand displaces one oleate ligand during the transfer to DMF, and that the exchange proceeds quantitatively with respect to displacement of oleate (within measurement error). Once transferred to water, both 31P and 1H NMR spectra show that PPA is in fast exchange (relative to the NMR timescale) on and off the QD surface, so quantification of its surface coverage is not straightforward. Addition of excess PPA to aqueous dispersions of PPA-capped QDs shifts the signals from PPA nuclei toward their chemical shifts in freely diffusing PPA; extraction of some excess PPA (by an alumina sorbent) from the dispersion shifts those signals in the opposite direction, see the Supporting Information.
To transfer the QDs from DMF into water, we diluted the suspension of PPA-capped QDs in DMF with 4 mL of water containing 10 mM KOH (a molar ratio KOH:PPA of 5:1) that had been sparged with nitrogen for 5 minutes (Step 4). The KOH serves to neutralize the PPA and maintains the slightly basic pH necessary to avoid aggregation of QDs at this point in the procedure. We observed that removing oxygen from the KOH solution improved the clarity of the QD suspension obtained in this step because, we suspect, oxygen competes with PPA for binding at Cd2+ sites. Once the QDs had been diluted in water, exposure to oxygen did not induce any degradation. We discuss the stability of the aqueous dispersions of PPA-capped QDs in various environments in detail below.
Finally, we removed DMF from the water/DMF mixture by washing it with chloroform (Step 5). After three washes with 40 mL chloroform each, we reduced the concentration of DMF to single-digits mM (< 5 mM, a molar ratio DMF:QDs of ~1000:1) concentration, as measured by NMR (see the Supporting Information, Figure S2). Washing resulted in a slightly cloudy solution, the clarity of which was restored by brief bubbling with nitrogen to evaporate the remaining chloroform. The average yield of the phase transfer from hexanes to water was 43±9% for CdS QDs (for n=7 samples measured) and 48±12% for CdSe QDs (n=7), measured using absorption spectroscopy and the previously determined extinction coefficients for CdS and CdSe QDs.44 The vast majority of the loss of QDs occurs during the transfer from DMF to water, and specifically during the washing steps to eliminate residual DMF, upon which we observe visible precipitation of the QDs and cannot resuspend them. Transmission electron micrograph images of the CdSe QDs show that their average diameter decreased from 3.9 nm to 3.8 nm, and their size dispersity increased from ±10.6% to ±25.2%, on going from hexanes to water, see the Supporting Information, Figure S3.
We have not explicitly proven that PPA binds through the phosphonate group, but we have three pieces of circumstantial evidence for this binding mode. First, we are not able to transfer the QDs to water using an excess of dicarboxylate, but, as discussed above, we are able to quantitatively displace a carboxylate ligand (oleate) from the QD surface and transfer the QDs to water without a large excess of PPA (only 500 PPA per QD, approximately two PPA ligands per binding site). This result relies on the binding group of PPA being of much higher affinity for the QD surface than a carboxylate, so we conclude it is the phosphonate. Second, it has been shown experimentally40 that the binding affinity of phosphonates for the surfaces of CdSe QDs greatly exceeds that of carboxylates, and that, while phosphonates will effectively displace carboxylates from the QD, the reverse process requires elevated temperatures (our ligand exchange procedures are carried out at room temperature). Finally, we require basic water to accomplish the phase transfer, probably to ensure that the population of groups interfacing with the solvent (the carboxylates) are as deprotonated, statistically speaking, as possible. If the phosphonate were interfacing with the solvent, we would be able to accomplish the phase transfer at lower pH because the phosphonate becomes negatively charged at lower pH (the pKa of the first acidic proton of the phosphonate is ~1), and the carboxylate would adsorb through proton transfer with the oleate.
Figure 2 shows absorbance and PL spectra of the CdS and CdSe QDs (and photographs of CdSe QDs) capped with oleate (in hexanes), capped with PPA (in water), and capped with MPA, the thiol analogue of PPA (in water). The optical bandgaps of PPA-capped QDs are slightly smaller (~30 meV smaller for CdS and ~38 meV for CdSe) than those of oleate- and MPA-capped QDs, as reflected in their absorption and PL spectra. The origin of this spectral shift and broadening is the formation of a population of larger QDs during the exchange through Ostwald ripening, as can be seen in the TEM images, Figure S1.
Figure 2.
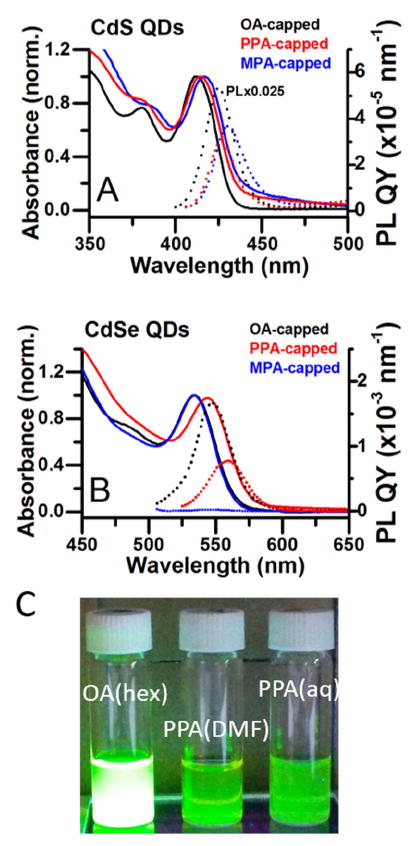
A) Normalized absorption spectra (solid lines, left axis) and PL spectra (dotted lines, right axis) of oleate-capped CdS QDs in hexanes (black), PPA-capped CdS QDs in water (red), and MPA-capped QDs in water (blue). The small scattering baseline in the spectrum of the PPA-capped CdS QDs is due to the formation of small QD aggregates during the phase transfer. The PL spectrum of oleate-capped CdS QDs has been multiplied by 0.025. The pH of the aqueous CdS samples is 8. B) Analogous spectra for CdSe QDs. The pH of the aqueous CdSe samples is 11. All PL spectra have been scaled by the respective absorbances of the sample at the excitation wavelength (350 nm for CdS and 500 nm for CdSe), and by the integrated fluorescence intensity of a standard (anthracene for CdS or Rhodamine B for CdSe), to convert the y-axis to quantum yield in nm-1. C) Photographs of oleate-capped CdSe QDs in hexanes, PPA-capped CdSe QDs in DMF, and PPA-capped CdSe QDs in water, under illumination with a UV lamp.
Table 1 summarizes the PL QYs from the band-edge exciton (the exciton where both the electron and hole are delocalized in the core) of oleate-, MPA-, and PPA-capped QDs. As synthesized, the QY of emission from the band-edge exciton of the oleate-capped CdSe QDs (the sharp emission peak centered at 550 nm - 560 nm, depending on the capping group) was 10.1% in hexanes, measured using rhodamine B as a fluorescence standard, which is comparable to previously reported values for unshelled CdSe QDs in non-polar solvent.46-48 The best single band-edge QY observed for PPA-capped CdSe QDs in water was 2.9% at pH 11, with an average of 1.6%±1.0 % (n=6) at this pH. The average band-edge QY of PPA-capped CdSe QDs at pH 7 was 0.60%±0.34% (n=6). The pH-dependence of the QY is also related to the protonation equilibrium of the phosphonate group, and is discussed elsewhere.49 Although transfer of CdSe QDs to water using PPA was accompanied by a significant decrease in PL, aqueous MPA-capped CdSe QDs had PL QYs of only 0.017% ± 0.001% (n= 3) at pH 11 and 0.15% ± 0.01% (n= 3) at pH 7, Table 1, presumably due to efficient hole transfer from the QD valence band to the adsorbed thiolate.8, 26-30
Table 1.
Photoluminescence Quantum Yields of OA-, MPA-, and PPA-Capped CdS and CdSe QDs.a
| OA-capped (hexanes) | MPA-capped (aq, pH 7) | PPA-capped (aq, pH 7) | MPA-capped (aq, pH 11) | PPA-capped (aq, pH 11) | |
|---|---|---|---|---|---|
| CdS QDs | 8.1% | 0.09±0.02% | 0.024±0.016% | 0.011±0.001% | 0.058±0.047% |
| CdSe QDs | 10.1% | 0.15±0.01% | 0.60±0.34% | 0.017±0.001% | 1.6±1.0% |
Relative QYs were determined by comparison with a anthracene standard (CdS) or Rhodamine B standard (CdSe), and, for the aqueous samples, were measured within one day of phase transfer.
The band-edge QY of oleate-capped CdS in hexanes was 8.7%, measured using anthracene as a fluorescence standard. PPA-capped QDs were not, on average, brighter than MPA-capped QDs in water. The best single observed band-edge QY for the PPA-capped CdS QDs was 0.11% at pH 9.6, with an average of 0.058%±0.047% (n=6) at this pH. The average QY of PPA-capped CdS QDs was 0.024%±0.016% (n=4) at pH 7. The band-edge QY of the MPA-capped CdS QDs was 0.011% ± 0.001% at pH 11 and 0.09% ± 0.02% at pH 7, Table 1. The emission from trapped excitons, which produces a broad peak in the PL spectra of CdS QDs centered at 570 nm – 600 nm is similar in intensity for PPA- and MPA-capped QDs in water (see the Supporting Information for details on QY calculations).
We suspect, but have not proven, that the decrease in PL QY upon transferring the QDs to water using PPA is primarily due to rearrangement of the QD surfaces resulting in undercoordinated surface atoms and carrier trapping,50 and, in some cases, the formation of small aggregates of QDs as reflected in a slight scattering baseline most obvious in Figure 2A, during the ligand exchange. Importantly, however, the PL QYs of aqueous PPA-capped CdSe QDs are, on average, a factor of 100 higher than those of aqueous MPA-capped CdSe QDs under basic conditions, and a factor of four higher at neutral pH. We believe that CdSe QDs benefit from PPA capping more than do CdS QDs because trapping on thiolate groups is a known non-radiative exciton decay mechanism for CdSe QDs,51 and the PPA eliminates this pathway, whereas thiolates can both form and eliminate trap states on CdS QDs, and so their effects on PL, aside from those attributable to surface reconstruction, are typically small.52
Oxidative Stability of the PPA-Capped QDs
To evaluate the relative stabilities of the QDs capped with PPA and MPA to oxidation, we performed cyclic voltammetry (CV) on films of PPA-capped CdS QDs and MPA-capped QDs (Figure 3), and compared them to CVs of films of Cd-PPA and Cd-MPA molecular complexes, prepared as described in the Supporting Information. We prepared films by drop-casting 30 μL of a 200 μM aqueous suspension of Cd-PPA or Cd-MPA molecular complexes, or 30 μL of a 1-μM aqueous dispersion of QDs, onto a glassy carbon electrode. None of the oxidative reactions (with QDs or molecular complexes) are reversible, probably due to desorption of oxidized species from the electrode surface.
Figure 3.
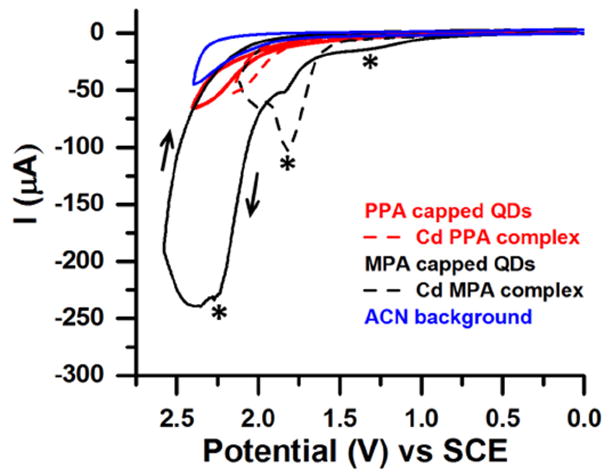
CVs of films of Cd-bound PPA (dashed red), Cd-bound MPA (dashed black), PPA-capped CdS QDs (solid red), and MPA-capped CdS QDs (solid black). The solvent background is in blue. All measurements were done with 0.1 M NBu4PF6 as the supporting electrolyte, at a scan rate of 0.1 V/s, with a glassy carbon working electrode, a Pt wire counter electrode and Ag wire reference electrode. CVs were shifted to the SCE scale using Fc/Fc+ (= 0.342 V vs. SCE) as a standard, see the Supporting Information. The black arrows show the scan direction used for all samples.
The CVs of both the PPA-capped CdS QDs and the Cd-PPA complex showed no discernible oxidative peaks and only small current increases above the background upon applying up to +2.5 V. Given that the reported oxidation potential of QD- or Cd-bound phosphonates is +2.8 V vs. SCE,31 we believe that this current, which has an onset of ~+1.7 – +2.0 V, arises from capacitance at the electrode surface or impurities, and not from oxidation of the ligand. In contrast, we measured an anodic peak potential of +1.7 V vs. SCE (with a current of -60 μA) for MPA-capped QDs, which matches well with the position of the oxidation peak of the Cd-MPA complex, and can therefore be assigned to the oxidation of the ligand headgroup. The CV of MPA-capped QDs also has a peak at +1.2 V vs. SCE, which we attribute to oxidation of free MPA that was deposited along with the QDs on the electrode (see the Supporting Information, Figure S4), and a broad peak or set of peaks at ~+2.2 V vs. SCE. We suspect that the current at +2.2 V and higher is due to oxidation of MPA ligands in a second binding geometry (we have shown previously that thiolates bind in several geometries to cadmium chalcogenide QDs)53,54 that then facilitates oxidation of sulfur on the QD surface. Other sulfur compounds, in particular disulfides, may also contribute to the larger current of the MPA-capped QD sample at the highest applied voltages.55
Colloidal and Chemical Stability of the PPA-Capped QDs
We tested the chemical and colloidal stability of the aqueous dispersions of PPA-capped CdS and CdSe QDs by monitoring their absorption and PL spectra for five days, stored in the dark or in room light, under Ar or in air, and in the prescence or absence of triethanolamine (TEOA) as sacrificial reductant. We focused on two characteristics of the absorption spectrum, the intensity and the linewidth (plotted as full-width-at-half-max, FWHM) of the first excitonic peak, because they are sensitive to both etching of the QD surfaces and aggregation of the QDs, whereas changes in the energy of the absorption peak are not always straightforwardly related to these degradation processes because etching results in a hypsochromic shift of optical spectra while aggregation results in a bathochromic shift. When stored in the dark, with or without exposure to O2, the surfaces of both CdS and CdSe QDs were chemically very stable; after 5 days, the absorbance and FWHM of the first excitonic peak had not changed, although the PL intensity had decreased by 10% for CdS and 40% for CdSe (see the Supporting Information, Figure S5). One can therefore store the QDs in the aqueous dispersion for long periods of time without degradation (etching, oxidation) or precipiation.
If we are to use the PPA-capped QDs for photocatalysis and biological imaging, however, the stability under continuous illumination is also important. Figure 4 summarizes our results for samples stored under room light. When stored in room light and in air for five days, we observed a decrease in absorbance by 75% for CdS and 50% for CdSe of the starting value (A0), Figure 4A,B, and concomitant broadening of the first excitonic peak (increase in FWHM), Figure 4C,D, which is characteristic of etching of the QD surface. Storing the QDs under Ar almost completely suppresses the photocorrosion process in PPA-capped CdS QDs; over five days there is only a 20% decrease in absorbance and no broadening. Storing the CdSe QDs under Ar also significantly slowed the etching process for those QDs. Since the samples do not etch in the dark, even in the presence of O2, we conclude that light is the primary cause of the etching, but it is facilitated by exposure to O2. We postulate that the etching is caused by the oxidation of the chalcogenide component of the QDs by photogenerated holes and subsequent formation of SO42- in the case of CdS56 and and Se0 and SeO32- in the the case of CdSe57, as reported previously. The de-passivation of chalcogenide surface sites during the phase transfer probably makes these sites vulnerable to oxidation (and leads to the observed decreases in PL QY, Table 1), as we did not observe any changes in the optical properties of oleate-capped CdS QDs stored in room light in air (see the Supporting Information, Figure S9).
Figure 4.
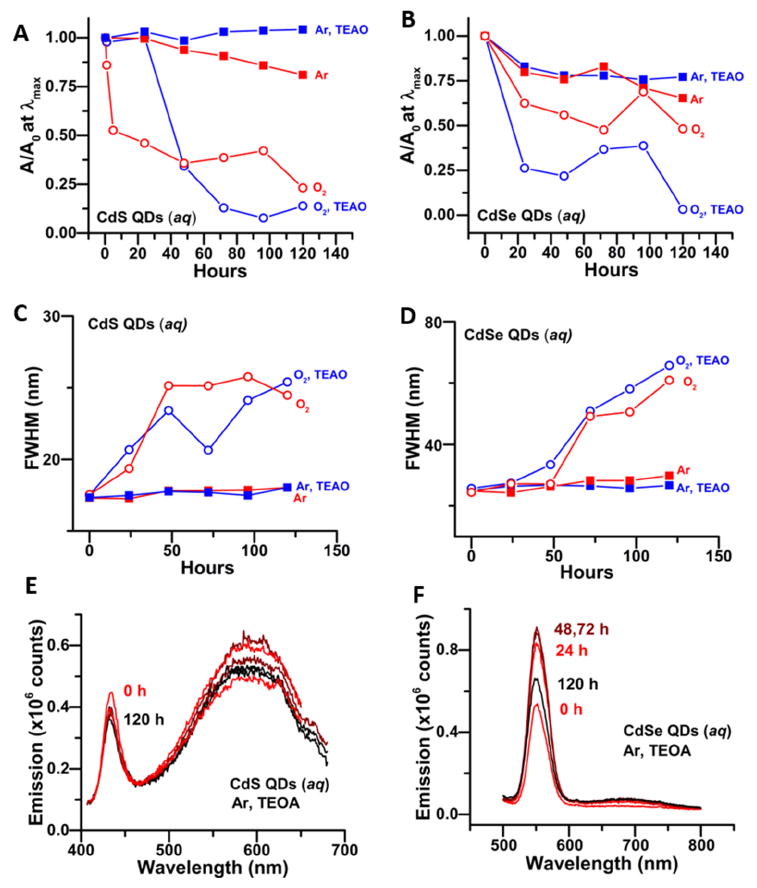
A, B) Absorbance at the energy of the first excitonic peak as a function of time over 5 days, scaled by its value at time zero (A0), for PPA-capped CdS (A) and CdSe (B) QDs. The QDs were stored in room light under Ar or air, and in the prescence or absence of 50 mM TEAO as sacrificial reductant, as labeled. C,D) FWHM of the first excitonic peak in the absorbance spectra of the same samples of PPA-capped CdS (C) and CdSe (D) QDs under the same set of conditions as in A,B. E,F) PL spectra, acquired over 5 days, of the same samples of PPA-capped CdS (E, excited at 350 nm) and CdSe (F, excited at 450 nm) stored under Ar with 50 mM TEOA. The quantities in A-D were obtained by fitting the absorbance spectrum with a set of Gaussian functions, see the Supporting Information, Figure S10-S17. The PL spectra are scaled by their absorbances at the excitation wavelength, relative to that at 0 h.
To inhibit the etching process further, as complete exlusion of O2 is difficult without freeze-pump-thawing of samples, we added 100,000 equivalents of a sacrificial reductant, triethanolamine (TEOA) to the PPA-capped QDs upon their transfer to water. For samples stored in air, the prescence of TEOA delayed the etching of CdS QDs for 24 h (Figure 4A,C), but did not improve the long-term stability of the QDs. The absorbance of the CdSe QDs stored in air with TEAO decreased quickly due to some precipitation of QDs but, again, the TEAO did not inhibit etching over five days (Figure 4B,D). The stability of the CdS QDs stored under Ar was however improved further by addition of TEOA: we observed no decrease in absorbance, no broadening of the absorbance peak, and only a ~15% decrease in the band-edge emission intensity (after scaling for any changes in absorbance), Figure 4E, in these samples after five days of illumination by room light. The stability of the PPA-capped CdSe QDs stored under Ar also benefited from the addition of the hole scavenger: we observed only a 20% decrease in absorbance, no peak broadening, and an increase in the PL intensity of the sample (Figure 4F) after storage in room light for five days. Increased PL intensity concomitant with mild photo-etching of QD surfaces, a process called photobrightening, is well-documented.58, 59 The addition of TEAO therefore helps to stabilize the QDs by abstracting the photogenerated hole before it reacts with surface chalcogenide. The Supporting Information (Figures S5 – S9) shows plots of (i) absorbance spectra of the PPA-capped QD samples stored in the dark or light, and in Ar or air over five days, (ii) change in PL intensity over 5 days for CdS and CdSe QDs stored in the light with TEOA, (iii) and absorbance and PL spectra of oleate-capped CdS QDs stored in air and light for five days.
CONCLUSIONS
We have introduced a method to use phosphonopropionic acid for transfer of CdS and CdSe QDs from non-polar organic solvent into water. This method is important for application of QDs for both biological imaging and photocatalysis applications because the coating of the water-soluble QDs is thin (a 3-carbon chain) and does not contain a thiolate group. Water soluble QDs capped with PPA have greater oxidative stability than their thiolate-capped analogs because the ligands themselves do not oxidize at applied potentials up to +2.5 V vs. SCE. PPA-capped CdSe QDs have higher photoluminescence quantum yields than their thiolate-capped analogs because thiolates act as traps for excitonic holes in CdSe QDs, but fluorescence imaging applications of these QDs will probably still require the growth of a high-bandgap semiconductor shell between the QD core and the PPA layer. The PPA-capped QDs remain colloidally and chemically stable in aqueous dispersion for at least five days when stored in the dark. When stored in room light, one can prevent photoetching and precipitation, and retain the original intensity and linewidth of the QDs’ optical spectra, by minimizing the samples’ exposure to oxygen and by adding a sacrificial reductant (we used triethanolamine) to the dispersion.
Supplementary Material
Acknowledgments
Research primarily supported as part of the Argonne-Northwestern Solar Energy Research (ANSER) Center, an Energy Frontier Research Center funded by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences (BES), under Award # DE-SC0001059 (ligand exchange optimization, electrochemistry, and stability testing), by the Northwestern Materials Research Science and Engineering Center (MRSEC), funded by the National Science Foundation (NSF) under Award # DMR-1121262 (ligand exchange development). This work made use of the IMSERC at Northwestern University, which has received support from the NIH (1S10OD012016-01/1S10RR019071-01A1), the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF NNCI-1542205), the State of Illinois, and the International Institute for Nanotechnology (IIN).
Footnotes
Supporting Information. Description of CdS and CdSe QD synthesis, 1H NMR spectra of the supernatant from the exchange in C6D6 and of the QDs in water, TEM images and histogram of sizes of oleate-capped and PPA-capped QDs, and additional stability data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interests.
References
- 1.Sukhanova A, Devy J, Venteo L, Kaplan H, Artemyev M, Oleinikov V, Klinov D, Pluot M, Cohen JHM, Nabiev I. Biocompatible Fluorescent Nanocrystals for Immunolabeling of Membrane Proteins and Cells. Anal Biochem. 2004;324(1):60–67. doi: 10.1016/j.ab.2003.09.031. [DOI] [PubMed] [Google Scholar]
- 2.Rosenthal SJ, Tomlinson I, Adkins EM, Schroeter S, Adams S, Swafford L, McBride J, Wang Y, DeFelice LJ, Blakely RD. Targeting Cell Surface Receptors with Ligand-conjugated Nanocrystals. J Am Chem Soc. 2002;124(17):4586–4594. doi: 10.1021/ja003486s. [DOI] [PubMed] [Google Scholar]
- 3.Dubertret B, Skourides P, Norris DJ, Noireaux V, Brivanlou AH, Libchaber A. In vivo Imaging of Quantum Dots Encapsulated in Phospholipid Micelles. Science (New York, NY) 2002;298(5599):1759–1762. doi: 10.1126/science.1077194. [DOI] [PubMed] [Google Scholar]
- 4.Chan WCW, Nie S. Quantum Dot Bioconjugates for Ultrasensitive Nonisotopic Detection. Science. 1998;281(5385):2016–2018. doi: 10.1126/science.281.5385.2016. [DOI] [PubMed] [Google Scholar]
- 5.Bruchez M, Moronne M, Gin P, Weiss S, Alivisatos AP. Semiconductor Nanocrystals as Fluorescent Biological Labels. Science (New York, NY) 1998;281(5385):2013–2016. doi: 10.1126/science.281.5385.2013. [DOI] [PubMed] [Google Scholar]
- 6.Biju V, Itoh T, Anas A, Sujith A, Ishikawa M. Semiconductor Quantum Dots and Metal Nanoparticles: Syntheses, Optical Properties, and Biological Applications. Anal Bioanal Chem. 2008;391(7):2469–2495. doi: 10.1007/s00216-008-2185-7. [DOI] [PubMed] [Google Scholar]
- 7.Akerman ME, Chan WCW, Laakkonen P, Bhatia SN, Ruoslahti E. Nanocrystal Targeting In vivo. Proc Natl Acad Sci USA. 2002;99(20):12617–12621. doi: 10.1073/pnas.152463399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu WW, Chang E, Drezek R, Colvin VL. Water-soluble Quantum Dots for Biomedical Applications. Biochem Biophys Res Commun. 2006;348(3):781–786. doi: 10.1016/j.bbrc.2006.07.160. [DOI] [PubMed] [Google Scholar]
- 9.Jensen SC, Homan SB, Weiss EA. Photocatalytic Conversion of Nitrobenzene to Aniline through Sequential Proton-Coupled One-Electron Transfers from a Cadmium Sulfide Quantum Dot. J Am Chem Soc. 2016;(5):1591–1600. doi: 10.1021/jacs.5b11353. [DOI] [PubMed] [Google Scholar]
- 10.Chen XB, Shen SH, Guo LJ, Mao SS. Semiconductor-based Photocatalytic Hydrogen Generation. Chem Rev. 2010;110(11):6503–6570. doi: 10.1021/cr1001645. [DOI] [PubMed] [Google Scholar]
- 11.Yehezkeli O, de Oliveira DRB, Cha JN. Electrostatically Assembled CdS-Co3O4 Nanostructures for Photo-assisted Water Oxidation and Photocatalytic Reduction of Dye Molecules. Small. 2015;11(6):668–674. doi: 10.1002/smll.201401490. [DOI] [PubMed] [Google Scholar]
- 12.Harris LA, Wilson RH. Semiconductors for Photoelectrolysis. Annu Rev Mater Sci. 1978;8:99–134. [Google Scholar]
- 13.Greene BL, Joseph CA, Maroney MJ, Dyer RB. Direct Evidence of Active-Site Reduction and Photodriven Catalysis in Sensitized Hydrogenase Assemblies. J Am Chem Soc. 2012;134(27):11108–11111. doi: 10.1021/ja3042367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown KA, Dayal S, Ai X, Rumbles G, King PW. Controlled Assembly of Hydrogenase-CdTe Nanocrystal Hybrids for Solar Hydrogen Production. J Am Chem Soc. 2010;132(28):9672–9680. doi: 10.1021/ja101031r. [DOI] [PubMed] [Google Scholar]
- 15.Brown KA, Wilker MB, Boehm M, Dukovic G, King PW. Characterization of Photochemical Processes for H2 Production by CdS Nanorod-[FeFe] Hydrogenase Complexes. J Am Chem Soc. 2012;134(12):5627–5636. doi: 10.1021/ja2116348. [DOI] [PubMed] [Google Scholar]
- 16.Larson DR, Zipfel WR, Williams RM, Clark SW, Bruchez MP, Wise FW, Webb WW. Water-soluble Quantum Dots for Multiphoton Fluorescence Imaging In vivo. Science (New York, NY) 2003;300(5624):1434–1436. doi: 10.1126/science.1083780. [DOI] [PubMed] [Google Scholar]
- 17.Mei BC, Susumu K, Medintz IL, Mattoussi H. Polyethylene Glycol-based Bidentate Ligands to Enhance Quantum Dot and Gold Nanoparticle Stability in Biological Media. Nat Protocols. 2009;4:412–423. doi: 10.1038/nprot.2008.243. [DOI] [PubMed] [Google Scholar]
- 18.Uyeda HT, Medintz IL, Mattoussi H. Synthesis of Surface Ligands to Prepare Hydrophilic and Biologically Compatible Quantum Dots. Proc Soc Photo-Opt Ins. 2005;5705:131–138. [Google Scholar]
- 19.Susumu K, Uyeda HT, Medintz IL, Mattoussi H. Design of Biotin-functionalized Luminescent Quantum Dots. J Biomed Biotechnol. 2007;2007:90651/1–7. doi: 10.1155/2007/90651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhan NQ, Palui G, Safi M, Ji X, Mattoussi H. Multidentate Zwitterionic Ligands Provide Compact and Highly Biocompatible Quantum Dots. J Am Chem Soc. 2013;135(37):13786–13795. doi: 10.1021/ja405010v. [DOI] [PubMed] [Google Scholar]
- 21.Gill R, Zayats M, Willner I. Semiconductor Quantum Dots for Bioanalysis. Angew Chem Intl Ed. 2008;47(40):7602–7625. doi: 10.1002/anie.200800169. [DOI] [PubMed] [Google Scholar]
- 22.Zhao CZ, Bai ZL, Liu XY, Zhang YJ, Zou BS, Zhong HZ. Small GSH-Capped CuInS2 Quantum Dots: MPA-Assisted Aqueous Phase Transfer and Bioimaging Applications. ACS Appl Mater Interf. 2015;7(32):17623–17629. doi: 10.1021/acsami.5b05503. [DOI] [PubMed] [Google Scholar]
- 23.Uyeda HT, Medintz IL, Jaiswal JK, Simon SM, Mattoussi H. Synthesis of Compact Multidentate Ligands to Prepare Stable Hydrophilic Quantum Dot Fluorophores. J Am Chem Soc. 2005;127(11):3870–3878. doi: 10.1021/ja044031w. [DOI] [PubMed] [Google Scholar]
- 24.Zhan NQ, Palui G, Mattoussi H. Preparation of Compact Biocompatible Quantum Dots using Multicoordinating Molecular-scale Ligands Based on a Zwitterionic Hydrophilic Motif and Lipoic Acid Anchors. Nat Protocols. 2015;10(6):859–874. doi: 10.1038/nprot.2015.050. [DOI] [PubMed] [Google Scholar]
- 25.Liu WH, Choi HS, Zimmer JP, Tanaka E, Frangioni JV, Bawendi M. Compact Cysteine-coated CdSe(ZnCdS) Quantum Dots for In vivo Applications. J Am Chem Soc. 2007;129(47):14530–14531. doi: 10.1021/ja073790m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bullen C, Mulvaney P. The Effects of Chemisorption on the Luminescence of CdSe Quantum Dots. Langmuir. 2006;22(7):3007–3013. doi: 10.1021/la051898e. [DOI] [PubMed] [Google Scholar]
- 27.Kalyuzhny G, Murray RW. Ligand Effects on Optical Properties of CdSe Nanocrystals. J Phys Chem B. 2005;109(15):7012–7021. doi: 10.1021/jp045352x. [DOI] [PubMed] [Google Scholar]
- 28.Peterson MD, Cass LC, Harris RD, Edme K, Sung K, Weiss EA. The Role of Ligands in Determining the Exciton Relaxation Dynamics in Semiconductor Quantum Dots. Annu Rev Phys Chem. 2014;65:317–339. doi: 10.1146/annurev-physchem-040513-103649. [DOI] [PubMed] [Google Scholar]
- 29.Hines DA, Kamat PV. Recent Advances in Quantum Dot Surface Chemistry. ACS Appl Mater Interf. 2014;6(5):3041–3057. doi: 10.1021/am405196u. [DOI] [PubMed] [Google Scholar]
- 30.Baker DR, Kamat PV. Tuning the Emission of CdSe Quantum Dots by Controlled Trap Enhancement. Langmuir. 2010;26(13):11272–11276. doi: 10.1021/la100580g. [DOI] [PubMed] [Google Scholar]
- 31.Gronbeck H, Curioni A, Andreoni W. Thiols and Disulfides on the Au(111) Surface: The Headgroup-gold Interaction. J Am Chem Soc. 2000;122(16):3839–3842. [Google Scholar]
- 32.Zhu H, Hu MZ, Shao L, Yu K, Dabestani R, Zaman MB, Liao S. Synthesis and Optical Properties of Thiol Functionalized CdSe/ZnS (Core/Shell) Quantum Dots by Ligand Exchange. J Nanomater. 2014;2014:324972/1–14. [Google Scholar]
- 33.Aldana J, Wang YA, Peng XG. Photochemical Instability of CdSe Nanocrystals Coated by Hydrophilic Thiols. J Am Chem Soc. 2001;123(36):8844–8850. doi: 10.1021/ja016424q. [DOI] [PubMed] [Google Scholar]
- 34.Das A, Han ZJ, Haghighi MG, Eisenberg R. Photogeneration of Hydrogen from Water using CdSe Nanocrystals Demonstrating the Importance of Surface Exchange. Proc Natl Acad Sci USA. 2013;110(42):16716–16723. doi: 10.1073/pnas.1316755110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang CW, Moffitt MG. Surface-tunable Photoluminescence from Block Copolymer-stabilized Cadmium Sulfide Quantum Dots. Langmuir. 2004;20(26):11784–11796. doi: 10.1021/la048390g. [DOI] [PubMed] [Google Scholar]
- 36.Pellegrino T, Manna L, Kudera S, Liedl T, Koktysh D, Rogach AL, Keller S, Radler J, Natile G, Parak WJ. Hydrophobic Nanocrystals Coated with an Amphiphilic Polymer Shell: A General Route to Water Soluble Nanocrystals. Nano Lett. 2004;4(4):703–707. [Google Scholar]
- 37.Lin CAJ, Sperling RA, Li JK, Yang TY, Li PY, Zanella M, Chang WH, Parak WGJ. Design of an Amphiphilic Polymer for Nanoparticle Coating and Functionalization. Small. 2008;4(3):334–341. doi: 10.1002/smll.200700654. [DOI] [PubMed] [Google Scholar]
- 38.Gerion D, Pinaud F, Williams SC, Parak WJ, Zanchet D, Weiss S, Alivisatos AP. Synthesis and Properties of Biocompatible Water-soluble Silica-coated CdSe/ZnS Semiconductor Quantum Dots. J Phys Chem B. 2001;105(37):8861–8871. [Google Scholar]
- 39.Nann T, Mulvaney P. Single Quantum Dots in Spherical Silica Particles. Angew Chem Intl Ed. 2004;43(40):5393–5396. doi: 10.1002/anie.200460752. [DOI] [PubMed] [Google Scholar]
- 40.Gomes R, Hassinen A, Szczygiel A, Zhao QA, Vantomme A, Martins JC, Hens Z. Binding of Phosphonic Acids to CdSe Quantum Dots: A Solution NMR Study. J Phys Chem Lett. 2011;2(3):145–152. [Google Scholar]
- 41.Li Z, Ji YJ, Xie RG, Grisham SY, Peng XG. Correlation of CdS Nanocrystal Formation with Elemental Sulfur Activation and Its Implication in Synthetic Development. J Am Chem Soc. 2011;133(43):17248–17256. doi: 10.1021/ja204538f. [DOI] [PubMed] [Google Scholar]
- 42.Yu WW, Peng XG. Formation of High-quality CdS and Other II-VI Semiconductor Nanocrystals in Noncoordinating Solvents: Tunable Reactivity of Monomers. Angew Chem Intl Ed. 2002;41(13):2368–2371. doi: 10.1002/1521-3773(20020703)41:13<2368::AID-ANIE2368>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 43.Chen O, Chen X, Yang YA, Lynch J, Wu HM, Zhuang JQ, Cao YC. Synthesis of Metal-Selenide Nanocrystals Using Selenium Dioxide as the Selenium Precursor. Angew Chem Intl Ed. 2008;47(45):8638–8641. doi: 10.1002/anie.200804266. [DOI] [PubMed] [Google Scholar]
- 44.Yu WW, Qu L, Guo W, Peng X. Experimental Determination of the Extinction Coefficient of CdTe , CdSe , and CdS Nanocrystals. Chem Mater. 2003;125(17):2854–2860. [Google Scholar]
- 45.Cass LC, Malicki M, Weiss EA. The Chemical Environments of Oleate Species within Samples of Oleate-Coated PbS Quantum Dots. Anal Chem. 2013;85(14):6974–6979. doi: 10.1021/ac401623a. [DOI] [PubMed] [Google Scholar]
- 46.Nan WN, Niu YA, Qin HY, Cui F, Yang Y, Lai RC, Lin WZ, Peng XG. Crystal Structure Control of Zinc-Blende CdSe/CdS Core/Shell Nanocrystals: Synthesis and Structure-Dependent Optical Properties. J Am Chem Soc. 2012;134(48):19685–19693. doi: 10.1021/ja306651x. [DOI] [PubMed] [Google Scholar]
- 47.Dabbousi BO, RodriguezViejo J, Mikulec FV, Heine JR, Mattoussi H, Ober R, Jensen KF, Bawendi MG. (CdSe)ZnS Core-shell Quantum Dots: Synthesis and Characterization of a Size Series of Highly Luminescent Nanocrystallites. J Phys Chem B. 1997;101(46):9463–9475. [Google Scholar]
- 48.Peng XG, Schlamp MC, Kadavanich AV, Alivisatos AP. Epitaxial Growth of Highly Luminescent CdSe/CdS Core/Shell Nanocrystals with Photostability and Electronic Accessibility. J Am Chem Soc. 1997;119(30):7019–7029. [Google Scholar]
- 49.Thompson CM, Kodaimati M, Westmoreland DE, Weiss EA. Electrostatic Control of Excitonic Energies and Dynamics in a CdS Quantum Dot through Reversible Protonation of its Ligands. doi: 10.1021/acs.jpclett.6b01899. submitted. [DOI] [PubMed] [Google Scholar]
- 50.Klimov VI, McBranch DW, Leatherdale CA, Bawendi MG. Electron and Hole Relaxation Pathways in Semiconductor Quantum Dots. Phys Rev B. 1999;60(19):13740–13749. [Google Scholar]
- 51.Konstantatos G. Colloidal Quantum Dot Optoelectronics and Photovoltaics. Cambridge University Press; New York: 2013. p. 314. [Google Scholar]
- 52.Bel Haj Mohamed N, Haouari M, Zaaboub Z, Nafoutti M, Hassen F, Maaref H, Ben Ouada H. Time Resolved and Temperature Dependence of the Radiative Properties of Thiol-capped CdS Nanoparticles Films. J Nanopart Res. 2014;16(2):1–17. doi: 10.1007/s11051-013-2242-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Amin VA, Aruda KO, Lau B, Rasmussen AM, Edme K, Weiss EA. Dependence of the Band Gap of CdSe Quantum Dots on the Surface Coverage and Binding Mode of an Exciton-Delocalizing Ligand, Methylthiophenolate. J Phys Chem C. 2015;119(33):19423–19429. [Google Scholar]
- 54.Aruda KO, Amin VA, Thompson CM, Lau B, Nepomnyashchii AB, Weiss EA. Description of the Adsorption and Exciton Delocalizing Properties of p-Substituted Thiophenols on CdSe Quantum Dots. Langmuir. 2016;32(14):3354–3364. doi: 10.1021/acs.langmuir.6b00689. [DOI] [PubMed] [Google Scholar]
- 55.Terashima C, Rao TN, Sarada BV, Kubota Y, Fujishima A. Direct Electrochemical Oxidation of Disulfides at Anodically Pretreated Boron-doped Diamond Electrodes. Anal Chem. 2003;75(7):1564–1572. doi: 10.1021/ac020583q. [DOI] [PubMed] [Google Scholar]
- 56.van Dijken A, Vanmaekelbergh D, Meijerink A. Size Selective Photoetching of Nanocrystalline CdS Particles. Chem Phys Lett. 1997;269(5):494–499. [Google Scholar]
- 57.Allongue P, Tenne R. Primary Reactions in the Photocorrosion of CdSe through Photocapacitance Measurements. J Electrochem Soc. 1991;138(1):261–268. [Google Scholar]
- 58.Asami H, Abe Y, Ohtsu T, Kamiya I, Hara M. Surface State Analysis of Photobrightening in CdSe nanocrystal thin films. J Phys Chem B. 2003;107(46):12566–12568. [Google Scholar]
- 59.Wang Y, Tang ZY, Correa-Duarte MA, Liz-Marzan LM, Kotov NA. Multicolor Luminescence Patterning by Photoactivation of Semiconductor Nanoparticle Films. J Am Chem Soc. 2003;125(10):2830–2831. doi: 10.1021/ja029231r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.