

Abstract
Background
Major pulmonary manifestations associated with microscopic polyangiitis (MPA) include diffuse alveolar hemorrhage (DAH) and interstitial pneumonia (IP).We previously showed bronchiectasis (BE) was one of the pulmonary complications of MPA. However, clinical features of BE patients with MPA are not fully understood. We investigated the characteristics and prognosis of BE patients with MPA.
Methods
Forty-five MPA patients were retrospectively studied. The patients were divided into two groups: patients with BE and those without BE.
Results
Thirty-one of 45 patients (69%) had pulmonary involvement including IP (23/45, 51%), BE (7/45, 16%), and DAH (5/45, 11%). There were no differences between the patients with BE versus those without with regard to clinical characteristics and initial treatments. However, the prognosis for patients with BE was better than those without BE during the first year after diagnosis, but it was worse between 1 and 5 years, which was statistically significant. Two BE patients died between 1 and 5 years as a result of pneumonia.
Conclusions
BE as a complication of MPA might be related to lower mortality in the acute phase and higher mortality in the chronic phase compared to other pulmonary manifestations. More attention to pulmonary infection is needed for patients with BE during the chronic phase.
Keywords: Microscopic polyangiitis (MPA), bronchiectasis (BE)
Introduction
Anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) is a group of heterogeneous diseases including microscopic polyangiitis (MPA), granulomatosis with polyangiitis (GPA), eosinophilic granulomatosis with polyangiitis (EGPA), and renal-limited AAV (1). MPA is histologically characterized by inflammation and fibrinoid necrosis of systemic small vessel walls, which induces the dysfunction of various organs including the kidneys, lungs, skin, digestive system, and nerves (2). Pulmonary manifestations, such as interstitial pneumonia (IP) (3,4), diffuse alveolar hemorrhage (DAH) (2,5), pulmonary infiltrations, and pleural effusions, affect the prognosis of patients with MPA (6). The Japanese patients in the MPO-ANCA-associated Vasculitis (JMAAV) study showed that the rate of pulmonary involvement with MPA was 45.8%, and the mean survival rate of all MPA patients at 1 year was 89.6% (7). Pulmonary involvement was a predictive factor of disease relapse in AAV patients (8), and DAH was associated with a poorer prognosis in MPA compared to other manifestations (9,10).
Bronchiectasis (BE) is radiologically defined by irreversible dilatation of the bronchus rendering it more than 1.5 times as wide as a nearby vessel as well as by lack of bronchial narrowing in the lung periphery (11,12). Clinical symptoms of BE include cough, sputum, and hemoptysis. Progression of BE is related to neutrophilic inflammation induced by bacterial infection, and 84% of BE patients have sputum cultures positive for pathogens (13). Common etiologies of BE have been reported to be idiopathic, sinobronchial syndrome, and non-tuberculous mycobacteriosis in Japan, in contrast to immune dysfunction in the US (14). We previously reported that BE defined by chest radiography was found to occur in 35% of MPA patients, indicating that BE was one of the pulmonary complications (15). Lhote et al. reported that establishment of BE was related to the production of bactericidal/permeability–increasing protein (BPI)-ANCA, which is one of the target antigen of ANCA (16). These data suggested that BE may be an important pulmonary complication rather than just a co-existing sign of MPA (15).
In the present study, we investigate the clinical characteristics of MPA patients with BE, including the prognosis, compared to patients without BE, and we discuss the appropriate management of MPA patients with BE.
Methods
This data in this study were analyzed retrospectively. All patients’ data were collected exhaustively from medical record which registered disease name of MPA including out and administrative data. We reviewed 45 patients diagnosed with MPA at Saga University Hospital between 2004 and 2011 and analyzed 31 patients with MPA who had pulmonary involvement. As the first step after diagnosis, GPA patients were excluded. Patients who had sinusitis, even if GPA was not diagnosed, were also excluded. Diagnosis of MPA was based on the Japanese Ministry of Health, Labor and Welfare Report 1999. The criteria consisted of three symptoms, four laboratory findings, and histological findings. Symptoms included: (I) rapidly progressive glomerulonephritis; (II) DAH or IP; and (III) others such as subcutaneous hemorrhage and gastrointestinal bleeding. Laboratory and histological findings included: (I) a high titer of MPO-ANCA; (II) increased C-reactive protein (CRP); (III) proteinuria and hematuria; and (IV) pulmonary consolidations or interstitial shadows on radiological examinations. Histological findings included invasion with inflammatory cells and necrosis of the microvasculature. Disease activity of MPA at diagnosis was evaluated by the Birmingham Vasculitis Activity Score (BVAS), version 3 (17), and prognostic factor of MPA was evaluated by the Five-Factor Score (FFS), revised (18,19).
In the present study, high-resolution CT (HRCT) of the chest before treatment was evaluated by two pulmonologists who referenced official reports from a radiologist. We divided the dominant pulmonary findings into three types, based on HRCT, including IP, DAH, and BE. All patients received induction therapy with various doses of corticosteroid as prescribed by their attending physicians. Cyclophosphamide and other immunosuppressive therapies were administered according to their physicians’ judgment. Administration of vaccination against influenza virus and pneumococcal pathogens was decided by the attending physician. The radiological severity of BE was evaluated using the Bhalla score (20). This score consisted of (I) severity of BE; (II) peribronchial thickening; (III) extent of BE (number of lung segments); (IV) extent of mucus plugs (number of lung segments); (V) abscesses or sacculations (number of lung segments); (VI) generalities of the bronchial division involved (BE/plug); (VII) number of bubbles; (VIII) emphysema (number of lung segments); and (IX) collapse/consolidation. The total score for each patient was obtained by summing the scores for each morphological change, which were assigned on the basis of the severity/extent of the abnormality. The total score could range from zero (absence of abnormalities) to 25 (all abnormalities present and severe). The patients were followed over a median of 52.9 months (range, 1–125 months) at either Saga University Hospital or affiliated hospitals and follow-up information was collected in December 2014 to maintain consistency. This study was approved by the ethics committee of Saga University Hospital (approval number: 2015-11-09) and therefore performed in accordance with the 1964 Declaration of Helsinki.
For statistical analysis, quantitative data are presented as mean ± standard deviation (SD). The significance of the differences between the two groups was analyzed using the Mann-Whitney U test and Fisher’s exact test for nonparametric variables. The significance of the differences among frequency data were evaluated using Fisher’s exact test. Survival probabilities were estimated using the Kaplan-Meier technique. Differences in survival between the two groups were analyzed using the log-rank test. The two-tailed significance level was set at P<0.05.
Results
Characteristics of patients with MPA manifested by pulmonary involvements
Among 45 patients diagnosed with MPA, 31 (69%) had pulmonary involvements. We divided them into two groups: with BE (7/45, 16%) and without BE (24/45, 53%) (Table 1). The latter included patients with IP (23/45, 51%), DAH (5/45, 11%). Four patients had coexisting IP and DAH. The patients with BE did not have other coexisting pulmonary manifestation (Figure 1). The average Bhalla score in patients with BE was 10.0 (range, 6–21). There were no differences in demographic data, symptoms, other organ complications, laboratory data or disease activity as investigated by BVAS and FFS between the patients with or without BE. In microbial testing of seven patients with BE, two patients (28.6%) were colonized by Pseudomonas aeruginosa (P. aeruginosa). Initial treatment is shown in Table 2. Glucocorticoid pulse therapy was given to three patients (43%) with BE and 12 patients (50%) without BE. Cyclophosphamide plus glucocorticoid therapy was not given to the patients with BE, but it was given to four patients (17%) without BE. There were no statistically significant differences between the groups.
Table 1. Comparative characteristics of patients with or without bronchiectasis.
| Variables | with BE (n=7) | without BE (n=24) | P value |
|---|---|---|---|
| Demographic data | |||
| Age | 75.0±6.3 | 72.0±9.2 | 0.14 |
| Gender (M/F) | 1/6 | 12/12 | 0.19 |
| Symptoms and complications [%] | |||
| Fever (>37.5 °C) | 5 [71] | 12 [50] | 0.41 |
| Myalgia | 0 | 5 [21] | 0.31 |
| Skin lesion | 0 | 6 [25] | 0.29 |
| Renal involvement | 6 [86] | 20 [83] | 1.00 |
| Permanent dialysis | 1 [14] | 4 [17] | 1.00 |
| Peripheral polymyoneuropathy | 2 [29] | 5 [21] | 0.64 |
| Laboratory data | |||
| MPO-ANCA* (range) (IU/mL) | 337.6 (116.0–585.0) | 222.5 (40.0–640.0) | 0.19 |
| C-reactive protein (range) (mg/dL) | 6.6 (0.02–12.6) | 9.4 (0.07–28.7) | 0.51 |
| Neutrophilia (>8,000/µL) [%] | 5 [71] | 14 [58] | 0.68 |
| Anemia (Hb <10 mg/mL) [%] | 4 [57] | 11 [46] | 0.69 |
| Serum creatinine (>1.2 mg/dL) [%] | 5 [71] | 12 [50] | 0.41 |
| Activity (BVAS) (range) | 5.3 (2.0–10.0) | 7.2 (4.0–14.0) | 0.25 |
| Prognostic factor (FFS) (range) | 2.6 (2.0–3.0) | 2.4 (0–4.0) | 0.69 |
Continuous variables with normal distribution are expressed as mean ± SD. *, all 31 patients who had pulmonary involvements were positive in serum MPO-ANCA. MPO-ANCA, myeloperoxidase anti-neutrophil cytoplasmic antibody; Hb, hemoglobin; BVAS, Birmingham Vasculitis Activity Score version 3; FFS, The Five-Factor-Score revised.
Figure 1.
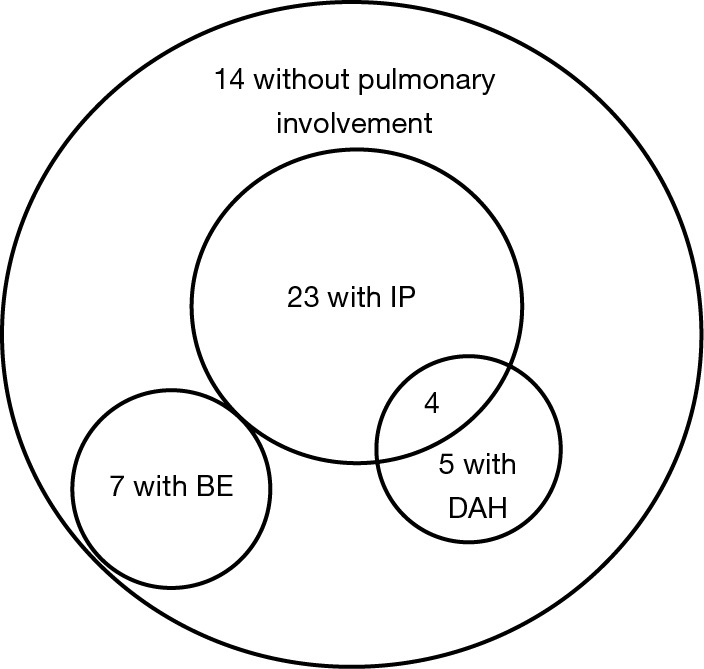
Pulmonary manifestations of 45 patients with MPA. Venn diagram of 45 patients with MPA including 23 patients with IP, 7 patients with BE, 5 patients with DAH, and 14 patients without pulmonary involvement. MPA, microscopic polyangiitis; IP, interstitial pneumonia; BE, bronchiectasis; DAH, diffuse alveolar hemorrhage.
Table 2. Initial treatment and prognosis of microscopic polyangiitis patients with or without bronchiectasis.
| Variables | With BE (n=7) [%] | Without BE (n=24) [%] | P value |
|---|---|---|---|
| Initial treatment | |||
| Methylprednisolone pulse therapy | 3 [43] | 12 [50] | 1.00 |
| Prednisolone with cyclophosphamide* | 0 [0] | 4 [17] | 0.55 |
| Prognosis | |||
| Total of death | 2 [29] | 7 [29] | 1.00 |
| <1 year | 0 [0] | 7 [29] | 0.03 |
| ≥1 and <5 years | 2 [29] | 0 [0] | – |
*, the dosages of prednisolone were ranged between 0.5 and 1.2 mg/kg/day. Prognosis between less than 1 year and more than 1 year to 5 years were analyzed using Fisher’s exact test.
Prognosis of MPA patients with or without BE
As for prognosis of the patients with pulmonary complications, two patients (29%) with BE and seven patients (29%) without BE died over the 5 years after diagnosis (Table 2). Kaplan-Meier analysis showed no statistically significant differences between the patients with or without BE (log-rank, P=0.90, Figure 2). We compared the results of patients between two different time periods, i.e., 1 year from diagnosis and 1–5 years after diagnosis. None of the patients with BE and seven patients (9.1%) without BE died in the first year. However, 2 patients (28.6%) with BE, who were not colonized by any pathogens, and none of the patients without BE died over the next 1 to 5 years. These data were statistically significant (Fisher’s exact test P=0.03, Table 2). Causes of death in patients with MPA are shown in Table 3. Nine patients, including five with IP, two with DAH, and two with BE, died within 5 years after diagnosis. These patients had no significantly differences in treatment. Both patients with BE died from bacterial pneumonia (Table 3).
Figure 2.
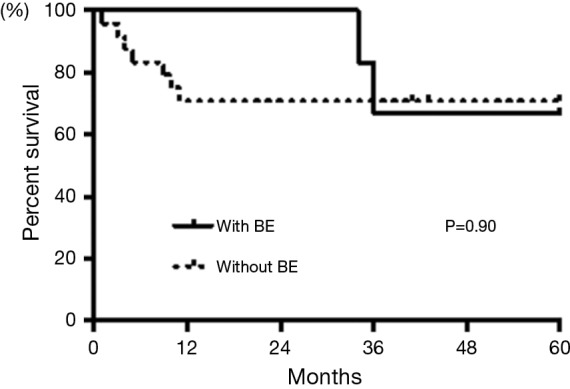
Survival curve of MPA patients with BE and without BE. Kaplan-Meier curve of survival of patients with BE (n=7, solid line) and patients without BE (n=24, dashed line) over 60 months. MPA, microscopic polyangiitis; BE, bronchiectasis.
Table 3. Causes of death in patients with microscopic polyangiitis.
| Pt | Lung involvement | Treatment | Overall survival (months) | Cause of death |
|---|---|---|---|---|
| 1 | IP | Steroid pulse | 1 | Pneumonia, ARDS |
| 2 | IP | Oral steroid | 4 | Exacerbation of DAH |
| 3 | IP | Steroid pulse, CP | 5 | Multiple organ failure |
| 4 | IP | Oral steroid | 10 | Sudden death (unknown) |
| 5 | IP | Steroid pulse, CP | 11 | Exacerbation of IP |
| 6 | DAH | Steroid pulse | 3 | Exacerbation of DAH |
| 7 | DAH | Steroid pulse | 9 | Heart failure |
| 8 | BE | Oral steroid | 34 | Pneumonia |
| 9 | BE | Steroid pulse | 36 | Pneumonia |
IP, interstitial pneumonia; ARDS, acute respiratory distress syndrome; DAH, diffuse alveolar hemorrhage; CP, cyclophosphamide; BE, bronchiectasis.
Discussion
In the present study, we showed that pulmonary complications were found in 31 patients (69%) among 45 patients with MPA, in which IP (23/45, 51%), BE (7/45, 16%), and DAH (5/45, 11%) were represented. Comparing clinical characteristics between the patients with BE and without BE, the prognosis of patients with BE was better than patients without BE during the first year after diagnosis, whereas it was worse between 1 and 5 years. All patients died from pneumonia with acute respiratory distress syndrome.
Incidence of pulmonary manifestations among MPA patients has been reported to be 60–70%, which includes pulmonary infiltrates, pleural effusion, DAH, and pulmonary fibrosis (7,9). IP has been shown to be present in 7.2–36% of MPA patients (3). DAH has also been shown to occur in 29–36% of MPA patients (2,5). However, the complication rate of BE is reported to be from 29–35% of MPA patients (15,21). In the present study, IP was the most frequent pulmonary manifestation with MPA, and BE was the second most frequent complication. These results suggest that BE is one of the major pulmonary involvements of MPA, in agreement with previous reports (15,21).
BE is a chronic inflammatory lung disease that is characterized by various respiratory symptoms and repeated respiratory infection. Chronic airway inflammation is speculated as a possible trigger of ANCA production. Indeed, B cells continuously stimulated by Staphylococcus aureus in the lungs may result in the generation of ANCA (22). Persistent infection induces pro-inflammatory cytokines, such as TNF-a, IL-1b, and IFN-g, and is related to the production of ANCA (23). In regard to mechanisms of ANCA production, BPI, which is also a target antigen of ANCA, is reported to be an important molecule. BPI has an amino-terminal domain that binds with lipopolysaccharide, and the amino acid sequence of BPI epitopes has homology with the membranes of Escherichia coli and P. aeruginosa (24). It has been speculated that bacterial infections stimulate the production of BPI autoantibodies, such as ANCA, and the molecular mimicry of BPI and infectious pathogens could be one of the mechanisms of initiation of BPI auto-antibody development (24). In the present study, it is unclear whether BE is an important complication or just a coincidental pulmonary finding with MPA. However, BE was frequently seen in MPA patients in our present study, and chronic airway inflammation related to bacterial infection as a feature of BE is reported to be the pathogenesis of MPA. Accordingly, we believe that BE may be an important complication of rather than just a coincidental finding with MPA.
Pulmonary involvement has been reported to be a poor prognostic factor for survival in patients with MPA (10) and with ANCA-associated rapidly progressive glomerulonephritis (25). In this study, four patients with IP, two patients with DAH, and no patients with BE died in the first year. DAH is the most serious pulmonary manifestation associated with MPA, and the relative risk of patient death is 8.65 times greater in patients with DAH (5). Other studies have reported that DAH and IP are related to 1-year mortality (3,9). These results suggest that IP and DAH with MPA are associated with worse survival rates than BE with MPA in the early clinical phase. On the other hand, two patients with BE died over the period of 1 to 5 years. The cause of death of those two patients was respiratory infection. Infectious complications are associated with mortality in patients with MPA over the first year after diagnosis (26), and maintenance therapy with corticosteroids increases the incidence rate of infections (27). Furthermore, BE is closely related to respiratory infection, and the exacerbation rate is 1.94 times per year (28). In the present study, two patients (28.6%) with BE were colonized by P. aeruginosa. However, no colonization with pathogens was detected in either of the patients with BE who died. McDonnell and colleagues reported that there was no relationship between colonization of P. aeruginosa and exacerbation rate in patients with BE (29). According to these data, MPA patients with BE had higher mortality rates due to respiratory infection in the late phase, even if the patients had not been colonized by any pathogens. Moreover, the reason why BE patients had a higher mortality rate in the late phase may have been related to an increasing incidence rate of respiratory infection induced by the accumulation of immunosuppressive treatment.
There are possible limitations to the present study. As part of the initial treatment, the administration rate of cyclophosphamide was low in all of the MPA patients. That may have affected the mortality rate of IP and DAH patients with MPA in the acute phase. Moreover, this study was performed on a small number of patients at a single university hospital. To ensure the validity of our findings, multicenter studies involving a large number of patients should be performed.
In conclusion, BE is one of the major pulmonary complications of MPA but is related to better survival in the acute phase than IP or DAH. However, BE is more highly associated with complications of critical respiratory infection during the chronic phase than IP or DAH. Physicians should pay more attention to infections in patients with BE during the chronic phase of MPA, even in those patients who have not been colonized by any pathogens.
Acknowledgements
None.
Ethical Statement: This study was approved by the ethics committee of Saga University Hospital (approval number: 2015-11-09) and therefore performed in accordance with the 1964 Declaration of Helsinki.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013;65:1-11. 10.1002/art.37715 [DOI] [PubMed] [Google Scholar]
- 2.Guillevin L, Durand-Gasselin B, Cevallos R, et al. Microscopic polyangiitis: clinical and laboratory findings in eighty-five patients. Arthritis Rheum 1999;42:421-30. [DOI] [PubMed] [Google Scholar]
- 3.Arulkumaran N, Periselneris N, Gaskin G, et al. Interstitial lung disease and ANCA-associated vasculitis: a retrospective observational cohort study. Rheumatology (Oxford) 2011;50:2035-43. 10.1093/rheumatology/ker236 [DOI] [PubMed] [Google Scholar]
- 4.Tzelepis GE, Kokosi M, Tzioufas A, et al. Prevalence and outcome of pulmonary fibrosis in microscopic polyangiitis. Eur Respir J 2010;36:116-21. 10.1183/09031936.00110109 [DOI] [PubMed] [Google Scholar]
- 5.Hogan SL, Nachman PH, Wilkman AS, et al. Prognostic markers in patients with antineutrophil cytoplasmic autoantibody-associated microscopic polyangiitis and glomerulonephritis. J Am Soc Nephrol 1996;7:23-32. [DOI] [PubMed] [Google Scholar]
- 6.Gómez-Puerta JA, Hernández-Rodríguez J, López-Soto A, et al. Antineutrophil cytoplasmic antibody-associated vasculitides and respiratory disease. Chest 2009;136:1101-11. 10.1378/chest.08-3043 [DOI] [PubMed] [Google Scholar]
- 7.Ozaki S, Atsumi T, Hayashi T, et al. Severity-based treatment for Japanese patients with MPO-ANCA-associated vasculitis: the JMAAV study. Mod Rheumatol 2012;22:394-404. 10.3109/s10165-011-0525-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pagnoux C, Hogan SL, Chin H, et al. Predictors of treatment resistance and relapse in antineutrophil cytoplasmic antibody-associated small-vessel vasculitis: comparison of two independent cohorts. Arthritis Rheum 2008;58:2908-18. 10.1002/art.23800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oh JS, Lee CK, Kim YG, et al. Clinical features and outcomes of microscopic polyangiitis in Korea. J Korean Med Sci 2009;24:269-74. 10.3346/jkms.2009.24.2.269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uezono S, Sato Y, Hara S, et al. Outcome of ANCA-associated primary renal vasculitis in Miyazaki Prefecture. Intern Med 2007;46:815-22. 10.2169/internalmedicine.46.6371 [DOI] [PubMed] [Google Scholar]
- 11.Barker AF. Bronchiectasis. N Engl J Med 2002. 346:1383-93. 10.1056/NEJMra012519 [DOI] [PubMed] [Google Scholar]
- 12.Desai SR, Wells AU, Cheah FK, et al. The reproducibility of bronchial circumference measurements using computed tomography. Br J Radiol 1994;67:257-62. 10.1259/0007-1285-67-795-257 [DOI] [PubMed] [Google Scholar]
- 13.Kadowaki T, Yano S, Wakabayashi K, et al. An analysis of etiology, causal pathogens, imaging patterns, and treatment of Japanese patients with bronchiectasis. Respir Investig 2015;53:37-44. 10.1016/j.resinv.2014.09.004 [DOI] [PubMed] [Google Scholar]
- 14.McShane PJ, Naureckas ET, Strek ME. Bronchiectasis in a diverse US population: effects of ethnicity on etiology and sputum culture. Chest 2012;142:159-67. 10.1378/chest.11-1024 [DOI] [PubMed] [Google Scholar]
- 15.Takahashi K, Hayashi S, Ushiyama O, et al. Development of microscopic polyangiitis in patients with chronic airway disease. Lung 2005;183:273-81. 10.1007/s00408-004-2540-1 [DOI] [PubMed] [Google Scholar]
- 16.Lhote R, Theodore C, Issoufaly T, et al. Successful treatment of antineutrophil cytoplasmic antibody-associated bronchiectasis with immunosuppressive therapy. Eur Respir J 2015;46:554-7. 10.1183/09031936.00031115 [DOI] [PubMed] [Google Scholar]
- 17.Mukhtyar C, Lee R, Brown D, et al. Modification and validation of the Birmingham Vasculitis Activity Score (version 3). Ann Rheum Dis 2009;68:1827-32. 10.1136/ard.2008.101279 [DOI] [PubMed] [Google Scholar]
- 18.Guillevin L, Lhote F, Gayraud M, et al. Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome. A prospective study in 342 patients. Medicine (Baltimore) 1996;75:17-28. 10.1097/00005792-199601000-00003 [DOI] [PubMed] [Google Scholar]
- 19.Guillevin L, Pagnoux C, Seror R, et al. The Five-Factor Score revisited: assessment of prognoses of systemic necrotizing vasculitides based on the French Vasculitis Study Group (FVSG) cohort. Medicine (Baltimore) 2011;90:19-27. 10.1097/MD.0b013e318205a4c6 [DOI] [PubMed] [Google Scholar]
- 20.Bhalla M, Turcios N, Aponte V, et al. Cystic fibrosis: scoring system with thin-section CT. Radiology 1991;179:783-8. 10.1148/radiology.179.3.2027992 [DOI] [PubMed] [Google Scholar]
- 21.Kadowaki T, Yano S, Yamadori I, et al. A case of sinobronchial syndrome complicated with myeloperoxidase antineutrophil cytoplasmic antibody associated vasculitis: review of the literature. Intern Med 2012;51:763-7. 10.2169/internalmedicine.51.5957 [DOI] [PubMed] [Google Scholar]
- 22.Chen M, Kallenberg CG. The environment, geoepidemiology and ANCA-associated vasculitides. Autoimmun Rev 2010;9:A293-8. 10.1016/j.autrev.2009.10.008 [DOI] [PubMed] [Google Scholar]
- 23.de Lind van Wijngaarden RA, van Rijn L, Hagen EC, et al. Hypotheses on the etiology of antineutrophil cytoplasmic autoantibody associated vasculitis: the cause is hidden, but the result is known. Clin J Am Soc Nephrol 2008;3:237-52. 10.2215/CJN.03550807 [DOI] [PubMed] [Google Scholar]
- 24.Schultz H. From infection to autoimmunity: a new model for induction of ANCA against the bactericidal/permeability increasing protein (BPI). Autoimmun Rev 2007;6:223-7. 10.1016/j.autrev.2006.08.005 [DOI] [PubMed] [Google Scholar]
- 25.Yamagata K, Hirayama K, Mase K, et al. Apheresis for MPO-ANCA-associated RPGN-indications and efficacy: lessons learned from Japan nationwide survey of RPGN. J Clin Apher 2005;20:244-51. 10.1002/jca.20035 [DOI] [PubMed] [Google Scholar]
- 26.Kitagawa K, Furuichi K, Sagara A, et al. Risk factors associated with relapse or infectious complications in Japanese patients with microscopic polyangiitis. Clin Exp Nephrol 2016;20:703-11. 10.1007/s10157-015-1199-7 [DOI] [PubMed] [Google Scholar]
- 27.Stuck AE, Minder CE, Frey FJ. Risk of infectious complications in patients taking glucocorticosteroids. Rev Infect Dis 1989;11:954-63. 10.1093/clinids/11.6.954 [DOI] [PubMed] [Google Scholar]
- 28.Goeminne PC, Scheers H, Decraene A, et al. Risk factors for morbidity and death in non-cystic fibrosis bronchiectasis: a retrospective cross-sectional analysis of CT diagnosed bronchiectatic patients. Respir Res 2012;13:21. 10.1186/1465-9921-13-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McDonnell MJ, Jary HR, Perry A, et al. Non cystic fibrosis bronchiectasis: A longitudinal retrospective observational cohort study of Pseudomonas persistence and resistance. Respir Med 2015;109:716-26. 10.1016/j.rmed.2014.07.021 [DOI] [PubMed] [Google Scholar]