

Abstract
Background
The lung cancer mortality rate in Xuanwei city is among the highest in China and adenocarcinoma is the major histological type. Lung cancer has been associated with exposure to indoor smoky coal emissions that contain high levels of polycyclic aromatic hydrocarbons; however, the pathogenesis of lung cancer has not yet been fully elucidated.
Methods
We performed whole genome sequencing with lung adenocarcinoma and corresponding non‐tumor tissue to explore the genomic features of Xuanwei lung cancer. We used the Molecule Annotation System to determine and plot alterations in genes and signaling pathways.
Results
A total of 3 428 060 and 3 416 989 single nucleotide variants were detected in tumor and normal genomes, respectively. After comparison of these two genomes, 977 high‐confidence somatic single nucleotide variants were identified. We observed a remarkably high proportion of C·G‐A·T transversions. HECTD4, RCBTB2, KLF15, and CACNA1C may be cancer‐related genes. Nine copy number variations increased in chromosome 5 and one in chromosome 7. The novel junctions were detected via clustered discordant paired ends and 1955 structural variants were discovered. Among these, we found 44 novel chromosome structural variations. In addition, EGFR and CACNA1C in the mitogen‐activated protein kinase signaling pathway were mutated or amplified in lung adenocarcinoma tumor tissue.
Conclusion
We obtained a comprehensive view of somatic alterations of Xuanwei lung adenocarcinoma. These findings provide insight into the genomic landscape in order to further learn about the progress and development of Xuanwei lung adenocarcinoma.
Keywords: Lung adenocarcinoma, single nucleotide variants, somatic mutations, whole genome sequencing
Introduction
Lung cancer is the leading cause of cancer‐related death globally. Based on GLOBOCAN estimates, 14.1 million new cancer cases and 8.2 million deaths occurred in 2012 worldwide,1, 2, 3 more than 40% of which were lung adenocarcinomas. Despite improvement in molecular diagnosis and targeted therapies, most tumors are only discovered at advanced stage. The overall five‐year survival rate is approximately 15% worldwide, mainly because of late‐stage detection and a paucity of late‐stage treatments.4 In China, lung cancer is the fastest increasing cancer and the leading cause of all cancer death since 2004. In some regions, such as Xuanwei, the incidence of lung cancer is among the highest in China and the world.5 Recently, a retrospective sampling survey reported mortality rates of lung cancer in Xuanwei of 98.10/105 in men and 83.21/105 in women.6 Lung cancer in Xuanwei has four remarkable characteristics: higher incidence, higher mortality, adenocarcinoma is the major histological type, and similar incidence rates between men and women. Lan et al. 7 found that the higher incidence was most likely a result of the use of smoky coal in unvented stoves in this area. Despite large‐scale improvement in stoves, the lung cancer mortality rate remains very high in Xuanwei (91/105 compared with China's average of 31/105 in 2004–2005). We believe that other factors may contribute to the mechanisms of lung cancer development and progress in this area. Thus, the molecular study of lung cancer, especially genetic mechanisms, is of great importance.
The development of next generation sequencing technology has greatly facilitated the detection and characterization of genetic variations, including single nucleotide variations (SNVs), chromosome structural variations (SVs), and copy number variations (CNVs) in human genomes, especially in the field of cancer research.8 Recently, cancer sequencing efforts based on next generation sequencing technologies have provided a genome‐wide view of mutations in leukemia, melanoma, lung cancer, and others.9, 10, 11 In addition, somatic mutation events can reveal specific and novel information about the fundamental genetic mechanisms that may be involved in the development and progression of lung adenocarcinoma in Xuanwei. Studies have validated the various genomic aberrations that may act as therapeutic targets. In recent years, new molecular targeted drugs in the pharmacological treatment of adenocarcinoma have been introduced, and their effectiveness is closely dependent on the presence of specific genetic mutations in the tumor.12 Somatic mutations in the tyrosine kinase domain of the epidermal growth factor receptor (EGFR) gene are one of the most relevant targets for lung cancer treatment.13, 14 To date, research of next generation sequencing has discovered most of the relevant mutations that contribute to the pathogenesis of adenocarcinoma; however, there is limited genomic data of lung adenocarcinoma in Xuanwei. Therefore, we believe that unbiased whole genome sequencing (WGS) is required to screen more mutations of lung adenocarcinoma in Xuanwei.
Herein, we present a detailed analysis of paired tumor and normal tissues from Xuanwei by WGS. Our findings present insights into the frequent somatic alterations that may be associated with the adenocarcinoma pathogenesis. Our results show a significant advance toward a comprehensive annotation of somatic alterations of lung adenocarcinoma in Xuanwei, and demonstrate the need for unbiased whole genome approaches to discover all mutations associated with cancer pathogenesis.
Methods
Sample description and preparation
Lung adenocarcinoma and corresponding non‐tumor tissues were collected from a 41‐year‐old, non‐smoking male patient from Xuanwei County at the First Affiliated Hospital of Kunming Medical University, China. The patient had been exposed to coal smoke for 10 years. Sections that underwent curative resection were stained with hematoxylin and eosin and examined by a pathologist to verify the diagnosis and evaluate tumor stage. Lung adenocarcinoma and corresponding non‐tumor tissues were stored in liquid nitrogen until genomic DNA extraction. The Committee on Ethics in Research on Humans of the First Affiliated Hospital of Kunming Medical University approved the study. Informed consent was obtained from the patient.
Genomic DNA isolation and qualification
An experienced pathologist examined the lung adenocarcinoma and corresponding non‐tumor tissues to confirm the presence (>80%) or absence of cancer cells. Lung adenocarcinoma and non‐tumor tissues (25 mg each) were placed in liquid nitrogen and grinded thoroughly using a mortar and pestle. Decanted tissue powder and liquid nitrogen were placed into a 1.5 mL microcentrifuge tube containing 180 μL of buffer animal tissue lysis. Genomic DNA (gDNA) was extracted using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol. gDNA was quantified using a NanoDrop ND‐1000 spectrophotometer (CapitalBio Nano Q, Beijing, China). The integrity of the gDNA was tested using 1.0% agarose gel.
Whole genome sequencing (WGS)
Whole genome sequencing was performed by unchained combinatorial probe anchor ligation (cPAL) and DNA Nanoball (DNB) arrays. Sequencing substrates were generated by means of gDNA fragmentation and recursive cutting with type IIS restriction enzymes and directional adapter insertion. cPAL was based on unchained hybridization and ligation technology, using degenerate anchors to read up to 10 bases from each eight adapter sites. DNB production was conducted using Phi29 DNA polymerase and nanoarray. WGS was performed using the Complete Genomics sequencing platform (Mountain View, CA, USA), resulting in a total of 259 218 Gb mapped sequences (86× average coverage) for the lung adenocarcinoma and 242 639 Gb (81× average coverage) for the corresponding non‐tumor tissue samples (Table 1). Reads were aligned to the reference genome (NCBI Build 37) to reach a 97% matching rate. Somatic variations in lung adenocarcinoma tissue were identified by comparing variations with the corresponding non‐tumor tissue. A Circos plot of somatic variation between the lung adenocarcinoma and non‐tumor tissue was created, containing information about SNP, CNV, and SV (Fig S1).
Table 1.
Sequence coverage summary for normal and tumor genomes
| Sequence coverage | Normal | Tumor |
|---|---|---|
| Gross mapping yield (Gb) | 307.288 | 324.531 |
| Both mates mapped yield (Gb) | 242.639 | 259.218 |
| Average haploid coverage | 81× | 86× |
| Fully called genome fraction | 0.975 | 0.976 |
| SNVs | 3 416 989 | 3 428 060 |
| Substitution | 89 536 | 90 455 |
SNV, single nucleotide variant.
Coverage percentage and variations are with respect to National Center for Biotechnology Information Build 37 of the human genome reference assembly.
Mapping reads and calling variations
Genomic reads were initially mapped to the reference genome using a fast algorithm. These initial mappings were both expanded and refined by a form of local de novo assembly in all regions of the genome that appeared to contain variations based on these initial mappings. The de novo assembly fully leverages mate‐pair information, allowing reads to be recruited into variants with higher sensitivity than genome‐wide mapping methods alone typically provide. Assemblies were diploid, and sequencing produced two separate result sequences for each locus in diploid regions. Variants were called by independently comparing each of the diploid assemblies to the reference. The original computational methods for small variant detection are available at: http://online.liebertpub.com/doi/full/10.1089/cmb.2011.0201. These methods have evolved over the development of further Analysis Pipeline versions.
Single nucleotide variant (SNV) analysis
The algorithmic details of the small variant caller used to identify and score small variants (SNVs, insertions, deletions, and block substitutions) is available on the Complete Genomics website: www.completegenomics.com/customersupport/documentation. All variants were annotated for their presence in dbSNP 137 and the 1000 Genomes Project dataset. We evaluated the potential impact on protein function for somatic missense mutations identified through WGS using SIFT, a computational method. SIFT scores, ranging from 0 to 1, were obtained from the program output. The SIFT score represents the normalized probability that a particular amino acid substitution is tolerated, and a score below the cut‐off value 0.05 is generally considered deleterious.
Copy number variation (CNV) region analysis
The processing steps and algorithmic details of the CNV pipeline used to identify and score regions of genomic copy number variation are available at: www.completegenomics.com/customersupport/documentation. The determination of CNV calling for normal and tumor samples consisted of the following steps: (i) computation of sequence coverage; (ii) estimation and correction of bias in coverage (modeling of coverage bias, correction of modeled bias, and coverage smoothing); and (iii) normalization of coverage by comparison to a baseline sample or set of samples.
Following normalization of coverage, both normal and tumor samples were segmented using Hidden Markov Models (HMM), but with a different model for each sample type: HMM segmentation, scoring, and output. Finally, normal samples were subjected to a “no‐calling” process that identified suspect CNV calls.
Structural variation (SV) analysis
Structural variation is generally defined as deletions, insertions, duplications, inversions, translocations, or CNV in large DNA segments (>1 kb), represented by one or more junctions. The process of detailed description of how an SV event was deduced from junction data involved the generation of an undirected graph of related junctions. We annotated each event with biological information: (i) with the list of all potentially disrupted genes – the genes that overlapped at least one of the junction side positions for any of the junctions that were grouped into the event; (ii) any events that may indicate a copy number change of a stretch of sequence (e.g. deletion, tandem‐duplication, and distal‐duplication events), including all of the genes that were completely contained in the affected sequence; and (iii) when a junction appeared to connect two different genes in a strand consistent manner, it was considered a possible gene fusion.
Molecule annotation of variations
Predicted single nucleotide polymorphisms (SNPs) were compared using National Center for Biotechnology Information (NCBI) dbSNP version 137 to annotate known SNP information. Each SNP was mapped on the genomic features of the UCSC gene table, such as coding region, untranslational region, and intron. Non‐synonymous SNP information was extracted by comparing UCSC reference gene information (http://genome.Ucsc.edu/). Information on cancer‐related mutations was obtained from the cosmic cancer information database (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/). The Ingenuity Molecule Annotation System (MAS) (http://bioinfo.capitalbio.com/mas3/) was used for functional interaction analysis of the genes affected. The MAS is a whole data‐mining and function annotation solution used to extract and analyze biological molecule relationships from a public knowledge base of biological molecules and signification.
Results
Identification and distribution of somatic SNVs
Called single‐nucleotide variations were filtered to obtain a list of candidate somatic mutations (Fig 1). A total of 3 428 060 and 3 416 989 SNPs were called independently from lung adenocarcinoma and corresponding non‐tumor tissue, respectively. After comparing the SNPs presented in the non‐tumor tissue, 55 142 somatic SNVs were identified in the lung adenocarcinoma tissue, reaching 11.4 mutations per megabase throughout the genome. Among them, 977 were high confident somatic SNVs and 964 were potentially novel somatic mutations, compared with lung adenocarcinoma tissue. Of these SNVs, about 32% (309 SNVs) were located in intronic regions, 65% (631 SNVs) in intergenic regions, 1% (6 SNVs) in upstream or downstream regions of a gene, and 1% (5 SNVs) were located in 3′ or 5′ untranslated regions (Fig 2). The transition (Ts, 2 383 204) and transversion (Tv, 1 154 590) ratio was 2.127, and the homozygosity (1383596) and heterozygosity (1890004) proportions were 42% and 58%, respectively.
Figure 1.
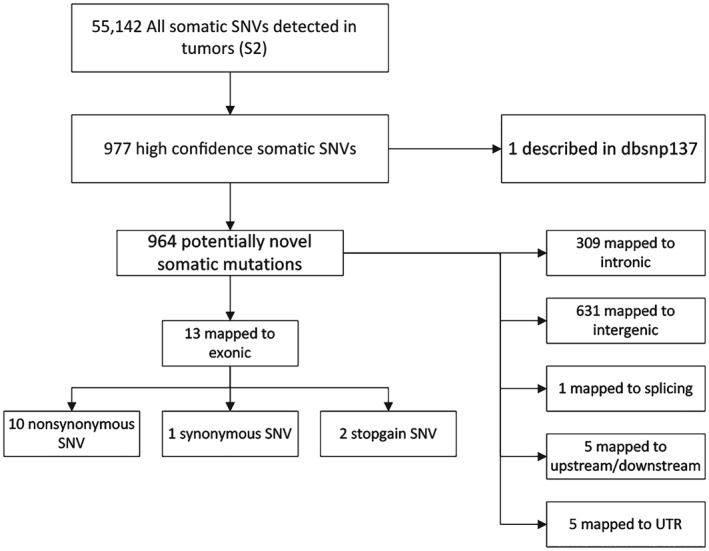
Flow chart describing the methodology used in filtering single nucleotide variants (SNVs) to obtain candidate novel somatic mutations.
Figure 2.

Distribution of the 964 novel somatic single nucleotide variants (SNVs) based on their genomic location. Of the 964 potentially novel somatic mutations, 32% were in an intronic region, 65% in an intergenic region, 1% in upstream or downstream regions of a gene, and an additional 1% were in an untranslated (UTR) 3′ or 5′ region.
HECTD4, RCBTB2B , KLF15, and CACNA1C may be cancer‐related genes
We obtained a list of candidate somatic mutations, of which 12 are identified in coding regions with 10 non‐synonymous and two stopgain SNVs (Table 2). We further analyzed the SNVs in exonic regions by comparing them with the cancer‐related genes in the Catalogue of Somatic Mutations in Cancer database. A SIFT score for amino acid substitutions below the cut‐off value 0.05 is generally considered deleterious. Finally, our results showed that HECTD4, RCBTB2, KLF15, and CACNA1C may be cancer‐related genes, with SIFT scores of 0.00, 0.00, 0.01, and 0.05, respectively.
Table 2.
Details of predicted non‐synonymous single nucleotide variations
| Chromosome | Start | End | Reference | Alt | Gene symbol | Gene function impaction | SIFT |
|---|---|---|---|---|---|---|---|
| 12 | 112 673 325 | 112 673 325 | G | A | HECTD4 | Nonsynonymous SNV | 0 |
| 13 | 49 075 918 | 49 075 918 | C | A | RCBTB2 | Stopgain SNV | 0 |
| 3 | 126 071 687 | 126 071 687 | C | A | KLF15 | Nonsynonymous SNV | 0.01 |
| 12 | 2 721 085 | 2 721 085 | G | T | CACNA1C | Nonsynonymous SNV | 0.05 |
| 14 | 92 482 047 | 92 482 047 | T | A | TRIP11 | Nonsynonymous SNV | 0.11 |
| 14 | 105 405 385 | 105 405 385 | T | A | AHNAK2 | Nonsynonymous SNV | 0.17 |
| 22 | 21 330 800 | 21 330 800 | G | T | AIFM3 | Nonsynonymous SNV | 0.27 |
| 5 | 94 764 399 | 94 764 399 | C | T | FAM81B | Nonsynonymous SNV | 0.32 |
| 9 | 136 421 040 | 136 421 040 | C | A | ADAMTSL2 | Nonsynonymous SNV | 0.34 |
| 16 | 309 499 | 309 499 | G | T | ITFG3 | Nonsynonymous SNV | 0.39 |
| 2 | 189 904 214 | 189 904 214 | T | G | COL5A2 | Nonsynonymous SNV | 0.69 |
| 6 | 47 847 014 | 47 847 014 | G | C | PTCHD4 | Stopgain SNV | 1 |
SNV, single nucleotide variant.
C·G‐A·T transversion was a common mutation in lung adenocarcinoma tissue
The composition of somatic variations in this lung adenocarcinoma sample was distinct from the germline variations. Across the entire genome, somatic variations occurred predominantly at C·G base pairs (75.31%), and the most prevalent change was C·G‐A·T transversion (56.86%). Germline variations occurred predominantly also at C·G base pairs (51.84%), whereas the most frequent transitions were A·T‐G·C (33.50%) and C·G‐T·A (34.80%). Interestingly, enrichment of the C·G‐A·T transversion was found to be a genome‐wide trend for somatic variations, a sharp contrast to the 8.31% occurrence of germline variations (Fig 3a).
Figure 3.
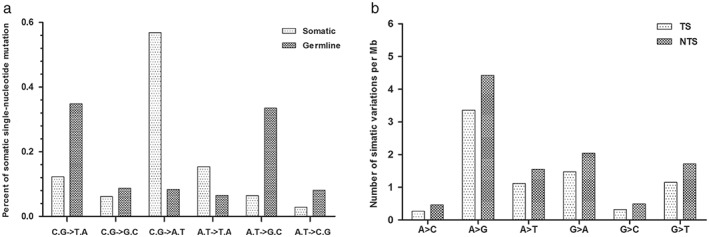
(a) Somatic single nucleotide mutation trends. Somatic mutations were primarily C·G‐A·T transversions. Distribution of specific nucleotide changes among germline and somatic variations in the lung adenocarcinoma genome. C·G‐A·T transversions accounted for 56% of high confidence somatic mutations, whereas most germline variations were A·T‐G·C and C·G‐T·A transitions. (b) Somatic single‐nucleotide mutations of all types occurred at a lower frequency on the transcribed strand (TS). TS represent single nucleotide somatic mutation rates, with different types of base substitutions on the coding regions. The non‐transcribed strand (NTS) represents the same classes of mutations occurring on the NTS.
The distribution of somatic mutations in the genome is highly non‐uniform, as protein‐coding exons have a substantially lower rate. In our sample, there were lower rates of mutation in the transcribed strand compared with the non‐transcribed strand (Fig 3b). The ratio of the overall non‐synonymous substitution rate (Ka) to synonymous substitution rate (Ks) in this sample was 0.95, suggesting that most mutations are “passengers.”
Nine CNVs increased in chromosome 5 and one in chromosome 7
Copy number variation is caused by the rearrangement of the genome. A DNA segment of 1 Kb or larger presents genome large copies that are increased or decreased compared with a reference genome. CNV of more than two was defined as a CNV increase, while less than two was defined as a CNV decrease. We found that 14 CNVs increased in chromosome 5 and one CNV increased in chromosome 7. When the exact loci of tumor suppressors and oncogenes were analyzed, we identified nine CNVs increased in chromosome 5 and one in chromosome 7 involving gene expression (Table 3). Interestingly, tumor oncogenes EGFR at chromosome 7 and TERT at chromosome 5 were highly increased on chromosomal regions.
Table 3.
Copy number variation increased in chromosomes 5 and 7
| Chromosome | Start | End | Called score | CNV type score | Overlapping gene |
|---|---|---|---|---|---|
| chr5 | 9 590 000 | 9 632 000 | 3 | 32 | TAS2R1 |
| chr5 | 1 156 000 | 1 228 000 | 3 | 33 | SLC6A18; SLC6A19 |
| chr5 | 1 478 000 | 1 498 000 | 3 | 6 | LPCAT1 |
| chr5 | 1 532 000 | 2 582 000 | 3 | 50 |
IRX4; LOC100506843;
LOC100506858;
LOC728613; MIR4277; MRPL36; NDUFS6; SDHAP3 |
| chr5 | 15 934 000 | 16 328 000 | 3 | 31 | FBXL7; LOC100505959; LOC401176; MARCH11; MIR887 |
| chr5 | 15 122 000 | 15 890 000 | 3 | 43 | FBXL7 |
| chr5 | 1 248 000 | 1 410 000 | 3 | 52 | CLPTM1L; LOC100506791; SLC6A3; TERT |
| chr5 | 19 856 000 | 20 046 000 | 3 | 22 | CDH18 |
| chr5 | 2 720 000 | 4 094 000 | 3 | 42 | C5orf38; IRX1; IRX2; LOC285577 |
| chr5 | 2 582 000 | 2 720 000 | 4 | 69 | |
| chr5 | 4 194 000 | 4 382 000 | 3 | 26 | |
| chr5 | 4 412 000 | 4 486 000 | 3 | 32 | |
| chr5 | 18 718 000 | 18 852 000 | 3 | 19 | |
| chr5 | 19 256 000 | 19 346 000 | 3 | 4 | |
| chr7 | 55 018 000 | 55 776 000 | 3 | 54 | EGFR; FKBP9L; LANCL2; LOC100507500; VOPP1 |
CNV, copy number variation.
Forty‐four novel SVs were identified
We screened for SVs based on discordant paired end genomic sequencing data. Normal SVs called from non‐tumor tissue were used as a filter to remove somatic SVs and technical artifacts, resulting in a list of candidate SVs. The SV analysis resulted in 1955 junction counts. During SV detection, 44 were not present with the required coverage in the normal sample and were designated as somatic SVs: 16 deletions, eight tandem‐duplication, four distal‐duplication, five inversions, and 11 complex (Table S1). The majority of rearrangements cannot be ascribed to classical SV patterns, because of the considerably greater complexity of somatically acquired rearrangements compared to germline events.
Molecule annotation of variations
The MAS was used to determine and plot the alterations of genes and signaling pathways. Indeed, two genes in the mitogen‐activated protein kinase (MAPK) signaling pathway, EGFR and CACNA1C, were found either mutated or amplified in lung adenocarcinoma tissue (Fig S2) and other cancer‐related pathways also harbored multiple mutations (Fig S3). Activation of the MAPK pathway can result in a multitude of physiological effects, including apoptosis, cell proliferation, mitosis, and the transcription of several classes of genes. EGFR was found in telomerase, cellular aging, and immortality pathways and plays a role in nicotinic acetylcholine receptors in the regulation of cellular apoptosis (Fig 4).
Figure 4.
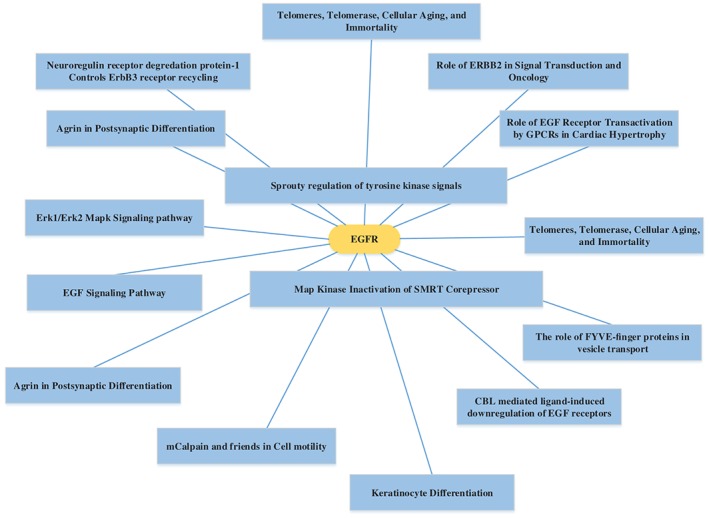
Gene pathway network BioCarta analysis. EGFR was found to play an important role in telomerase, cellular aging, and immortality.
Discussion
Research has shown that lung cancer is caused by the accumulation of genomic alterations. Until recently, it has been difficult to interpret these various changes within a single tumor that may work together to fulfill the hallmark traits of the malignant phenotype. The development of parallel sequencing technologies has provided a powerful tool for the study of lung cancer genomes. In this paper, we conducted high‐depth coverage (~80×) and detailed analysis of the whole genome sequence to a case of lung adenocarcinoma in Xuanwei. The sequencing analysis proved that data quality was high (only 5% of the total sequence was filtered out) and the reads almost completely covered the reference human genome. Our results showed that Xuanwei lung adenocarcinoma could have a large number of new mutations, ranging from the single nucleotide to chromosome alterations. We also found that some of these mutation genes participated in several well‐described pathways that were known to contribute to cancer pathogenesis; however, most genes were unknown in our current understanding of the development and progression of disease. Therefore, WGS facilitated the investigation of the biological and clinical implications of such variations.
In this study, the SNVs exhibited a highly distinctive pattern, showing a high proportion of C·G‐A·T transversions and a low fraction of A·T‐C·G transversions. Studies have indicated that C·G to A·T transversions are the most common substitution in non‐small cell lung cancer associated with smoking, and our experimental results were consistent with tobacco exposure‐related mutation signatures.15, 16 Non‐small cell lung tumors from non‐smokers are dominated by C·G‐T·A transitions compared with C·G‐A·T transversions in lung tumors from smokers. Lui et al. identified that C·G‐T·A transitions were decreased in a progressive manner with cumulative exposure to tobacco.17 Interestingly, our patient was a non‐smoker who had been exposed to coal smoke for 10 years, and the C·G‐T·A transitions (12%) were of relatively lower frequency compared with C·G‐A·T transversions (56%), which may be associated with the coal smoke. However, our hypothesis requires further study for confirmation.
A previous study showed that compositions of germline and somatic variants were different in lung cancer genomes, with enriched C·G‐A·T transversions in the somatic mutation group.18 A direct comparison between germline and somatic variations highlighted the strong influence of smoking‐related DNA damage.19 Furthermore, G·C‐C·G somatic changes were strongly enriched at GpA/TpC dinucleotides, which accounted for 52% of the nucleotide G·C variations. Lower rates of mutation have been found in a transcribed strand compared with a non‐transcribed strand in a small cell cancer cell line.15 We observed a similar trend, with a pattern that would result from transcription‐coupled DNA repair processes.
In addition to commonly mutated genes, we also identified distinct mutational signatures and signaling pathways in Xuanwei lung adenocarcinoma. Some somatic SNVs and CNVs have been already reported as either oncogenes or tumor suppressors in the COSMIC database, including EGFR and CACNA1C. In addition, we identified 13 lung adenocarcinoma specific genes in our study. Twelve non‐synonymous somatic mutations and two mutations known to be associated with lung adenocarcinoma were identified, including two SNVs located in exon 18 and exon 20 of the EGFR gene. EGFR, a cell surface protein, binds to the epidermal growth factor. Binding of the protein to a ligand‐induced receptor dimerization and tyrosine autophosphorylation leads to cell proliferation.14 Both of these mutations are common in lung cancers, and both were thought to contribute to progression and development of the disease. This cancer genome had few mutated genes in the lung adenocarcinoma pathway, but showed statistically meaningful genetic changes in the MAPK signaling pathways. MAPK signaling pathways can contribute to carcinogenesis in lung adenocarcinoma.
This comparative analysis provides a complex mutation landscape as a reference data set in understanding lung adenocarcinoma genome variation and was an initial step in exploring lung adenocarcinoma genomic features in Xuanwei. The observed mutation landscape was probably shaped by many different processes, including how to generate the original mutations, how to affect DNA repair mechanisms, and how to select during tumor evolution.20 Selection could act in two directions: by retaining mutations that will benefit tumor growth, while also limiting mutations in key functional regions of the genome, such as promoters and expressed genes.
As a tumor from only one case was sequenced, we could not determine whether these somatic variations were derived from driver or passenger mutations. Our hypotheses ideally need to be tested in an in vitro model. The mutated target genes and their cellular signaling mechanisms indicated aberrations in DNA repair mechanisms, which may be related to the lung adenocarcinoma progression in our patient. Our study and other studies of individual cancer genomes have shown a trend in mutations, but identification of the recurrent driver mutations will require many more samples to sequence. Nevertheless, we believe the data provides a cornerstone for such studies and enriches the analysis of Xuanwei lung adenocarcinoma in genomic variation.
Disclosure
No authors report any conflict of interest.
Supporting information
Figure S1 The genomic landscape of somatic alterations.
Figure S2 Mitogen‐activated protein kinase (MAPK) signaling pathway analysis.
Figure S3 Gene Pathway Network_KEGG analysis.
Table S1 Details of somatic structural variations.
Acknowledgments
The Natural Science Foundation of China (Grant number: 81160292), the Health Science and Technology Project in Yunnan Province (2014NS152), and the leading talent project of the health system in Yunnan province (L‐201202) supported this study. We also wish to thank the study participant.
References
- 1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin 2015; 65: 87–108. [DOI] [PubMed] [Google Scholar]
- 2. Travis WD. Pathology of lung cancer. Clin Chest Med 2002; 23: 65–81. [DOI] [PubMed] [Google Scholar]
- 3. Imielinski M, Berger AH, Hammerman PS et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012; 150: 1107–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ettinger DS, Akerley W, Bepler G et al. Non‐small cell lung cancer. J Natl Compr Canc Netw 2010; 8: 740–801. [DOI] [PubMed] [Google Scholar]
- 5. Barone‐Adesi F, Chapman RS, Silverman DT et al. Risk of lung cancer associated with domestic use of coal in Xuanwei, China: Retrospective cohort study. BMJ 2012; 345: e5414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wu H, Meng S, Xu Q et al. Gene expression profiling of lung adenocarcinoma in Xuanwei, China. Eur J Cancer Prev 2016; 25: 508–17. [DOI] [PubMed] [Google Scholar]
- 7. Lan Q, He X, Shen M et al. Variation in lung cancer risk by smoky coal subtype in Xuanwei, China. Int J Cancer 2008; 123: 2164–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second‐generation sequencing. Nat Rev Genet 2010; 11: 685–96. [DOI] [PubMed] [Google Scholar]
- 9. Mardis ER, Ding L, Dooling DJ et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 2009; 361: 1058–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Turajlic S, Furney SJ, Lambros MB et al. Whole genome sequencing of matched primary and metastatic acral melanomas. Genome Res 2012; 22: 196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Suzuki A, Mimaki S, Yamane Y et al. Identification and characterization of cancer mutations in Japanese lung adenocarcinoma without sequencing of normal tissue counterparts. PLoS One 2013; 8: e73484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pao W, Girard N. New driver mutations in non‐small‐cell lung cancer. Lancet Oncol 2011; 12: 175–80. [DOI] [PubMed] [Google Scholar]
- 13. Maemondo M, Inoue A, Kobayashi K et al. Gefitinib or chemotherapy for non‐small‐cell lung cancer with mutated EGFR. N Engl J Med 2010; 362: 2380–8. [DOI] [PubMed] [Google Scholar]
- 14. Marchetti A, Del Grammastro M, Filice G et al. Complex mutations & subpopulations of deletions at exon 19 of EGFR in NSCLC revealed by next generation sequencing: Potential clinical implications. PLoS One 2012; 7: e42164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pleasance ED, Stephens PJ, O'Meara S et al. A small‐cell lung cancer genome with complex signatures of tobacco exposure. Nature 2010; 463: 184–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Govindan R, Ding L, Griffith M et al. Genomic landscape of non‐small cell lung cancer in smokers and never‐smokers. Cell 2012; 150: 1121–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu J, Lee W, Jiang Z et al. Genome and transcriptome sequencing of lung cancers reveal diverse mutational and splicing events. Genome Res 2012; 22: 2315–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee W, Jiang Z, Liu J et al. The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature 2010; 465: 473–7. [DOI] [PubMed] [Google Scholar]
- 19. Hecht SS. Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst 1999; 91: 1194–210. [DOI] [PubMed] [Google Scholar]
- 20. Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature 2009; 458: 719–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 The genomic landscape of somatic alterations.
Figure S2 Mitogen‐activated protein kinase (MAPK) signaling pathway analysis.
Figure S3 Gene Pathway Network_KEGG analysis.
Table S1 Details of somatic structural variations.