

Abstract
An elevation of fatty acid delivery amplifies the TCA cycle flux with a rise in anaplerosis/cataplerosis, leading to a proportional rise in oxidative stress and inflammation in liver.
Ectopic fat accumulation in insulin‐target organs leads to the development of insulin resistance in each organ by altering oxidative stress1, 2 and gene expression profiles3, 4. Specifically, the liver functions as a center to maintain whole‐body energy homeostasis by sensing nutrient stimuli, and producing a variety of nutrients and bioactive substances. Liver fat is associated with not only enhanced hepatic glucose production, but also skeletal muscle insulin resistance, supporting a central role of fatty liver in systemic insulin resistance in patients with non‐alcoholic fatty liver disease (NAFLD)5. Of the triacylglycerol accumulated in the liver of patients with NAFLD, 59.0% arises from non‐esterified fatty acids (NEFAs), mainly derived from adipose tissue, whereas 14.9% arises from diet and 26.1% from de novo lipogenesis6. Triglyceride (TG) itself is not a toxic lipid, but might rather protect against the toxic effects of free fatty acids. However, some free fatty acids are regarded as toxic lipids that cause hepatic insulin resistance through oxidative stress7. In an in vitro fatty liver system using H4IIEC3 hepatocytes, a saturated fatty acid, palmitate, and not an unsaturated fatty acid, oleate, inhibits insulin‐stimulated tyrosine phosphorylation of insulin receptor substrate 2 and serine phosphorylation of Akt through c‐Jun NH2‐terminal kinase activation8. In that model, mitochondrial β oxidation‐derived reactive oxygen species (ROS) play a causal role in the palmitate‐induced c‐Jun NH2‐terminal kinase activation8. Therefore, toxic lipid‐induced mitochondrial ROS might underlie the link between steatosis and insulin resistance in the liver. Indeed, in humans, genes involved in mitochondrial oxidative phosphorylation (OXPHOS)/electron transport chain are more coordinately upregulated and positively correlated with those involved in a ROS‐related pathway in the livers of obese type 2 diabetic patients compared with those of non‐obese type 2 diabetic patients4. These findings suggest that mitochondria play a key role in governing energy homeostasis in the liver, and might be a potential therapeutic target for type 2 diabetes and NAFLD.
Recently, Burgess et al.9, 10 proposed that an enhanced OXPHOS pathway in the liver causes elevated hepatic glucose production, oxidative stress and inflammation. They focused on substrate fluxes in the mitochondrial tricarboxylic acid (TCA) cycle and OXPHOS. Mitochondria play a central role in energy production in whole tissues, mostly in the form of adenosine triphosphate, through the TCA cycle. As the TCA cycle is the major source of electrons for adenosine triphosphate production, a small percentage of electrons can ‘leak’ from the electron transport chain, composed of respiratory chain complex I–IV, to induce the formation of ROS, even during normal electron transport. For liver in an overnutrition state, lipid and glucose are insufficiently oxidized, leading to a decrease in the adenosine triphosphate production/oxygen consumption ratio, which leads to further ROS production in the mitochondria. Such excess ROS production can contribute to further hepatic mitochondrial dysfunction, and the development of hepatic insulin resistance and NAFLD. In addition to excess ROS formation, genetic factors, aging and reduced mitochondrial biogenesis also contribute to further mitochondrial dysfunction. Mitochondrial function in hepatic metabolic processes involves anaplerosis and cataplerosis pathways. The pathways are metabolic processes that allow 4‐carbon intermediates to move into (anaplerosis) and out of (cataplerosis) the TCA cycle. Anaplerosis and cataplerosis pathways should operate at the same flux to avoid accumulation of the intermediates in the TCA cycle (Figure 1).
Figure 1.
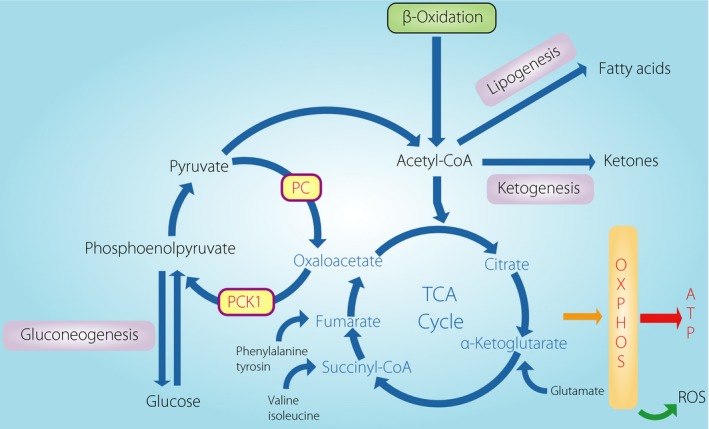
Schematic of the hepatic tricarboxylic acid (TCA) cycle with the major anaplerotic and cataplerotic pathways associated with glucogenonesis, lipogenesis, and ketogeneis. An elevation of fatty acid delivery amplifies the TCA cycle flux with a rise in anaplerosis/cataplerosis, leading to a proportional rise in oxidative stress and inflammation in liver. The redox state and intermetabolites in the TCA cycle amplify or suppress the flux. Oxidative stress is causal for further mitochondrial dysfunction. ATP, adenosine triphosphate; OXPHOS, oxidative phosphorylation; PC, pyruvate carboxylase; PCK1, phosphoenolpyruvate carboxykinase; ROS, reactive oxygen species.
Sunny et al.9 quantitated intrahepatic TG content (IHTG) using 1H magnetic resonance spectroscopy, and assayed anaplerotic substrate flux and cataplerotic substrate efflux in the mitochondrial TCA cycle by a tracer method using 2H and 13C in participants with NAFLD. Hepatic oxidative flux in the TCA cycle was doubly increased in participants with high IHTG compared with those with low IHTG; 50% higher rates of lipolysis increased mitochondrial anaplerosis, which increased cataplerotic efflux, leading to 30% higher rates of gluconeogenesis. There was a strong positive correlation between IHTG content and anaplerotic flux. Unexpectedly, ketone production assessed by tracer dilution of β‐hydroxybutylate was not different between participants with high and low IHTG, suggesting a relatively suppressed ketogenic pathway even under enhanced TCA cycle flux in human NAFLD.
Next, Satapati et al.10 examined the molecular mechanisms underlying increased hepatic anaplerotic flux in diet‐induced mouse and rat models of obesity. In concert with the aforementioned human data, ketogenesis, oxidative flux in the TCA cycle, anaplerosis, cataplerosis and gluconeogenesis were all increased in the livers from fed mice perfused with high NEFA (0.8 mmol/L) compared with those perfused with low NEFA (0.2 mmol/L). Although insulin administration to the liver perfused with high NEFA suppressed glycogenolysis, it did not prevent the elevation in anaplerosis and gluconeogenesis. Therefore, NEFA overload is considered sufficient to increase ex vivo hepatic oxidative metabolism, anaplerosis and gluconeogenesis. In agreement with the ex vivo liver perfusion, in the liver from rats in which circulating NEFA was increased by infusing intralipid for 6 h, ketone turnover, oxidative flux in the TCA cycle, and gluconeogenesis were increased together with increased circulating levels of TG and insulin. Thus, increased lipid delivery is sufficient to amplify anaplerosis and gluconeogenesis in vivo. In that in vivo model, messenger ribonucleic acid levels for mitochondrial superoxide dismutase 2, inflammatory cytokines, such as tnfa and Il6, and lipid peroxidation were elevated in proportion to the rise in oxygen consumption that is strongly associated with anaplerosis/cataplerosis flux and gluconeogenesis during intralipid infusion.
In an in vitro study using H4IIE hepatocytes10, NEFA increased ROS production, which was attenuated with an inhibitor for phosphoenolpyruvate carboxykinase (PEPCK), which links cataplerosis efflux from the TCA cycle and gluconeogenesis. Based on this in vitro finding, they investigated the therapeutic role of PEPCK in NAFLD in vivo. Systemic knockdown of Pck1 (PEPCK‐C) retained the ability to suppress hepatic glucose production during a hyperinsulinemic–euglycemic clamp study. Also, insulin‐induced phosphorylation of Akt was reduced in the liver of mice fed a high fat diet (HFD), which was rescued by Pck1 knockdown. Interestingly, Pck1 knockdown sufficiently prevented a fatty acid‐induced rise in anaplerosis/cataplerosis in mice fed a HFD, but not in mice fed a control diet. Why does Pck1 knockdown decrease anaplerosis/cataplerosis? TCA cycle oxidation is regulated by redox states and product inhibition11. Pck1 knockdown reduced the hepatic mitochondrial and cytosolic nicotinamide adenine dinucleotide/reduced nicotinamide adenine dinucleotide ratio estimated from the plasma acetoacetate/β‐hydroxybutylate and liver lactate/pyruvate ratio, respectively. Because nicotinamide adenine dinucleotide serves as a coenzyme for dehydrogenases in the TCA cycle, a reduced nicotinamide adenine dinucleotide/reduced nicotinamide adenine dinucleotide ratio suppresses TCA flux. Such a redox state is known to inhibit the forward reaction of the TCA cycle11. In addition, several TCA cycle metabolites are known to inhibit upstream enzymes, which is known as product inhibition11. Pck1 knockdown markedly increased oxaloacetate, one of the intermediate products produced during the TCA cycle, which inhibits upstream enzyme succinate dehydrogenase. As the TCA cycle is the major source of electrons for OXPHOS, a Pck1 knockdown‐mediated decrease in anaplerosis/cataplerosis might decrease ROS production through OXPHOS. Indeed, hepatic expression of the genes involved in oxidative stress, peroxidase and peroxiredoxins were upregulated by a HFD, which was canceled by Pck1 knockdown. Also, HFD‐induced lipid peroxidation was canceled by Pck1 knockdown. Pck1 knockdown resulted in a more oxidized redox state of mitochondrial inner membranes. Furthermore, a reduced cytosolic redox state is favorable for the reduced form of glutathione and the clearance of peroxide by its anti‐oxidant system. Pck1 knockdown accumulated some intermediates in the TCA cycle that activate anti‐oxidant transcription factors, such as nuclear respiratory factor 1 and nuclear factor, erythroid‐derived 2, like 2. Collectively, Pck1 knockdown reduces electron transport and activates anti‐oxidant systems, and thus reduces ROS levels in the liver. In agreement with low oxidative stress, Pck1 knockdown also protected mice against HFD‐induced inflammation.
Metformin suppresses hepatic glucose production, and is used as a first‐line oral antidiabetic agent for type 2 diabetes. Metformin might prevent a rise in anaplerosis/cataplerosis, as it suppresses Pck1 messenger ribonucleic acid levels in a concentration‐ and time‐dependent manner12. Indeed, Satapati et al.10 showed that metformin treatment reduces fasting hepatic gluconeogenesis by suppressing TCA flux and anaplorosis/cataplorosis in mice fed a HFD. Also, metformin reduced the hepatic expression of Tnfa and IL6 expressions in mice fed a HFD.
Finally, Satapati et al.10 examined the significance of oxidative metabolism in human NAFLD pathology. The histological NAFLD activity score of the human liver biopsy specimens correlated with oxygen consumption, indicating that activation of oxidative metabolism is associated with liver pathology in human NAFLD.
The Burgess et al.9, 10 linked mitochondrial oxidative metabolism, including amplified TCA cycle flux and anaplorosis/cataplerosis, to hepatic oxidative stress, inflammation and/or insulin resistance in mice infused with NEFA. Notably, substrate flux into ketogenesis is differently regulated in humans9 and mice10. How substrate flux is regulated still remains unclear. Metformin protects mice fed a HFD from oxidative stress and inflammation by reducing mitochondrial oxidative metabolism10. However, in a clinical setting, metformin seems unsatisfactory in ameliorating human NAFLD pathology13. Instead, they have proposed another candidate therapeutic target for reducing mitochondrial oxidative metabolism. Unexpectedly, inhibition of PEPCK protects HFD‐fed mice from oxidative stress and inflammation by reducing mitochondrial oxidative metabolism. As anaplerosis is elevated in people with NAFLD9, further studies are required to provide evidence of whether a PEPCK inhibitor is effective in ameliorating the pathology of NAFLD, including liver histology and insulin resistance, in humans.
References
- 1. Ota T, Takamura T, Kurita S, et al Insulin Resistance Accelerates a Dietary Rat Model of Nonalcoholic Steatohepatitis. Gastroenterology 2007; 132: 282–293. [DOI] [PubMed] [Google Scholar]
- 2. Matsuzawa N, Takamura T, Kurita S, et al Lipid‐induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatology 2007; 46: 1392–1403. [DOI] [PubMed] [Google Scholar]
- 3. Mootha VK, Lindgren CM, Eriksson KF, et al PGC‐1alpha‐responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003; 34: 267–273. [DOI] [PubMed] [Google Scholar]
- 4. Takamura T, Misu H, Matsuzawa‐nagata N, et al Obesity Upregulates Genes Involved in Oxidative Phosphorylation in Livers of Diabetic Patients. Obesity 2008; 16: 2601–2609. [DOI] [PubMed] [Google Scholar]
- 5. Kato KI, Takamura T, Takeshita Y, et al Ectopic fat accumulation and distant organ‐specific insulin resistance in Japanese people with nonalcoholic fatty liver disease. PLoS One 2014; 9: e92170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Donnelly KL, Smith CI, Schwarzenberg SJ, et al Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 2005; 115: 1343–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Takamura T, Misu H, Ota T, et al Fatty liver as a consequence and cause of insulin resistance: lessons from type 2 diabetic liver. Endocr J 2012; 59: 745–763. [DOI] [PubMed] [Google Scholar]
- 8. Nakamura S, Takamura T, Matsuzawa‐nagata N, et al Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. J Biol Chem 2009; 284: 14809–14818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sunny NE, Parks EJ, Browning JD, et al Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab 2011; 14: 804–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Satapati S, Kucejova B, Duarte JAG, et al Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J Clin Invest 2015; 125: 4447–4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Williamson JR, Cooper RH. Regulation of the citric acid cycle in mammalian systems. FEBS Lett 1980; 117:Suppl(S1) K73–K85. [DOI] [PubMed] [Google Scholar]
- 12. Takayama H, Misu H, Iwama H, et al Metformin suppresses expression of the Selenoprotein P gene via an AMP‐activated kinase (AMPK)/FoxO3a pathway in H4IIEC3 hepatocytes. J Biol Chem 2014; 289: 335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li Y, Liu L, Wang B, et al Metformin in non‐alcoholic fatty liver disease: a systematic review and meta‐analysis. Biomed Rep 2013; 1: 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]