

Abstract
Aims/Introduction
Dipeptidyl peptidase‐4 inhibitors might have pleiotropic protective effects on cardiovascular disease (CVD), in contrast to sulfonylureas. Therefore, we compared various CVD risk factors between vildagliptin and glimepiride.
Materials and Methods
We carried out a randomized, prospective and crossover trial. A total of 16 patients with type 2 diabetes whose glycated hemoglobin was >7% were randomized to add vildagliptin or glimepiride. After 12‐week treatment, each drug was replaced with the other for another 12 weeks. Before and after each treatment, glucose homeostasis and CVD risk factors were assessed, and the continuous glucose monitoring system was applied to calculate glycemic variability.
Results
The mean age of the participants was 60 years, 31% were men, body mass index 25.5 kg/m2 and HbA1c 8.41%. Both vildagliptin and glimepiride significantly decreased glycated hemoglobin and glycemic variability indices. Despite the improved glucose homeostasis, favorable change of CVD markers was not prominent in both the arms, along with significant weight gain. Only plasma stromal cell‐derived factor (SDF)‐1α decreased by 30% in the vildagliptin arm. According to regression analyses, the reduction of SDF‐1α was independently associated with vildagliptin usage and serum interleukin‐6 changes, but white blood cells were not related with the SDF‐1α changes.
Conclusion
Compared with glimepiride, vildagliptin arrestingly decreased plasma SDF‐1α, and its clinical implications should be further investigated.
Keywords: Cardiovascular risk, Stromal cell‐derived factor‐1α, Vildagliptin
Introduction
The most significant cause of mortality in diabetes mellitus is cardiovascular diseases (CVDs). Not only glycated hemoglobin (HbA1c) representing 3‐month mean blood glucose, but also postprandial hyperglycemia and hypoglycemia comprising glycemic variability (GV) have been reported to be independently associated with CVD1, 2. Additionally, dyslipidemia, inflammation, and oxidative stress can affect the development and prognosis of CVD3. Therefore, these factors should be taken into account in choosing treatment options for diabetes mellitus.
Dipeptidyl peptidase‐4 (DPP‐4) inhibitors inhibit degradation of incretin hormones and induce postprandial insulin secretion through augmented incretin effects4. Therefore, they would be preferable to the traditional insulin secretagogue, sulfonylureas, in terms of postprandial hyperglycemia and GV5, 6. In addition, DPP‐4 inhibitors are suggested to have various pleiotropic protective effects on the cardiovascular system7, whereas some sulfonylureas were reported to increase CVD compared with metformin8. However, the large clinical trials, Trial Evaluating Cardiovascular Outcomes with Sitagliptin (TECOS), Examination of Cardiovascular Outcomes with Alogliptin vs Standard of Care (EXAMINE) and The Saxagliptin Assessment of Vascular Outcomes Recorded in Patients with Diabetes Mellitus (SAVOR)–Thrombolysis in Myocardial Infarction (TIMI) 53, for CVD outcome of DPP‐4 inhibitors did not show their superiority compared with conventional antidiabetic agents.
There has been no report from prospective trials comparing CVD outcomes between DPP‐4 inhibitors and sulfonylureas: a head‐to‐head trial comparing linagliptin and glimepiride (CARdiovascular Outcome Trial of LINAgliptin vs Glimepiride in Type 2 Diabetes [CAROLINA] trial) has been ongoing since 2010, with a total of 6,041 patients9. A retrospective analysis using the Korean national health insurance claims database has shown an increased hazard ratio for sulfonylureas plus metformin compared with a DPP‐4 inhibitor plus metformin for total CVD10. Regarding CVD risk factors, DPP‐4 inhibitors have been reported to be favorable to body mass index (BMI), insulin resistance and triglyceride levels compared with sulfonylureas, whereas they were comparable in low‐density lipoprotein cholesterol (LDL‐C), high‐density lipoprotein cholesterol (HDL‐C), arterial stiffness, blood pressure, oxidative stress and high‐sensitivity C‐reactive protein6, 11, 12. Therefore, although DPP‐4 inhibitors logically have more favorable influences on CVD than sulfonylureas, clinical evidence is currently lacking. In the present study, we carried out a prospective and crossover study comparing various CVD risk factors between vildagliptin and glimepiride in patients with type 2 diabetes mellitus taking metformin.
Materials and Methods
Study design
We designed a prospective, open‐labeled, crossover trial (NCT01812122). Participants were recruited at Seoul National University Hospital from May 2013 through November 2014 by the staff of the diabetes clinic. Enrollment criteria were as follows: patients with type 2 diabetes mellitus aged 20–75 years, receiving metformin monotherapy for >3 months and HbA1c >7%. We excluded patients who had liver function abnormality (threefold higher than normal range), decreased kidney function (estimated glomerular filtration rate [eGFR] <60 mL/min/1.73 m2) and pancreatic diseases. Patients with malignancies, recent history of operation and medical treatment that could affect blood glucose levels were also excluded.
We calculated the sample size according to a hypothesis that the differences of CVD markers between glimepiride and vildagliptin would come from the differences of GV. For a significant difference (a two‐sided P‐value <0.05) of the mean amplitude of glycemic excursions (MAGE) by 1.1 mmol/L between the agents in a crossover trial, at least 14 participants were required to provide a power of at least 90%, according to previous reports13, 14. Including 20% dropout, we collected 18 participants.
Serial numbers of the participants were determined in order of enrollment consecutively, and odd‐number patients were to add glimepiride 1 mg twice daily for 12 weeks, and then switch to vildagliptin 50 mg twice daily for 12 weeks. Even‐number patients were vice versa: vildagliptin first, and then glimepiride. If hypoglycemia occurs with typical symptoms and self‐measured blood glucose <4.4 mmol/L, the dose of glimepiride or vildagliptin was reduced by half. At baseline and at each end of 12‐week treatment, anthropometric examinations and laboratory tests were carried out. The continuous glucose monitoring system (CGMS‐gold; Medtronic Minimed, Northridge, California, USA) was applied for three consecutive days, too (Figure S1).
Primary end‐points were traditional, and novel CVD risk factors (presented in Table 3) and secondary end‐points were composite CVD risk scores.
Clinical and laboratory parameters
Medical history, concomitant drugs and anthropometric measures, including blood pressure, heart rate, body weight, height and waist circumferences, were investigated by a trained coordinator at each visit.
After 12‐h overnight fasting, venous blood was collected and the following were measured: complete blood cell count with differential white blood cell (WBC) types (XE‐2100 Hematology Analyzer; Sysmex Corporation, Kobe, Japan) differentiates leukocytes by simultaneously measuring volume, structure and fluorescence), HbA1c (Variant tm II TURBO HbA1c kit 2.0; BIO‐RAD laboratories, Inc., Hercules, California, USA), 1,5‐anhydroglucitol (an enzymatic colorimetric assay kit; Kyowa Medex, Tokyo, Japan), insulin (DIAsource INS‐IRMA kit; Diasource Immuno Assays, Ottignies‐Louvain‐la‐Neuve, Belgium), fasting plasma glucose, aspartate aminotransferase, alanine aminotransferase (Shinyang Diagnostics, Seoul, Korea), creatinine (Jaffe method; Roche Crea, Roche Diagnostic, Basel, Switzerland), LDL‐C (RANDOX direct LDL cholesterol kit; Randox Laboratories Ltd, Crumlin, UK), HDL‐C (HDL‐C plusGen.3; Roche Diagnostic), high‐sensitivity C‐reactive protein (CRP‐latex(II)X2; latex‐enhanced turbidometric immunoassay; Denka Seiken Co., Ltd., Tokyo, Japan), lipoprotein(a) (Lp[a]; immunoturbidmetry; Roche Diagnostic), B‐type natriuretic peptide (chemiluminescent microparticle immunoassay using Abbot reagent and i2000 Architect analyzer; Abbott, Abbott Park, Illinois, USA), plasminogen activator inhibitor‐1 (enzyme‐linked immunosorbent assay using Asserachrom PAI‐1 kit; STAGO, Paris, France), interleukin (IL)‐6 and stromal cell‐derived factor‐1 alpha (SDF‐1α; enzyme‐linked immunosorbent assay kits; R&D System, Minneapolis, Minnesota, USA). Albumin and creatinine in the morning spot urine were also measured (immunoturbimetric assay, ALBT2; Roche, Basel, Switzerland; and Jaffe method, CREJ2; Roche, respectively) to calculate the albumin/creatinine ratio. The homeostasis model assessment of insulin resistance and homeostasis model assessment of β‐cell function were calculated by an equation using fasting glucose and insulin15. The eGFR was calculated by the Modification of Diet in Renal Disease method. Skewed variables were logarithmically converted for statistical analyses.
Calculation of CVD risk scores
We calculated Z‐scores16 of changes of each CVD marker, and summed them to create a compound CVD risk score. In the case of favorable factors (HDL‐C and eGFR), their Z‐scores were not added, but subtracted. Markers whose relationships with CVD in diabetes mellitus were not clear (Lp[a] and SDF‐1α) were not included in the calculation of the composite scores.
Calculation of GV from CGMS data
The standard deviation (SD), MAGE, continuous overall net glycemic action (CONGA)‐6 h, M100 and the area under the curve for blood glucose level ≥180 mg/L were calculated using the initial 48 h of the CGMS data2.
Statistical analysis
Data are expressed as mean ± standard deviation or median (range) or n (%) according to the variable's nature. Parametric and non‐parametric paired test was carried out to compare the values before and after treatment in each treatment arm. Serial changes of serum SDF‐1α were examined by the repeated measures anova. The Student's t‐test, Mann–Whitney test and χ2‐tests were used between the treatment arms, to compare continuous and categorical variables, respectively. The relationship of CVD markers and GV indices were identified using partial correlation analysis. Multiple linear regression analysis was carried out to identify variables that best predicted the change of SDF‐1α. Statistical analysis was carried out using Spss 20 (IBM Corp., Armonk, New York, USA) and GraphPad Prism 5 (GraphPad Software, La Jolla, California, USA). P < 0.05 was considered statistically significant.
Ethics
The study was approved by the institutional review board of Seoul National University Hospital (IRB number H‐1212‐042‐448), and was carried out according to the Declaration of Helsinki. All patients provided written informed consent.
Results
A total of 18 patients were enrolled; two dropped out due to follow‐up loss, and a total of 16 participants completed the study and were included in the final analysis. The baseline characteristics are shown in Table 1. Men constituted 31% of the sample, the mean age was 60.0 ± 9.6 years, BMI was 25.5 ± 4.1 kg/m2, duration of diabetes mellitus was 7.4 ± 5.2 years and the dose of metformin was 1,360 ± 490 mg/day. Although we did not intentionally exclude patients with CVD, there was no history of clinical CVD according to history taking and the medical records.
Table 1.
Baseline characteristic of the participants
| Variables | Values |
|---|---|
| Age (years) | 60.0 ± 9.6 |
| Men (%) | 31 |
| BMI (kg/m2) | 25.5 ± 4.1 |
| Diabetes duration (years) | 7.4 ± 5.2 |
| Hypertension (%) | 60 |
| History of CVD (%) | 0 |
| Diabetic retinopathy (%) | 13 |
| Urine albumin/creatinine (mg/g) | 58.3 ± 112.4 |
| eGFR (mL/min/1.73 m2) | 91.0 ± 21.4 |
| AST (IU/L) | 27 ± 17 |
| ALT (IU/L) | 30 ± 22 |
| Metformin dose (mg/day) | 1,360 ± 490 |
| ACE inhibitors and ARB use (%) | 53 |
| Statin use (%) | 67 |
Data are presented as mean value ± SD or number (%); n = 16. ACE, angiotensin‐converting enzyme; ALT, alanine transaminase; ARB, angiotensin II receptor blocker; AST, aspartate transaminase; BMI, body mass index; CVD, cardiovascular disease; eGFR, estimated glomerular filtration rate.
Because of hypoglycemic episodes, the mean dose of glimepiride became 1.45 ± 0.34 mg/day, and that of vildagliptin 80.4 ± 9.2 mg/day after 12‐week treatment. As a result, there was no statistical difference in the occurrence of hypoglycemia between the arms (Table 2). Both agents significantly decreased fasting plasma glucose and HbA1c, and increased 1,5‐anhydroglucitol and homeostasis model assessment of β‐cell function, but there was no difference between the agents, either. Mean blood glucose (MBG) and GV indices calculated from CGMS data also improved in both arms, except CONGA‐6 h; glimepiride did not change CONGA‐6 h significantly (Table 2). Even though the GV indices seemed better after the vildagliptin treatment, there was no statistical significance compared with the glimepiride. Although GV improved by both treatments, duration of hypoglycemia (glucose less than 4.4 mmol/L) increased regardless of the agents, suggesting the improved GV was mainly caused by a reduction of hyperglycemic surges.
Table 2.
Changes in glycemic control by glimepiride and vildagliptin
| Baseline | Glimepiride | Vildagliptin | P † | |
|---|---|---|---|---|
| Dose (mg/day) | NA | 1.45 ± 0.34 | 80.4 ± 9.2 | NA |
| Symptomatic hypoglycemia, n (12 weeks) | NA | 0.75 ± 1.24 | 0.44 ± 1.26 | 0.381 |
| SMBG at hypoglycemic episodes (mmol/L) | NA | 4.3 ± 0.7 | 3.8 ± 0.1 | 0.464 |
| FPG (mmol/L) | 9.6 ± 1.4 | 8.2 ± 1.8‡ | 7.7 ± 2.4‡ | 0.496 |
| HbA1c, % (mmol/mol) | 8.4 ± 0.9 (68.0 ± 5.6) | 6.7 ± 0.4§ (50.0 ± 2.5) | 6.6 ± 0.9§ (49.0 ± 5.6) | 0.799 |
| 1,5‐AG (μmol/L) | 34.8 ± 23.8 | 74.0 ± 56.6§ | 85.4 ± 42.2§ | 0.569 |
| HOMA‐B | 25.8 ± 14.4 | 53.0 ± 30.5§ | 61.5 ± 39.6§ | 0.499 |
| CGMS data | ||||
| Log(MBG) (mmol/L) | 1.03 ± 0.11 | 0.93 ± 0.09‡ | 0.91 ± 0.12‡ | 0.547 |
| Log(MAGE) (mmol/L) | 0.75 ± 0.17 | 0.64 ± 0.18‡ | 0.62 ± 0.19‡ | 0.768 |
| Log(SD) (mmol/L) | 0.38 ± 0.15 | 0.29 ± 0.15‡ | 0.25 ± 0.14‡ | 0.456 |
| CONGA‐6 (mmol/L) | 67.8 ± 31.5 | 54.3 ± 23.3 | 46.9 ± 23.2‡ | 0.373 |
| Log(M100) | 1.41 ± 0.44 | 0.99 ± 0.46§ | 0.87 ± 0.51§ | 0.493 |
| AUC180 (mmol/L∙min) | 6,103 ± 6,206 | 1,913 ± 2,566§ | 2,137 ± 4,129§ | 0.855 |
| Duration of glucose <4.4 mmol/L (min) | 8.14 ± 16.72 | 68.75 ± 184.81‡ | 86.56 ± 190.56‡ | 0.752 |
Data are presented as mean ± standard deviation. †Student's t‐test or Mann–Whitney test between glimepiride and vildagliptin. ‡ P < 0.05 vs baseline by paired t‐test or Wilcoxon matched‐pairs signed rank test. § P < 0.01 vs baseline by paired t‐test or Wilcoxon matched‐pairs signed rank test. 1,5‐AG, 1,5‐anhydroglucitol; AUC180, area under the curve for glucose above 180 mg/dL; CONGA‐6, continuous overlapping net glycemic action calculated with 6‐h time intervals; FPG, fasting plasma glucose; HOMA‐B, homeostasis model assessment for β‐cell function; M100, weighted average of glucose values; MAGE, mean amplitude glycemic excursion; MBG, mean blood glucose; NA, not applicable; SD, standard deviation; SMBG, self‐measured blood glucose.
Despite the significantly improved HbA1c and GV, favorable change of CVD risk factors was not prominent in both the arms (Table 3). When we analyzed the changes of traditional CVD risk factors, bodyweights significantly increased in both the arms. Among novel biomarkers recently observed to be related with CVD17, 18, 19, 20, 21, 22, resting heart rates increased in the glimepiride arm and Lp(a) increased in the vildagliptin arm, but the final measures were not different between the arms. The most remarkable finding was a reduction of SDF‐1α from 188.1 ± 31.2 to 133.8 ± 30.8 pmol/L, by 30% in the vildagliptin arm, causing a significant difference between the two agents (P = 0.005). The composite risk scores calculated excluding Lp(a) and SDF‐1α were comparable between the arms, too.
Table 3.
Changes in cardiovascular risk factors
| Baseline | Glimepiride | Vildagliptin | P † | |
|---|---|---|---|---|
| Traditional risk factors | ||||
| Weight (kg) | 65.8 ± 12.3 | 68.1 ± 12.7§ | 67.6 ± 12.8§ | 0.907 |
| Waist (cm) | 90.6 ± 8.7 | 92.0 ± 9.6‡ | 91.8 ± 9.4 | 0.946 |
| SBP (mmHg) | 125 ± 14 | 129 ± 18 | 126 ± 14 | 0.687 |
| DBP (mmHg) | 80 ± 12 | 82 ± 11 | 79 ± 10 | 0.481 |
| LDL‐C (mmol/L) | 2.36 ± 0.53 | 2.41 ± 0.38 | 2.38 ± 0.59 | 0.844 |
| HDL‐C (mmol/L) | 1.37 ± 0.35 | 1.43 ± 0.42 | 1.35 ± 0.30 | 0.551 |
| Log(HOMA‐IR) | 0.44 ± 0.22 | 0.58 ± 0.33 | 0.43 ± 0.33 | 0.215 |
| Novel biomarkers | ||||
| Pulse pressure (mmHg) | 45 ± 8 | 47 ± 14 | 47 ± 11 | 0.945 |
| Heart rate (b.p.m.) | 78 ± 9 | 82 ± 8‡ | 80 ± 11 | 0.495 |
| eGFR (mL/min/1.73 m2) | 91.0 ± 21.4 | 89.8 ± 21.7 | 88.0 ± 18.9 | 0.796 |
| Log(Lp[a]) (μmol/L¶) | −0.27 ± 0.46 | −0.21 ± 0.36 | −0.13 ± 0.32‡ | 0.514 |
| Log(IL‐6) (pmol/L) | −3.01 ± 1.48 | −3.31 ± 1.46 | −3.41 ± 1.48 | 0.842 |
| Log(hsCRP) (nmol/L) | −0.42 ± 0.72 | −0.61 ± 0.51 | −0.42 ± 0.56 | 0.323 |
| Log(PAI‐1) (pmol/L) | 2.56 ± 0.35 | 2.63 ± 0.20 | 2.63 ± 0.14 | 0.981 |
| Log(BNP) (pmol/L) | 1.44 ± 1.18 | 1.47 ± 0.88 | 1.33 ± 1.18 | 0.703 |
| SDF‐1α (pmol/L¶) | 188.1 ± 31.2 | 180.8 ± 47.7 | 133.8 ± 40.8§ | 0.005 |
| Risk scores | ||||
| Traditional factors | 0.46 ± 2.34 | −0.46 ± 2.78 | 0.317 | |
| Novel markers | 0.04 ± 2.73 | −0.04 ± 1.44 | 0.925 | |
| Entire markers | 0.50 ± 4.58 | −0.50 ± 2.80 | 0.464 | |
Data are presented as mean ± standard deviation. †Student's t‐test between glimepiride and vildagliptin. ‡ P < 0.05 vs baseline by paired t‐test. § P < 0.01 vs baseline by paired t‐test. ¶These variables were excluded in the calculation of risk scores. BNP, B‐type natriuretic peptide; DBP, diastolic blood pressure; HDL‐C, high‐density lipoprotein cholesterol; HOMA‐IR, homeostasis model assessment for insulin resistance; hsCRP, high‐sensitive C‐reactive protein; IL‐6, interleukin‐6; LDL‐C, low‐density lipoprotein cholesterol; Lp(a), lipoprotein(a); PAI‐1, plasminogen activator inhibitor‐1; SBP, systolic blood pressure; SDF‐1α, stromal cell‐derived factor‐1α.
When we showed the SDF‐1α levels separately according to the treatment order, a significant decrease in SDF‐1α was found only after vildagliptin, and then a subsequent switch to glimepiride recovered the levels (Figure 1). Therefore, lowering of SDF‐1α was a specific and reversible effect by vildagliptin. In partial correlation analyses using all the variables (presented in Tables 2 and 3), a change of SDF‐1α by either agent was associated with changes of LDL‐C (r = 0.361, P = 0.046), log(IL‐6) (r = 0.366, P = 0.043) and log(CONGA‐6) (r = 0.302, P = 0.098) after adjustment by the treatment of vildagliptin. Because SDF‐1α is a regulator of immune cells and platelets23, changes in total WBC counts, differential composition and platelet counts were analyzed with regard to SDF‐1α, but there was no significant result. In the multiple linear regression analyses with several independent variables (Table 4), vildagliptin use was the most powerful determinant of SDF‐1α change, and the change of IL‐6 was also a significant factor.
Figure 1.
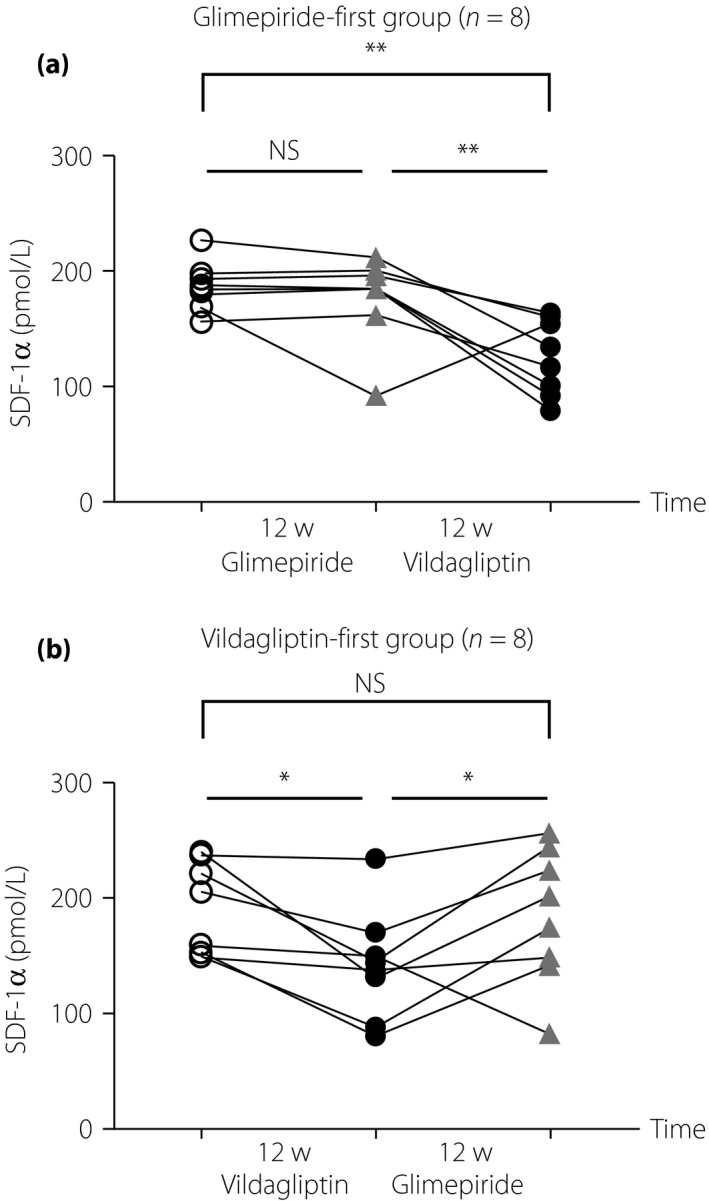
Changes of stromal cell‐derived factor‐1α (SDF‐1α) according to the treatment order. Changes of serum SDF‐1α are shown separately according to the first‐administered agent. (a) Glimepiride‐first group (n = 8). (b) Vildagliptin‐first group (n = 8). Open circles show the levels before any treatment, grey triangles, after glimepiride, and solid circles, after vildagliptin treatment. Means of the levels were significantly different by the repeated measures anova, and then post‐hoc Tukey's multiple comparison test was carried out. *P < 0.05; **P < 0.005. 12w, 12 weeks; NS, no significant difference.
Table 4.
Multiple linear regression analyses determining plasma stromal cell‐derived factor‐1α changes
| Independent variables | β | P‐value |
|---|---|---|
| Model 1† | ||
| Vildagliptin | −0.532 | <0.001 |
| ΔLDL‐C | 0.218 | 0.125 |
| ΔLog(IL‐6) | 0.327 | 0.018 |
| ΔLog(CONGA‐6) | 0.214 | 0.141 |
| Model 2‡ | ||
| Vildagliptin | −0.528 | <0.001 |
| ΔLDL‐C | 0.211 | 0.146 |
| ΔLog(IL‐6) | 0.319 | 0.024 |
| ΔLog(CONGA‐6) | 0.239 | 0.125 |
| 1,5‐AG | −0.073 | 0.604 |
| Model 3§ | ||
| Vildagliptin | −0.572 | <0.001 |
| ΔLDL‐C | 0.317 | 0.027 |
| ΔLog(IL‐6) | 0.352 | 0.027 |
| ΔLog(BNP) | −0.114 | 0.465 |
| Model 4¶ | ||
| Vildagliptin | −0.534 | 0.001 |
| ΔLDL‐C | 0.221 | 0.135 |
| ΔLog(IL‐6) | 0.345 | 0.027 |
| ΔLog(CONGA‐6) | 0.195 | 0.224 |
| Age | 0.010 | 0.947 |
| Sex | −0.049 | 0.752 |
†Adjusted R 2 = 0.489, F = 8.418; P < 0.001. ‡Adjusted R 2 = 0.475, F = 6.609; P < 0.001. §Adjusted R 2 = 0.457, F = 7.510; P < 0.001. ¶Adjusted R 2 = 0.451, F = 5.241; P = 0.001. 1,5‐AG, 1,5‐anhydroglucitol; BNP, B‐type natriuretic peptide; CONGA‐6, continuous overlapping net glycemic action calculated with 6‐h time intervals; IL‐6, interleukin‐6; LDL‐C, low‐density lipoprotein cholesterol.
Next, because GV has been suggested to increase oxidative stress and CVD risk, we examined the relationships of GV indices with CVD markers. Changes of the five indices – MAGE, SD, CONGA‐6, M100 and area under the curve for blood glucose level ≥180 mg/L – by either agent were positively correlated with not only each other, but also change of MBG (P < 0.05; data not shown). The changes of GV indices also showed a positive correlation with changes of fasting plasma glucose, and a negative correlation with changes of 1,5‐anhydroglucitol (P < 0.05, data not shown), as expected. Among the CVD risk factors, changes of HbA1c, plasminogen activator inhibitor‐1, heart rates and the composite risk scores were positively correlated with the changes of MBG, but change of Lp(a) was negatively correlated with it (Table S1). Among the GV indices, M100 and area under the curve for blood glucose level ≥180 mg/L showed similar patterns with MBG, and adjustment with the change of MBG left nothing significant. In the case of MAGE, the association between changes of MAGE and of LDL‐C was left significant after adjustment with the change of MBG. A change of SD was positively associated with novel score independently from the change of MBG. A negative correlation between SD change and eGFR change was presumed to result from the increase of weight and body surface area; further adjustment with weight change removed the statistical significance.
Discussion
In the present study, we could observe that there was a significant difference in the plasma levels of SDF‐1α between glimepiride and vildagliptin, even though their favorable effects on glycemia were comparable. Except for SDF‐1α levels, other clinical and laboratory factors related with CVD were not significantly different between the agents.
SDF‐1α/C‐X‐C motif chemokine 12 is a highly‐conserved chemokine, and the biological effects are mediated by the chemokine receptor, C‐X‐C chemokine receptor type 4. SDF‐1α is a major regulator of stem/progenitor cell trafficking in the bone marrow and tissues, suggesting its role in tissue regeneration, although it is also a potent platelet agonist highly expressed in atherosclerotic plaques, suggesting its contribution to atherogenesis23. SDF‐1 governs the homing of endothelial progenitor cells from bone marrow to areas of vascular injury for angiogenesis and repair. There is also an association among CXCL12 genetic variation, circulating SDF‐1 levels and circulating endothelial progenitor cells. Therefore, an understanding of SDF‐1α–CXCR4 signaling and associated biological functions with respect to CVD seems complicated now. Recently, in the 3,359 Framingham Heart Study participants, high plasma SDF‐1α was reported to be associated with older age, lower levels of HDL‐C, cigarette smoking and lower CD34+ cell frequency. Cox regression (median 9.3 years) showed that high plasma SDF‐1α was associated with heart failure and all‐cause mortality risk, but not with new‐onset CVD and myocardial infarction20. As the SDF‐1α was negatively correlated with circulating CD34+ frequency in the study, we could infer that constitutively high plasma SDF‐1α might reflect the reactive response to low circulating CD34+ cells, and the impaired regenerative capacity might induce heart failure rather than new‐onset coronary heart disease. Emerging data show that diabetes is associated with impaired bone marrow structure and function, attenuating vascular regenerative cells and contributing to vascular disease24. Delayed stem cell mobilization and/or impaired differentiation towards the endothelial phenotype in type 2 diabetes mellitus might increase plasma SDF‐1α; however, it has not been established.
Incretin‐based antidiabetic agents, DPP‐4 inhibitors’ effects on SDF‐1α have been examined, because SDF‐1α is one of the substrates of DPP‐4. DPP‐4 specifically cleaves dipeptides from substrates containing a penultimate proline or alanine residue at the NH2‐terminus. SDF‐1α and B‐type natriuretic peptide are regarded as important substrates of DPP‐4, inactivated by DPP‐4. As a result, they are supposed to mediate favorable effects of DPP‐4 inhibitors on CVD7. DPP‐4 inhibition around acute ischemic injury, such as hind limb ischemia and cardiac ischemia/reperfusion in animal models, has consistently enhanced recruitment of SDF‐1α and endothelial progenitor cells in the damaged tissue, promoting tissue regeneration25. Therefore, administration of DPP‐4 inhibitors in the case of chronic subclinical ischemia, such as diabetes, would also be expected to increase plasma SDF‐1α levels. However, in our participants, SDF‐1α levels were rather decreased by vildagliptin (Table 3). In the case of non‐diabetic HIV‐positive patients, sitagliptin treatment up to 24 weeks also decreased serum SDF‐1a levels compared with a placebo26. More recently, Aso et al.27 also found that sitagliptin to type 2 diabetes mellitus lowered plasma SDF‐1α levels compared with glimepiride. Long‐term administration of DPP‐4 inhibitors might induce such contradictory reduction, because active SDF‐1α levels acutely increased by 4‐day treatment of linagliptin28. However, SDF‐1α reduction could result from the assay method of the SDF‐1α levels. Most researchers including the present authors used an enzyme‐linked immunosorbent assay kit for total SDF‐1α from the same company, including both active and inactive forms. It is possible that DPP‐4 inhibitors increased active SDF‐1α causing a reactive reduction in total form, which is not clear now.
Anyway, several clinical studies in type 2 diabetes mellitus consistently showed that DPP‐4 inhibitors increased circulating stem/progenitor cell numbers27, 28, 29, 30, although causal relationships with SDF‐1α change were controversial. We could not check stem cell frequency, having only total WBC, lymphocytes and monocytes as available data. Vildagliptin decreased neutrophil frequency and increased monocyte frequency without effects on total WBC counts (data not shown), but any changes of these were not related to SDF‐1α change. We can speculate that decreased circulating stem cells in type 2 diabetes mellitus might induce plasma SDF‐1α20, and administration of DPP‐4 inhibitors decreased it by stem cell mobilization. There is a report that different DPP‐4 inhibitors had different effects on plasma SDF‐1α levels in type 2 diabetes mellitus31. If some DPP‐4 inhibitors differentially increase the risk of heart failure32, it would be related to the different effects on SDF‐1α involving tissue protection and angiogenesis, and progenitor cell recruitment might mediate the mechanisms33.
According to multiple regression analyses, plasma SDF‐1α was associated with serum IL‐6 independently from the use of vildagliptin (Table 4). IL‐6 is one of the inflammatory markers associated with CVD risk and mortality, and it has been recently suggested as the key causal cytokine compared with CRP and fibrinogen in the pathogenesis of CVD, by large‐scale human genetic and biomarker data34, 35. Specific interactions between IL‐6 and SDF‐1α have not been established in this setting, while there were some reports on the interrelationships between IL‐6 and SDF‐1α36, 37.
There were several unexpected findings for the CVD markers in the present study. Another study comparing high‐dose glimepiride (6 mg/day) and vildagliptin (100 mg/day) reported a better profile of weight and insulin resistance in the vildagliptin arm11. Unlike that study, we observed significant weight gain in both the arms similarly (Table 3). Although we did not quantitate food intake and physical activity in the present study, we presume that the weight gain was caused mainly from the improvement in hyperglycemia, because we did not reinforce concurrent lifestyle modification during the study, which can induce weight gain. The reason why the participants in the vildagliptin arm also gained weight seems to come from the crossover design in part; increased weight by glimepiride would not be easily lost by a switch to vildagliptin. In addition, there was no washout period in the study design, and the dose of glimepiride was much smaller than previous studies, which could induce no difference with vildagliptin in weight gain. This unexpected weight gain in the vildagliptin arm could have interfered with improvements in other CVD markers.
In addition, according to the literature, an increase of heart rate by sulfonylurea (Table 3) is not a usual finding, but enhanced sympathetic activity by glibenclamide has been described38. The small sample size could have also influenced this unexpected finding.
Another unexpected finding was the increase of Lp(a) after vildagliptin treatment (Table 3). Lp(a) changes were also negatively correlated with MBG changes (Table S1). Lp(a) is an LDL‐like particle consisting of an apolipoprotein. A moiety linked to one molecule of apolipoprotein B(100), and there has been highly suggestive evidence for a potentially causal role of Lp(a) in affecting CVD risk in general populations18, 39. However, plasma Lp(a) levels were observed to be inversely associated with type 2 diabetes mellitus, prediabetes and insulin resistance in several recent studies, and epidemiological studies of Lp(a) and CVD risk in diabetes mellitus generated inconsistent results. Lp(a) might differentially affect CVD risk between patients with diabetes mellitus and the general population39, 40.
Finally, contrary to previous reports between DPP‐4 inhibitors and sulfonylureas5, 6, there was no significant difference in the GV indices between glimepiride and vildagliptin (Table 2). Indeed, a study examining 5‐day effects of glimepiride and vildagliptin in well‐controlled patients (HbA1c 7.6%) also failed to show statistically significant differences in MAGE and SD between them41. Glucose fluctuation is composed of both hyperglycemia and hypoglycemia. We tried to prevent recurrent hypoglycemia by dose reduction in patients who had complained of it, as should be done in real‐world practice. As a result, the dose of glimepiride was lower than that usually used in clinical studies, and there was no significant difference in the degree and frequency of hypoglycemia between glimepiride and vildagliptin (Table 2). We speculate that the attenuated risk of hypoglycemia in the glimepiride arm could have improved GV. Among the GV indices, changes in MAGE and SD were associated with changes in LDL‐C and compound risk score by novel biomarkers, respectively (Table S1). MAGE has also been shown to be positively associated with oxidized LDL‐C in adolescents42. Therefore, among the indices examined in the present study, these two most popular GV indices seemed to be able to provide additional information about MBG with respect to CVD risk. However, because of the limitation of the small sample size of the present study, further investigation would be required for this issue.
There were several limitations to the present study; as for the study design, the participants were not blinded, not really randomized and did not undergo a washout period, although the duration of each treatment seemed long enough to countervail most effects of previous treatment. The small sample size seemed to contribute to the failure in obtaining statistically significant differences in most CVD markers between the agents. Another weak point is that there was no mechanistic study of the SDF‐1α change, leaving the clinical implications unclear.
In conclusion, in poorly‐controlled patients with type 2 diabetes mellitus without established CVD, vildagliptin decreased SDF‐1α, which has been reported to be positively associated with heart failure and mortality, whereas glimepiride did not. This change would be especially meaningful, because other CVD markers were not so significantly different in the present small study. Plasma SDF‐1α change was not related with glycemic control including GV, but serum IL‐6 change was closely and independently associated.
Disclosure
The authors declare no conflict of interest.
Supporting information
Table S1 ¦ Correlation analyses between glycemic variability indices and cardiovascular disease risk factors.
Figure S1 ¦ Diagram of the study design. CGMS, continuous glucose monitoring system; CVD, cardiovascular disease; HbA1c, glycated hemoglobin; T2DM, type 2 diabetes mellitus.
Acknowledgments
This study was supported by a grant from the Innovative Research Institute for Cell Therapy (A062260), and by a grant (KSP, 2013) from the Korean Diabetes Association. Clinical Trial Registration NCT01812122.
J Diabetes Investig 2017; 8: 218–226
References
- 1. Desouza CV, Bolli GB, Fonseca V. Hypoglycemia, diabetes, and cardiovascular events. Diabetes Care 2010; 33: 1389–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jung HS. Clinical implications of glucose variability: chronic complications of diabetes. Endocrinol Metab (Seoul) 2015; 30: 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ross R. Atherosclerosis–an inflammatory disease. New Engl J Med 1999; 340: 115–126. [DOI] [PubMed] [Google Scholar]
- 4. Drucker DJ. Dipeptidyl peptidase‐4 inhibition and the treatment of type 2 diabetes: preclinical biology and mechanisms of action. Diabetes Care 2007; 30: 1335–1343. [DOI] [PubMed] [Google Scholar]
- 5. Monnier L, Colette C, Comenducci A, et al Add‐on therapies to metformin in type 2 diabetes: what modulates the respective decrements in postprandial and basal glucose? Diabetes Technol Ther 2012; 14: 943–950. [DOI] [PubMed] [Google Scholar]
- 6. Kim HS, Shin JA, Lee SH, et al A comparative study of the effects of a dipeptidyl peptidase‐IV inhibitor and sulfonylurea on glucose variability in patients with type 2 diabetes with inadequate glycemic control on metformin. Diabetes Technol Ther 2013; 15: 810–816. [DOI] [PubMed] [Google Scholar]
- 7. Aroor AR, Sowers JR, Jia G, et al Pleiotropic effects of the dipeptidylpeptidase‐4 inhibitors on the cardiovascular system. Am J Physiol Heart Circ Physiol 2014; 307: H477–H492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schramm TK, Gislason GH, Vaag A, et al Mortality and cardiovascular risk associated with different insulin secretagogues compared with metformin in type 2 diabetes, with or without a previous myocardial infarction: a nationwide study. Eur Heart J 2011; 32: 1900–1908. [DOI] [PubMed] [Google Scholar]
- 9. Marx N, Rosenstock J, Kahn SE, et al Design and baseline characteristics of the CARdiovascular Outcome Trial of LINAgliptin Versus Glimepiride in Type 2 Diabetes (CAROLINA(R)). Diab Vasc Dis Res 2015; 12: 164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Seong JM, Choi NK, Shin JY, et al Differential cardiovascular outcomes after dipeptidyl peptidase‐4 inhibitor, sulfonylurea, and pioglitazone therapy, all in combination with metformin, for type 2 diabetes: a population‐based cohort study. PLoS One 2015; 10: e0124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Derosa G, Bonaventura A, Bianchi L, et al Vildagliptin compared to glimepiride on post‐prandial lipemia and on insulin resistance in type 2 diabetic patients. Metabolism 2014; 63: 957–967. [DOI] [PubMed] [Google Scholar]
- 12. Koren S, Shemesh‐Bar L, Tirosh A, et al The effect of sitagliptin versus glibenclamide on arterial stiffness, blood pressure, lipids, and inflammation in type 2 diabetes mellitus patients. Diabetes Technol Ther 2012; 14: 561–567. [DOI] [PubMed] [Google Scholar]
- 13. Irace C, Fiorentino R, Carallo C, et al Exenatide improves glycemic variability assessed by continuous glucose monitoring in subjects with type 2 diabetes. Diabetes Technol Ther 2011; 13: 1261–1263. [DOI] [PubMed] [Google Scholar]
- 14. Rizzo MR, Barbieri M, Marfella R, et al Reduction of oxidative stress and inflammation by blunting daily acute glucose fluctuations in patients with type 2 diabetes: role of dipeptidyl peptidase‐IV inhibition. Diabetes Care 2012; 35: 2076–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Matthews DR, Hosker JP, Rudenski AS, et al Homeostasis model assessment: insulin resistance and beta‐cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985; 28: 412–419. [DOI] [PubMed] [Google Scholar]
- 16. Barbieri M, Rizzo MR, Marfella R, et al Decreased carotid atherosclerotic process by control of daily acute glucose fluctuations in diabetic patients treated by DPP‐IV inhibitors. Atherosclerosis 2013; 227: 349–354. [DOI] [PubMed] [Google Scholar]
- 17. Di Angelantonio E, Chowdhury R, Sarwar N, et al B‐type natriuretic peptides and cardiovascular risk: systematic review and meta‐analysis of 40 prospective studies. Circulation 2009; 120: 2177–2187. [DOI] [PubMed] [Google Scholar]
- 18. Emerging Risk Factors Collaboration , Di Angelantonio E, Gao P, et al Lipid‐related markers and cardiovascular disease prediction. JAMA 2012; 307: 2499–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kodama S, Horikawa C, Fujihara K, et al Meta‐analysis of the quantitative relation between pulse pressure and mean arterial pressure and cardiovascular risk in patients with diabetes mellitus. Am J Cardiol 2014; 113: 1058–1065. [DOI] [PubMed] [Google Scholar]
- 20. Subramanian S, Liu C, Aviv A, et al Stromal cell‐derived factor 1 as a biomarker of heart failure and mortality risk. Arterioscler Thromb Vasc Biol 2014; 34: 2100–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tousoulis D, Papageorgiou N, Androulakis E, et al Diabetes mellitus‐associated vascular impairment: novel circulating biomarkers and therapeutic approaches. J Am Coll Cardiol 2013; 62: 667–676. [DOI] [PubMed] [Google Scholar]
- 22. Wulsin LR, Horn PS, Perry JL, et al Autonomic imbalance as a predictor of metabolic risks, cardiovascular disease, diabetes, and mortality autonomic imbalance predicts CVD, DM, Mortality. J Clin Endocrinol Metab 2015; 100: 2443–2448. [DOI] [PubMed] [Google Scholar]
- 23. Doring Y, Pawig L, Weber C, et al The CXCL12/CXCR4 chemokine ligand/receptor axis in cardiovascular disease. Front Physiol 2014; 5: 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fadini GP, Avogaro A. Dipeptidyl peptidase‐4 inhibition and vascular repair by mobilization of endogenous stem cells in diabetes and beyond. Atherosclerosis 2013; 229: 23–29. [DOI] [PubMed] [Google Scholar]
- 25. Matheeussen V, Jungraithmayr W, De Meester I. Dipeptidyl peptidase 4 as a therapeutic target in ischemia/reperfusion injury. Pharmacol Ther 2012; 136: 267–282. [DOI] [PubMed] [Google Scholar]
- 26. Goodwin SR, Reeds DN, Royal M, et al Dipeptidyl peptidase IV inhibition does not adversely affect immune or virological status in HIV infected men and women: a pilot safety study. J Clin Endocrinol Metab 2013; 98: 743–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aso Y, Jojima T, Iijima T, et al Sitagliptin, a dipeptidyl peptidase‐4 inhibitor, increases the number of circulating CD34(+)CXCR4(+) cells in patients with type 2 diabetes. Endocrine 2015; 50: 659–664. [DOI] [PubMed] [Google Scholar]
- 28. Fadini GP, Bonora BM, Cappellari R, et al Acute effects of linagliptin on progenitor cells, monocyte phenotypes, and soluble mediators in type 2 diabetes. J Clin Endocrinol Metab 2016; 101: 748–756. [DOI] [PubMed] [Google Scholar]
- 29. Fadini GP, Boscaro E, Albiero M, et al The oral dipeptidyl peptidase‐4 inhibitor sitagliptin increases circulating endothelial progenitor cells in patients with type 2 diabetes: possible role of stromal‐derived factor‐1alpha. Diabetes Care 2010; 33: 1607–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nakamura K, Oe H, Kihara H, et al DPP‐4 inhibitor and alpha‐glucosidase inhibitor equally improve endothelial function in patients with type 2 diabetes: EDGE study. Cardiovasc Diabetol 2014; 13: 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fujita H, Taniai H, Murayama H, et al DPP‐4 inhibition with alogliptin on top of angiotensin II type 1 receptor blockade ameliorates albuminuria via up‐regulation of SDF‐1alpha in type 2 diabetic patients with incipient nephropathy. Endocrine J 2014; 61: 159–166. [DOI] [PubMed] [Google Scholar]
- 32. Son JW, Kim S. Dipeptidyl peptidase 4 inhibitors and the risk of cardiovascular disease in patients with type 2 diabetes: a tale of three studies. Diabetes Metab J 2015; 39: 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shigeta T, Aoyama M, Bando YK, et al Dipeptidyl peptidase‐4 modulates left ventricular dysfunction in chronic heart failure via angiogenesis‐dependent and ‐independent actions. Circulation 2012; 126: 1838–1851. . [DOI] [PubMed] [Google Scholar]
- 34. Lowe G, Woodward M, Hillis G, et al Circulating inflammatory markers and the risk of vascular complications and mortality in people with type 2 diabetes and cardiovascular disease or risk factors: the ADVANCE study. Diabetes 2014; 63: 1115–1123. [DOI] [PubMed] [Google Scholar]
- 35. Collaboration IRGCERF , Sarwar N, Butterworth AS, et al Interleukin‐6 receptor pathways in coronary heart disease: a collaborative meta‐analysis of 82 studies. Lancet 2012; 379:1205–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Odemis V, Moepps B, Gierschik P, et al Interleukin‐6 and cAMP induce stromal cell‐derived factor‐1 chemotaxis in astroglia by up‐regulating CXCR4 cell surface expression. Implications for brain inflammation. J Biol Chem 2002; 277: 39801–39808. [DOI] [PubMed] [Google Scholar]
- 37. Chen HT, Tsou HK, Hsu CJ, et al Stromal cell‐derived factor‐1/CXCR4 promotes IL‐6 production in human synovial fibroblasts. J Cell Biochem 2011; 112: 1219–1227. [DOI] [PubMed] [Google Scholar]
- 38. Yosefy C, Magen E, Kiselevich A, et al Rosiglitazone improves, while Glibenclamide worsens blood pressure control in treated hypertensive diabetic and dyslipidemic subjects via modulation of insulin resistance and sympathetic activity. J Cardiovasc Pharmaco. 2004; 44: 215–222. [DOI] [PubMed] [Google Scholar]
- 39. Qi Q, Qi L. Lipoprotein(a) and cardiovascular disease in diabetic patients. Clin Lipidol 2012; 7: 397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ding L, Song A, Dai M, et al Serum lipoprotein (a) concentrations are inversely associated with T2D, prediabetes, and insulin resistance in a middle‐aged and elderly Chinese population. J Lipid Res 2015; 56: 920–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. He YL, Foteinos G, Neelakantham S, et al Differential effects of vildagliptin and glimepiride on glucose fluctuations in patients with type 2 diabetes mellitus assessed using continuous glucose monitoring. Diabetes Obes Metab 2013; 15: 1111–1119. [DOI] [PubMed] [Google Scholar]
- 42. Dasari PS, Gandomani BS, Teague AM, et al Glycemic variability is Aassociated with markers of vascular stress in adolescents. J Pediatr 2016; 172: 47–55. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 ¦ Correlation analyses between glycemic variability indices and cardiovascular disease risk factors.
Figure S1 ¦ Diagram of the study design. CGMS, continuous glucose monitoring system; CVD, cardiovascular disease; HbA1c, glycated hemoglobin; T2DM, type 2 diabetes mellitus.