

Abstract
Necroptosis is a caspase 8-independent cell death that requires co-activation of receptor-interacting protein (RIP) 1 and RIP 3 kinases. The necrosome is a complex consisting of RIP1, RIP3 and Fas-associated protein with death domain (FADD) leading to activation of the pseudokinase mixed lineage kinase like (MLKL) followed by a rapid plasma membrane rupture and inflammatory response through the release of damage-associated molecular patterns (DAMPs) and cytokines. The necrosome has been shown to be relevant in multiple tumor types, including pancreatic adenocarcinoma, melanoma and several hematological malignancies. Preclinical data suggest that targeting this complex can have differential impact on tumor progression and that the effect of necroptosis on oncogenesis is cell type- and context-dependent. The emerging data suggest that targeting the necrosome may lead to immunogenic reprogramming in the tumor microenvironment in multiple tumors and that combining therapies targeting the necrosome with either conventional chemotherapy or immunotherapy may have beneficial effects. Thus, understanding the interplay of necroptotic cell death, transformed cells, and the immune system may enable the development of novel therapeutic approaches.
Background
Apoptosis and necrosis are classically understood processes of cell death that, respectively, denote either an organized caspase 8-dependent programmed cell death or non-programmed haphazard cellular death, the latter of which commonly results from ischemic or traumatic injury. Apoptosis produces cellular fragments, denoted apoptotic bodies, which phagocytic cells are able to engulf before the contents of the cell spill into the interstitium and activate the innate immune system (1). Tumor cells are widely believed to die via caspase 8-dependant apoptotic programmed cell death (2). Chemotherapeutic agents are thought to further promote apoptosis in tumor cells (3, 4). As such, apoptosis is considered to be an essential regulatory mechanism, which delimits the rate of neoplasia and tumor progression. Conversely, progressive disruption of the apoptotic pathway via acquired mutations in genes such as p53, p16, or c-myc promotes tumor viability and precipitates accelerated oncogenesis (5, 6). By contrast, necrosis leads to the release of damage-associated molecular patterns (DAMPs) and cytokines and hence sustains inflammation (7). Necroptosis is a more recently described caspase 8-independent mode of cell death, which implies organized cellular necrosis and requires complex formation of the key signaling molecules receptor-interacting protein 1 (RIP1) and RIP3 kinases, called necrosome (8) (Figure 1).
Figure 1.
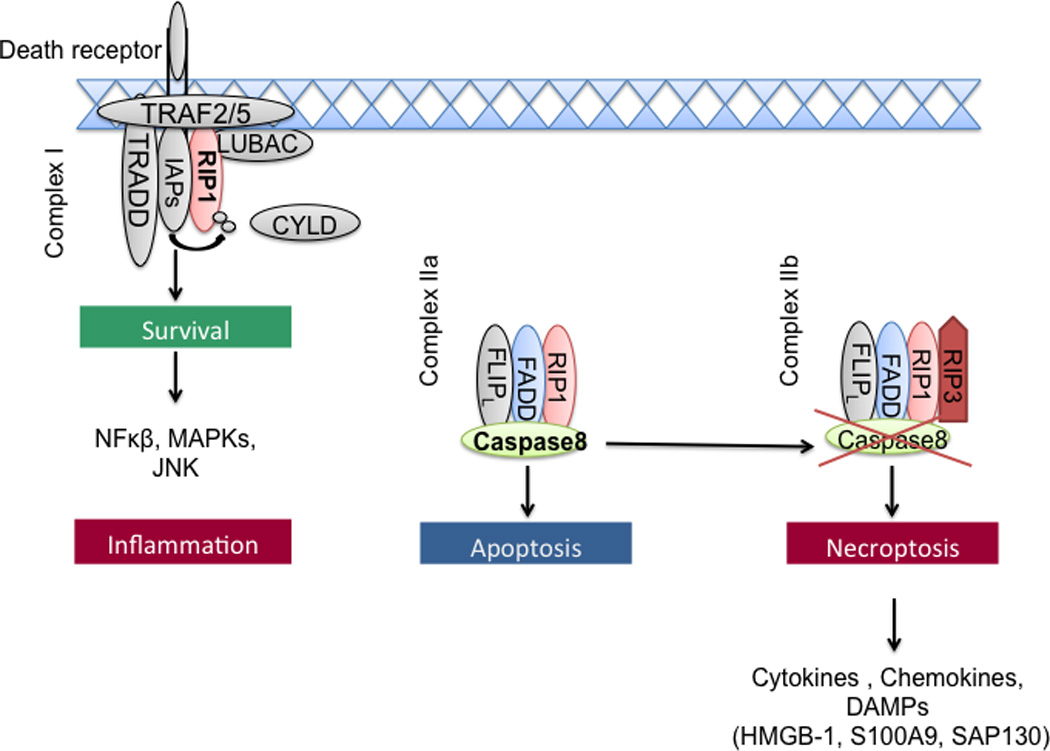
The necrototic pathway
Necroptotic pathway
Necroptotic cell death can be initiated by the ligation of death receptors (DRs) in the TNFR superfamily, including Fas (CD95), TNF receptor 1 (TNFR1), TNFR2, TNF-related apoptosis-inducing ligand receptor (TRAILR) 1 and TRAILR 2 as well as DR6. Members of the pathogen recognition receptor (PRR) family including Toll-like receptors (e.g. TLR3, TLR4) as well as NOD-like receptors (NLR) and several viral- or bacterial-associated pathogen-associated molecular patterns (PAMPs) have also been suggested to induce necrosome formation (9–13). Furthermore, a diversity of cellular stress signals can precipitate necroptosis, including reactive oxygen species, ischemia–reperfusion injury, anti-cancer drugs and chemotherapy including DNA-damaging agents, ionizing radiation, photodynamic therapy, and metabolic imbalances leading to glutamate or calcium overload (14). TNFR1 stimulation induced by cellular stress, damage and infection can either result in cell survival, apoptosis or necroptosis. Activation of TNFR1 induces ubiquitylation of RIP1 and facilitates pro-inflammatory signaling through the formation of the prosurvival complex (complex I) containing TNF receptor–associated death domain (TRADD), cellular inhibitor of apoptosis 1 (cIAP1), cIAP2, TNF receptor–associated factor 2 (TRAF2), TRAF5 and the linear ubiquitin chain assembly complex (LUBAC). This membrane-associated complex prevents cell death through activation of nuclear factor κB (NF-kB), mitogen–activated protein kinase (MAPK) or c-Jun N-terminal kinase (JNK) leading to inflammation (15). Upon deubiquitylation of RIP1 by the enzymes deubiquitinase cylindromatosis (CYLD) or A20, RIP1 kinase is recruited to a complex in the cytoplasm that includes Fas-associated death domain protein (FADD), caspase 8 and RIP3 (complex IIa/b), which results in necroptosis (16). In contrast, activation of Fas, TRAILR1 or TRAIL2 induces the death-inducing signaling complex (DISC) directly and consecutively leads to apoptotic cell death. The precise mechanisms determining the decision whether a cell will die by apoptosis or necroptosis a poorly understood. Caspase 8 is a crucial factor for preventing necroptosis and induces the apoptotic pathway by controlling the RIP1-RIP3 cleavage. RIP3 contains an N terminal kinase domain and a C terminal homotypic interaction domain (RHIM), which allows complex formation with RIP1 and is required for induction of necroptosis (17). The catalytical activity of caspase 8 requires high levels of FADD-like interleukin (IL)-1β-converting enzyme (FLICE)-inhibitory protein (FLIPL) (18). However, when caspase 8 activity is reduced or absent, the cellular response switches from apoptosis to necroptosis through the accumulation and RHIM-dependent phosphorylation of RIP3 (19). Further, the pseudokinase mixed lineage kinase like (MLKL) has been identified as a critical substrate of RIP3 during the induction of necroptosis. The phosphorylation of RIP3 results in the recruitment and phosphorylation of MLKL, which mediates the sodium influx through Ca2+ and Na+ ion channels. The increased osmotic pressure of necroptotically dying cells is leading to a rapid plasma membrane rupture and promotes inflammation by the release of substantial amounts of DAMPs, interleukin-1β (IL-1β) and other cytokines (20). Furthermore, the mitochondrial phosphatase phosphoglycerate mutase family member 5 (PGAM5), a regulator of dynamic-related protein-1 (DRP1), has been identified as another, but dispensable downstream substrate of necroptosis (21).
Clinical-Translational Advances
Recent studies have shown that necroptosis plays an important role in several inflammatory diseases, including acute pancreatitis, inflammatory bowel disease (IBD), inflammatory skin diseases, and liver injury (22–25). The significance of the necroptotic pathway in cancer biology has been investigated in preclinical studies of several hematological malignancies and solid tumors. Depending on the cancer cell-type and the tumor microenvironment (TME) necroptosis has differential impact on tumor progression. In several subtypes of acute myeloid leukemia (AML) the expression of RIP3 and MLKL is reduced in patients. A preclinical study showed that RIP3 inhibits malignant myeloproliferation in AML by promoting cell death of transformed progenitor cells and that the release of IL-1β by dying cells and subsequent the activation of the inflammasome promotes differentiation of leukemia-initiating cells (26). Further, in chronic lymphocytic leukemia (CLL) RIP3 was markedly downregulated through a transcription repressor of CYLD, called lymphoid enhancer-binding factor 1 (LEF1) (27). Knockdown of LEF1 sensitized CLL cells to TNFα-induced necroptosis. In T cell lymphomas, deletion or perturbation of RIP3 signaling promoted cellular growth and disease progression, a consequence of cells failing to undergo necroptosis (28). Moreover, in non-Hodgkin lymphoma, single nucleotide polymorphisms (SNPs) in the RIP3 gene were identified in a cohort of 458 patients and correlated with increased risk of lymphomagenesis, suggesting that genetic variations in the RIP3 gene may contribute to the etiology of this cancer (29). Based on these studies, triggering necroptosis seems to be effective in tumor cells in hematopoietic malignancies. In human and murine pancreatic adenocarcinoma (PDA) RIP1 and RIP3 were widely expressed (30). The necrosome complex formation was inducible by environmental stimuli or by chemotherapeutics. Blockade of the necrosome in PDA in a cell autonomous manner accelerated cellular proliferation and the acquisition of oncogenic changes in transformed pancreatic ductal epithelial cells. However, the in vivo effects were diametrically reversed with RIP3 deletion or RIP1 blockade resulting in marked tumor protection. The dichotomy between the in vivo and in vitro results suggests that the micro-environmental milieu resulting from RIP1/RIP3 signaling is likely responsible for its pro-tumorigenic effects. These results contrast with the aforementioned studies implicate defective RIP3-mediated necroptosis in both AML and CLL as well as in T cell lymphoma progression. Further, RIP3 inhibited hepatocarcinogenesis by limiting hepatocyte proliferation and mutagenesis (31). While these data ostensibly conflict with the in vivo results in PDA, unlike other extra-pancreatic malignancies, whose respective courses are dominated primarily by tumor cell biology, in PDA tumor progression is equally driven by inflammation within the TME (32). In humans, chronic pancreatitis is the most prevalent risk factor for the development of PDA and tumorigenesis is invariably accompanied by peri-pancreatic inflammatory changes (33, 34). Similarly, in Kras-driven murine models of PDA, ablation of the inflammatory mechanism abrogated oncogenic progression suggesting that inflammation not only accompanies oncogenesis but also is necessary for malignant degeneration (32). A recent study showed that human and murine melanoma cells induce necroptosis of endothelial cells, which promoted tumor cell extravasation and metastasis via amyloid precursor protein (APP) and its receptor DR6. Targeting DR6-mediated necroptosis inhibited tumor cell-induced necroptosis and the development of metastasis in melanoma (35). Additionally, RIP1 expression in melanoma was reported to promote tumor cell proliferation via NF-κB dependent pathways (36). In the context of PDA, RIP3 deletion led to immunogenic reprogramming of innate and adaptive inflammatory entities as evidenced by an increase in tumor infiltrating CD8+ T cells and Th1-polarized CD4+ T cells as well as a reduction in myeloid derived suppressor cells (MDSCs) and tumor associated macrophages (TAMs) (Figure 2). Furthermore, TAMs exhibited a shift toward an M1-like immunogenic phenotype. Cellular depletion experiments suggested that whereas inhibitory macrophages promote tumorigenesis, they lose their immune-suppressive effects in the context of RIP3 deletion leading to the generation of immunogenic T cells, which have powerful anti-tumor properties (30). Necroptosis has been linked to immune suppression and tumorigenesis in PDA via expression of CXCL1, a potent chemoattractant for myeloid cells that was highly expressed in a RIP1 and RIP3 dependent manner. CXCL1 has a complex role in extra-pancreatic malignancies. In breast cancer, metastatic cells over-expressing CXCL1 exhibited chemo-resistance via a paracrine loop by attracting Gr1+CD11b+ myeloid cells, which enhance cancer survival by secretion of S100A8/9 (37). Further, in ovarian cancer CXCL1 was found to induce epithelial cell proliferation by transactivation of EGFR (38). Similarly, in melanoma CXCL1 has a role in the genesis of primary melanocytic lesions when coupled with the loss of INK-4a/ARF (39). Further, CXCL1-related cytokines recruited immune-suppressive MDSC to the pre-metastatic niche in the liver of colorectal carcinoma bearing hosts (40). Chemotherapies upregulated the expression of CXCL1, whereas blocking CXCL1 in mice diminished MDSC infiltration and tumor progression. These data provide a rationale for targeting CXCL1 in combination with cytotoxic therapy. A recent study demonstrated an increased recruitment of T cells to the TME after blocking the CXCL1 receptor CXCR2, which is expressed on neutrophils and MDSC and whose expression correlates with poor prognosis in PDA (41). Further, blocking CXCR2 sensitized the tumor cells to gemcitabine and anti-programmed cell death protein 1 (anti-PD-1) (41). Thus, insights into immunological mechanisms can lead to the development of combinatorial therapeutic approaches that are effective against advanced cancers. Necroptosis is also accompanied by the release of SAP-130, a nuclear protein, that was recently identified as an endogenous ligand recognized by Mincle, a C-type lectin receptor necessary for immunity to mycobacterial pathogens, following necrotic cell death (42). However, Mincle is upregulated in tumor-infiltrating myeloid cells and promotes sterile inflammation by ligating SAP-130. Furthermore, Mincle ligation accelerates oncogenesis and is associated with myeloid cell mediated adaptive immune suppression by increased MDSC and M2-like macrophage infiltration, which vetoes T cell expansion and immunogenicity (30). Both SAP130 levels and Mincle signaling are reduced upon necrosome deletion or blockade. More broadly, the modifiable pro-tumorigenic role for Mincle activation may have implications for novel therapeutics in malignancies in which necroptosis and inflammation modulate tumorigenesis. It is important to realize that whereas Mincle ligation is a novel avenue, which is sufficient to modulate pancreatic oncogenesis, it is likely not the only pro-inflammatory pathway induced by necroptotic cell death. Notably, HMGB1 is elevated in multiple tumor types and ligation of its cognate receptor TLR4 or TLR9 are potently pro-tumorigenic (43–45). Hence, it is conceivable, if not likely, that RIP1/RIP3-mediated cell death results in the release of an array of DAMPs, which influence tumorigenesis by a multitude of mechanisms, which relate to activation of innate inflammatory signaling.
Figure 2.
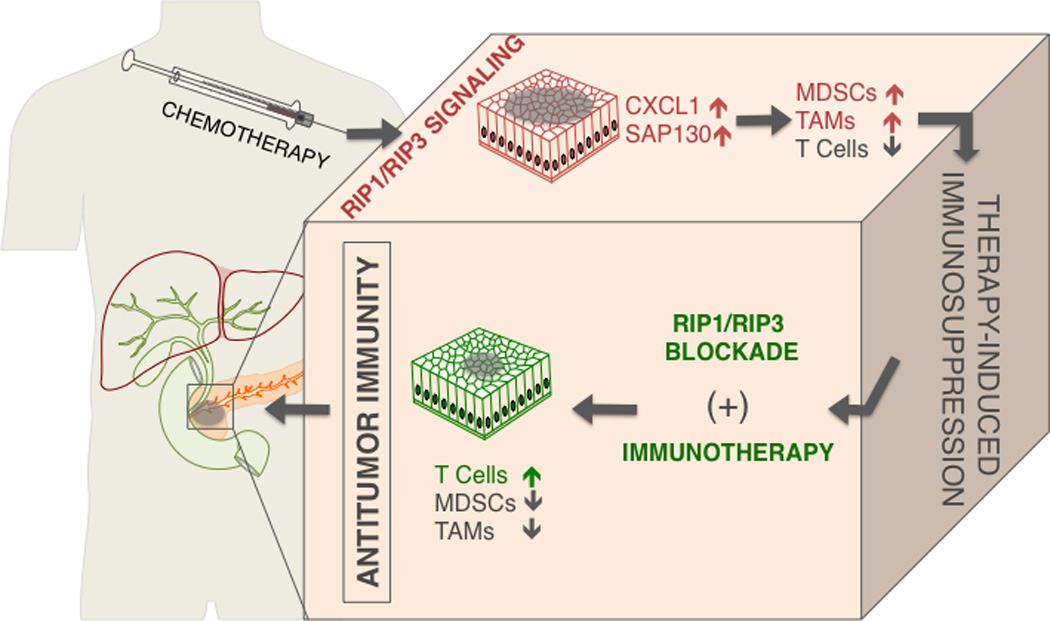
The necrosome may herald an exciting new era in cancer therapy. There is ample rationale for potential synergistic efficacy of targeting the necrosome in combination with available check-point-directed immunotherapy
Future Perspectives
The advent of clinical grade therapeutics targeting components of the necrosome may herald an exciting new era in cancer therapy. Further, there is ample rationale for potential synergistic efficacy of targeting the necrosome in combination with available check-point-directed immunotherapy. Ligation of T cell checkpoint receptors, including cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and PD-1 are known to dampen the activation of T cells and deliver inhibitory signals to the T cells (46–49). Monoclonal antibodies directed at T cell checkpoint receptors have shown significant antitumor effects in several solid tumors particularly melanoma, bladder, kidney and lung cancer (50–53). However, application of T cell checkpoint blockade in many other cancers, including PDA, has had minimal impact in early clinical studies (54, 55). The poor efficacy of checkpoint-based immunotherapeutics as single agents likely relates to the scarcity and poor activation of T cells at baseline in the TME. Thus, expansion and activation of T cells via blockade of necroptosis is an exciting potential avenue to increase T cell activity and enable efficacy of immunotherapeutics.
Acknowledgments
Funding: The Pancreatic Cancer Action Network (GM), and National Institute of Health Awards CA155649 (GM), and CA168611 (GM).
Footnotes
The authors have no conflicts of interest
References
- 1.Poon IK, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nature reviews Immunology. 2014;14:166–180. doi: 10.1038/nri3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lowe SW, Lin AW. Apoptosis in cancer. Carcinogenesis. 2000;21:485–495. doi: 10.1093/carcin/21.3.485. [DOI] [PubMed] [Google Scholar]
- 3.Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108:153–164. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 4.Pajak B, Orzechowski A. Overview how adenocarcinoma cancer cells avoid immune- and chemotherapy-induced apoptosis. Adv Med Sci. 2006;51:39–45. [PubMed] [Google Scholar]
- 5.Westermarck J. Regulation of transcription factor function by targeted protein degradation: an overview focusing on p53, c-Myc, and c-Jun. Methods Mol Biol. 2010;647:31–36. doi: 10.1007/978-1-60761-738-9_2. [DOI] [PubMed] [Google Scholar]
- 6.Gukovsky I, Li N, Todoric J, Gukovskaya A, Karin M. Inflammation, autophagy, and obesity: common features in the pathogenesis of pancreatitis and pancreatic cancer. Gastroenterology. 2013;144:1199 e4–1209 e4. doi: 10.1053/j.gastro.2013.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bustin M. At the crossroads of necrosis and apoptosis: signaling to multiple cellular targets by HMGB1. Science's STKE : signal transduction knowledge environment. 2002;2002:pe39. doi: 10.1126/stke.2002.151.pe39. [DOI] [PubMed] [Google Scholar]
- 8.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 9.Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013;288:31268–31279. doi: 10.1074/jbc.M113.462341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kalai M, Van Loo G, Vanden Berghe T, Meeus A, Burm W, Saelens X, et al. Tipping the balance between necrosis and apoptosis in human and murine cells treated with interferon and dsRNA. Cell Death Differ. 2002;9:981–994. doi: 10.1038/sj.cdd.4401051. [DOI] [PubMed] [Google Scholar]
- 11.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 12.Wilson NS, Dixit V, Ashkenazi A. Death receptor signal transducers: nodes of coordination in immune signaling networks. Nat Immunol. 2009;10:348–355. doi: 10.1038/ni.1714. [DOI] [PubMed] [Google Scholar]
- 13.Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–495. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 14.Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15:135–147. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- 15.Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell. 2009;36:831–844. doi: 10.1016/j.molcel.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 16.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 17.Sun X, Yin J, Starovasnik MA, Fairbrother WJ, Dixit VM. Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J Biol Chem. 2002;277:9505–9511. doi: 10.1074/jbc.M109488200. [DOI] [PubMed] [Google Scholar]
- 18.Pop C, Oberst A, Drag M, Van Raam BJ, Riedl SJ, Green DR, et al. FLIP(L) induces caspase 8 activity in the absence of interdomain caspase 8 cleavage and alters substrate specificity. Biochem J. 2011;433:447–457. doi: 10.1042/BJ20101738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16:55–65. doi: 10.1038/ncb2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moriwaki K, Farias Luz N, Balaji S, De Rosa MJ, O'Donnell CL, Gough PJ, et al. The Mitochondrial Phosphatase PGAM5 Is Dispensable for Necroptosis but Promotes Inflammasome Activation in Macrophages. J Immunol. 2016;196:407–415. doi: 10.4049/jimmunol.1501662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dannappel M, Vlantis K, Kumari S, Polykratis A, Kim C, Wachsmuth L, et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature. 2014;513:90–94. doi: 10.1038/nature13608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deutsch M, Graffeo CS, Rokosh R, Pansari M, Ochi A, Levie EM, et al. Divergent effects of RIP1 or RIP3 blockade in murine models of acute liver injury. Cell Death Dis. 2015;6:e1759. doi: 10.1038/cddis.2015.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nunes T, Bernardazzi C, de Souza HS. Cell death and inflammatory bowel diseases: apoptosis, necrosis, and autophagy in the intestinal epithelium. Biomed Res Int. 2014;2014:218493. doi: 10.1155/2014/218493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He S, Wang L, Miao L, Wang T, Du F, Zhao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 26.Hockendorf U, Yabal M, Herold T, Munkhbaatar E, Rott S, Jilg S, et al. RIPK3 Restricts Myeloid Leukemogenesis by Promoting Cell Death and Differentiation of Leukemia Initiating Cells. Cancer Cell. 2016;30:75–91. doi: 10.1016/j.ccell.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 27.Liu P, Xu B, Shen W, Zhu H, Wu W, Fu Y, et al. Dysregulation of TNFalpha-induced necroptotic signaling in chronic lymphocytic leukemia: suppression of CYLD gene by LEF1. Leukemia. 2012;26:1293–1300. doi: 10.1038/leu.2011.357. [DOI] [PubMed] [Google Scholar]
- 28.Kikuchi M, Kuroki S, Kayama M, Sakaguchi S, Lee KK, Yonehara S. Protease activity of procaspase-8 is essential for cell survival by inhibiting both apoptotic and nonapoptotic cell death dependent on receptor-interacting protein kinase 1 (RIP1) and RIP3. J Biol Chem. 2012;287:41165–41173. doi: 10.1074/jbc.M112.419747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cerhan JR, Ansell SM, Fredericksen ZS, Kay NE, Liebow M, Call TG, et al. Genetic variation in 1253 immune and inflammation genes and risk of non-Hodgkin lymphoma. Blood. 2007;110:4455–4463. doi: 10.1182/blood-2007-05-088682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seifert L, Werba G, Tiwari S, Giao Ly NN, Alothman S, Alqunaibit D, et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature. 2016;532:245–249. doi: 10.1038/nature17403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vucur M, Reisinger F, Gautheron J, Janssen J, Roderburg C, Cardenas DV, et al. RIP3 inhibits inflammatory hepatocarcinogenesis but promotes cholestasis by controlling caspase-8- and JNK-dependent compensatory cell proliferation. Cell Rep. 2013;4:776–790. doi: 10.1016/j.celrep.2013.07.035. [DOI] [PubMed] [Google Scholar]
- 32.Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 33.Hart AR, Kennedy H, Harvey I. Pancreatic cancer: a review of the evidence on causation. Clin Gastroenterol Hepatol. 2008;6:275–282. doi: 10.1016/j.cgh.2007.12.041. [DOI] [PubMed] [Google Scholar]
- 34.Lowenfels AB, Maisonneuve P, DiMagno EP, Elitsur Y, Gates LK, Jr, Perrault J, et al. Hereditary pancreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. J Natl Cancer Inst. 1997;89:442–446. doi: 10.1093/jnci/89.6.442. [DOI] [PubMed] [Google Scholar]
- 35.Strilic B, Yang L, Albarran-Juarez J, Wachsmuth L, Han K, Muller UC, et al. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature. 2016;536:215–218. doi: 10.1038/nature19076. [DOI] [PubMed] [Google Scholar]
- 36.Liu XY, Lai F, Yan XG, Jiang CC, Guo ST, Wang CY, et al. RIP1 Kinase Is an Oncogenic Driver in Melanoma. Cancer Res. 2015 doi: 10.1158/0008-5472.CAN-14-2199. [DOI] [PubMed] [Google Scholar]
- 37.Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris PG, et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell. 2012;150:165–178. doi: 10.1016/j.cell.2012.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bolitho C, Hahn MA, Baxter RC, Marsh DJ. The chemokine CXCL1 induces proliferation in epithelial ovarian cancer cells by transactivation of the epidermal growth factor receptor. Endocrine-related cancer. 2010;17:929–940. doi: 10.1677/ERC-10-0107. [DOI] [PubMed] [Google Scholar]
- 39.Yang J, Luan J, Yu Y, Li C, DePinho RA, Chin L, et al. Induction of melanoma in murine macrophage inflammatory protein 2 transgenic mice heterozygous for inhibitor of kinase/alternate reading frame. Cancer Res. 2001;61:8150–8157. [PubMed] [Google Scholar]
- 40.Connolly MK, Mallen-St Clair J, Bedrosian AS, Malhotra A, Vera V, Ibrahim J, et al. Distinct populations of metastases-enabling myeloid cells expand in the liver of mice harboring invasive and preinvasive intra-abdominal tumor. J Leukoc Biol. 2010;87:713–725. doi: 10.1189/jlb.0909607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steele CW, Karim SA, Leach JD, Bailey P, Upstill-Goddard R, Rishi L, et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2016;29:832–845. doi: 10.1016/j.ccell.2016.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamasaki S, Ishikawa E, Sakuma M, Hara H, Ogata K, Saito T. Mincle is an ITAM-coupled activating receptor that senses damaged cells. Nat Immunol. 2008;9:1179–1188. doi: 10.1038/ni.1651. [DOI] [PubMed] [Google Scholar]
- 43.Ochi A, Nguyen AH, Bedrosian AS, Mushlin HM, Zarbakhsh S, Barilla R, et al. MyD88 inhibition amplifies dendritic cell capacity to promote pancreatic carcinogenesis via Th2 cells. J Exp Med. 2012;209:1671–1687. doi: 10.1084/jem.20111706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zambirinis CP, Levie E, Nguy S, Avanzi A, Barilla R, Xu Y, et al. TLR9 ligation in pancreatic stellate cells promotes tumorigenesis. J Exp Med. 2015;212:2077–2094. doi: 10.1084/jem.20142162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Apetoh L, Ghiringhelli F, Tesniere A, Criollo A, Ortiz C, Lidereau R, et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol Rev. 2007;220:47–59. doi: 10.1111/j.1600-065X.2007.00573.x. [DOI] [PubMed] [Google Scholar]
- 46.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 47.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 48.Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, et al. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 2004;64:1140–1145. doi: 10.1158/0008-5472.can-03-3259. [DOI] [PubMed] [Google Scholar]
- 49.Schneider H, Downey J, Smith A, Zinselmeyer BH, Rush C, Brewer JM, et al. Reversal of the TCR stop signal by CTLA-4. Science. 2006;313:1972–1975. doi: 10.1126/science.1131078. [DOI] [PubMed] [Google Scholar]
- 50.Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–144. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515:558–562. doi: 10.1038/nature13904. [DOI] [PubMed] [Google Scholar]
- 53.McDermott DF, Drake CG, Sznol M, Choueiri TK, Powderly JD, Smith DC, et al. Survival, Durable Response, and Long-Term Safety in Patients With Previously Treated Advanced Renal Cell Carcinoma Receiving Nivolumab. J Clin Oncol. 2015;33:2013–2020. doi: 10.1200/JCO.2014.58.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother. 2010;33:828–833. doi: 10.1097/CJI.0b013e3181eec14c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]