

Abstract
Moebius syndrome is characterized by congenital unilateral or bilateral facial and abducens nerve palsies (sixth and seventh cranial nerves) causing facial weakness, feeding difficulties, and restricted ocular movements. Abnormalities of the chest wall such as Poland anomaly and variable limb defects are frequently associated with this syndrome. Most cases are isolated; however, rare families with autosomal dominant transmission with incomplete penetrance and variable expressivity have been described. The genetic basis of this condition remains unknown. In a cohort study of nine individuals suspected to have Moebius syndrome (six typical, three atypical), we performed whole-exome sequencing to try to identify a commonly mutated gene. Although no such gene was identified and we did not find mutations in PLXND1 and REV3L, we found a de novo heterozygous mutation, p.E410K, in the gene encoding tubulin beta 3 class III (TUBB3), in an individual with atypical Moebius syndrome. This individual was diagnosed with near-complete ophthalmoplegia, agenesis of the corpus callosum, and absence of the septum pellucidum. No substantial limb abnormalities were noted. Mutations in TUBB3 have been associated with complex cortical dysplasia and other brain malformations and congenital fibrosis of extraocular muscles type 3A (CFEOM3A). Our report highlights the overlap of genetic etiology and clinical differences between CFEOM and Moebius syndrome and describes our approach to identifying candidate genes for typical and atypical Moebius syndrome.
Keywords: aplasia/hypoplasia involving bones of the lower limbs, aplasia/hypoplasia involving bones of the upper limbs, aplasia/hypoplasia of the extremities, congenital extraocular muscle anomaly, congenital fibrosis of extraocular muscles
INTRODUCTION
Moebius syndrome was first described in the 1880s by Moebius and von Graefe, with cardinal features of facial and abducens nerve palsies (Möbius 1888; von Graefe 1880). Subsequently, additional clinical features were recognized within the diagnostic spectrum, including other cranial nerve (CN) defects, orofacial malformations, and skeletal abnormalities such as limb defects and chest wall abnormalities (Sprofkin and Hillman 1956; Wishnick et al. 1981; Stabile et al. 1984). Stromland et al. (2002) studied 25 Swedish individuals with Moebius syndrome and reported abnormalities of abducens and facial nerve involvement in all the affected individuals (100%). Speech was impaired in 17/22 (77%) patients, and malformations of the tongue including tongue atrophy, impaired motility, and/or hypoglossal nerve involvement occurred in about two-thirds of cases. Limb malformations were seen in 10/25 (40%) individuals, such as talipes equinovarus deformity, syndactyly, and oligodactyly. Poland anomaly was noted in 2/25 cases. Periodontal disease was observed frequently in the patients because of impaired motility of facial and tongue muscles. Less frequent abnormalities included cleft lip/palate and sensorineural hearing loss.
Several features associated with Moebius syndrome are considered to derive from abnormal development of the rhombencephalon or hindbrain, which develops into the medulla, pons, and cerebellum (Verzijl et al. 2003). Although the pathogenesis remains unknown, an intrauterine vascular event linked to subclavian artery disruption or a toxic injury in the critical period during the first trimester has been proposed (Dunham and Spellacy 1967). Misoprostol, thalidomide, and maternal cocaine exposure have all been implicated in vascular events affecting the sixth and seventh cranial nerves and limb defects (Kankirawatana et al. 1993; Puvabanditsin et al. 2005; Miller et al. 2009). In most cases, Moebius syndrome occurs as an isolated event. Autosomal dominant transmission in rare pedigrees (Krueger and Friedrich 1963; Dotti et al. 1989; Rojas-Martinez et al. 1991; Verzijl et al. 1999) and chromosomal abnormalities reported in singleton and familial cases (Ziter et al. 1977; Slee et al. 1991; Borck et al. 2001) imply genetic factors that likely contribute to disease causation. MacDermot et al. (1991) reported clinical findings in 31 affected individuals and concluded that, in cases with combined cranial nerve palsies and skeletal abnormalities, no familial transmission was observed. On the contrary, a mother and son were reported to have facial and ocular involvement with skeletal features (Graziadio et al. 2010). These examples highlight the clinical variability, potentially reflecting genetic heterogeneity in this disorder.
Several genetic loci have been implicated in Moebius syndrome based on cytogenetic anomalies described in rare families. A reciprocal translocation t(1;13)(p34;q13) was reported to segregate with congenital facial diplegia and flexion contractures of the fingers in multiple affected members spanning three generations (Ziter et al. 1977). A case report of a female with a deletion in 13q12.2 seemed to provide further evidence of a critical locus in the 13q12.2-13 region (Slee et al. 1991). A reciprocal translocation t(1;2)(p22.3;q21.1) was identified in a male with a Moebius-like syndrome, presenting with bilateral facial nerve palsy, ptosis, anteverted nostrils, and malformed and low-set ears (Nishikawa et al. 1997). Other chromosomal aberrations reported in rare cases include paracentric inversion of Chromosome 8 (inv(8) (q21.3q24.13)) (Kersey et al. 2006) and complex chromosomal rearrangement t(7;8;11;13) (Borck et al. 2001). The three Moebius syndrome loci have been defined as MBS1 (MIM 157900) at 13q12.2-q13 identified by Slee et al. (1991), MBS2 or HCFP1 (hereditary congenital facial paresis 1) (MIM 601471) at 3q21-q22 after a report by Kremer et al. (1996), and MBS3 or HCFP2 (hereditary congenital facial paresis 2) (MIM 604185) at 10q21.3-q22.1 from a report by Verzijl et al. (1999) of dominant Moebius syndrome in a Dutch family.
Despite numerous investigations, the molecular etiology of Moebius syndrome remains elusive. We performed an exome-sequencing study in a cohort of nine affected individuals because we hypothesized we could thus identify a commonly mutated gene. We describe an individual with a heterozygous de novo mutation in TUBB3 diagnosed with “atypical” Moebius syndrome. His phenotype included bilateral ocular anomalies with near-complete ophthalmoplegia, agenesis of the corpus callosum, and absence of the septum pellucidum. We also describe the clinical features of our cohort of Moebius syndrome and report whole-exome-sequencing (WES) findings in our study cohort.
RESULTS
Clinical Description of Cohort
Our study cohort included three females and six males, predominantly of European and Hispanic ethnicity ascertained by clinicians of the Baylor College of Medicine from May 2009 to April 2014 (pictures in Fig. 1 and clinical details in Table 1). Clinical details were compiled by the clinicians, and blood was drawn for DNA extraction.
Figure 1.

Pictures of the nine patients described in this manuscript: (A) individual 3, (B) individual 4, (C) individual 2, (D) individual 1, (E) individual 5, (F) individual 6, (G) individual 7, (H) individual 8, and (I) individual 9.
Table 1.
Clinical features of patients enrolled for exome sequencing
| Family and patient number | Figure 1 | Ethnicity and gender | Typical/atypical Moebius syndrome | FH of Moebius | Cranial nerve palsy | Comments (cranial nerves) | Hypoplastic tongue | Feeding problems | Brain MRI abnormalities |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1D | Hispanic female | Typical | Father has strabismus | VI, VII | + | + | Normally formed brain without any significant brain parenchymal abnormalities | |
| 2 | 1C | Hispanic male | Typical | − | VI, VII | + | + | Mild brainstem thinning, nonvisualization of the cisternal abducens and facial nerves | |
| 3 | 1A | Caucasian male | Typical | − | VI, VII | + | + | Absent bilateral cranial nerves VI and VII, absent facial colliculi, and pontine tegmental hypoplasia | |
| 4 | 1B | Hispanic male | Typical | Mother has congenital left thumb hypoplasia | VI, VII | Aberrant tearing, gaze palsy | Ankyloglossia | + | Diminutive pons |
| 5 | 1E | Hispanic female | Typical | − | VI, VII | Unknown | Unknown | Within normal limits, right vertebral artery hypoplastic | |
| 6 | 1F | Hispanic male | Atypical | − | All EOMs except the lateral rectus muscles, are nonfunctional, fibrotic, and thinner on MRI | Facial paralysis. Aberrant innervation of levator bilaterally with bilateral upper lid ptosis | − | + | Agenesis of corpus callosum and septum pellucidum, hypoplastic olfactory nerves. The other cranial nerves including the optic nerve, fifth, sixth, seventh, and eighth nerves have a within normal limits appearance, mild splaying of the cerebral peduncles, concerning for mild brainstem hypoplasia, simplified gyral pattern |
| 7 | 1G | Caucasian/Indian male | Atypical | − | VI, VII | Unknown | + | Not done | |
| 8 | 1h | Caucasian male | Atypical | − | VI, VII | Bilateral gaze palsy, decreased blinking rate, nasolacrimal duct obstruction | + | + | Facial colliculi present within the brainstem, abducence nerves not well-defined in the prepontine cisterns |
| 9 | 1I | Caucasian female | Typical | − | VI, VII | − | + | Bilateral facial nerves unremarkable, normal caliber of the right cranial nerve VI is seen. No visualization of the left cranial nerve VI is identified. Left vertebral artery is hypoplastic. |
| Family and patient number | Developmental delay/intellectual disability | Facial dysmorphisms | Short stature | Digits | Upper extremities | Lower extremities | Other anomalies |
|---|---|---|---|---|---|---|---|
| 1 | + | Bilateral facial nerve palsy and abduction deficits, epicanthal folds, flat nasal bridge, anteverted nares, broad nose, small mouth and micrognathia | − | Truncation anomaly of lower extremity | Normal | Limb length discrepancy, oligodactyly with only two digits present likely fourth and fifth toes, with cutaneous syndactyly | |
| 2 | + | Bilateral epicanthal folds, eyes are directed inferonasally, low-set ears lacking lobules, bulbous nose, malar hypoplasia, small mandible, small mouth | − | Right fourth digit camptodactyly | Right upper extremity and chest wall smaller than the left, Poland's Anomaly | Clubfeet | |
| 3 | + | Bilateral facial nerve palsy and abduction deficits, telecanthus, downslanting palpebral fissures, bilateral facial palsy, flat nasal bridge, hypoplastic alae nasi | + | Truncation anomaly of lower extremity | Normal | Truncation of lower extremity involving the right leg with complete absence of foot, also has club foot deformity involving the left foot | |
| 4 | + | Flat facies, hemangioma of nasal root, bilateral facial nerve palsy and abduction deficits, telecanthus, wide nasal bridge, bulbous nasal tip, small mouth, tented upper lip micrognathia | + | Oligodactyly | Left hand has only two distinct digits | Bilateral talipes equinovarus | Patent foramen ovale; sensorineural hearing loss |
| 5 | + | Facial asymmetry, bilateral abduction deficits, epicanthal folds, small mouth | − | Left second, third, and fourth syndactyly | Left hand syndactyly with Poland anomaly | Normal | |
| 6 | + | Flat facies, absent nasolabial folds, complete paresis of extraocular and facial muscles, downturned corners of the mouth, small jaw | − | Normal | Normal | Normal | High hyperopia |
| 7 | − | Decreased lateral and medial gaze,midface hypoplasia, small jaw | − | Normal | Normal | Normal | Submucosal cleft, atrial septal defect |
| 8 | − | Facial asymmetry, midface hypoplasia, small jaw | + | Normal | Normal | Normal | |
| 9 | + | Flat facies, tented open mouth | − | Normal | Normal | Bilateral talipes equinovarus |
Trio exomes (both parents and child) were conducted for families 1 and 3.
FH, family history; EOMs, extraocular muscles; MRI, magnetic resonance imaging; +, yes; –, no.
Unilateral or bilateral facial and abducens nerve palsies (sixth and seventh cranial nerves) were observed in all nine patients and was the eligibility criteria to be included in this study. Limb abnormalities were confirmed in 6/9 (66%) cases, seen as lower extremity truncating defects, oligodactyly, syndactyly, and/or talipes equinovarus. Poland anomaly, with ipsilateral aplasia or hypoplasia of the pectoralis muscle plus hand and digit anomalies, was present in 2/9 (22%) patients. No substantial limb/skeletal defects were identified in 3/9 patients (individuals 6, 7, and 8). Brain magnetic resonance imaging (MRI) studies were completed in 8/9 cases. Although many patients were noted to have defects of cranial nerves VI and VII on brain imaging, as typically seen in Moebius syndrome, patient 6 was found to have agenesis of corpus callosum, absent septum pellucidum, hypoplastic olfactory nerves, mild brainstem hypoplasia, and simplified gyral patterns. This patient also had atypical ocular findings with near-complete ophthalmoplegia. Two patients had magnetic resonance angiogram studies completed and were noted to have unilateral hypoplastic vertebral arteries (5 and 9).
Genetic Analyses
We first assessed novel and rare variants (minor allele frequency [MAF] < 1% in population databases) present in known related disease genes. When comparing the phenotypes associated with the genes containing candidate rare or novel variants and the phenotypes of the patients in our cohort, we identified a mutation in TUBB3 in patient 6 (g.Chr16:90,002,087(G>A); NM_006086.3:c.1228G>A; p.Glu410Lys). Trio analysis of proband 6 and his parents identified this mutation to be de novo in the affected patient (Fig. 2). Subsequent polymerase chain reaction (PCR) amplification and Sanger sequencing of this variant confirmed the WES results. This mutation has been shown previously to cause congenital fibrosis of the extraocular muscles type 3 (CFEOM3A; MIM# 600638), either isolated or in combination with other neurological disorders (Tischfield et al. 2010). A comparison between this condition, patient 6, and Moebius syndrome is shown in Table 2.
Figure 2.
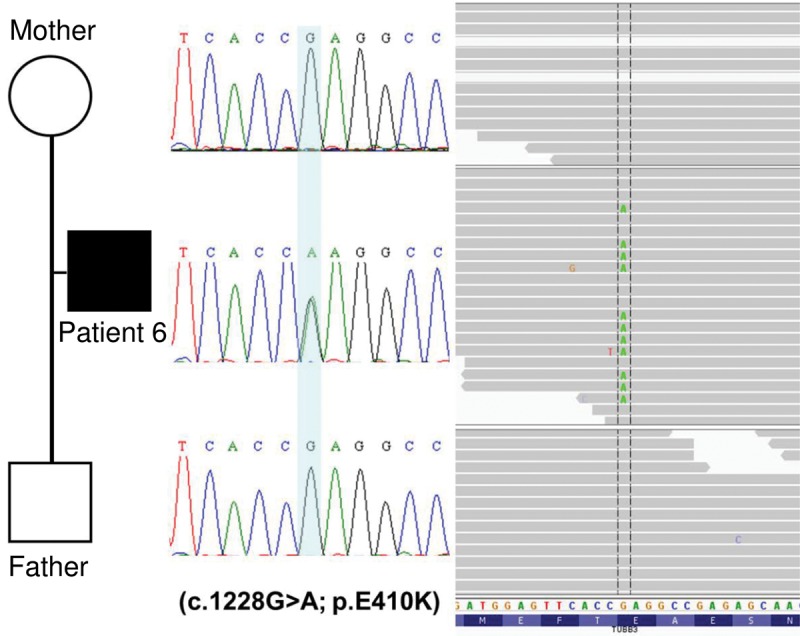
Exome sequencing revealed a mutation in TUBB3. Shown here is the Sanger confirmation and the exome-sequencing visualization of the de novo mutation identified in patient 6 in the gene TUBB3 (g.Chr16:90,002,087(G>A); NM_006086.3:c.1228G>A; p.Glu410Lys).
Table 2.
Comparison of disease features between Moebius syndrome, CDCBM/CFEOM3, and patient 6
| Moebius syndrome | CDCBM/CFEOM3 | TUBB3 E410K phenotype | Patient 6 with CFEOM3 | |
|---|---|---|---|---|
| Gene | Unknown | TUBB3 | TUBB3 | TUBB3 |
| Inheritance | Variable | De novo dominant | De novo dominant | De novo dominant |
| Neurological | Intellectual disability | Intellectual disability, spasticity, axial hypotonia | Intellectual disability, Kallmann Syndrome, vocal cord paralysis, hypotonia | Intellectual disability |
| MRI | Brainstem hypoplasia | Thin corpus callosum | Thinning of corpus callosum, anterior commissure, and internal capsule, hypoplastic/absent olfactory sulci/bulbs | Agenesis of corpus callosum, absence of septum pellucidum; hypoplastic olfactory nerves; possible mild brainstem hypoplasia and simplified gyral pattern |
| Ocular | CN VI, VII, XII palsy | Strabismus | CN III, VII palsy | CN VII palsy, near-complete ophthalmoplegia, exotropia |
| Skeletal (Kumar 1990) | Clubfoot, syndactyly, brachydactyly | Short stature, wrist/finger contractures | Short stature | Normal stature |
| Craniofacial (Kumar 1990) | Small palpebral fissures, epicanthal folds, hypertelorism, external ear defects, microstomia, micrognathia | Midface hypoplasia, short nose, short and smooth philtrum | Midface hypoplasia, short nose, short/smooth philtrum, low-set ears | Flat facies, absent nasolabial folds, downturned corners of the mouth, small jaw |
CDCBM, cortical dysplasia complex with other brain malformations; CFEOM3, congenital fibrosis of the extraocular muscles; CN, cranial nerve.
Searching for common causative genes in the other families, we considered genes in the previously proposed Moebius loci and also analyzed shared genes with rare or novel variants. We focused on genes with rare or novel mutations in at least two affected individuals. Considering genes with a single novel or very rare heterozygous mutation in three individuals, four genes were identified: HECW2, SIM1, BCDIN3D, and MRPL28 (details on all variants are shown in Table 3). The variants in all genes were inherited from an unaffected parent, and were thus considered less likely to be involved in the disease. Considering genes with one single novel or very rare heterozygous mutation in two individuals, there were seven genes: CDH11, KBTBD7, FAM71A, PLCB2, PTCH2, AMH, and HSPB7. The variants were also inherited from an unaffected parent, and were thus considered less likely to be disease-causing. Of note, the analysis of rare shared variants in two or more individuals was confounded by the Hispanic ethnicity of some of the patients. Many of these shared variants were determined to be polymorphisms as verified in an ethnicity matched Hispanic control sample during variant confirmation and segregation. For the analyzed trios, we did not identify de novo variants occurring in genes shared by at least two individuals; candidate genes with de novo variants were ADAMTS8, BSN, INTS6L, ZNF787, KDM6B, and NPIPA5. We did not identify mutations in PLXND1 and REV3L (Tomas-Roca et al. 2015), although coverage for the first exon of PLXND1 was incomplete because of high GC content. Although none of the genes with identified de novo variants replicated in other individuals, one variant that appears particularly interesting is a 12-bp deletion in BSN (Bassoon: NM_003458) in individual 9, which we confirmed to be de novo and which is predicted to be deleterious by bioinformatic algorithms like MutationTaster and PROVEAN (Protein Variation Effect Analyzer). Bassoon is a presynaptic scaffolding protein that seems to play an important role in cytoskeleton organization and recruitment of channels and other proteins relevant to presynaptic plasticity and regulated release of neurotransmitters (Kononenko et al. 2013; Davydova et al. 2014). However, BSN is considered as a gene with frequent rare mutations in exome-sequencing studies (Shyr et al. 2014), and this specific variant was found in the ExAC (Exome Aggregation Consortium) database (exac.broadinstitute.org) (Lek et al. 2016).
Table 3.
All variants referred to in the text
| Gene | Chromosome | HGVS coding DNA reference | HGVS Protein Reference | Predicted effect | Variant type | dbSNP ID if available | Genotype | ClinVar ID | ClinVar submission accession | ExAC highest MAF | Inheritance |
|---|---|---|---|---|---|---|---|---|---|---|---|
| TUBB3 | 16 | NM_006086.3:c.1228G>A | NP_006077.2 | p.(Glu410Lys) | Substitution | rs267607165 | Heterozygous | 6867 | SCV000299173.1 | Not found | De novo |
| HECW2 | 2 | NM_020760.1:c.3394G>A | NP_065811.1 | p.(Asp1132Asn) | Substitution | rs552109642 | Heterozygous | 254098 | SCV000299174.1 | 0.001911 | Inherited from a healthy parent |
| HECW2 | 2 | NM_020760.1:c.2270_2272del | NP_065811.1 | p.(Glu757del) | Deletion | rs757981529 | Heterozygous | 254099 | SCV000299175.1 | 9.81E-05 | Inherited from a healthy parent |
| HECW2 | 2 | NM_020760.1:c.1249_1251delAAT | NP_065811.1 | p.(Asn417del) | Deletion | rs774571391 | Heterozygous | 254100 | SCV000299176.1 | 0.000864 | Inherited from a healthy parent |
| SIM1 | 6 | NM_005068.2:c.2119G>C | NP_005059.2 | p.(Asp707His) | Substitution | rs74726213 | Heterozygous | 254101 | SCV000299177.1 | 0.000854 | Inherited from a healthy parent |
| SIM1 | 6 | NM_005068.2:c.1994G>A | NP_005059.2 | p.(Arg665His) | Substitution | rs146866401 | Heterozygous | 254102 | SCV000299178.1 | 0.05045 | Inherited from a healthy parent |
| BCDIN3D | 12 | NM_181708.2:c.23A>G | NP_859059.1 | p.(Asp8Gly) | Substitution | rs143608766 | Heterozygous | 254103 | SCV000299179.1 | 0.07137 | Inherited from a healthy parent |
| MRPL28 | 16 | NM_006428.4:c.610G>A | NP_006419.2 | p.(Val204Met) | Substitution | rs181590179 | Heterozygous | 254104 | SCV000299180.1 | 0.009075 | Inherited from a healthy parent |
| MRPL28 | 16 | NM_006428.4:c.176G>C | NP_006419.2 | p.(Arg59Pro) | Substitution | rs149440376 | Heterozygous | 254105 | SCV000299181.1 | 0.001542 | Inherited from a healthy parent |
| CDH11 | 16 | NM_001797.2:c.95G>A | NP_001788.2 | p.(Arg32Gln) | Substitution | rs757142171 | Heterozygous | 254106 | SCV000299182.1 | Not found | Inherited from a healthy parent |
| CDH11 | 16 | NM_001797.2:c.1247C>T | NP_001788.2 | p.(Pro416Leu) | Substitution | rs200234049 | Heterozygous | 254107 | SCV000299183.1 | 0.000867 | Inherited from a healthy parent |
| KBTBD7 | 13 | NM_032138.4:c.1496C>G | NP_115514.2 | p.(Pro499Arg) | Substitution | rs754048481 | Heterozygous | 254108 | SCV000299184.1 | Not found | Inherited from a healthy parent |
| KBTBD7 | 13 | NM_032138.4:c.1208A>C | NP_115514.2 | p.(Lys403Thr) | Substitution | rs748092018 | Heterozygous | 254109 | SCV000299185.1 | 0.000302 | Inherited from a healthy parent |
| FAM71A | 1 | NM_153606.3:c.1465T>C | NP_705834.2 | p.(Ser489Pro) | Substitution | rs767737768 | Heterozygous | 254110 | SCV000299186.1 | 0.003196 | Inherited from a healthy parent |
| FAM71A | 1 | NM_153606.3:c.1475G>A | NP_705834.2 | p.(Gly492Asp) | Substitution | rs886037883 | Heterozygous | 254111 | SCV000299187.1 | Not found | Inherited from a healthy parent |
| PLCB2 | 15 | NM_004573.2:c.1154A>G | NP_004564.2 | p.(Lys385Arg) | Substitution | rs769251460 | Heterozygous | 254112 | SCV000299188.1 | 1.56E-05 | Inherited from a healthy parent |
| PLCB2 | 15 | NM_004573.2:c.2585C>A | NP_004564.2 | p.(Thr862Lys) | Substitution | rs779256250 | Heterozygous | 254113 | SCV000299189.1 | Not found | Inherited from a healthy parent |
| PTCH2 | 1 | NM_003738.4:c.2018G>T | NP_003729.3 | p.(Arg673Leu) | Substitution | rs760548568 | Heterozygous | 254114 | SCV000299190.1 | 0.000347 | Inherited from a healthy parent |
| PTCH2 | 1 | NM_003738.4:c.1156A>T | NP_003729.3 | p.(Ile386Phe) | Substitution | rs775127172 | Heterozygous | 254115 | SCV000299191.1 | 8.64E-05 | Inherited from a healthy parent |
| AMH | 19 | NM_000479.3:c.350G>A | NP_000470.2 | p.(Arg117Gln) | Substitution | rs185020288 | Heterozygous | 254116 | SCV000299192.1 | 0.01899 | Inherited from a healthy parent |
| HSPB7 | 1 | NM_014424.4:c.442A>G | NP_055239.1 | p.(Thr148Ala) | Substitution | rs530970423 | Heterozygous | 254117 | SCV000299193.1 | 0.01699 | Inherited from a healthy parent |
| ADAMTS8 | 11 | NM_007037.4:c.2109C>A | NP_008968.4 | p.(Tyr703*) | Nonsense | rs199760382 | Heterozygous | 254118 | SCV000299194.1 | 0.01592 | De novo |
| BSN | 3 | NM_003458.3:c.7366_ 7377delCAGCAGCTGCAG | NP_003449.2 | p.(Gln2463_Leu2466del) | Deletion | rs759806020 | Heterozygous | 254119 | SCV000299195.1 | 0.000853 | De novo |
| INTS6L | X | NM_182540.4:c.352delT | NP_872346.3 | p.(Leu120*) | Nonsense | rs782242788 | Heterozygous | 254120 | SCV000299196.1 | Not found | De novo |
| ZNF787 | 19 | NM_001002836.2:c.1086G>C | NP_001002836.2 | p.(Glu362Asp) | Substitution | rs202243737 | Heterozygous | 254121 | SCV000299197.1 | 0.000173 | De novo |
| KDM6B | 17 | NM_001080424.1:c.768_769insCCACCA | NP_001073893.1 | p.(Pro263_Pro264dup) | Duplication | rs768799563 | Heterozygous | 254122 | SCV000299198.1 | 5.51E-05 | De novo |
| NPIPA5 | 16 | XM_003118698.1:c.962C>T | XP_003118746.1 | p.(Pro321Leu) | Substitution | rs886037884 | Heterozygous | 254123 | SCV000299199.1 | Not found | De novo |
HGVS, Human Genome Variation Society; dbSNP, Database for Short Genetic Variations; ExAC, Exome Aggregation Consortium; MAF, minor allele frequency.
DISCUSSION
Mutations in TUBB3 are known to cause diseases such as CFEOM (Tischfield et al. 2010) and complex cortical dysplasia with other brain malformations type 1 (CDCBM1, MIM# 614039) without major ocular findings (Poirier et al. 2010).
Moebius syndrome and CFEOM are classified as cranial nerve dysinnervation disorders (Bosley et al. 2013). CFEOM is characterized by bilateral blepharoptosis and ophthalmoplegia, with eyes infraducted at 20°–30° below the midline. Involvement of the horizontal extraocular muscles is variable. Bilateral involvement in all affected family members defines CFEOM1 (MIM# 135700) and is caused by dominant mutations in KIF21A (type 1A) or TUBB3 (type 1B). CFEOM2 (MIM# 602078) is caused by recessive PHOX2A mutations and is characterized by congenital incomitant strabismus secondary to orbital dysinnervation. There are poorly reactive pupils, ptosis, large angle exotropia, and ophthalmoplegia. CFEOM3 shows phenotypic overlap with CFEOM1, but with more variability (e.g., unilateral involvement, ability to raise eyes above midline, no ptosis). CFEOM3 is autosomal dominant with mutations in TUBB3 (type 3A, MIM# 600638) or KIF21A (type 3B, MIM# 135700).
Chew et al. (2013) described a cohort of individuals with the E410K mutation in TUBB3, thus defining the “TUBB3 E410K syndrome.” The syndrome is characterized by CFEOM, facial weakness, Kallmann syndrome, vocal cord paralysis, tracheomalacia, and later-onset progressive sensorimotor polyneuropathy and cyclic vomiting (Chew et al. 2013). The affected individuals often have perturbed social, behavioral, and intellectual function. Brain imaging studies often demonstrate dysgenesis of the corpus callosum, anterior commissure, and internal capsule (Tischfield et al. 2010) and thin corpus callosum, hypoplastic or absent olfactory sulci/olfactory bulbs, and small or absent oculomotor and facial nerves (Chew et al. 2013). Patient 6, whom we describe with the E410K mutation, exhibited facial weakness and near-complete ophthalmoplegia. He was found to have agenesis of the corpus callosum, absence of the septum pellucidum, hypoplastic olfactory nerves, mild brainstem hypoplasia, and simplified gyral pattern, highlighting the variability of TUBB3 mutations.
TUBB3 plays a role in axon guidance and normal brain development (Tischfield et al. 2010). The β-tubulins compose one of two core protein families (α- and β-tubulins) that heterodimerize to form microtubules. Microtubules perform essential functions especially during mitosis, intracellular transport, and both ciliary and flagellar motility. Mutations in TUBB3 perturb microtubule structure and function, which lead to aberrations in axonal guidance and cortical organization (Poirier et al. 2010; Tischfield et al. 2010; Chew et al. 2013). Functional studies of the E410K amino acid substitution indicate impaired tubulin dimerization and/or disruption of microtubule–kinesin interactions (Tischfield et al. 2010).
Poirier et al. (2010) further expanded the TUBB3 mutation spectrum to include CDCBM1. Although no individuals in their cohort had the E410K mutation, features similar to the individual we describe were noted, such as hypoplastic corpus callosum, strabismus, and developmental delay. Other brain abnormalities included dysmorphic and hypertrophic basal ganglia structures, dysplasia of the cerebellar vermis, and hypoplastic brainstem. Interestingly, none of the individuals in this study presented with external opthalmoplegia or facial weakness. More severely affected individuals displayed global gyral disorganization, agenesis of corpus callosum, and cerebellar dysplasia. On the contrary, less severely affected individuals with mild intellectual disability presented with frontoparietal gyral disorganization, but without corpus callosum and basal ganglia involvement.
The expansion of TUBB3-related phenotypes may provide clues into the function of this gene. Individuals with the E410K mutation in TUBB3 have CFEOM but can also have a number of additional characteristics. Our cohort of atypical Moebius individuals had many overlapping features. Further study of TUBB3 may help explain whether this mutation exhibits variable expressivity. Because these mutations can cause a phenotype resembling Moebius syndrome, clinicians should be aware of atypical presentations in the presence of classic facial weakness and ocular abnormalities. In our cohort, we considered findings such as agenesis of corpus callosum and septum pellucidum, hypoplastic olfactory nerves, brainstem hypoplasia, simplified gyral pattern, normal tongue, and normal development/intellect to be atypical of Moebius syndrome. Typical Moebius syndrome is classic for facial and abducens nerve palsy, orofacial malformations, and limb/chest wall abnormalities. The variation in the clinical findings makes the distinction between atypical and typical difficult. As more is learned about the spectrum of cranial nerve dysinnervation syndromes, the diagnosis criteria will hopefully become better defined. These investigations and reports will allow targeted management and guidance for individuals diagnosed with atypical Moebius syndrome.
The approach of WES did not identify a common genetic basis of Moebius syndrome in our study. The reasons could be multiple, such as technical artifacts in which our methods were unable to capture the coding regions of the responsible gene at all or sequencing was not achieved at a desirable coverage. Genetic heterogeneity, deletion of a single exon, mutations in a promoter region, deep intronic mutations, or other noncoding, nonexonic variants could be responsible for Moebius syndrome, as well as balanced translocations, which would be missed by the chromosome microarrays performed in a majority of the patients in our study. Epigenetic factors and other nongenetic or environmental events such as vascular disruption during gestation cannot be discarded. With the more common application and availability of genomic technologies and the data sharing among the scientific community of whole genomes and exomes of patients affected by rare disorders such as Moebius syndrome, we could begin to aggregate these data and dissect the possible causes for this significant disorder of as yet, unclear etiology.
METHODS
We performed whole-exome sequencing on peripheral blood DNA from all nine affected patients as described previously (Campeau et al. 2013). Case–parent trios were completed for six of nine families: 1, 3, 6, 7, 8, and 9. Using 1 µg of DNA an Illumina paired-end precapture library was constructed according to the manufacturer's protocol (Illumina Multiplexing_SamplePrep_Guide_1005361_D) with modifications as described in the BCM-HGSC Illumina Barcoded Paired-End Capture Library Preparation protocol. Precaptured libraries were pooled and then hybridized in solution to either the HGSC VCRome design (42 Mb, NimbleGen) or the HGSC CORE design (Bainbridge et al. 2011) (52 Mb, NimbleGen) according to the manufacturer's protocol NimbleGen SeqCap EZ Exome Library SR User's Guide (Version 2.2) with minor revisions. The sequencing run was performed in paired-end mode using the Illumina HiSeq 2000 platform, and with a sequencing yield of 10.3 Gb, the samples achieved 93% of the targeted exome bases covered to a depth of 20× or greater. Read mapping and alignment to the human genome reference assembly GRCh37 (hg19) was performed using the HGSC in-house developed Mercury pipeline (Reid et al. 2014) (https://www.hgsc.bcm.edu/content/mercury), achieving an average coverage of 116× read depth (Supplemental Table 1). Variant calling was performed using the Atlas2 (Shen et al. 2010) and SAMtools (Li et al. 2009) algorithms. Variants were annotated using intramurally developed pipelines for their coding potential (exonic, intronic, or intragenic) and anticipated functional effects (synonymous, missense, frameshift, or nonsense variants), as well as their frequency in different population-scale databases such as the Database for Short Genetic Variations (dbSNP) (Sherry et al. 2001), the 1000 Genomes Project (The 1000 Genomes Project 2015), and the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project (Exome Sequencing Project 2015). Novel and rare coding variants (MAF < 1% in population databases) were preferentially explored in order to filter out common polymorphisms and high frequency, probably benign variants. Algorithms for bioinformatic prediction of functional effects of variants, such as PolyPhen-2 (Adzhubei et al. 2010), SIFT (Kumar et al. 2009), and MutationTaster (Schwarz et al. 2014), along with conservation scores GERP (Gene Evolutionary Rate Profiling) (Davydov et al. 2010) and PhyloP and PhastCons (Hubisz et al. 2011), were incorporated as part of the variant annotation pipeline and used to inform the potential deleterious effects of identified candidate variants in order to prioritize them for additional functional testing. Candidate de novo variants and indels were inspected visually using the Integrative Genomics Viewer (IGV) in case–parent trios to discriminate false-positive calls (Thorvaldsdottir et al. 2013). Candidate variants were PCR-amplified using specific primers for the exons containing the variants of interest and further confirmed by orthogonal Sanger sequencing of the PCR products. For Sanger sequencing, amplicons were generated with 0.5 ng/µL of genomic DNA and TaqMan polymerase (ABI, Life Technologies) by the manufacturer's protocol. Primers sequences are shown in Supplemental Table 2. Products were sequenced at Beckman Coulter Genomics (Danvers, MA). In addition, a majority of the patients had a chromosomal microarray performed to rule out a chromosomal deletion or duplication (details in Supplemental Table 3).
ADDITIONAL INFORMATION
Data Deposition and Access
The families consented to the genetic study and the publication of genetic results, clinical details, and pictures. However, they did not specifically consent to the deposition of exome data in public repositories. The variants described in this study have been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) under accession numbers SCV000299173.1 through SCV000299199.1 (see Table 3 for full list).
Ethics Statement
Informed, written consent for the study was obtained from the families. The study and consent form was approved by the Institutional Review Board of the Baylor College of Medicine and Affiliated Hospitals.
Acknowledgments
The authors thank the families involved in this study for their participation. We thank Yuqing Chen, Xueqing Wang, Gladys Zapata, and Patricia Hernandez for technical assistance and Mahshid S. Azamian and Alyssa Tran for clinical research support.
Funding
This work was supported in part by the U.S. National Human Genome Research Institute/National Heart Blood and Lung Institute (NHGRI/NHBLI) grant U54 HG006542 to the Baylor-Hopkins Center for Mendelian Genomics. Funding was also provided by National Institutes of Health (NIH) grants PO1 HD22657 and PO1 HD070394 (B.H.L.) and U54 HG003273-09 (R.A.G.). Funding was also provided by The Rolanette and Berdon Lawrence Bone Disease Program of Texas (B.H.L.). P.M.C. is supported by a Canadian Institutes of Health Research (CIHR) clinician-scientist award. J.T.L. is supported by Ruth L. Kirschstein National Research Service Award F30 MH098571-01. R.A.L. is a Senior Scientific Investigator of Research to Prevent Blindness, New York.
Competing Interest Statement
The authors have declared no competing interest.
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. 2010. A method and server for predicting damaging missense mutations. Nat Methods 7: 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainbridge MN, Wang M, Wu Y, Newsham I, Muzny DM, Jefferies JL, Albert TJ, Burgess DL, Gibbs RA. 2011. Targeted enrichment beyond the consensus coding DNA sequence exome reveals exons with higher variant densities. Genome Biol 12: R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borck G, Wirth J, Hardt T, Tönnies H, Brøndum-Nielsen K, Bugge M, Tommerup N, Nothwang HG, Ropers HH, Haaf T. 2001. Molecular cytogenetic characterisation of a complex 46,XY,t(7;8;11;13) chromosome rearrangement in a patient with Moebius syndrome. J Med Genet 38: 117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosley TM, Abu-Amero KK, Oystreck DT. 2013. Congenital cranial dysinnervation disorders: a concept in evolution. Curr Opin Ophthalmol 24: 398–406. [DOI] [PubMed] [Google Scholar]
- Campeau PM, Lenk GM, Lu JT, Bae Y, Burrage L, Turnpenny P, Román Corona-Rivera J, Morandi L, Mora M, Reutter H, et al. 2013. Yunis–Varón syndrome is caused by mutations in FIG4, encoding a phosphoinositide phosphatase. Am J Hum Genet 92: 781–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew S, Balasubramanian R, Chan WM, Kang PB, Andrews C, Webb BD, MacKinnon SE, Oystreck DT, Rankin J, Crawford TO, et al. 2013. A novel syndrome caused by the E410K amino acid substitution in the neuronal β-tubulin isotype 3. Brain 136: 522–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. 2010. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol 6: e1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydova D, Marini C, King C, Klueva J, Bischof F, Romorini S, Montenegro-Venegas C, Heine M, Schneider R, Schroder MS, et al. 2014. Bassoon specifically controls presynaptic P/Q-type Ca2+ channels via RIM-binding protein. Neuron 82: 181–194. [DOI] [PubMed] [Google Scholar]
- Dotti MT, Federico A, Palmeri S, Guazzi GC. 1989. Congenital oculo-facial paralysis (Moebius syndrome): evidence of dominant inheritance in two families. Acta Neurol (Napoli) 11: 434–438. [PubMed] [Google Scholar]
- Dunham C, Spellacy W. 1967. Pregnancy in patients with osteogenesis imperfecta. A case report and a review of the literature. J Lancet 87: 293–295. [PubMed] [Google Scholar]
- Exome Sequencing Project. 2015. Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP). http://evs.gs.washington.edu/EVS/. Accessed 11/11/2015. [Google Scholar]
- Graziadio C, Lorenzen MB, Rosa RF, Pinto LL, Zen PR, Travi GM, Valiatti F, Paskulin GA. 2010. New report of a familial case of Moebius syndrome presenting skeletal findings. Am J Med Genet A 152A: 2134–2138. [DOI] [PubMed] [Google Scholar]
- Hubisz MJ, Pollard KS, Siepel A. 2011. PHAST and RPHAST: phylogenetic analysis with space/time models. Brief Bioinform 12: 41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kankirawatana P, Tennison MB, D'Cruz O, Greenwood RS. 1993. Möbius syndrome in infant exposed to cocaine in utero. Pediatr Neurol 9: 71–72. [DOI] [PubMed] [Google Scholar]
- Kersey JP, Vivian AJ, Reid E. 2006. A report of paracentric inversion of chromosome 8 in Moebius syndrome. Ophthalmic Genet 27: 29–31. [DOI] [PubMed] [Google Scholar]
- Kononenko N, Pechstein A, Haucke V. 2013. Synaptic requiem: a duet for Piccolo and Bassoon. EMBO J 32: 920–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer H, Kuyt LP, van den Helm B, van Reen M, Leunissen JA, Hamel BC, Jansen C, Mariman EC, Frants RR, Padberg GW. 1996. Localization of a gene for Möbius syndrome to chromosome 3q by linkage analysis in a Dutch family. Hum Mol Genet 5: 1367–1371. [DOI] [PubMed] [Google Scholar]
- Krueger K, Friedrich D. 1963. Familiäre kongenitale Motilitätsstörungen der Augen. Klin Monatsbl Augenheilkd 142: 101–117. [PubMed] [Google Scholar]
- Kumar D. 1990. Moebius syndrome. J Med Genet 27: 122–126. [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. 2009. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4: 1073–1081. [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536: 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDermot KD, Winter RM, Taylor D, Baraitser M. 1991. Oculofacialbulbar palsy in mother and son: review of 26 reports of familial transmission within the ‘Möbius spectrum of defects’. J Med Genet 28: 18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MT, Ventura L, Stromland K. 2009. Thalidomide and misoprostol: ophthalmologic manifestations and associations both expected and unexpected. Birth Defects Res A Clin Mol Teratol 85: 667–676. [DOI] [PubMed] [Google Scholar]
- Möbius P. 1888. Ueber angeborene doppelseitige Abducens-facialis-lähmung. Munch Med Wochenschr 35: 91–94. [Google Scholar]
- Nishikawa M, Ichiyama T, Hayashi T, Furukawa S. 1997. Möbius-like syndrome associated with a 1; 2 chromosome translocation. Clin Genet 51: 122–123. [DOI] [PubMed] [Google Scholar]
- Poirier K, Saillour Y, Bahi-Buisson N, Jaglin XH, Fallet-Bianco C, Nabbout R, Castelnau-Ptakhine L, Roubertie A, Attie-Bitach T, Desguerre I, et al. 2010. Mutations in the neuronal β-tubulin subunit TUBB3 result in malformation of cortical development and neuronal migration defects. Hum Mol Genet 19: 4462–4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puvabanditsin S, Garrow E, Augustin G, Titapiwatanakul R, Kuniyoshi KM. 2005. Poland–Möbius syndrome and cocaine abuse: a relook at vascular etiology. Pediatr Neurol 32: 285–287. [DOI] [PubMed] [Google Scholar]
- Reid JG, Carroll A, Veeraraghavan N, Dahdouli M, Sundquist A, English A, Bainbridge M, White S, Salerno W, Buhay C, et al. 2014. Launching genomics into the cloud: deployment of Mercury, a next generation sequence analysis pipeline. BMC Bioinformatics 15: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas-Martinez A, Garcia-Cruz D, Rodriguez Garcia A, Sanchez-Corona J, Rivas F. 1991. Poland–Moebius syndrome in a boy and Poland syndrome in his mother. Clin Genet 40: 225–228. [DOI] [PubMed] [Google Scholar]
- Schwarz JM, Cooper DN, Schuelke M, Seelow D. 2014. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 11: 361–362. [DOI] [PubMed] [Google Scholar]
- Shen Y, Wan Z, Coarfa C, Drabek R, Chen L, Ostrowski EA, Liu Y, Weinstock GM, Wheeler DA, Gibbs RA, et al. 2010. A SNP discovery method to assess variant allele probability from next-generation resequencing data. Genome Res 20: 273–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. 2001. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29: 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyr C, Tarailo-Graovac M, Gottlieb M, Lee JJ, van Karnebeek C, Wasserman WW. 2014. FLAGS, frequently mutated genes in public exomes. BMC Med Genomics 7: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slee J, Smart R, Viljoen D. 1991. Deletion of chromosome 13 in Moebius syndrome. J Med Genet 28: 413–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprofkin BE, Hillman JW. 1956. Moebius's syndroma—congenital oculofacial paralysis. Neurology 6: 50–54. [DOI] [PubMed] [Google Scholar]
- Stabile M, Cavaliere M, Scarano G, Fels A, Valianl R, Ventruto V. 1984. Abnormal BAEP in a family with Moebius syndrome: evidence for supranuclear lesion. Clin Genet 25: 459–463. [DOI] [PubMed] [Google Scholar]
- Stromland K, Sjogreen L, Miller M, Gillberg C, Wentz E, Johansson M, Nylen O, Danielsson A, Jacobsson C, Andersson J, et al. 2002. Möbius sequence—a Swedish multidiscipline study. Eur J Paediatr Neurol 6: 35–45. [DOI] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, et al. 2015. A global reference for human genetic variation. Nature 526: 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14: 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischfield MA, Baris HN, Wu C, Rudolph G, Van Maldergem L, He W, Chan WM, Andrews C, Demer JL, Robertson RL, et al. 2010. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell 140: 74–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomas-Roca L, Tsaalbi-Shtylik A, Jansen JG, Singh MK, Epstein JA, Altunoglu U, Verzijl H, Soria L, van Beusekom E, Roscioli T, et al. 2015. De novo mutations in PLXND1 and REV3L cause Möbius syndrome. Nat Commun 6: 7199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verzijl HT, van den Helm B, Veldman B, Hamel BC, Kuyt LP, Padberg GW, Kremer H. 1999. A second gene for autosomal dominant Möbius syndrome is localized to chromosome 10q: a in a Dutch family. Am J Hum Genet 65: 752–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verzijl HT, van der Zwaag B, Cruysberg JR, Padberg GW. 2003. Möbius syndrome redefined: a syndrome of rhombencephalic maldevelopment. Neurology 61: 327–333. [DOI] [PubMed] [Google Scholar]
- von Graefe A. 1880. Handbuch der Gesammten Augenheilkunde (ed. von Graefe A, Saemisch T), Vol. 6, p. 60 Engelmann, Leipzig. [Google Scholar]
- Wishnick M, Nelson L, Reich E, Hubbard L. 1981. Moebius syndrome with dominant inheritance. Am J Hum Genet 33: 96A. [Google Scholar]
- Ziter FA, Wiser WC, Robinson A. 1977. Three-generation pedigree of a Möbius syndrome variant with chromosome translocation. Arch Neurol 34: 437. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.