

Abstract
Objective:
ATP1A3-related neurologic disorders encompass a broad range of phenotypes that extend well beyond initial phenotypic criteria associated with alternating hemiplegia of childhood (AHC) and rapid-onset dystonia parkinsonism.
Methods:
In 2014, the Alternating Hemiplegia of Childhood Foundation hosted a multidisciplinary workshop intended to address fundamental challenges surrounding the diagnosis and management of individuals with ATP1A3-related disorders.
Results:
Workshop attendees were charged with the following: (1) to achieve consensus on expanded diagnostic criteria to facilitate the identification of additional patients, intended to supplement existing syndrome-specific diagnostic paradigms; (2) to standardize definitions for the broad range of paroxysmal manifestations associated with AHC to disseminate to families; (3) to create clinical recommendations for common recurrent issues facing families and medical care providers; (4) to review data related to the death of individuals in the Alternating Hemiplegia of Childhood Foundation database to guide future efforts in identifying at-risk subjects and potential preventative measures; and (5) to identify critical gaps where we most need to focus national and international research efforts.
Conclusions:
This report summarizes recommendations of the workshop committee, highlighting the key phenotypic features to facilitate the diagnosis of possible ATP1A3 mutations, providing recommendations for genetic testing, and outlining initial acute management for common recurrent clinical conditions, including epilepsy.
Na+/K+ adenosine triphosphatases (sodium/potassium pumps) are P-type cation transport proteins that establish and maintain electrochemical gradients for Na+ and K+ across the plasma membrane. Mutations in the α3 catalytic subunit, encoded by ATP1A3, which in the nervous system is exclusively expressed in neurons, are associated with at least 3 distinct, yet overlapping, neurologic syndromes: rapid-onset dystonia parkinsonism (RDP); alternating hemiplegia of childhood (AHC); and cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss (CAPOS). From the past descriptions of each entity, distinct phenotypes have emerged, demonstrating that classic AHC, classic RDP, and CAPOS syndrome constitute clinical prototypes in a continuous and still expanding phenotypic spectrum of ATP1A3-related disorders.
In June of 2014, an international consortium of experts consisting of neurologists, epileptologists, clinical geneticists, genetic counselors, physiatrists, physical therapists, psychologists, basic scientists, and foundation representatives convened in Minneapolis, MN, in conjunction with the Alternating Hemiplegia of Childhood Foundation (AHCF) International Family Meeting. A series of quarterly teleconferences performed in the year preceding the conference, including representatives from the AHCF parent advisory board and supplemented by online queries to parents via online discussion boards, were used to identify key issues and points of concern from the parent/caregiver perspective. An initial expert panel discussion and multidisciplinary workshop were followed by the selection of a task force assigned to specific key areas to further refine and revise materials over the course of the subsequent 18 months. The following article serves as a research conference summary initiated at the AHCF International Family Meeting.
Workshop attendees focused efforts on the following aims: (1) to reach consensus on an expanded diagnostic paradigm for identifying patients with an ATP1A3-related neurologic disorder to supplement existing syndrome-based diagnostic criteria; (2) to develop standardized definitions for types of episodic manifestations associated with AHC, the most complex of the ATP1A3-related neurologic phenotypes and to disseminate these among families; (3) to create initial clinical recommendations for recurrent issues facing families and medical care providers; (4) to review deaths in the AHCF database to guide future efforts in identifying at-risk subjects and potential preventative measures; and (5) to identify critical gaps where the AHC research community most needs to focus national and international efforts. The primary goal of these efforts is to promote additional reviews and discussions among the broader international community of clinicians, scientists, and family support organizations in anticipation of the robust infrastructure needed for future collaborative clinical studies and trials. In particular, natural longitudinal history data are critically needed to identify end points necessary to assess the benefit of new treatments. Our work here admittedly focused on those on the more severe end of the spectrum of ATP1A3-related disorders within developed countries. However, ATP1A3-related disorders clearly affect patients of all ages, races, and ethnicities. Continued efforts to enroll patients in registries worldwide to better understand both phenotypes and outcomes remains an important goal.
CONSENSUS TOWARD AN EXPANDED NON–SYNDROME-BASED DIAGNOSTIC ALGORITHM FOR ATP1A3-RELATED NEUROLOGIC DISORDERS
Numerous publications over the past 4 decades have described and subsequently refined clinical diagnostic criteria for what are now recognized to be ATP1A3-related disorders, beginning with the original description of AHC in 19711 followed by RDP in 19932 and of CAPOS in 1996.3 Mutations in ATP1A3 were identified as the genetic cause of RDP in 2004.4 It was not until 2012 that de novo mutations in ATP1A3 were found in the majority of patients with AHC.5,6 In 2014, a novel recurrent mutation in ATP1A3 was identified in patients with CAPOS.7 Since then, an increasing number of atypical cases with ATP1A3 mutations have been identified spanning the phenotypic spectrum between AHC, RDP, and CAPOS syndromes, creating controversy as to whether these disorders should be considered discrete syndromes or points along a broadly heterogeneous spectrum.8–13
The identification and description of distinct neurologic syndromes has served as a critical point of reference to help clinicians identify and study rare patient populations in the absence of a clear molecular etiology. Prior to 2004 in RDP and 2012 in AHC, expert consensus–developed clinical criteria served as the primary pathway to diagnosis. More recently, important genotype-phenotype correlations relating to individual mutations have begun to emerge in ATP1A3-related neurologic disorders. We await to see whether genotype-phenotype correlations hold up over time with the accrual of new patient cohorts identified in the context of an unbiased next-generation sequencing–based approach.
Members of the task force attempted to provide a more comprehensive diagnostic approach than was afforded under previous clinical criteria. This would allow for clinicians to go beyond existing core phenotypic components in identifying ATP1A3-related disorders. As a starting point, we compiled diagnostic criteria for AHC and RDP and clinical features of CAPOS, inclusive of previously suggested supporting features based on the available literature (table 1). Next, we sought consensus on a new diagnostic paradigm that focused on major and minor neurologic symptoms or signs that broadly supported an association with mutations in ATP1A3 in the absence of a context of a specific syndrome (table 2). A brief summary of each disorder is provided below, highlighting important features both shared and distinct among the 3 syndromes. Table 3 reviews the differential diagnosis of disorders with overlapping signs and symptoms.
Table 1.
Compilation of previously published diagnostic criteria for AHC, RDP, and CAPOS syndromes

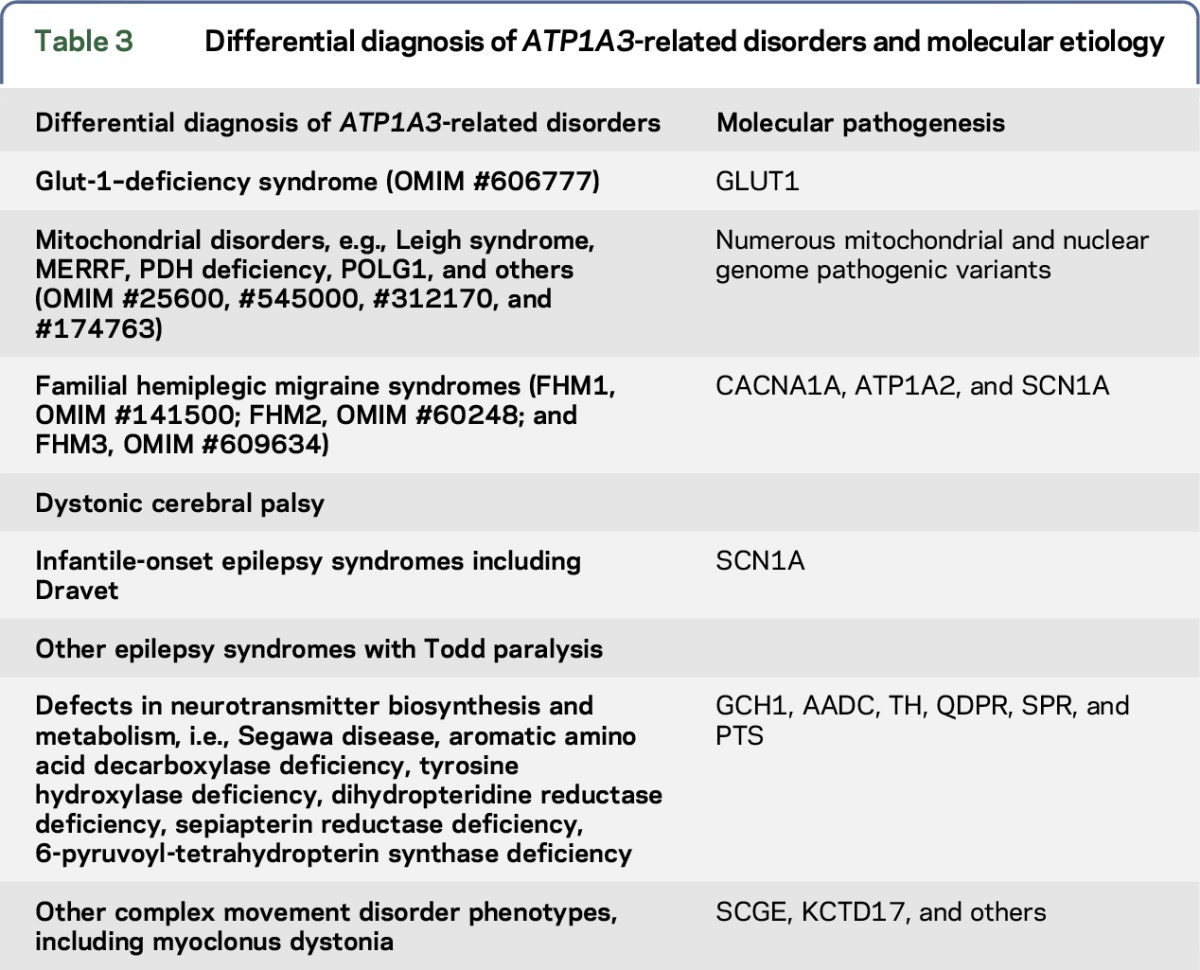
Table 2.
Major and minor features supporting ATP1A3 molecular analysis
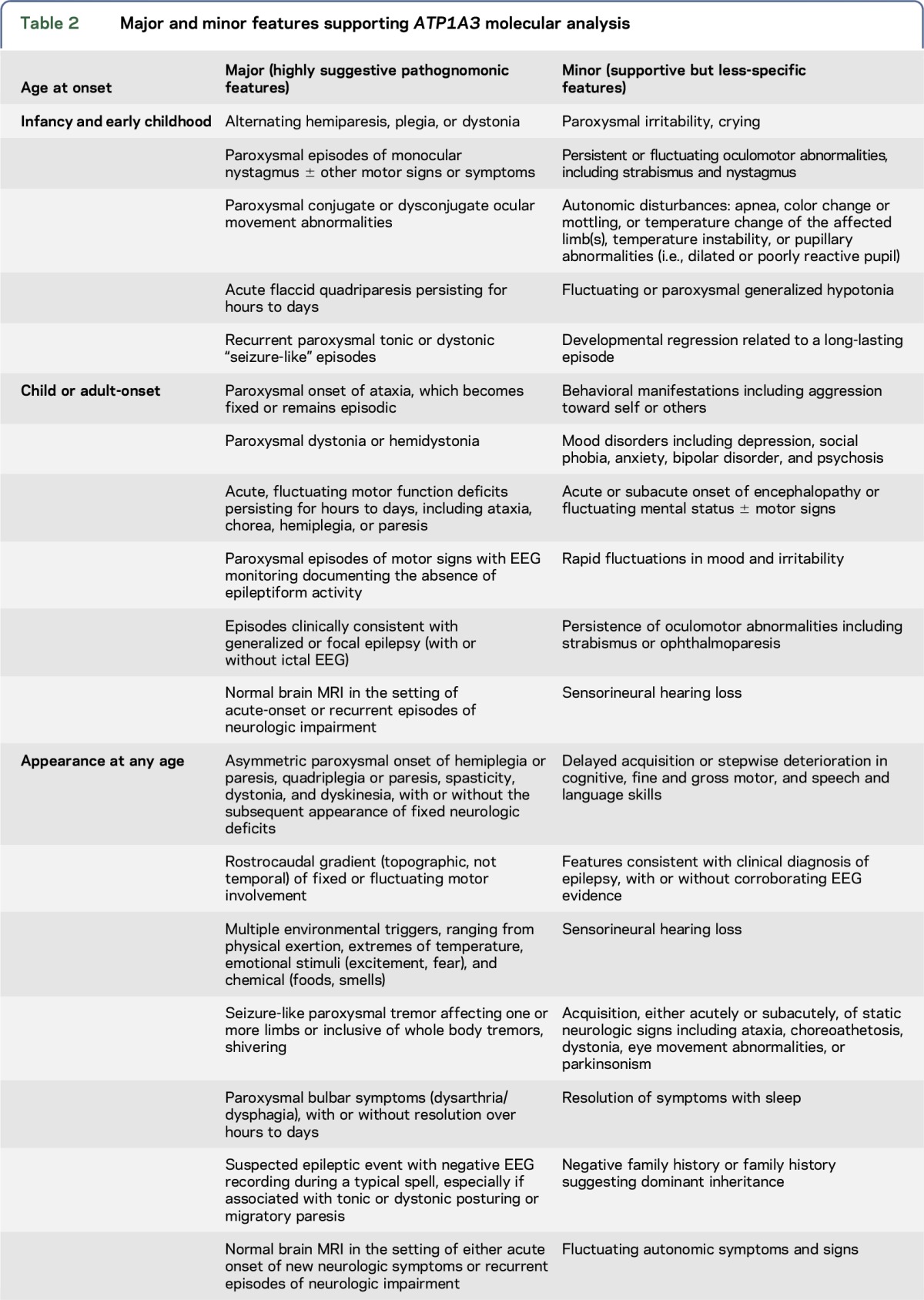
Table 3.
Differential diagnosis of ATP1A3-related disorders and molecular etiology
Classic AHC.
Since the discovery of ATP1A3 as the causative mutation in the majority of AHC cases,5,6 at least 34 unique mutations have been identified.14 AHC is characterized by recurrent episodes of paroxysmal hemiplegia, dystonia, ocular movement abnormalities (monocular nystagmus and deviation), autonomic dysfunction, as well as developmental delay. With increasing age, many patients ultimately manifest epilepsy; however, the characteristic presentation in infants includes a variety of episodic, nonepileptic tonic, dystonic, or plegic (weakness) spells that often are refractory to treatment with most therapeutic agents. The early onset and rapidly evolving clinical signs and symptoms frequently contribute to diagnostic delays. The relatively broad differential diagnosis (table 3) and the lack of a specific genetic test prior to 2012 resulted in a widely variable approach to attempts to treat paroxysmal events (often presumed to be seizures even in the absence of electrophysiologic evidence). While the most pathognomonic events include hemiplegic or hemidystonic episodes that affect either side of the body, children also frequently have bilateral involvement, including generalized fluctuating dystonia, quadriplegia, or paresis of variable duration.
A variety of nonparoxysmal symptoms and signs become increasingly evident with age, including global developmental delays; behavioral, attentional, and executive dysfunction with variable intellectual disability; dysarthria, ataxia, chorea, dystonia, and, less frequently, pyramidal tract signs such as spasticity and hyperreflexia. To date, we have limited data on the full spectrum of the burden of acquired neurologic deficits in this population; more robust longitudinal data are needed.
Rapid-onset dystonia parkinsonism.
In 2004, it was demonstrated that heterozygous mutations in the ATP1A3 gene are causative of RDP.4 Since then, at least 14 mutations have been identified.4,9,15–19 Clinical hallmarks of RDP include the abrupt onset of bulbar symptoms and limb dystonia with features of parkinsonism (bradykinesia and postural instability) that exhibit limited progression over weeks and little change thereafter. Dystonic symptoms typically show minimal response to l-dopa or dopaminergic agonists. The severity of acquired neurologic signs and symptoms demonstrates a rostrocaudal gradient of involvement (face > arm > leg). Disease onset has been reported between the ages of 4 and 58 years. Symptoms can be triggered by a broad range of physical, emotional, or chemical triggers. Beyond dystonia and parkinsonism, several patients with classic RDP display other symptoms including depression and anxiety/social phobia. A minority of patients develop seizures years after the disease onset. The diagnostic criteria for RDP were revised in 2007.20 Major and minor diagnostic criteria for RDP are summarized in table 1.
CAPOS syndrome.
The constellation of CAPOS was first described as a unique clinical entity in 1996.3 To date, very few patients have been described with this syndrome and no information about prevalence and incidence is available. The combination of the aforementioned symptoms is characteristic of this syndrome, although these do not constitute diagnostic criteria, given the limited number of cases. A single recurrent missense mutation in ATP1A3 (c.2452G>A;Glu818Lys) has been identified in all cases to date.21 Of note, only 5 of 18 identified patients exhibit pes cavus, and a full spectrum of manifestations is likely broader.7,21–23
The identification of an increasing number of patients with mutations in the ATP1A3 gene and phenotypic overlap of the above-mentioned diseases demonstrates that classic AHC, classic RDP, and CAPOS syndrome constitute clinical prototypes of a continuous phenotypic spectrum from ATP1A3-related disorders.24
Updating the current syndrome-based diagnostic algorithm using criteria outlined in table 1, with one emphasizing the presence of the key major (pathognomonic) and minor (supporting) signs and symptoms (table 2), is intended to focus attention on the rapidly evolving spectrum of ATP1A3-related disorders for clinicians and families. Ideally, this dual approach will facilitate the earliest possible identification of the primary genetic defect in patients presenting a wider phenotypic spectrum. This will reduce the often prolonged diagnostic odyssey and facilitate prospective enrollment into clinical studies designed to help better understand the natural history of the disorder.
STANDARDIZED DEFINITIONS FOR PAROXYSMAL EVENTS IN AHC
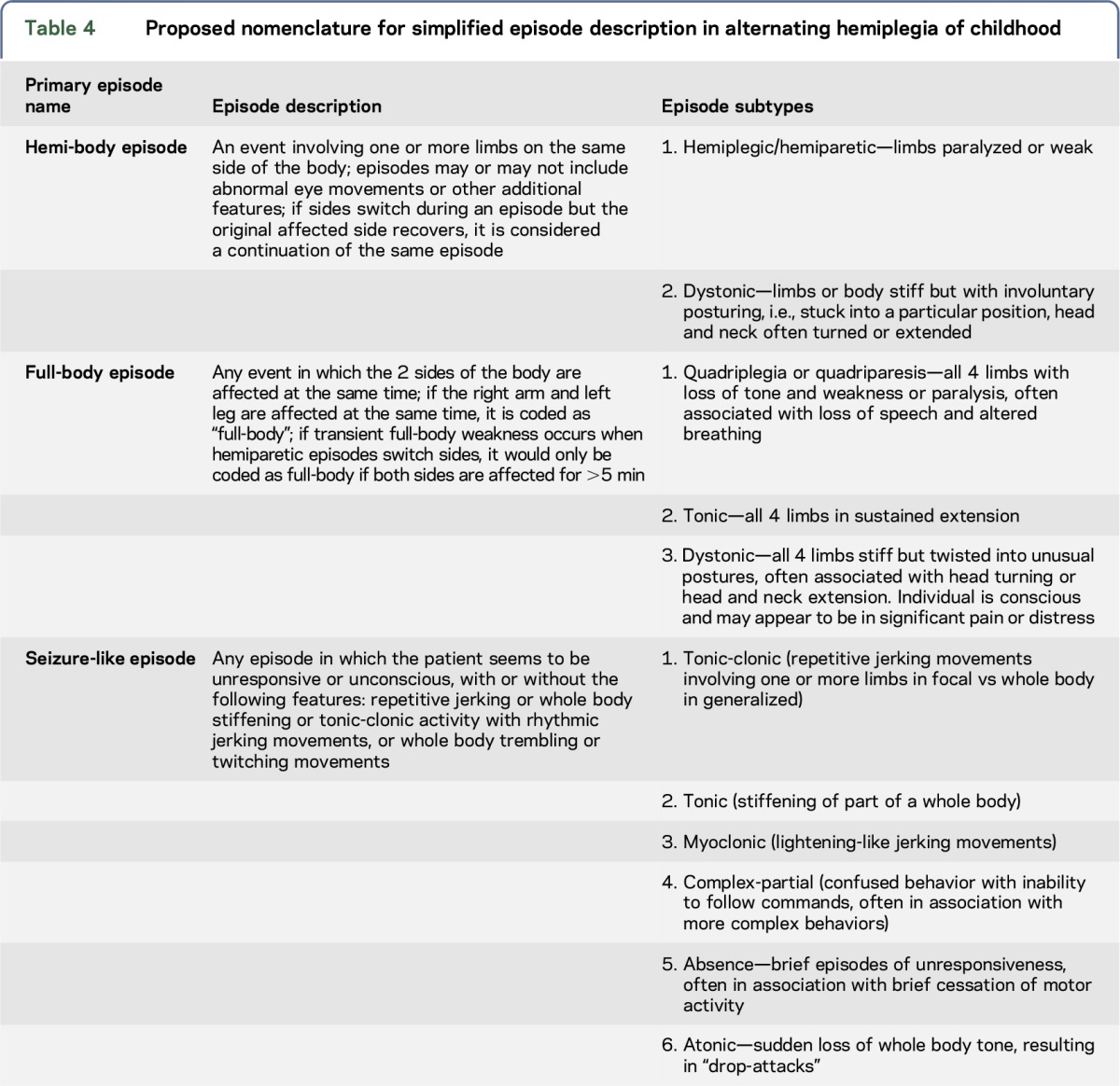
The complex and variable paroxysmal events in AHC result in challenges in defining aspects of the ATP1A3-related disorders. Currently, neither families nor clinicians use a unified set of definitions to guide them when they attempt to communicate the frequency, duration, and overall effect of types of episodes their child may be experiencing. It is often not clear to families or their clinicians how they should categorize episodes that may or may not be seizures or some other complex movement disorders. Not only does this result in an inconsistent approach among both caregivers and clinicians, it makes it more challenging for parents to determine what constitutes an emergency for which they should seek additional urgent medical assistance. Table 4 presents a simplified nomenclature using a language more accessible for caregivers to document events in a way that might be valuable in the context of longitudinal studies or clinical trials.
Table 4.
Proposed nomenclature for simplified episode description in alternating hemiplegia of childhood
CATASTROPHIC EVENTS IN ATP1A3-RELATED DISORDERS
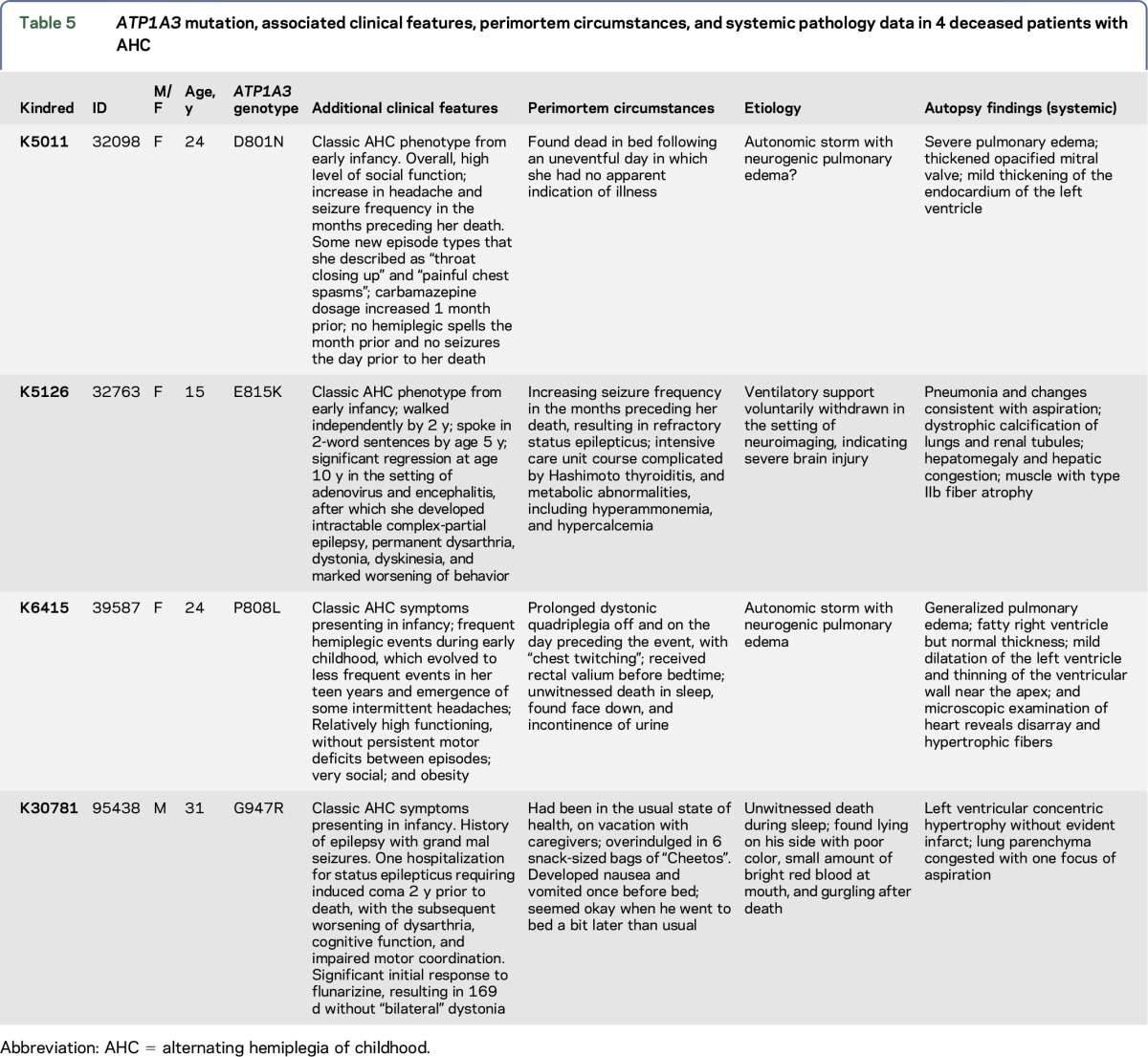
Over the course of the past 2 decades, 4 of 187 (2%) prospectively enrolled patients in the US/International AHCF registry with documented pathogenic mutations in ATP1A3 and clinical features consistent with AHC have died, many of them unexpectedly, with or without an apparent antecedent stressor. Three scenarios have emerged that deserve additional attention in terms of focused research, prevention strategies, and clinical management recommendations: (1) the sudden acquisition of newly acquired, nonparoxysmal severely disabling neurologic deficits, representing a substantial lasting change in the neurologic status in a previously stable patient, (2) the appearance of increasingly refractory epilepsy or bilateral dystonia and status epilepticus or status dystonicus, and (3) sudden unexpected death, with or without an obvious antecedent stressor. Admittedly, the literature regarding these scenarios, as well as potential causes and/or interventions, is essentially nonexistent. Table 5 lists age, ATP1A3 mutation, associated clinical features, perimortem circumstances, and systemic pathology data for the 4 patients with AHC.
Table 5.
ATP1A3 mutation, associated clinical features, perimortem circumstances, and systemic pathology data in 4 deceased patients with AHC
The sudden acquisition of new and severely disabling neurologic deficits in patients with AHC, particularly dystonia, has many parallels with the acute or subacute acquisition of evolving neurologic deficits initially described in patients with RDP.20 However, in patients with AHC, these deficits occur in the context of an already established syndrome of paroxysmal AHC symptoms and neurodevelopmental deficits, making it initially less obvious that any episode may ultimately prove irreversible. Our understanding of the critical trigger(s) for such irreversible transformations remains unclear, since antecedent events often appear no different from hundreds of prior episodes these children may have already experienced.
Sudden unexpected death in epilepsy (SUDEP) has demanded increasing attention on the part of families and practitioners within the epilepsy community and is the leading cause of death in the absence of status epilepticus or seizure-related trauma.25 A widely accepted definition for SUDEP is included in table e-1 at Neurology.org/ng.26 Proposed risk factors include the use of multiple anticonvulsants, poor medication compliance, uncontrolled seizures, and the presence of additional neurologic deficits, in addition to epilepsy, among others.27
In light of the heightened awareness that certain molecular etiologies may predispose to an increased risk of sudden death,28 it should be emphasized that ATP1A3-related disorders may have a similar association, both in the presence and absence of comorbid epilepsy. ATP1A3 is expressed in human cardiac tissue,29 and faulty cardiac repolarization has been demonstrated in patients with AHC.30 Repetitive brief asystole was documented in an individual with a confirmed ATP1A3 mutation and AHC.31 Finally, as illustrated in table 5, 3 patients prospectively enrolled in the AHCF registry who died suddenly had documented evidence of cardiac structural abnormalities, including ventricular hypertrophy, on autopsy studies. In trying to understand and prevent sudden death in ATP1A3-related neurologic disorders, it is important to create appropriate evidence-based recommendations regarding initial evaluation and long-term monitoring of cardiac status.
CRITICAL GAPS IN OUR CURRENT KNOWLEDGE OF ATP1A3-RELATED DISORDERS
Children who present acutely with dystonia, encephalopathy, or ataxia in the absence of pathognomonic features of classic AHC (i.e., alternating weakness or dystonia) account for an increasing proportion of ATP1A3 mutation–positive cases. The prognosis for children with variants of uncertain significance or for those who have a variant with known low frequency is uncertain, resulting in increased concern for both parents and clinicians.
Even in patients with AHC presenting with a classic classic presentation in the context of one of the common mutations, we have very limited information on long-term outcome with these disorders. This is due to (1) lack of rigorous longitudinal natural history follow-up, (2) the relatively recent identification of the primary genetic defect in AHC and CAPOS, (3) delays in clinical awareness/implementation of molecular testing, and (4) the enormous variability of episodes and the absence of standardized measurement tools. The development of such tools is critical in quantifying disease burden and ideally would allow us to accurately represent episode type, frequency, and severity and the subsequent influence on cognition, mood and behavior, language, bulbar function, gross and fine motor function, activities of daily living, and quality of life.
Disease progression in subsets of ATP1A3-related disorders associated with fluctuating neurologic signs and symptoms is often hard to appreciate, in that symptoms follow a waxing/waning course that may result in increasing difficulty (and parental fatigue) in complying with routine clinic visits. Children may gradually fall behind in development relative to their peers, or repeatedly lose then regain skills, making acquired accumulated and progressive neurologic deficits not immediately obvious. As children get older, caregivers often become increasingly reluctant to use the urgent care setting when new events occur because knowledge and tools are ineffective and lasting solutions to understand and manage these complex events are not yet available.
Over time, documenting the frequency and course of events may seem overwhelming and futile to primary caregivers. Physicians, likewise, become discouraged when imaging tests, EEGs, and laboratory testing repeatedly fail to provide answers or guidance for management. As an example, when a new type of paroxysmal event appears, it may initially be considered a variation of a previously documented complex nonepileptic movement disorder rather than a possible seizure. Appropriate intervention may, thus, be delayed when overt seizures finally manifest, resulting in potentially catastrophic outcomes such as death or acquisition of new permanent neurologic deficits in the setting of status epilepticus.
There may be important variations in phenotypic severity among patients with the most common ATP1A3 mutations affecting “hot spots” in exons 17, 18, and 21.9 Initial data from patients with AHC with an E815K mutation demonstrated more severe impairment in motor function, intellectual disability, and epilepsy.14,32 Moving forward, development of standardized multicenter databases would allow rigorous monitoring, and documentation of clinical disease evolution should help permit us to more accurately capture mutation-specific differences with prognostic implications.
In many disorders, tools such as motor function scales, timed tests, and neuropsychological tools, including age-appropriate assessments of executive and cognitive function, mood, and behavior, can be standardized for use across patient cohorts. Unfortunately, the fluctuating nature of AHC symptoms in many children makes standardized motor and cognitive function tests more challenging to use than in other more static or progressive neurologic disorders. Therefore, a focus on caregiver-reported outcomes or a measure of overall functional assessment of patients in performing daily self-care tasks may be more practical in this setting.
SUGGESTED ACUTE MANAGEMENT RECOMMENDATIONS FOR COMMONLY ENCOUNTERED ADVERSE EVENTS IN PATIENTS WITH ATP1A3-RELATED DISORDERS
During the workshop, members attempted to identify the most common and challenging clinical scenarios encountered in the acute care setting. As yet, these issues are largely underaddressed in the existing literature. It was agreed that action plans for certain commonly encountered issues could help standardize approaches in the acute setting. These recommendations could be an invaluable tool to facilitate communication between primary caregivers and physicians when they seek urgent care for their child with an ATP1A3-related neurologic disorder. They may help streamline discussions with health care providers and minimize the risk of adverse complications. We have created initial drafts for action recommendations (see supplemental materials) to address 3 recurring clinical scenarios frequently involving affected children or adults: (1) increasingly frequent and painful dystonic crises or status dystonicus, (2) new signs or symptoms thought to be clinically consistent with epilepsy or status epilepticus, and (3) recurring behavioral crises constituting a danger to self or others.
RECOMMENDATIONS FOR MOLECULAR DIAGNOSTIC TESTING
Both AHC and RDP phenotypes can be associated with de novo mutations or dominant inheritance within families, although the vast majority of patients with AHC identified to date have de novo mutations. Because neurologic symptoms and signs associated with ATP1A3-related disorders overlap with many other neurologic syndromes, the ATP1A3 gene has been increasingly included in next-generation sequencing (NGS) panels for epilepsy, ataxia, and dystonia/parkinsonism. Patients identified by NGS techniques account for a growing percentage of newly identified patients, whose signs and symptoms may or may not fit within the spectrum of previously described phenotypes for AHC and RDP.
Despite the important observation that mutations in the ATP1A3 gene are a frequent cause of both AHC and RDP, some individuals who meet clinical diagnostic criteria for these syndromes will not have evident pathogenic mutations in ATP1A3. Studies continue to identify alternate molecular etiologies in patients with well-defined AHC and RDP phenotypes and for potentially valuable biomarkers that can distinguish between patients with or without pathogenic mutations in ATP1A3. If AHC is suspected clinically and an ATP1A3 mutation cannot be identified, consideration could be given to then sequence individual genes or NGS panels that include complete coverage for genes associated with overlapping neurologic phenotypes (table 3).
A medical genetics consultation is strongly recommended before pursuing whole-exome sequencing (WES), a complex test with the potential for incidental findings that requires detailed pretest and posttest counseling. When a diagnosis of AHC or RDP is not previously suspected by the clinician, extra caution should be used in the interpretation of previously unreported or rare pathogenic mutations or variants of unknown significance (VUSs), with care taken to evaluate both the specific mutation and the symptoms of the patient for proper diagnosis along the spectrum of ATP1A3-related disorders. Caution is warranted because VUSs are very common in panel testing and extremely common in WES. Consultation with a medical genetics professional familiar with the evaluation of a VUS and with family studies can aid in results interpretation, testing of family members, and genetic counseling.
ATP1A3-RELATED DISORDERS AND EPILEPSY
Approximately 50% of children with AHC also carry a diagnosis of epilepsy,32–34 although the certainty of this diagnosis can remain elusive because of similarity of epileptic and nonepileptic paroxysmal events such as dystonia, tremor, or paresis. By contrast, patients with RDP have seizures as a rare comorbidity,20 although this manifestation may be underrecognized as the number of patients with RDP is far less than AHC.
Recently, the International League Against Epilepsy revised the Operational Clinical Definition of Epilepsy to allow for the diagnosis of epilepsy following a first seizure in the context of increased recurrence risk.35 The challenge in accurately assessing the recurrence risk in adults and children with ATP1A3-related disorders is significant and may ultimately be dependent on specific genetic mutations.
Reviews of ATP1A3-related disorders have emphasized the potentially catastrophic course of epilepsy in patients with AHC.32 For this reason, we attempted to more precisely address seizure and epilepsy-related clinical practice issues. It was agreed that, when feasible, video-EEG characterization should be obtained when new, distinct, and stereotyped events are noted. However, the abundance and variability of paroxysmal events in AHC makes video-EEG characterization impractical for some instances. Broad input from family members, caretakers, and other medical personnel, as well as the use of personal video capabilities, may aid in more accurately characterizing the events in question.
Routine EEG may provide limited benefit early in the course of AHC evolution because classic epileptiform features are absent during most episodes.33 It is unknown whether EEG may help to better assess further risk of seizure later in the course of the disorder, when epilepsy is more typically present. Ultimately, workshop participants agreed that if clinical suspicion is high and EEG features suggestive of an underlying seizure tendency are noted, initiation of an anticonvulsant medication is warranted. If the EEG is unrevealing, then further clinical assessment and video-EEG monitoring is reasonable.
DISCUSSION AND FUTURE DIRECTIONS
ATP1A3-related disorders have become increasingly recognized with the advent of more potent molecular diagnostic tools. However, substantial knowledge gaps remain in understanding how specific genetic mutations and other epigenetic phenomena might influence physiologic mechanisms to produce the variable phenotypes. There is no specific treatment for ATP1A3-related diseases other than symptomatic, although flunarizine is widely used.36,37 There is no approved medication or device that alters the underlying deficit in the Na+/K+ pump.
During the course of workshop proceedings, it became clear that considerable challenges remain. A critical impediment to effective clinical trial design is a lack of standardized language to capture and measure disease burden for paroxysmal symptoms in AHC. Clinical trial designs typically implemented in epilepsy are inadequate. Patients with ATP1A3-related disorders also demonstrate variable impairments in cognition, language, mood, behavior, and gross motor and fine motor functions, and these outcomes will also be important to effectively document and assess reliably over time. Finally, development or use of caregiver and patient-reported outcome tools will likely prove a critical part of this process, especially if broader quality of life trends can be incorporated in addition to typical event counting. This combination may in the end prove the most effective way to measure improvement in response to specific targeted interventions. Given the variable phenotype, a multifaceted approach will be needed in which outcome measures are carefully chosen to reflect the most likely benefit of any intervention.
Supplementary Material
GLOSSARY
- AHC
alternating hemiplegia of childhood
- AHCF
Alternating Hemiplegia of Childhood Foundation
- CAPOS
cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss
- NGS
next-generation sequencing
- RDP
rapid-onset dystonia parkinsonism
- SUDEP
sudden unexpected death in epilepsy
- WES
whole-exome sequencing
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR CONTRIBUTIONS
Hendrik Rosewich: study concept and design, analysis and interpretation, study supervision, and critical revision of manuscript for important intellectual content. Matthew T. Sweney, Suzanne DeBrosse, Kevin Ess, and Laurie Ozelius: study concept and design, analysis and interpretation, and critical revision of manuscript for important intellectual content. Eva Andermann, Frederick Andermann, Gene Andrasco, and Alice Belgrade: study concept and design and critical revision of manuscript for important intellectual content. Allison Brashear: study concept and design, analysis and interpretation, and critical revision of manuscript for important intellectual content. Sharon Ciccodicola and Lynn Egan: study concept and design and critical revision of manuscript for important intellectual content. Alfred L. George: study concept and design, analysis and interpretation, and critical revision of manuscript for important intellectual content. Aga Lewelt, Joshua Magelby, Mario Merida, Tara Newcomb, Vicky Platt, Dominic Poncelin, Sandra Reyna, Masayuki Sasaki, Marcio Sotero de Menezes, Kathleen Sweadner, Louis Viollet, Mary Zupanc, and Kenneth Silver: study concept and design and critical revision of manuscript for important intellectual content. Kathryn Swoboda: study concept and design, analysis and interpretation, study supervision, and critical revision of manuscript for important intellectual content.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
Hendrik Rosewich reports no disclosures. Matthew T. Sweney has received a speaker honorarium from LivaNova. Suzanne DeBrosse has received travel funding from the AHC Foundation; has received publishing royalties from Decision Support in Medicine LLC and Elsevier; and has received research support from NIH, Philanthropic Account for Pyruvate Dehydrogenase Complex Deficiency Research, and University Hospitals Case Medical Center. Kevin Ess has served on a scientific advisory board for Novartis Pharmaceuticals; has served on the editorial boards of the Journal of Child Neurology and Epilepsy Currents; receives research funding through the Alternating Hemiplegia of Childhood Foundation, NIH/National Institute of Neurological Disorders and Stroke, and the Alternating Hemiplegia Childhood Foundation; and has provided expert witness for defense (unrelated to the present study). Laurie Ozelius holds patents related to DYT1 and DYT6 gene tests; has received research support from NIH and the Icahn School of Medicine at Mount Sinai; and receives royalty payments from Athena Diagnostics. Eva Andermann has served on the editorial boards of the Turkish Journal of Neurology, Epilepsy and Behavior, and the Journal of Epileptology. Frederick Andermann has served on the editorial boards of Brain and Development, Epilepsy and Behavior, the Journal of Epileptology, and the Turkish Journal of Neurology. Gene Andrasco and Alice Belgrade report no disclosures. Allison Brashear has served on the scientific advisory boards of Ipsen and Allergan; has salary support from the National Institute of Neurological Disorders and Stroke; performs research for Allergan, Merz USA, Revance, and Ipsen; consults for Ispsen, Allegan, Revance, and WorldMeds (all grants paid directly to the Wake Forest School of Medicine); is on the Board of Directors of the American Board of Psychiatry and Neurology and of the Association of University Professors of Neurology; reviews for the National Institute of Neurological Disorders and Stroke; has received publishing royalties from Demos; holds a patent (pending) for Application for care management; has received research support from the National Institute of Neurological Disorders and Stroke and NIH; and has been an expert reviewer for a patient during the last year. Sharon Ciccodicola and Lynn Egan report no disclosures. Alfred L. George has served on the scientific advisory board of Amgen; has received travel funding from EpiPM Therapeutics (now called Praxis Therapeutics); has served on the editorial boards of the Journal of Clinical Investigation, Heart Rhythm Journal, and the Journal of General Physiology; and has received research support from Merck Xenon Pharmaceuticals, NIH, and the Alternating Hemiplegia of Childhood Foundation. Aga Lewelt, Joshua Magelby, and Mario Merida report no disclosures. Tara Newcomb has received research support from NICHD. Vicky Platt is an employee of Willamette Management Associates. Dominic Poncelin, Sandra Reyna, and Masayuki Sasaki report no disclosures. Marcio Sotero de Menezes is employed by a nonprofit organization; has served on the speakers' bureau of Cyberonics; and is an investigator for Zogenix (not related to the study in question). Kathleen Sweadner has served on the scientific advisory board of Aarhus University; has received travel funding/speaker honoraria from the Weizmann Institute, Gordon Research Conferences, the University of Lausanne, the Harvard Club of Australia, and the Royal North Shore Hospital (University of New South Wales, Australia); and has received research support from NIH. Louis Viollet reports no disclosures. Mary Zupanc has served on the scientific advisory boards of Mallinckrodt Pharma and Insys Pharma; has received travel funding/speaker honoraria from Insys Pharma, Mallinckrodt Pharma, and the Penry Epilepsy Minifellowship Program; has served on the editorial board of Pediatric Neurology; and has received research support from Lundbeck and Insys Pharma. Kenneth Silver has served on the scientific advisory boards of the Childhood Foundation, Marathon Pharmaceuticals, PTC Therapeutics, and Retrophin Inc and has received travel funding/speaker honoraria from Munster Hospital Grand Rounds. Kathryn Swoboda has served on the scientific advisory board of AveXis; has served on the editorial board of the Journal of Neuromuscular Disorders; has been a consultant for the Biogen Nurture Advisory Board; and has received research support from Ionis Pharmaceuticals, Biogen, NIH, and the University of Utah. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Verret S, Steele JC. Alternating hemiplegia in childhood: a report of eight patients with complicated migraine beginning in infancy. Pediatrics 1971;47:675–680. [PubMed] [Google Scholar]
- 2.Dobyns WB, Ozelius LJ, Kramer PL, et al. Rapid-onset dystonia-parkinsonism. Neurology 1993;43:2596–2602. [DOI] [PubMed] [Google Scholar]
- 3.Nicolaides P, Appleton RE, Fryer A. Cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss (CAPOS): a new syndrome. J Med Genet 1996;33:419–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Carvalho Aguiar P, Sweadner KJ, Penniston JT, et al. Mutations in the Na+/K+ -ATPase alpha3 gene ATP1A3 are associated with rapid-onset dystonia parkinsonism. Neuron 2004;43:169–175. [DOI] [PubMed] [Google Scholar]
- 5.Heinzen EL, Swoboda KJ, Hitomi Y, et al. De novo mutations in ATP1A3 cause alternating hemiplegia of childhood. Nat Genet 2012;44:1030–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosewich H, Thiele H, Ohlenbusch A, et al. Heterozygous de-novo mutations in ATP1A3 in patients with alternating hemiplegia of childhood: a whole-exome sequencing gene-identification study. Lancet Neurol 2012;11:764–773. [DOI] [PubMed] [Google Scholar]
- 7.Demos MK. A novel recurrent mutation in ATP1A3 causes CAPOS syndrome. Orphanet J Rare Dis 2014;9:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heinzen EL, Arzimanoglou A, Brashear A, et al. Distinct neurological disorders with ATP1A3 mutations. Lancet Neurol 2014;13:503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosewich H, Ohlenbusch A, Huppke P, et al. The expanding clinical and genetic spectrum of ATP1A3-related disorders. Neurology 2014;82:945–955. [DOI] [PubMed] [Google Scholar]
- 10.Sweney MT, Newcomb TM, Swoboda KJ. The expanding spectrum of neurological phenotypes in children with ATP1A3 mutations, Alternating Hemiplegia of Childhood, Rapid-onset Dystonia-Parkinsonism, CAPOS and beyond. Pediatr Neurol 2015;52:56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosewich H, Baethmann M, Ohlenbusch A, Gärtner J, Brockmann K. A novel ATP1A3 mutation with unique clinical presentation. J Neurol Sci 2014;341:133–135. [DOI] [PubMed] [Google Scholar]
- 12.Dard R, Mignon C, Durr A, et al. Relapsing encephalopathy with cerebellar ataxia related to an ATP1A3 mutation. Dev Med Child Neurol 2015;57:1183–1186. [DOI] [PubMed] [Google Scholar]
- 13.Paciorkowski AR, McDaniel SS, Jansen LA, et al. Novel mutations in ATP1A3 associated with catastrophic early life epilepsy, episodic prolonged apnea, and postnatal microcephaly. Epilepsia 2015;56:422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Viollet L, Glusman G, Murphy KJ, et al. Alternating hemiplegia of childhood: retrospective genetic study and genotype-phenotype correlations in 187 subjects from the US AHCF registry. PLoS One 2015;10:e0127045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brashear A, Mink JW, Hill DF, et al. ATP1A3 mutations in infants: a new rapid-onset dystonia-Parkinsonism phenotype characterized by motor delay and ataxia. Dev Med Child Neurol 2012;54:1065–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blanco-Arias P, Einholm AP, Mamsa H, et al. A C-terminal mutation of ATP1A3 underscores the crucial role of sodium affinity in the pathophysiology of rapid-onset dystonia-parkinsonism. Hum Mol Genet 2009;18:2370–2377. [DOI] [PubMed] [Google Scholar]
- 17.Kamm C, Fogel W, Wachter T, et al. Novel ATP1A3 mutation in a sporadic RDP patient with minimal benefit from deep brain stimulation. Neurology 2008;70(16 pt 2):1501–1503. [DOI] [PubMed] [Google Scholar]
- 18.Svetel M, Ozelius LJ, Buckley A, et al. Rapid-onset dystonia-parkinsonism: case report. J Neurol 2010;257:472–474. [DOI] [PubMed] [Google Scholar]
- 19.Zanotti-Fregonara P, Vidailhet M, Kas A, et al. [123I]-FP-CIT and [99mTc]-HMPAO single photon emission computed tomography in a new sporadic case of rapid-onset dystonia-parkinsonism. J Neurol Sci 2008;273:148–151. [DOI] [PubMed] [Google Scholar]
- 20.Brashear A, Dobyns WB, de Carvalho Aguiar P, et al. The phenotypic spectrum of rapid-onset dystonia-parkinsonism (RDP) and mutations in the ATP1A3 gene. Brain 2007;130(pt 3):828–835. [DOI] [PubMed] [Google Scholar]
- 21.Maas RP, Schieving JH, Schouten M, Kamsteeg EJ, van de Warrenburg BP. The genetic homogeneity of CAPOS syndrome: four new cases with the c.2452G>A (p.Glu818Lys) mutation in the ATP1A3 gene. Pediatr Neurol 2016;59:71–75.e1. [DOI] [PubMed] [Google Scholar]
- 22.Potic A, Nmezi B, Padiath QS. CAPOS syndrome and hemiplegic migraine in a novel pedigree with the specific ATP1A3 mutation. J Neurol Sci 2015;358:453–456. [DOI] [PubMed] [Google Scholar]
- 23.Heimer G, Sadaka Y, Israelian L, et al. CAOS-episodic cerebellar ataxia, areflexia, optic atrophy, and sensorineural hearing loss: a third allelic disorder of the ATP1A3 gene. J Child Neurol 2015;30:1749–1756. [DOI] [PubMed] [Google Scholar]
- 24.Rosewich H, Weise D, Ohlenbusch A, Gärtner J, Brockmann K. Phenotypic overlap of alternating hemiplegia of childhood and CAPOS syndrome. Neurology 2014;83:861–863. [DOI] [PubMed] [Google Scholar]
- 25.Tomson T, Walczak T, Sillanpaa M, Sander JW. Sudden unexpected death in epilepsy: a review of incidence and risk factors. Epilepsia 2005;46(suppl 11):54–61. [DOI] [PubMed] [Google Scholar]
- 26.Nashef L, So E, Ryvlin P, Tomson T. Unifying the definitions of sudden unexpected death in epilepsy. Epilepsia 2012;53:227–233. [DOI] [PubMed] [Google Scholar]
- 27.Tomson T, Surges R, Delamont R, Haywood S, Hesdorffer DC. Who to target in sudden unexpected death in epilepsy prevention and how? Risk factors, biomarkers, and intervention study designs. Epilepsia 2016;57(suppl 1):4–16. [DOI] [PubMed] [Google Scholar]
- 28.Delogu AB, Spinelli A, Battaglia D, et al. Electrical and autonomic cardiac function in patients with Dravet syndrome. Epilepsia 2011;52(suppl 2):55–58. [DOI] [PubMed] [Google Scholar]
- 29.Wang J, Schwinger RH, Frank K, et al. Regional expression of sodium pump subunits isoforms and Na+-Ca++ exchanger in the human heart. J Clin Invest 1996;98:1650–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jaffer F, Avbersek A, Vavassori R, et al. Faulty cardiac repolarization reserve in alternating hemiplegia of childhood broadens the phenotype. Brain 2015;138(pt 10):2859–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Novy J, McWilliams E, Sisodiya SM. Asystole in alternating hemiplegia with de novo ATP1A3 mutation. Eur J Med Genet 2014;57:37–39. [DOI] [PubMed] [Google Scholar]
- 32.Sasaki M, Ishii A, Saito Y, et al. Genotype-phenotype correlations in alternating hemiplegia of childhood. Neurology 2014;82:482–490. [DOI] [PubMed] [Google Scholar]
- 33.Sweney MT, Silver K, Gerard-Blanluet M, et al. Alternating hemiplegia of childhood: early characteristics and evolution of a neurodevelopmental syndrome. Pediatrics 2009;123:e534–e541. [DOI] [PubMed] [Google Scholar]
- 34.Mikati MA, Kramer U, Zupanc ML, Shanahan RJ. Alternating hemiplegia of childhood: clinical manifestations and long-term outcome. Pediatr Neurol 2000;23:134–141. [DOI] [PubMed] [Google Scholar]
- 35.Fisher RS, Acevedo C, Arzimanoglou A, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia 2014;55:475–482. [DOI] [PubMed] [Google Scholar]
- 36.Sasaki M, Sakuragawa N, Osawa M. Long-term effect of flunarizine on patients with alternating hemiplegia of childhood in Japan. Brain Dev 2001;23:303–305. [DOI] [PubMed] [Google Scholar]
- 37.Chi LY, Zao XH, Liu XW, Jiang WJ, Chi ZF, Wang SJ. Alternating hemiplegia of childhood in chinese following long-term treatment with flunarizine or topiramate. Int J Neurosci 2012;122:506–510. [DOI] [PubMed] [Google Scholar]
- 38.Bourgeois M, Aicardi J, Goutieres F. Alternating hemiplegia of childhood. J Pediatr 1993;122:673–679. [DOI] [PubMed] [Google Scholar]
- 39.Panagiotakaki E, Gobbi G, Neville B, et al. Evidence of a non-progressive course of alternating hemiplegia of childhood: study of a large cohort of children and adults. Brain 2010;133:3598–3610. [DOI] [PubMed] [Google Scholar]
- 40.Anselm IA, Sweadner KJ, Gollamudi S, Ozelius LJ, Darras BT. Rapid-onset dystonia-parkinsonism in a child with a novel ATP1A3 gene mutation. Neurology 2009;73:400–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sweadner KJ, Toro C, Whitlow CT, et al. ATP1A3 mutation in adult rapid-onset ataxia. PLoS One 2016;11:e0151429. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.