

ABSTRACT
Transcription of type I interferon genes during RNA virus infection requires signal communication between several pattern recognition receptor (PRR)-adaptor complexes located at distinct subcellular membranous compartments and a central cytoplasmic TBK1-interferon regulatory factor 3 (IRF3) kinase-transcription factor module. However, how the cell integrates signal transduction through spatially distinct modules of antiviral signaling pathways is less defined. RIG-I is a major cytosolic PRR involved in the control of several RNA viruses. Here we identify ArfGAP domain-containing protein 2 (ADAP2) as a key novel scaffolding protein that integrates different modules of the RIG-I pathway, located at distinct subcellular locations, and mediates cellular antiviral type I interferon production. ADAP2 served to bridge the mitochondrial membrane-bound upstream RIG-I adaptor MAVS and the downstream cytosolic complex of NEMO (regulatory subunit of TBK1), TBK1, and IRF3, leading to IRF3 phosphorylation. Furthermore, independently, ADAP2 also functioned as a major orchestrator of the interaction of TBK1 with NEMO and IRF3. Mutational and in vitro cell-free reconstituted RIG-I signaling assay-based analyses identified that the ArfGAP domain of ADAP2 mediates the interferon response. TRAF3 acted as a trigger for ADAP2 to recruit RIG-I pathway component proteins into a single macromolecular complex. This study provides important novel insights into the assembly and integration of different modules of antiviral signaling cascades.
KEYWORDS: RIG-I, innate immunity, interferons
INTRODUCTION
RNA viruses cause a significant majority of acute and chronic human viral infections (1, 2). The innate immune system has evolved multiple strategies to effectively control RNA viruses (3–9). Type I interferons (TI-IFNs), potent antiviral proteins induced by the innate immune system, are critical for controlling viral infections (10–13).
The major antiviral TI-IFNs (e.g., IFN-β and IFN-α) are produced through a signaling cascade initiated upon the recognition of incoming viruses by several pattern recognition receptors (PRRs) located at different sites within host cells. RIG-I is a prominent PRR that detects RNA viruses in the cytoplasm and plays critical roles in the control of infections by several viruses (4–7, 9, 14–16). Upon viral sensing, cytoplasmic RIG-I interacts with the adaptor protein MAVS, which is localized on mitochondria, the mitochondrion-associated membrane system, and peroxisomes (17–22). Activated MAVS polymerizes to form prion-like structures and subsequently activates NEMO, the regulatory subunit of cytosolic Tank binding kinase 1 (TBK1), leading to phosphorylation of the latent cytoplasmic transcription factor interferon regulatory factor 3 (IRF3) (23, 24). Signals from multiple antiviral PRRs converge at the level of TBK1 activation (6–8, 25–28). Activated IRF3 dimerizes, migrates to the nucleus, binds to the interferon gene promoter, and then, along with cofactors such as CBP/p300, initiates TI-IFN gene transcription (29–34).
Although several molecules and mechanisms involved in the RIG-I-mediated TI-IFN response are well studied, many aspects of the regulation of this pathway have yet to be understood. It is of interest to determine how different upstream and downstream protein modules of RIG-I signaling are assembled, and understanding how these modules bridge each other and IRF3 needs further studies (35, 36). In particular, how mitochondrial membrane-bound MAVS transduces its activation to the cytoplasmic NEMO-TBK1-IRF3 complex and how activation of TBK1 by multiple antiviral PRRs is integrated with the recruitment and phosphorylation of IRF3 by TBK1 are not completely understood. Compartmentalized organization of various modules of antiviral PRRs and their integration at organelle membranes are widespread phenomena, as also seen in the case of many other antiviral PRRs, such as Toll-like receptors (TLRs) and cyclic GMP-AMP synthase, among others (4, 37–39). Therefore, it is logical to presume that several of these components need to be brought into spatial proximity for interferon gene transcription to initiate and that there may exist a scaffold protein platform(s) integrating multiple components and modules of PRR signaling and IRF3 activation.
MAVS is localized on membranous platforms, primarily mitochondria, along with distributions on peroxisomes and the mitochondrion-associated membrane system (17–22). MAVS is known to be part of a signaling complex involving cytosolic NEMO as well as TBK1 (39, 40). How activated MAVS that is localized to specific organelles communicates with cytosolic NEMO and TBK1, leading to IRF3 activation, is unclear. The role of ubiquitination in transducing signals from MAVS to IRF3 has been studied extensively (41–45). Some previous studies recorded important redundant roles for TRAF proteins 2, 5, and 6, as well as for TRAF3, in transducing signals from activated MAVS and other antiviral PRRs to IRF3, leading to IRF3 phosphorylation (36, 46–49). Although TRAFs are important in RIG-I signaling, current evidence is inconclusive regarding whether they directly mediate the association of the NEMO-TBK1 complex with MAVS as well as IRF3 (36). Recently, TRIM14-based ubiquitination was also determined to be essential for NEMO recruitment to MAVS (50). Unlike our understanding of the role of ubiquitination in RIG-I signaling, the identity and role of the scaffolding protein platforms on which activated membrane-bound MAVS transduces signals to cytosolic IRF3 are less well understood. Although TANK was determined to interact with MAVS, NEMO, TBK1, TRAF3, and IRF3, no information is available on whether it mediates the assembly of these signaling proteins (51, 52). Moreover, TANK was later found to be dispensable for TLR3-mediated interferon production in vivo (53). Another study reported that Hsp90 associates with TBK1 and IRF3 and that this complex is recruited to MAVS upon stimulation; however, Hsp90 was not demonstrated to directly bridge NEMO and TBK1 to MAVS (54). Yet another study identified that the tetratricopeptide domain-containing protein IFIT3 plays a role in the association of MAVS with TBK1 (40). A detailed previous study identified MITA, a mitochondrial protein, as playing an important role in bridging MAVS to TBK1 and IRF3 (37, 39). It was found that a constitutively formed MITA-IRF3 complex remained localized on the mitochondrial surface and that viral infection promoted MITA (with bound IRF3)-MAVS interaction. However, it is unclear how phosphorylation of the bulk of the free IRF3 present in the cytoplasm is mediated by MITA during RIG-I signaling. MITA was found to be nonessential for TRIF/TLR3-induced IRF3 activation, arguing that its role is associated only with MAVS-requiring pathways. Hence, how IRF3 activation is integrated with the primed TBK1 complex cannot be explained entirely by MITA. A very recent study identified that phosphorylated MAVS interacts with IRF3 and proposed that MAVS may act as a scaffold for bringing TBK1 and IRF3 into close proximity (55). All these studies indicated that the transduction of signals between the upstream membrane-bound and downstream cytosolic modules of RIG-I signaling requires complex regulatory processes, including specific protein scaffold platforms that may bring various components together. Discovery of the mechanisms involved in the coupling of membrane-anchored MAVS with its downstream cytosolic molecular complex will greatly enhance our understanding of interferon response regulation.
Another area of antiviral PRR signaling that needs further studies pertains to the protein(s) involved in mediating the binding of the activated TBK1 complex to IRF3 (56). Because IRF3 activation drives interferon gene transcription, this step has to be tightly regulated. Although NAP1 and SINTBAD were previously shown to bind TBK1, whether they help in the recruitment of IRF3 to TBK1 is not known (57–60). Similarly, whether TANK can act as a scaffold for TBK1-IRF3 association is unknown (51, 52). Because multiple antiviral PRRs (e.g., RIG-I and TLR3) converge to activate TBK1, it is likely that there exist unidentified unifying protein scaffolds that orchestrate the assembly and integration of TBK1 activation with IRF3.
In this study, we investigated the regulation of integration of different modules of interferon signaling by using RIG-I signaling as a model pathway. We report the identification of the protein ArfGAP domain-containing protein 2 (ADAP2) as a critical protein scaffold that couples different modules of RIG-I signaling, leading to interferon gene transcription.
RESULTS
ADAP2 is a positive regulator of RIG-I-mediated interferon gene transcription.
We previously reported the identification of over 200 proteins as novel regulators of RIG-I signaling by use of a human genome-wide RNA interference (RNAi) screen (61). Because our goal in the present study was to determine the scaffold proteins that bridge different modules of RIG-I signaling, we investigated whether any of these newly reported proteins could be candidate molecules serving this function. One of the genes identified in our published RNAi screen for RIG-I regulators, ADAP2, caught our attention because a previous proteomics study had reported the ADAP2 protein as an interacting partner of NEMO (62). In a related context, it is also worth noting another study which identified ADAP2 as one of the two proteins present in the isolated minimal cytoplasmic fraction that could support the DNA-mediated interferon response (63). Although these two earlier studies indirectly hinted at a possible role for ADAP2 in interferon production, neither performed any validation or functional and mechanistic studies on the role of ADAP2 in interferon gene transcription. Because ADAP2 was already reported to bind to NEMO, we specifically explored in this study whether ADAP2 could be a scaffolding platform mediating the assembly of the MAVS-NEMO-TBK1-IRF3 complex.
ADAP2 belongs to the ArfGAP (ADP ribosylation factor, GTPase-activating proteins) family of genes (64–66). We first validated and established the role of ADAP2 in RIG-I-mediated IFNB1 (the gene that encodes type I IFN-β) gene transcription in HEK293T cells, a model cell line that is widely used to dissect antiviral pathways. For this purpose, ADAP2 was knocked down by use of small interfering RNAs (siRNAs), and the RIG-I pathway was stimulated by transfection with the ligand poly(I·C). Silencing of the positive-control gene MAVS heavily abolished RIG-I-induced human IFN-β promoter-driven luciferase reporter activity. Similarly, silencing of ADAP2 by two independent siRNAs also led to significant (up to 9-fold; P < 0.01) reductions of the IFN-β signal. Gene silencing was confirmed by Western blotting (Fig. 1A). To ascertain the on-target specificity of ADAP2-targeting siRNAs, we silenced endogenous ADAP2 by using siRNAs against its 3′ untranslated region (UTR) in HEK293 cells, and we transfected an ADAP2 coding region-containing expression plasmid into the cells to rescue the IFN-β activation that was lost due to knockdown. As shown in Fig. 1B, this approach confirmed that the tested siRNAs were indeed targeting ADAP2 and that the observed defect in the interferon response was due to the lack of ADAP2 expression.
FIG 1.

ADAP2 is essential for the interferon response. (A) Knockdown of ADAP2 by use of two independent siRNAs reduced poly(I·C)-stimulated RIG-I-mediated IFN-β luciferase reporter activity in HEK293T cells. Gene knockdown efficiencies of the siRNAs as determined by Western blotting are shown in the inset. (B) Rescue of loss of IFN-β luciferase reporter activity in HEK293T cells caused by 3′-UTR-targeting siRNA-mediated endogenous ADAP2 silencing. The phenotype was rescued through overexpression of the ADAP2 ORF. (C) Knockdown of ADAP2 by use of two independent siRNAs reduced MDA5 overexpression-mediated IFN-β luciferase reporter activity in HEK293T cells. (D and E) ADAP2 knockdown reduced poly(I·C)-stimulated RIG-I-mediated IFN-α4 (D) and NF-κB (E) luciferase reporter activation. (F) ADAP2 knockdown attenuated ISG15 expression in Sendai virus-infected HEK293T cells as determined by qRT-PCR. (G) Knockdown of ADAP2 in human primary monocytes ablated SeV infection-induced IFNB1 transcript formation as determined by qRT-PCR analysis of viral RNA. (H) Knockdown of ADAP2 in HEK293T cells increased the VSV infection load as determined by plaque assay at 18 h postinfection. The luciferase values shown are means and SD for a representative experiment performed in triplicate. Reporter (firefly) luciferase values were normalized with a renilla luciferase internal control and expressed as fold induction values. RNA transcript loads were determined using qRT-PCR and expressed as fold changes. The qRT-PCR data were calculated by determining the relative threshold cycle (CT) values, based on the formula 2−(CT of target gene − CT of β-actin). The values for uninfected cells (F and G) were taken as 1. The statistical significance of differences in mean values was analyzed using unpaired two-tailed Student's t test, and P values of <0.05 were considered statistically significant (**, P < 0.01). si, siRNA; si-NT, nontargeting negative-control siRNA; EV, empty vector; UTR, untranslated region; ORF, open reading frame; VSV, vesicular stomatitis virus; SeV, Sendai virus; GAPDH, glyceraldehyde-3-phosphate dehydrogenase (cytoplasmic internal control marker).
ADAP2 regulates both interferon and NF-κB responses from PRRs.
MDA5 is another cytosolic antiviral PRR that signals through MAVS and induces IFN-β gene transcription. It was determined that similar to that of RIG-I, ADAP2 knockdown resulted in a reduced IFN-β response driven by MDA5 (Fig. 1C). In addition to IFN-β, RIG-I also induces transcription of genes encoding another class of interferons, IFN-α1 to -13. Additionally, proinflammatory pathways are also triggered by RIG-I, through the activation of NF-κB. To determine whether ADAP2 regulates multiple pathways downstream from RIG-I, we stimulated ADAP2-silenced HEK293T cells by use of poly(I·C) and determined the IFN-α4 promoter- and NF-κB target promoter-driven luciferase activities. The results identified that ADAP2 was indeed needed for multiple signaling pathways downstream from RIG-I. ADAP2-silenced cells showed reduced activation of the IFN-α4-driven (Fig. 1D) (2-fold; P < 0.05) and NF-κB-driven (Fig. 1E) (3.5-fold; P < 0.01) luciferase reporters. These results indicated that ADAP2 is central to several downstream effects of RIG-I signaling and likely acts at a step upstream of pathway bifurcation into interferon and NF-κB branches.
ADAP2 is needed to control viral infection.
We further proceeded to assess whether ADAP2 is involved in the host cell IFN-β response and resistance to actual viral infection of human cells. We used Sendai virus (SeV), a paramyxovirus that activates RIG-I, as the model virus (67). Consistent with the observed defect in IFN-β promoter-driven luciferase reporter activity upon RIG-I activation by poly(I·C), Sendai virus-infected ADAP2-silenced HEK293T cells also showed reduced (up to 2.4-fold; P < 0.01) expression of ISG15, an interferon-stimulated gene (Fig. 1F). We also investigated whether ADAP2 has any role in the interferon response in human primary cells during viral infection. For this purpose, ADAP2-silenced human primary monocytes were infected with Sendai virus and assessed for IFNB1 transcript formation. As revealed in Fig. 1G, silencing of ADAP2 attenuated IFNB1 transcript formation (up to 2.4-fold; P < 0.01) induced by SeV infection in primary immune cells.
Next, we assessed whether ADAP2 contributes to host control of viruses by assessing the effect of altered ADAP2 expression on the infectivity of vesicular stomatitis virus (VSV), a rhabdovirus known to be sensitive to interferon. The load of VSV as determined by plaque assay was significantly enhanced (up to 26-fold; P < 0.01) in ADAP2-silenced HEK293T cells (Fig. 1H).
The ArfGAP domain of ADAP2 regulates the interferon response.
Further experiments were performed to determine the structural regions of ADAP2 needed for RIG-I signaling by using mutagenesis and truncations. Complementing the results obtained from ADAP2 knockdown experiments, ectopic expression of full-length ADAP2 notably enhanced IFN-β production (up to 3.5-fold; P < 0.01) in a dose-dependent manner (Fig. 2A). We also examined which domain(s) of ADAP2 is needed to augment IFN-β reporter activity upon ectopic expression. ADAP2 has an N-terminal ArfGAP domain and two C-terminal PH domains (PH1 and PH2) (Fig. 2B). Subsequently, truncation experiments were performed to identify the domains of ADAP2 needed to modulate the interferon response. Ectopically expressed PH domains (PH1 alone, PH2 alone, or both domains) without the adjoining ArfGAP domain revealed that the PH domains are dispensable for the IFN-β response-modulating activity of ADAP2 (Fig. 2C). On the other hand, the ectopically expressed ArfGAP domain enhanced the interferon response comparably to the wild-type (WT) ADAP2 protein, conclusively identifying that the ArfGAP domain is essential and sufficient for the IFN-β-stimulating activity of ADAP2 (Fig. 2C).
FIG 2.

The ArfGAP domain of ADAP2 is needed for the antiviral response. (A) Ectopic expression of ADAP2 enhanced RIG-I overexpression-mediated IFN-β luciferase reporter activity in HEK293T cells. (B) Schematic showing the domain organization of the ADAP2 protein. (C) Effects of ectopic expression of different truncations of ADAP2 on RIG-I overexpression-mediated IFN-β luciferase reporter activity. The inset shows expression levels of truncation proteins determined by Western blotting (IB). Reporter (firefly) luciferase values were normalized with a renilla luciferase internal control and expressed as fold induction values. The values shown are means and SD for a representative experiment performed in triplicate. The statistical significance of differences in mean values was analyzed using unpaired two-tailed Student's t test, and P values of <0.05 were considered statistically significant (*, P < 0.05; **, P < 0.01). EV, empty vector; WT, wild type; Mut, truncation mutant; PH, pleckstrin homology domain; none, only the IFN-β promoter-driven luciferase reporter was present within the cells; GAPDH, glyceraldehyde-3-phosphate dehydrogenase (cytoplasmic internal control marker).
ADAP2 is needed for IRF3 phosphorylation.
After establishing the role of ADAP2 as a positive regulator of the antiviral response, we attempted to identify the precise stage of RIG-I signaling that is regulated by ADAP2. Ectopic expression of wild-type RIG-I, MAVS, and TBK1 and a constitutively active phosphomimetic IRF3 mutant (IRF3-D5) can induce IFN-β gene transcription without any ligand stimulation (61). By combining ADAP2 silencing with pathway component ectopic expression-based IFN-β activation, we identified that knockdown of ADAP2 attenuated IFN-β activation induced by ectopic expression of RIG-I, MAVS, and TBK1 but not IRF3-D5 (Fig. 3A). These data indicated that ADAP2 affects IFN-β gene transcription by regulating a step upstream of IRF3 activation. Accordingly, coexpressed ADAP2 was found to enhance IFN-β gene transcription induced by overexpression of MAVS and TBK1 (Fig. 3B). The detection of viruses by PRRs initiates a signaling event that activates the cytosolic latent transcription factor IRF3 through phosphorylation by the kinase TBK1. To further confirm that ADAP2 regulates RIG-I signaling upstream of IRF3 activation, we investigated whether the phosphorylation of IRF3 is intact within ADAP2-silenced cells. For this purpose, ADAP2-silenced HEK293T cells were stimulated with SeV, and phosphorylation of IRF3 was detected by Western blotting. The level of phosphorylated IRF3 within ADAP2 knockdown cells was found to be notably less than that in negative-control siRNA (siNT)-transfected cells (Fig. 3C). This experiment further confirmed that ADAP2 regulates RIG-I signaling upstream of IRF3 activation.
FIG 3.

ADAP2 is needed for IRF3 phosphorylation. (A) Knockdown of ADAP2 reduced IFN-β promoter-driven luciferase reporter activity induced by ectopic expression of MAVS, TBK1, and IRF3-D5 in HEK293T cells. (B) Coexpression of ADAP2 increased IFN-β promoter-driven luciferase reporter activity induced by overexpression of MAVS and TBK1. (C) ADAP2 silencing reduced SeV infection-induced IRF3 phosphorylation in HEK293T cells. (D) A cell-free in vitro RIG-I-mediated IRF3 phosphorylation assay established ADAP2 as a regulator of the interferon response. Western blotting was used to detect pIRF3 formation after stimulation of uninfected HEK293T cellular cytoplasm with mitochondria from infected HEK293T cells. The ADAP2-silenced cytoplasm was supplemented with either vehicle only or recombinant ADAP2 (full length, Mut1, and/or Mut3). Reporter (firefly) luciferase values were normalized with a renilla luciferase internal control and expressed as fold induction values. The values shown are means and SD for a representative experiment performed in triplicate. The statistical significance of differences in mean values was analyzed using unpaired two-tailed Student's t test, and P values of <0.05 were considered statistically significant (**, P < 0.01). Si, siRNA; Si-NT, nontargeting negative-control siRNA; EV, empty vector; ORF, open reading frame; h, hours postinfection; none, only the IFN-β promoter-driven luciferase reporter was present within the cells; mito, mitochondrial fraction; cyto, cytoplasmic fraction; rADAP2, purified recombinant ADAP2 protein; WT, wild-type full-length ADAP2; GAPDH, glyceraldehyde-3-phosphate dehydrogenase (cytoplasmic internal control marker); siNT Sev (Mito), mitochondrial fraction of siNT-treated Sendai virus-infected cells; siNT (Cyto), cytoplasmic fraction of siNT-treated uninfected cells; siADAP2 Sev (Mito), mitochondrial fraction of siADAP2-treated Sendai virus-infected cells; siADAP2 (Cyto), cytoplasmic fraction of siADAP2-treated uninfected cells.
Recombinant ADAP2 supports IRF3 phosphorylation in a cell-free IRF3 activation assay.
We recently reported the use of a cell-free IRF3 activation assay to characterize the regulation of RIG-I signaling, based on earlier studies (44, 61). As another approach to establish the critical requirement for ADAP2 in IRF3 activation, we used the cell-free IRF3 activation assay. In this assay, the purified mitochondrial fraction from Sendai virus-infected wild-type 293T cells was able to induce phosphorylation of IRF3 in the cytoplasm from unstimulated wild-type cells. Consistent with the cell-based assay results, it was found that cytoplasm from ADAP2-silenced cells was unable to support IRF3 phosphorylation when stimulated with the mitochondrial fraction from either negative-control siRNA (siNT)-treated or ADAP2 siRNA-treated Sendai virus-infected 293T cells (Fig. 3D). Addition of recombinant full-length ADAP2 (expressed and purified from HEK293T cells) to the cytoplasmic extract from ADAP2-silenced cells rescued IRF3 phosphorylation induced by the SeV-stimulated mitochondrial fraction (Fig. 3D). Using the cell-free assay, we also investigated the specific domain of ADAP2 needed for RIG-I signaling. It was observed that addition of the purified recombinant ArfGAP domain alone compensated for the defect in IRF3 phosphorylation in the endogenous ADAP2-deficient cytoplasm (Fig. 3D). On the other hand, the purified recombinant PH1 domain of ADAP2 did not rescue the defect in IRF3 phosphorylation in the cytoplasm of ADAP2-silenced cells (Fig. 3D). This result was also consistent with the data obtained from the mutagenesis studies described for Fig. 2C. These data provided direct evidence to definitively demonstrate that the ArfGAP domain of ADAP2 is sufficient and essential for RIG-I-induced IRF3 activation.
ADAP2 interacts with multiple proteins of the RIG-I signaling pathway.
Additional experiments were performed to delineate how ADAP2 regulates IRF3 activation. Because ADAP2 is not a catalytic protein, we reasoned that it might serve a scaffolding function. As ADAP2 was previously reported to interact with NEMO in a proteomics screen (62), our initial investigations were focused on assessing whether ADAP2 physically interacts with RIG-I pathway signaling component proteins, such as MAVS, NEMO, TBK1, and IRF3. For the assay, HEK293T cells were stimulated with Sendai virus (SeV) for different periods, and potential interactions of endogenous ADAP2 with endogenous RIG-I, MAVS, NEMO, TBK1, and IRF3 were analyzed by coimmunoprecipitation (co-IP) using antibodies detecting the respective proteins (Fig. 4A). There was no significant interaction between ADAP2 and MAVS, NEMO, TBK1, or IRF3 prior to SeV challenge. However, upon infection with SeV, strong interactions were observed between ADAP2 and MAVS, NEMO, TBK1, and IRF3. We also investigated whether ADAP2 interacts with RIG-I signaling complex proteins in human primary monocytes. ADAP2 was also found to interact with MAVS, NEMO, TBK1, and IRF3 in human primary monocytes during SeV challenge (Fig. 4B).
FIG 4.

The ArfGAP domain of ADAP2 interacts with RIG-I pathway proteins. Cells were infected with SeV, and endogenous protein interactions were detected using coimmunoprecipitation and Western blotting. (A) Immunoprecipitated ADAP2 from HEK293T cells showed interactions with MAVS, NEMO, TBK1, and IRF3 upon SeV infection. (B) Immunoprecipitated ADAP2 from human primary monocytes showed interactions with MAVS, NEMO, TBK1, and IRF3 upon SeV infection. (C to F) Coimmunoprecipitation of ectopically expressed full-length NEMO (C), TBK1 (D), IRF3 (E), and MAVS (F) in cells transfected with various truncation plasmids of ADAP2 identified that the ArfGAP domain interacts with RIG-I pathway proteins. hpi, hours postinfection; FL, full length; Mut, truncation mutant of ADAP2; SeV, Sendai virus; WCL, whole-cell lysate; IP, immunoprecipitation; IB, immunoblot; HA, hemagglutinin tag; GAPDH, glyceraldehyde-3-phosphate dehydrogenase (cytoplasmic internal control marker).
We then proceeded to map the domains of ADAP2 needed for binding to MAVS, NEMO, TBK1, and IRF3. The PH1 and PH2 domains were dispensable for ADAP2 to interact with all of these proteins. Interestingly, it was found that only the ArfGAP domain (Mut1) was needed and sufficient for ADAP2 to interact with NEMO, TBK1, IRF3, and MAVS (Fig. 4C to F). Intriguingly, although the ArfGAP domain alone interacted strongly with the pathway proteins, we observed that the ArfGAP-PH1 fragment (Mut2) did not show consistent interactions (or showed only a barely detectable weak interaction) with NEMO, TBK1, IRF3, and MAVS at lower expression levels, as shown in Fig. 4C to F. This anomaly may be due to instability or conformational alterations of the ArfGAP-PH1 fragment.
Furthermore, we designed experiments to identify the specific domains of MAVS, NEMO, TBK1, and IRF3 that interact with ADAP2. We identified that deletion of the N-terminal 150 amino acids of NEMO abolished its ability to interact with ADAP2 (Fig. 5A). Truncation experiments identified that the amino acids between residues 300 and 385 of TBK1 mediated its association with ADAP2 (Fig. 5B). This region corresponds to the ubiquitin-like domain (ULD) of TBK1. Using truncated IRF3, we determined that the N-terminal 131 amino acids were not essential for its interaction with ADAP2 (Fig. 5C). The domains of IRF3 between amino acids 253 and 325 enabled it to interact weakly with ADAP2. The region of IRF3 between amino acids 325 and 390 was needed for it to strongly interact with ADAP2. The N-terminal first 100 amino acids of MAVS were found to be essential and sufficient for interaction with ADAP2 (Fig. 5D). As this region corresponds to the caspase activation and recruitment domain (CARD), we reasoned that the CARD of MAVS is critical for its interaction with ADAP2.
FIG 5.

ADAP2 interacts with specific domains of RIG-I pathway proteins. Cells were transfected with plasmids for full-length HA-ADAP2 and various truncations of NEMO, TBK1, IRF3, and MAVS, and coimmunoprecipitation was performed using an antibody detecting the HA tag. (A) The N-terminal 150 amino acids of NEMO were needed to interact with ADAP2. (B) Amino acids between positions 300 and 385 of TBK1 were needed for its association with ADAP2. (C) The region of IRF3 between amino acids 325 and 390 was needed for strong interaction with ADAP2. (D) MAVS interacted with ADAP2 through its N-terminal 100 amino acids. FL, full length; SeV, Sendai virus; WCL, whole-cell lysate; IP, immunoprecipitation; IB, immunoblot; HA, hemagglutinin tag; GAPDH, glyceraldehyde-3-phosphate dehydrogenase (cytoplasmic internal control marker).
Collectively, these studies indicated that ADAP2 interacts with MAVS, TBK1, NEMO, and IRF3 at specific domains of these target proteins. This interaction was triggered by viral infection. Strikingly, the ArfGAP domain of ADAP2 was determined to be critical and sufficient for interacting with RIG-I pathway components; this is consistent with the results obtained from the IFN-β promoter activity assays and the in vitro reconstituted IRF3 activation assay, in which the isolated ArfGAP domain alone was found to support pathway activation.
ADAP2 mediates the interaction of MAVS with the downstream NEMO-TBK1 signaling complex.
The above-described experiments identified that ADAP2 interacts with multiple components of the RIG-I pathway, leading to interferon production. The critical question that now needs to be answered is the precise mechanism by which ADAP2 regulates RIG-I signaling. A key event in RIG-I signaling involves transduction of signals from membrane-bound MAVS to cytosolic NEMO and TBK1, leading to phosphorylation of IRF3. Activated MAVS is established as a component of a complex containing NEMO and TBK1. Because ADAP2 interacted with both MAVS and several downstream components (e.g., TBK1, NEMO, and IRF3), we first asked whether ADAP2 functions by linking MAVS to its downstream cytosolic components. To test this, we immunoprecipitated endogenous MAVS from SeV-infected 293T cells silenced for ADAP2 and assessed the status of the interaction of MAVS with various downstream proteins. Strikingly, it was observed that the ability of MAVS to interact with endogenous NEMO and TBK1 was notably attenuated in the absence of ADAP2 expression (Fig. 6A). Conversely, immunoprecipitated TBK1 was unable to bind efficiently to MAVS and to form a complex with NEMO in ADAP2-silenced cells (Fig. 6B). Similarly, ADAP2 silencing was found to impair the association of MAVS with NEMO and TBK1 in human primary monocytes challenged with SeV (Fig. 6C). These results demonstrated that ADAP2 is essential for MAVS to interact with the downstream cytosolic components NEMO and TBK1.
FIG 6.

ADAP2 mediates assembly of two modules of antiviral signaling pathway components. (A to C) ADAP2 silencing abolished interaction of MAVS with TBK1 and NEMO. (A) ADAP2 was silenced in HEK293T cells, endogenous MAVS was immunoprecipitated, and interactions with endogenous TBK1 and NEMO were probed by Western blotting. (B) ADAP2 was silenced in HEK293T cells, endogenous TBK1 was immunoprecipitated, and interactions with endogenous MAVS and NEMO were probed by Western blotting. (C) ADAP2 was silenced in human primary monocytes, endogenous MAVS was immunoprecipitated, and interactions with endogenous TBK1 and NEMO were probed by Western blotting. (D) ADAP2-mediated assembly of TBK1 with NEMO and IRF3. Overexpressed Myc-TBK1 was immunoprecipitated from ADAP2-silenced HEK293T cells, and interactions with endogenous NEMO and IRF3 were probed by Western blotting. (E) ADAP2 silencing attenuated TLR3-mediated IFN-β promoter-driven luciferase reporter activity. ADAP2-silenced TLR3-expressing HEK293T cells were stimulated by poly(I·C) added to the medium. *, P < 0.05. (F) ADAP2 silencing abolished TLR3 activation-induced interaction of endogenous TBK1 with NEMO and IRF3. Endogenous TBK1 was immunoprecipitated after poly(I·C) stimulation of TLR3, and interactions with endogenous NEMO and IRF3 were probed by Western blotting. Reporter (firefly) luciferase values were normalized with a renilla luciferase internal control and expressed as fold induction values. The values shown are means and SD for a representative experiment performed in triplicate. The statistical significance of differences was calculated by comparing the value for each test condition (gene silencing) with that for the corresponding stimulated siNT-treated control samples. si, siRNA; siNT, nontargeting negative-control siRNA; EV, empty vector; hpi, hours postinfection; SeV, Sendai virus; WCL, whole-cell lysate; IP, immunoprecipitation; IB, immunoblot; p-IRF3, phosphorylated IRF3; GAPDH, glyceraldehyde-3-phosphate dehydrogenase (cytoplasmic internal control marker).
ADAP2 is a scaffolding platform orchestrating the interaction between TBK1, NEMO, and IRF3.
The above-described data demonstrated that ADAP2 expression is needed for the interaction of activated MAVS with the NEMO and TBK1 complex. However, the data shown in Fig. 3A also showed that ADAP2 silencing attenuated IFN-β promoter-driven reporter activity induced by TBK1 overexpression. In addition, the data shown in Fig. 6B revealed that ADAP2 silencing reduced the interaction of TBK1 with NEMO. It is unclear how ADAP2 with an upstream role would also regulate the downstream TBK1-NEMO interaction. One potential explanation is that ADAP2 may serve another independent and parallel role, besides bridging from MAVS to NEMO and TBK1, by acting as a major scaffolding protein. Since TBK1 is known to interact with NEMO and IRF3 during RIG-I stimulation, we investigated whether ADAP2 is also needed for TBK1 to interact with NEMO and IRF3. Co-IP experiments in ADAP2-silenced, TBK1-overexpressing cells (for specifically activating the downstream steps of the pathway without any virus infection-based signals from upstream MAVS) revealed that the absence of ADAP2 caused a substantial reduction in the ability of TBK1 to interact with both NEMO and IRF3 (Fig. 6D). These data indicated that ADAP2 also has a role independent of MAVS, in which it also mediates the association of TBK1 with NEMO and IRF3. To further establish conclusively whether ADAP2 has a role in the interferon response separate from regulating MAVS, we investigated the potential contribution of ADAP2 to MAVS-independent antiviral pathways. We hypothesized that if ADAP2 has a MAVS-independent role in the antiviral response, then ADAP2 silencing will attenuate the IFN-β response from TLR3, an antiviral PRR that does not require MAVS. In fact, we found that ADAP2 knockdown caused a significant reduction of the IFN-β response from TLR3 stimulated with poly(I·C) (up to 6-fold; P < 0.01) (Fig. 6E). Consistent with this, ADAP2 silencing led to a prominent reduction in the interaction of TBK1 with NEMO and IRF3 during TLR3 signaling (Fig. 6F). These data demonstrate that ADAP2 also mediates the interaction between TBK1, NEMO, and IRF3 independently of its role in mediating the association of MAVS with TBK1 and NEMO.
TRAF3 is needed for ADAP2 to interact with RIG-I pathway proteins.
While the data described so far successfully established the interaction of ADAP2 with key proteins of the RIG-I-mediated interferon production pathway, the kinetics and interdependence of these interactions are not yet clear. We therefore investigated the temporal dynamics of the protein interactions happening during RIG-I signaling in relation to ADAP2. First, we investigated whether the ability of ADAP2 to bind NEMO, TBK1, and IRF3 is dependent on virus infection-induced signaling through MAVS. For this purpose, we silenced MAVS and investigated whether ADAP2 could interact with NEMO, TBK1, and IRF3 when cells were stimulated with SeV. We found that in the absence of MAVS expression, ADAP2 was still able to interact with NEMO, TBK1, and IRF3, similarly to the case in wild-type cells (Fig. 7A). These data were consistent with the results shown in Fig. 6F, where ADAP2 silencing attenuated MAVS-independent TLR3-induced IFN production. We further enquired whether the interactions of ADAP2 with other proteins (NEMO, TBK1, and IRF3) are interrelated (Fig. 7A). When NEMO was silenced, ADAP2 was able to interact with MAVS, TBK1, and IRF3 at levels comparable to those in wild-type cells in the context of viral infection. In TBK1-silenced cells, ADAP2 interacted with MAVS, NEMO, and IRF3 without any change. These experiments indicated that individual binding interactions of ADAP2 with MAVS, NEMO, and TBK1 are independent of each other during pathway activation.
FIG 7.

ADAP2 regulates the RIG-I pathway in concert with TRAF3. (A) Interactions of ADAP2 with MAVS, NEMO, and TBK1 are independent of each other. Coimmunoprecipitation was used to detect interactions of ADAP2 with various proteins at their endogenous levels after silencing of the indicated genes individually. (B) ADAP2 interacted with endogenous TRAF3 but not with TRAFs 2, 5, and 6. (C) Simultaneous silencing of TRAFs 2, 5, and 6 did not affect the interactions of ADAP2 with MAVS, NEMO, TBK1, and IRF3. The genes encoding TRAFs 2, 5, and 6 were simultaneously silenced using siRNAs, ADAP2 was immunoprecipitated, and interactions were probed by Western blotting. (D) TRAF3 silencing abolished the interactions of ADAP2 with MAVS, NEMO, TBK1, and IRF3. (E) ADAP2 silencing reduced the interaction of MAVS with TRAF3. All experiments were performed with SeV infection. (F) ADAP2 undergoes ubiquitination during SeV infection independently of TRAF3. Ubiquitination of ADAP2 was monitored by siNT or siTRAF3 treatment. si, siRNA; siNT, nontargeting negative-control siRNA; hpi, hours postinfection; SeV, Sendai virus; WCL, whole-cell lysate; IP, immunoprecipitation; IB, immunoblot; GAPDH, glyceraldehyde-3-phosphate dehydrogenase (cytoplasmic internal control marker); Ubi, ubiquitin.
We next investigated the potential signals that trigger ADAP2 to recruit various proteins of the RIG-I pathway. To address this, we first queried whether the signals originating from some of the key known signal transducers acting downstream of MAVS during antiviral signaling exhibit cross talk with ADAP2. Recently, TRAFs 2, 5, and 6 were identified as important proteins functioning redundantly to relay signals from MAVS to IRF3 activation (36). MAVS was previously established to interact with TRAFs 2, 5, and 6 (36). TRAF3 is another molecule previously reported to play roles in interferon gene transcription (48). Given that both ADAP2 and TRAFs regulate RIG-I signaling at overlapping sites in the signaling cascade, we asked whether ADAP2 and TRAFs exhibit cross talk in regulating the interferon response. We first investigated whether ADAP2 interacts with these TRAFs, and it was determined that TRAF3, but none of the other TRAFs, interacts with ADAP2 (Fig. 7B). We then asked whether the expression of these TRAF proteins is needed for ADAP2 to bind to MAVS, NEMO, TBK1, and IRF3. Simultaneous silencing of TRAFs 2, 5, and 6 was previously reported to attenuate RIG-I signaling-induced interferon gene transcription (36). We observed that simultaneous knockdown of TRAFs 2, 5, and 6 did not affect the binding of ADAP2 to MAVS, NEMO, TBK1, and IRF3 (Fig. 7C). However, silencing of TRAF3 alone led to a notable reduction in the ability of ADAP2 to bind MAVS, NEMO, TBK1, and IRF3 (Fig. 7D). These results highlighted that the ability of ADAP2 to bind to the components of the RIG-I signaling pathway is dependent on TRAF3. Conversely, we also asked whether ADAP2 is involved in the binding of MAVS to TRAFs 2, 3, 5, and 6. Interestingly, ADAP2 expression was found to be needed for TRAF3, but not TRAFs 2, 5, and 6, to interact with MAVS (Fig. 7E). Because TRAF3 is a ubiquitin ligase, we also investigated the possibility that TRAF3 regulates ADAP2 by ubiquitination. It was found that ADAP2 undergoes prominent ubiquitination upon SeV infection (Fig. 7F). However, this was independent of TRAF3 (Fig. 7F). These results showed that TRAF3 is a trigger needed for ADAP2 to recruit MAVS, NEMO, TBK1, and IRF3 during the antiviral response to infection.
Subcellular localization and expression of ADAP2 are modulated by viral infection.
We also investigated whether the subcellular localization of ADAP2 is sensitive to viral infection. Although plasma membrane localization of ADAP2 was reported earlier, we revisited this in our experimental system by adopting a highly sensitive approach involving the fractionation of various subcellular components through differential centrifugation and probing for the endogenous ADAP2 protein by Western blotting (64). Our results showed that ADAP2 was present in both the cytoplasmic and membrane fractions (Fig. 8A). In addition, infection led to the accumulation of a small amount of ADAP2 in the mitochondrial fraction. The data in Fig. 4A showed that viral infection induces interaction of ADAP2 with MAVS. Based on this, we predicted that the observed accumulation of ADAP2 in the isolated mitochondrial fraction during infection is most likely because of its association with MAVS. Consistent with this, we found that ADAP2 was unable to localize with mitochondria of cells in which MAVS gene expression was silenced upon virus infection (Fig. 8B).
FIG 8.
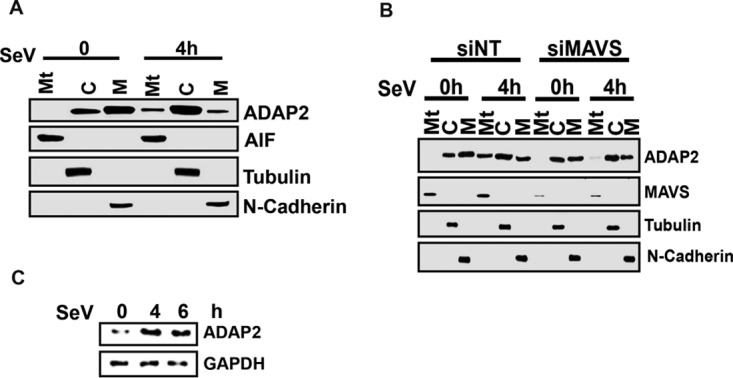
Subcellular distribution of ADAP2 during infection. (A) Subcellular distribution of ADAP2. Immunoblot of subcellular fractions for the presence of the indicated proteins are shown; (B) MAVS silencing abolished the mitochondrial localization of ADAP2 during SeV infection. Subcellular fractionation identifies a distribution of ADAP2 in cytosolic, plasma membrane and mitochondrial fractions of Sendai virus infected cells; (C) Sendai virus infection upregulated expression of ADAP2 protein in HEK293Tcells. si, siRNA; NT-si, nontargeting negative-control siRNA; h, hours; SeV, Sendai virus; Mt, mitochondrial fraction; M, membrane fraction; C, cytosolic fraction; AIF, apoptosis-inducing factor, a mitochondrial marker; tubulin, cytosolic marker; N-cadherin, plasma membrane marker.
Viral infection and interferon pathway stimulation are known to modulate the expression of several genes involved in innate immune pathways. To test whether immune activation alters the expression of ADAP2, HEK293T cells were infected with SeV for 24 h, and ADAP2 expression was detected by Western blotting. SeV infection was found to induce upregulation of ADAP2 protein expression (Fig. 8C).
DISCUSSION
We identified the ArfGAP domain-containing protein ADAP2 as an important positive regulator of RIG-I-, MDA5-, and TLR3-mediated interferon gene transcription. Specifically, our data identified the following two functions for ADAP2 in the interferon response: as a protein linking MAVS with its downstream key signaling molecules NEMO and TBK1 and as a scaffolding platform on which the crucial interaction of TBK1 with NEMO and IRF3 occurs. The latter function makes ADAP2 a positive regulator of multiple antiviral PRR signaling cascades that converge to TBK1-mediated IRF3 phosphorylation. Both gene knockdown and ectopic expression data, along with protein-protein interaction analyses, confirmed the role of ADAP2 in interferon production.
The ArfGAP family is less understood for its role in antiviral immune responses (65, 68). CENTB1, an ACAP subfamily ArfGAP, was previously shown to function as a negative regulator of bacterial immune receptor NOD1-mediated NF-κB signaling (69). There are about 31 members of the ArfGAP family, which is divided into 10 subfamilies (65). Among these, our recently published human genome-wide RNAi screen identified only ADAP2 as a novel regulator of the RIG-I-mediated interferon response (61). While these data potentially exclude other members from having any role in regulating the interferon response, it should be kept in mind that the RNAi screening study did not validate the silencing efficiency and on-target specificity of the siRNAs used against other members of the ArfGAP family. Other known functions of ADAP2 include regulation of heart development and stabilization of microtubules (66, 70, 71). Collectively, all the evidence provides an increased appreciation of the roles of ArfGAP family proteins in host-pathogen interactions.
The evidence generated in this study predicts that ADAP2 is a major scaffolding platform on which a RIG-I pathway-specific module (the MAVS-NEMO-TBK1 complex) is assembled. A key paradigm of RIG-I signaling is the need for a mechanism to bridge the spatial parity between the organizations of different modules of the RIG-I pathway. While both RIG-I and MAVS are found in association with organelles and membrane structures upon activation, their key downstream effectors, such as NEMO, TBK1, and IRF3, are present in the cytoplasm. Our identification of ADAP2 as an essential protein linking the transduction of signals from activated MAVS to NEMO and TBK1 to phosphorylate IRF3 thus has notable significance. The redundant contributions of TRAFs 2, 5, and 6 were recently identified as key requirements for RIG-I signaling (36). Our observation that the interaction of ADAP2 with MAVS, TBK1, and NEMO was independent of the expression of TRAFs 2, 5, and 6 hints at the existence of complex regulatory mechanisms underlying RIG-I signaling. It is possible that TRAFs 2, 5, and 6 and ADAP2 operate as parallel regulatory modules to transduce signals from MAVS to the downstream NEMO-TBK1-IRF3 complex. While ADAP2 plays a scaffolding role in bringing components spatially closer, TRAFs 2, 5, and 6 may regulate the RIG-I pathway through their ubiquitin ligase activity-mediated, posttranslational modification-based signals (36).
Determining the exact sequence of events by which ADAP2 regulates the interferon response needs further studies. Our interaction analyses coupled with gene silencing revealed that ADAP2 could bind to NEMO, TBK1, and IRF3 even in the absence of expression of MAVS. This indicated that the signal triggering the binding of ADAP2 to NEMO-TBK1-IRF3 acts independently of MAVS. This conclusion also fits well with the observed role of ADAP2 in the MAVS-independent TLR3 pathway. It is possible that MAVS binds to ADAP2 that is already in a complex with NEMO, TBK1, and IRF3. Moreover, individual silencing of NEMO and TBK1 did not affect the ability of ADAP2 to bind to any one of these proteins as well as MAVS. This observation also argues that there is a separate signal (signal 1), triggered by viral infection, which activates and readies ADAP2 for binding to these proteins. In the unstimulated state, ADAP2 did not show notable binding to MAVS, TBK1, NEMO, and IRF3. However, infection induced robust binding of ADAP2 to these proteins. This further indicates that specific signals that originate from PRR pathways are needed for ADAP2 to interact with the signaling component proteins needed for the interferon response. The observation that TRAF3 silencing abrogated the ability of ADAP2 to bind to MAVS, TBK1, NEMO, and IRF3 hinted that TRARF3 is either signal 1 or part of signal 1. However, the mechanism by which TRAF3 regulates ADAP2 functioning has yet to be discovered. Although we observed that ADAP2 undergoes ubiquitination upon RIG-I pathway stimulation, TRAF3 was found to be nonessential for this. It is also possible that physical interaction with TRAF3 alone drives ADAP2 to recruit other proteins of the interferon pathway.
This study also revealed that ADAP2 is a major protein scaffold required for assembly of the conserved core IRF3 activation machinery (NEMO-TBK1-IRF3 complex) of antiviral signaling pathways. Such scaffold proteins can serve to bring pathway-specific upstream components (e.g., adaptors of PRRs) in close proximity to downstream NEMO, TBK1, and IRF3. In the case of the RIG-I pathway, because ADAP2 can bind to both MAVS and the NEMO-TBK1-IRF3 complex, it is possible that ADAP2 brings all key components of RIG-I signaling (MAVS, NEMO, TBK1, and IRF3) together so that TBK1 can be activated and phosphorylate IRF3. However, the mechanism by which TLR3 signaling connects with ADAP2 to bridge that pathway to the NEMO-TBK1-IRF3 axis remains to be determined.
The mapping of the domains of ADAP2 and its interacting partners revealed several interesting insights. One finding was that ADAP2 requires only the ArfGAP domain to interact with many RIG-I pathway component proteins. Interestingly, mutation of the key amino acids required for the GTPase-activating function of ADAP2 reduced its ability to support the interferon response. Whether this was specifically because of the loss of the GTPase-activating function or due to mutation-induced structural changes of ADAP2 rendering it unable to physically associate with its interacting proteins is unclear. The ArfGAP domain of ADAP2 is approximately 131 amino acids long and is located at the N terminus. How multiple proteins converge to bind to a stretch of 131 amino acids can be revealed only through structural studies of protein complexes. ADAP2 interacted within the N-terminal 100-amino-acid CARD of MAVS, which is located adjacent to the sites for binding of TRAF proteins. TRAF2 and -5 bind to amino acids 143 to 147 of MAVS, while TRAF6 binds to amino acids 153 to 158 of MAVS (22, 48, 72). The N-terminal 150 amino acids of NEMO were needed for its interaction with ADAP2. This region of NEMO was reported as the kinase binding domain (44). Interestingly, ADAP2 was found to bind to the ubiquitin-like domain (ULD) of TBK1. This observation is notable because a previous study identified that ULD-deficient TBK1 was unable to activate IRF3 and interferon gene transcription (73, 74). Based on the truncation results, it can be concluded that ADAP2 binds to the C-terminal half of the IRF association domain (IAD) of IRF3. Whether ADAP2 also brings the IADs of multiple IRF3 molecules close together to promote its dimer formation upon phosphorylation is currently unknown. The binding of NEMO, TBK1, and a region of IRF3 between amino acids 253 and 390 to the short ArfGAP domain may bring the target phosphorylation sites of IRF3 (Ser396/Ser386) (29, 33) spatially closer to TBK1, facilitating the catalysis.
Based on our experimental evidence, we generated a model for the role played by ADAP2 in the interferon gene transcription from pattern recognition receptors (Fig. 9). In the resting state, there is no appreciable interaction of ADAP2 with MAVS, NEMO, TBK1, and IRF3. During viral infection, a TRAF3-dependent signal (signal 1) activates ADAP2 to interact with MAVS, NEMO, TBK1, and IRF3 to form a complex. This brings IRF3 into close proximity with TBK1. Subsequently, in the case of RIG-I signaling, ADAP2 that is likely in complex with NEMO, TBK1, and IRF3 binds to activated MAVS. This step activates TBK1 to phosphorylate IRF3. The recently reported phosphorylation of MAVS by TBK1 may also happen at this stage (55). Whether the binding of the ADAP2-NEMO-TBK1-IRF3 complex to MAVS requires a second signal is unclear. Thus, ADAP2 functions as a pivotal scaffold where different modules of PRR signaling are assembled and functionally integrated, resulting in IRF3 phosphorylation.
FIG 9.

Proposed model for the role of ADAP2 in antiviral response. A cartoon showing the proposed model to explain the mode of action of ADAP2 during RIG-I signaling. The proteins are shown with their domains labeled. The interactions of ADAP2 with individual protein are specifically marked at their experimentally identified sites of interaction, except for TRAF3. (A) In uninfected cells, ADAP2 is not in complex with any other protein of PRR signaling pathway. (B) Upon viral infection, two independent events will happen: (i) sensing of virus by RIG-I activates and induces polymerization of MAVS, and (ii) ADAP2 forms a complex with TRAF3, NEMO, TBK1 and IRF3, triggered by signal-1. This will bring IRF3 in proximity to TBK1. (C) In the later stages of viral infection, ADAP2-NEMO-TBK1-IRF3 complex will bind to polymerized MAVS, likely triggered by an unidentified signal-2. This will activate TBK1 to phosphorylate IRF3. KBD, kinase binding domain; KD, kinase domain; CC, coiled coil; ULD, ubiquitin like domain; IAD, IRF association domain; DBD, DNA binding domain; CARD, caspase activation and recruitment domain.
In summary, this study provided important mechanistic insights into how different modules of antiviral pattern recognition signaling pathways are assembled, leading to the activation of IRF3 and to interferon gene transcription. We anticipate that systematic profiling of the regulation of PRR signaling would enable the generation of a comprehensive functional understanding of antiviral innate immune responses during viral infections.
MATERIALS AND METHODS
Cells, antibodies, and reagents.
The human embryonic kidney cell line 293T (HEK293T; ATCC CRL-3216) and human primary monocytes (Stemcell Technologies) were used for the studies.
The antibodies and other reagents used were as follows: anti-pIRF3 (serine 396), -IRF3, -TBK1, -NEMO, -hemagglutinin (anti-HA) tag, and -pancadherin (all from Cell Signaling Technology); antiubiquitin, -TRAF5, -TRAF6, -TRAF2, -TRAF3, -MAVS, -RIG-I, -tubulin, and -NEMO (all from Santa Cruz Biotechnology); anti-ADAP2 antibody (Abcam and Sigma); anti-FLAG, anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH), and anti-FLAG affinity gel (all from Sigma); protein G-agarose (Pierce); poly(I·C) (Invivogen); Halt protease and phosphatase inhibitors (Pierce); and iQ SYBR green supermix (Bio-Rad).
The expression plasmids for epitope-tagged RIG, MAVS, TBK1, IRF3, and IRF3-D5 and promoter-luciferase reporter plasmids for IFN-β and NF-κB were described previously (61). Various mutants of NEMO, MAVS, and IRF3 were expressed from epitope-tagged mammalian expression vectors. TBK1 truncation was described earlier (74). The ADAP2 open reading frame (ORF) was purchased from the Dana Farber Cancer Institute and subcloned into pCDNA3.1-FLAG or pCMV-HA. Truncations of ADAP2 were generated by designing primers for various regions and subcloning the DNA regions into the pCDNA3.1-FLAG vector. The primers used for construction of the N-terminal truncations were as follows: Mut1F, 5′-ATAGGATCCGGCGATCGCGAGCGCAAC; Mut1R, 5′-ATACTCGAGTCAGAGCGAGATGGTTTCCCC; Mut2F, 5′-ATAGGATCCGGCGATCGCGAGCGCAAC; Mut2R, 5′-ATACTCGAGTCACCTGGTGAGGAATGGCAC; Mut3F, 5′-ATAGGATCCCCAGGTAACCGAGAAGGA; Mut3R, 5′-ATACTCGAGTCACCTGGTGAGGAATGGCAC; Mut4F, 5′-ATACTCGAGTCACCTGCTGCTGCGGC; Mut4R, 5′-ATAGGATCCCCAGGTAACCGAGAAGGA; Mut5F, 5′-ATAGGATCCAGGAACTACCTCAAACAA; and Mut5R, 5′-ATACTCGAGTCACCTGCTGCTGCGGCC. Site-directed mutagenesis was performed using a QuikChange kit (Stratagene, Agilent Technologies).
Gene silencing and interferon reporter assays.
The siRNAs for the ADAP2 coding region (sense strand) were Si-ADAP2-1 (5′-GGAAGAAGGUCCGCGUUAAdTdT-3′) and Si-ADAP2-2 (5′-GGACUAUGAAAUCCACGAUdTdT-3′). siRNAs targeting the ADAP2 3′-UTR were Si-ADAP2-UTR-1 (5′-CCAUACACACCUAGGCUUGUU-3′) and Si-ADAP2-UTR-2 (5′-GAAAAGGCACCCACAGCAUUU-3′). Other siRNAs included Si-MAVS (5′-UAGUUGAUCUCGCGGACGAdTdT-3′), Si-TRAF2-1 (5′-CAACCAGAAGGUGACCUUAdTdT-3′), Si-TRAF2-2 (5′-GAAUACGAGAGCUGCCACGdTdT-3′), Si-TRAF5-1 (5′-GGUCACACCUGUCCCUAUA-3′), Si-TRAF5-2 (5′-GGAUGUAAUGCCAAGGUUA-3′), Si-TRAF6-1 (5′-CCACGAAGAGAUAAUGGAUdTdT-3′), and Si-TRAF6-2 (5′-CAUUAAGGAUGAUACAUUATT-3′). The negative-control siRNA was purchased from SA Bio, Singapore. siRNAs (50 nM) were transfected into HEK293T-RIG-I cells by use of the lipid transfection reagent Dharmafect 1 (Dharmacon). siRNAs (200 nM) were introduced into human primary blood cells (1 × 106) by nucleofection methodology, using a 4D-Nucleofector system (Lonza) per the manufacturer's instructions.
For the reporter assays, HEK293T cells were transfected with expression plasmids for human PRRs (RIG-I, TLR3, and MDA5), pathway proteins (MAVS, NEMO, and TBK1), and IFN-β or IFN-α4 or NF-κB target promoter-driven luciferase reporters (pGL2 vector; Promega) along with a constitutively transcribed renilla luciferase reporter (p-RL-TK; Promega) for 24 h, and luciferase readings were performed using a Dual-Glow assay kit (Promega).
Viral infection and viral load determination.
Sendai virus (Cantell strain) was obtained from Charles River Laboratories. The cells were infected with 30 to 80 HA units of virus for the indicated treatments. For vesicular stomatitis virus (VSV) in vitro infection studies, ADAP2 siRNA-transfected cells were infected with VSV at a multiplicity of infection (MOI) of 0.05 for 1 h, unbound virus was removed by washing in culture medium and allowed to infect cells for 18 h, and the culture supernatant was collected for plaque assay. Plaque assay was performed on BHK-21 cells.
Co-IP experiments.
For co-IP experiments, 1 × 106 HEK293T cells per well in 6-well plates were harvested in ice-cold phosphate-buffered saline (PBS), washed twice to remove serum components, lysed in lysis buffer (150 mM NaCl, 1% NP-40, and 50 mM Tris-Cl, pH 7.0) containing protease inhibitors, and clarified by spinning for 15 min at 12,000 × g. The supernatants were incubated with primary antibodies (2 μg) overnight at 4°C, and using protein G-agarose, the antibody-antigen complexes were purified, coupled with stringent washing with lysis buffer. The protein interactions were assayed by Western blotting. For this purpose, the proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes, and immunodetected with appropriate primary antibodies by using an infrared detection system (LiCor) with IRDye 800CW- and 680RD-coupled secondary antibodies.
IRF3 phosphorylation assays.
For the detection of IRF3 phosphorylation, 1 × 106 siRNA-treated HEK293T cells were stimulated by transfection with 4 μg of poly(I·C) for 4 h and then lysed in cold RIPA buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 2 mM EDTA, 1% NP-40, 0.1% SDS) containing protease and phosphatase inhibitors. Following clarification by centrifugation for 15 min at 12,000 × g, the proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and probed with anti-pIRF3 and anti-IRF3 antibodies.
Subcellular fractionation.
HEK293T cells were lysed in buffer A (10 mM Tris-HCl [pH 7.5], 10 mM KCl, 0.5 mM EGTA, 1.5 mM MgCl2, and protease inhibitor cocktail). The lysate was centrifuged at 1,000 × g for 5 min, and the supernatant was subjected to further centrifugation at 5,000 × g for 10 min. The pellet contained the mitochondria. To obtain the cytosolic fraction, the latter supernatant was further centrifuged at 100,000 × g for 1 h, and the resultant supernatant contained the cytoplasm. The pellet (plasma membrane) was washed gently twice with buffer B (20 mM HEPES-KOH [pH 7.4], 10% glycerol, 0.5 mM EGTA, and protease inhibitor cocktail).
Expression and purification of recombinant ADAP2.
Five micrograms each of wild-type (WT) pcDNA3.1-Flag-ADAP2 and its mutant (MT1 or MT2) was transiently expressed in HEK293T cells for 48 h and purified using FLAG antibody (M2) beads (Sigma) overnight. The agarose beads were extensively washed with 10 mM Tris-HCl (pH 7.5), 150 mM NaCl, and 0.1% Triton X-100, and the protein was eluted with the FLAG peptide (200 μg/ml; Sigma) in a buffer containing 50 mM Tris-HCl (pH 7.5) and 0.1% Triton X-100. Eluted proteins were stored in a buffer containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 10% glycerol, and 0.1% Triton X-100.
In vitro reconstituted IRF3 activation assay.
The in vitro assay of IRF3 activation was performed as described previously (44, 61), with some modifications. Briefly, a cytoplasmic preparation (20 μg) isolated from siRNA-treated uninfected cells was mixed with purified mitochondria (5 μg) from Sendai virus-challenged cells in the presence or absence of 100 ng of purified full-length ADAP2 or its mutants and incubated at 30°C for 1 h in a 25-μl mixture containing 20 mM HEPES-KOH (pH 7.0), 2 mM ATP, 5 mM MgCl2, and 250 mM d-mannitol. After the incubation, the levels of the pIRF3 and IRF3 proteins were detected by Western blotting.
Ubiquitination assays.
Ubiquitination of ADAP2 was monitored in HEK293 cells challenged with SeV. For this purpose, endogenous ADAP2 was immunoprecipitated from siRNA-treated SeV- or mock-infected cells. The presence of ubiquitin linkage on ADAP2 was monitored by use of an antibody that detects ubiquitin (Santa Cruz).
Quantitative PCR.
Total RNA was extracted by use of an RNeasy kit (Qiagen), and cDNA was prepared using an iSCRIPT cDNA synthesis kit (Bio-Rad). Quantitative real-time PCR (qRT-PCR) was performed in triplicate for each sample. Gene-specific primers and SYBR green (Bio-Rad) were used to quantify the transcripts. The primers used were as follows (“h” stands for “human”): hIFNB1F, 5′-CTTGGATTCCTACAAAGAAGCAGC-3′; hIFNB1R, 5′-TCCTCCTTCTGGAACTGCTGCA-3′; hβ-actinF, 5′-CGTCCGCCCCGCGAGCAC-3′; hβ-actinR, 5′-GTTGAATAAAAGTGCACACC-3′; hISG15-F, 5′-CGCAGATCACCCAGAAGAT-3′; and hISG15-R, 3′-TCCTCACCAGGATGTTCAGA-5′.
Statistics.
Quantified data are expressed as means ± standard deviations (SD) for a representative experiment performed in triplicate, and experiments were performed at least three independent times. The statistical significance of differences in mean values was analyzed using unpaired two-tailed Student's t test, and P values of <0.05 were considered statistically significant.
ACKNOWLEDGMENTS
M.N.K. was funded by the Ministry of Education and the National Research Foundation of Singapore. R.L. was funded by Canadian Institutes of Health Research grant MOP42562.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Guzman MG, Halstead SB, Artsob H, Buchy P, Farrar J, Gubler DJ, Hunsperger E, Kroeger A, Margolis HS, Martinez E, Nathan MB, Pelegrino JL, Simmons C, Yoksan S, Peeling RW. 2010. Dengue: a continuing global threat. Nat Rev Microbiol 8:S7–S16. doi: 10.1038/nrmicro2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morens DM, Fauci AS. 2012. Emerging infectious diseases in 2012: 20 years after the Institute of Medicine report. mBio 3:e00494–12. doi: 10.1128/mBio.00494-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kumagai Y, Akira S. 2010. Identification and functions of pattern-recognition receptors. J Allergy Clin Immunol 125:985–992. doi: 10.1016/j.jaci.2010.01.058. [DOI] [PubMed] [Google Scholar]
- 4.Kumar H, Kawai T, Akira S. 2009. Pathogen recognition in the innate immune response. Biochem J 420:1–16. doi: 10.1042/BJ20090272. [DOI] [PubMed] [Google Scholar]
- 5.Patel JR, Garcia-Sastre A. 2014. Activation and regulation of pathogen sensor RIG-I. Cytokine Growth Factor Rev 25:513–523. doi: 10.1016/j.cytogfr.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 6.Ramos HJ, Gale M Jr. 2011. RIG-I like receptors and their signaling crosstalk in the regulation of antiviral immunity. Curr Opin Virol 1:167–176. doi: 10.1016/j.coviro.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seth RB, Sun L, Chen ZJ. 2006. Antiviral innate immunity pathways. Cell Res 16:141–147. doi: 10.1038/sj.cr.7310019. [DOI] [PubMed] [Google Scholar]
- 8.Yan N, Chen ZJ. 2012. Intrinsic antiviral immunity. Nat Immunol 13:214–222. doi: 10.1038/ni.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoneyama M, Fujita T. 2010. Recognition of viral nucleic acids in innate immunity. Rev Med Virol 20:4–22. doi: 10.1002/rmv.633. [DOI] [PubMed] [Google Scholar]
- 10.Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, Stark GR. 2007. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov 6:975–990. doi: 10.1038/nrd2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia-Sastre A, Biron CA. 2006. Type 1 interferons and the virus-host relationship: a lesson in detente. Science 312:879–882. doi: 10.1126/science.1125676. [DOI] [PubMed] [Google Scholar]
- 12.Schoggins JW, Rice CM. 2011. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol 1:519–525. doi: 10.1016/j.coviro.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stetson DB, Medzhitov R. 2006. Type I interferons in host defense. Immunity 25:373–381. doi: 10.1016/j.immuni.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 14.Rehwinkel J, Tan CP, Goubau D, Schulz O, Pichlmair A, Bier K, Robb N, Vreede F, Barclay W, Fodor E, Reis e Sousa C. 2010. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell 140:397–408. doi: 10.1016/j.cell.2010.01.020. [DOI] [PubMed] [Google Scholar]
- 15.Takeuchi O, Akira S. 2008. MDA5/RIG-I and virus recognition. Curr Opin Immunol 20:17–22. doi: 10.1016/j.coi.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 16.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 17.Dixit E, Boulant S, Zhang Y, Lee AS, Odendall C, Shum B, Hacohen N, Chen ZJ, Whelan SP, Fransen M, Nibert ML, Superti-Furga G, Kagan JC. 2010. Peroxisomes are signaling platforms for antiviral innate immunity. Cell 141:668–681. doi: 10.1016/j.cell.2010.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horner SM, Liu HM, Park HS, Briley J, Gale M Jr. 2011. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc Natl Acad Sci U S A 108:14590–14595. doi: 10.1073/pnas.1110133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. 2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol 6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 20.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. 2005. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 21.Seth RB, Sun L, Ea CK, Chen ZJ. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 22.Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. 2005. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell 19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 23.Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. 2011. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 146:448–461. doi: 10.1016/j.cell.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu H, He X, Zheng H, Huang LJ, Hou F, Yu Z, de la Cruz MJ, Borkowski B, Zhang X, Chen ZJ, Jiang QX. 2014. Structural basis for the prion-like MAVS filaments in antiviral innate immunity. eLife 3:e01489. doi: 10.7554/eLife.01489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. 2003. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol 4:491–496. [DOI] [PubMed] [Google Scholar]
- 26.Hiscott J. 2007. Convergence of the NF-kappaB and IRF pathways in the regulation of the innate antiviral response. Cytokine Growth Factor Rev 18:483–490. doi: 10.1016/j.cytogfr.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 27.Hiscott J, Pitha P, Genin P, Nguyen H, Heylbroeck C, Mamane Y, Algarte M, Lin R. 1999. Triggering the interferon response: the role of IRF-3 transcription factor. J Interferon Cytokine Res 19:1–13. doi: 10.1089/107999099314360. [DOI] [PubMed] [Google Scholar]
- 28.Servant MJ, Grandvaux N, Hiscott J. 2002. Multiple signaling pathways leading to the activation of interferon regulatory factor 3. Biochem Pharmacol 64:985–992. doi: 10.1016/S0006-2952(02)01165-6. [DOI] [PubMed] [Google Scholar]
- 29.Lin R, Heylbroeck C, Pitha PM, Hiscott J. 1998. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol Cell Biol 18:2986–2996. doi: 10.1128/MCB.18.5.2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin R, Mamane Y, Hiscott J. 1999. Structural and functional analysis of interferon regulatory factor 3: localization of the transactivation and autoinhibitory domains. Mol Cell Biol 19:2465–2474. doi: 10.1128/MCB.19.4.2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Panne D, McWhirter SM, Maniatis T, Harrison SC. 2007. Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. J Biol Chem 282:22816–22822. doi: 10.1074/jbc.M703019200. [DOI] [PubMed] [Google Scholar]
- 32.Suhara W, Yoneyama M, Iwamura T, Yoshimura S, Tamura K, Namiki H, Aimoto S, Fujita T. 2000. Analyses of virus-induced homomeric and heteromeric protein associations between IRF-3 and coactivator CBP/p300. J Biochem 128:301–307. doi: 10.1093/oxfordjournals.jbchem.a022753. [DOI] [PubMed] [Google Scholar]
- 33.Takahasi K, Horiuchi M, Fujii K, Nakamura S, Noda NN, Yoneyama M, Fujita T, Inagaki F. 2010. Ser386 phosphorylation of transcription factor IRF-3 induces dimerization and association with CBP/p300 without overall conformational change. Genes Cells 15:901–910. doi: 10.1111/j.1365-2443.2010.01427.x. [DOI] [PubMed] [Google Scholar]
- 34.Wathelet MG, Lin CH, Parekh BS, Ronco LV, Howley PM, Maniatis T. 1998. Virus infection induces the assembly of coordinately activated transcription factors on the IFN-beta enhancer in vivo. Mol Cell 1:507–518. doi: 10.1016/S1097-2765(00)80051-9. [DOI] [PubMed] [Google Scholar]
- 35.Jacobs JL, Coyne CB. 2013. Mechanisms of MAVS regulation at the mitochondrial membrane. J Mol Biol 425:5009–5019. doi: 10.1016/j.jmb.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu S, Chen J, Cai X, Wu J, Chen X, Wu YT, Sun L, Chen ZJ. 2013. MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. eLife 2:e00785. doi: 10.7554/eLife.00785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ishikawa H, Barber GN. 2008. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun L, Wu J, Du F, Chen X, Chen ZJ. 2013. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, Lei C, He X, Zhang L, Tien P, Shu HB. 2008. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 29:538–550. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 40.Liu XY, Chen W, Wei B, Shan YF, Wang C. 2011. IFN-induced TPR protein IFIT3 potentiates antiviral signaling by bridging MAVS and TBK1. J Immunol 187:2559–2568. doi: 10.4049/jimmunol.1100963. [DOI] [PubMed] [Google Scholar]
- 41.Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, Jung JU. 2007. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446:916–920. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- 42.Nakhaei P, Mesplede T, Solis M, Sun Q, Zhao T, Yang L, Chuang TH, Ware CF, Lin R, Hiscott J. 2009. The E3 ubiquitin ligase Triad3A negatively regulates the RIG-I/MAVS signaling pathway by targeting TRAF3 for degradation. PLoS Pathog 5:e1000650. doi: 10.1371/journal.ppat.1000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oshiumi H, Miyashita M, Inoue N, Okabe M, Matsumoto M, Seya T. 2010. The ubiquitin ligase Riplet is essential for RIG-I-dependent innate immune responses to RNA virus infection. Cell Host Microbe 8:496–509. doi: 10.1016/j.chom.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 44.Zeng W, Xu M, Liu S, Sun L, Chen ZJ. 2009. Key role of Ubc5 and lysine-63 polyubiquitination in viral activation of IRF3. Mol Cell 36:315–325. doi: 10.1016/j.molcel.2009.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao T, Yang L, Sun Q, Arguello M, Ballard DW, Hiscott J, Lin R. 2007. The NEMO adaptor bridges the nuclear factor-kappaB and interferon regulatory factor signaling pathways. Nat Immunol 8:592–600. doi: 10.1038/ni1465. [DOI] [PubMed] [Google Scholar]
- 46.Li S, Wang L, Berman M, Kong YY, Dorf ME. 2011. Mapping a dynamic innate immunity protein interaction network regulating type I interferon production. Immunity 35:426–440. doi: 10.1016/j.immuni.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mao AP, Li S, Zhong B, Li Y, Yan J, Li Q, Teng C, Shu HB. 2010. Virus-triggered ubiquitination of TRAF3/6 by cIAP1/2 is essential for induction of interferon-beta (IFN-beta) and cellular antiviral response. J Biol Chem 285:9470–9476. doi: 10.1074/jbc.M109.071043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saha SK, Pietras EM, He JQ, Kang JR, Liu SY, Oganesyan G, Shahangian A, Zarnegar B, Shiba TL, Wang Y, Cheng G. 2006. Regulation of antiviral responses by a direct and specific interaction between TRAF3 and Cardif. EMBO J 25:3257–3263. doi: 10.1038/sj.emboj.7601220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tang ED, Wang CY. 2010. TRAF5 is a downstream target of MAVS in antiviral innate immune signaling. PLoS One 5:e9172. doi: 10.1371/journal.pone.0009172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou Z, Jia X, Xue Q, Dou Z, Ma Y, Zhao Z, Jiang Z, He B, Jin Q, Wang J. 2014. TRIM14 is a mitochondrial adaptor that facilitates retinoic acid-inducible gene-I-like receptor-mediated innate immune response. Proc Natl Acad Sci U S A 111:E245–E254. doi: 10.1073/pnas.1316941111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gatot JS, Gioia R, Chau TL, Patrascu F, Warnier M, Close P, Chapelle JP, Muraille E, Brown K, Siebenlist U, Piette J, Dejardin E, Chariot A. 2007. Lipopolysaccharide-mediated interferon regulatory factor activation involves TBK1-IKKepsilon-dependent Lys(63)-linked polyubiquitination and phosphorylation of TANK/I-TRAF. J Biol Chem 282:31131–31146. doi: 10.1074/jbc.M701690200. [DOI] [PubMed] [Google Scholar]
- 52.Guo B, Cheng G. 2007. Modulation of the interferon antiviral response by the TBK1/IKKi adaptor protein TANK. J Biol Chem 282:11817–11826. doi: 10.1074/jbc.M700017200. [DOI] [PubMed] [Google Scholar]
- 53.Kawagoe T, Takeuchi O, Takabatake Y, Kato H, Isaka Y, Tsujimura T, Akira S. 2009. TANK is a negative regulator of Toll-like receptor signaling and is critical for the prevention of autoimmune nephritis. Nat Immunol 10:965–972. doi: 10.1038/ni.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu XY, Wei B, Shi HX, Shan YF, Wang C. 2010. Tom70 mediates activation of interferon regulatory factor 3 on mitochondria. Cell Res 20:994–1011. doi: 10.1038/cr.2010.103. [DOI] [PubMed] [Google Scholar]
- 55.Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu YT, Grishin NV, Chen ZJ. 2015. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347:aaa2630. doi: 10.1126/science.aaa2630. [DOI] [PubMed] [Google Scholar]
- 56.Chau TL, Gioia R, Gatot JS, Patrascu F, Carpentier I, Chapelle JP, O'Neill L, Beyaert R, Piette J, Chariot A. 2008. Are the IKKs and IKK-related kinases TBK1 and IKK-epsilon similarly activated? Trends Biochem Sci 33:171–180. doi: 10.1016/j.tibs.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 57.Fujita F, Taniguchi Y, Kato T, Narita Y, Furuya A, Ogawa T, Sakurai H, Joh T, Itoh M, Delhase M, Karin M, Nakanishi M. 2003. Identification of NAP1, a regulatory subunit of IkappaB kinase-related kinases that potentiates NF-kappaB signaling. Mol Cell Biol 23:7780–7793. doi: 10.1128/MCB.23.21.7780-7793.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ryzhakov G, Randow F. 2007. SINTBAD, a novel component of innate antiviral immunity, shares a TBK1-binding domain with NAP1 and TANK. EMBO J 26:3180–3190. doi: 10.1038/sj.emboj.7601743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sasai M, Oshiumi H, Matsumoto M, Inoue N, Fujita F, Nakanishi M, Seya T. 2005. Cutting edge: NF-kappaB-activating kinase-associated protein 1 participates in TLR3/Toll-IL-1 homology domain-containing adapter molecule-1-mediated IFN regulatory factor 3 activation. J Immunol 174:27–30. doi: 10.4049/jimmunol.174.1.27. [DOI] [PubMed] [Google Scholar]
- 60.Sasai M, Shingai M, Funami K, Yoneyama M, Fujita T, Matsumoto M, Seya T. 2006. NAK-associated protein 1 participates in both the TLR3 and the cytoplasmic pathways in type I IFN induction. J Immunol 177:8676–8683. doi: 10.4049/jimmunol.177.12.8676. [DOI] [PubMed] [Google Scholar]
- 61.Pulloor NK, Nair S, Kostic AD, Bist P, Weaver JD, Riley AM, Tyagi R, Uchil PD, York JD, Snyder SH, Garcia-Sastre A, Potter BV, Lin R, Shears SB, Xavier RJ, Krishnan MN. 2014. Human genome-wide RNAi screen identifies an essential role for inositol pyrophosphates in type-I interferon response. PLoS Pathog 10:e1003981. doi: 10.1371/journal.ppat.1003981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fenner BJ, Scannell M, Prehn JH. 2010. Expanding the substantial interactome of NEMO using protein microarrays. PLoS One 5:e8799. doi: 10.1371/journal.pone.0008799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li Y, Xie J, Wu S, Xia J, Zhang P, Liu C, Zhang P, Huang X. 2013. Protein kinase regulated by dsRNA downregulates the interferon production in dengue virus- and dsRNA-stimulated human lung epithelial cells. PLoS One 8:e55108. doi: 10.1371/journal.pone.0055108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hanck T, Stricker R, Sedehizade F, Reiser G. 2004. Identification of gene structure and subcellular localization of human centaurin alpha 2, and p42IP4, a family of two highly homologous, Ins 1,3,4,5-P4-/PtdIns 3,4,5-P3-binding, adapter proteins. J Neurochem 88:326–336. [DOI] [PubMed] [Google Scholar]
- 65.Kahn RA, Bruford E, Inoue H, Logsdon JM Jr, Nie Z, Premont RT, Randazzo PA, Satake M, Theibert AB, Zapp ML, Cassel D. 2008. Consensus nomenclature for the human ArfGAP domain-containing proteins. J Cell Biol 182:1039–1044. doi: 10.1083/jcb.200806041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Whitley P, Gibbard AM, Koumanov F, Oldfield S, Kilgour EE, Prestwich GD, Holman GD. 2002. Identification of centaurin-alpha2: a phosphatidylinositide-binding protein present in fat, heart and skeletal muscle. Eur J Cell Biol 81:222–230. doi: 10.1078/0171-9335-00242. [DOI] [PubMed] [Google Scholar]
- 67.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 68.Randazzo PA, Inoue H, Bharti S. 2007. Arf GAPs as regulators of the actin cytoskeleton. Biol Cell 99:583–600. doi: 10.1042/BC20070034. [DOI] [PubMed] [Google Scholar]
- 69.Yamamoto-Furusho JK, Barnich N, Xavier R, Hisamatsu T, Podolsky DK. 2006. Centaurin beta1 down-regulates nucleotide-binding oligomerization domains 1- and 2-dependent NF-kappaB activation. J Biol Chem 281:36060–36070. doi: 10.1074/jbc.M602383200. [DOI] [PubMed] [Google Scholar]
- 70.Venturin M, Carra S, Gaudenzi G, Brunelli S, Gallo GR, Moncini S, Cotelli F, Riva P. 2014. ADAP2 in heart development: a candidate gene for the occurrence of cardiovascular malformations in NF1 microdeletion syndrome. J Med Genet 51:436–443. doi: 10.1136/jmedgenet-2013-102240. [DOI] [PubMed] [Google Scholar]
- 71.Zuccotti P, Cartelli D, Stroppi M, Pandini V, Venturin M, Aliverti A, Battaglioli E, Cappelletti G, Riva P. 2012. Centaurin-alpha(2) interacts with beta-tubulin and stabilizes microtubules. PLoS One 7:e52867. doi: 10.1371/journal.pone.0052867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Paz S, Vilasco M, Werden SJ, Arguello M, Joseph-Pillai D, Zhao T, Nguyen TL, Sun Q, Meurs EF, Lin R, Hiscott J. 2011. A functional C-terminal TRAF3-binding site in MAVS participates in positive and negative regulation of the IFN antiviral response. Cell Res 21:895–910. doi: 10.1038/cr.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ikeda F, Hecker CM, Rozenknop A, Nordmeier RD, Rogov V, Hofmann K, Akira S, Dotsch V, Dikic I. 2007. Involvement of the ubiquitin-like domain of TBK1/IKK-i kinases in regulation of IFN-inducible genes. EMBO J 26:3451–3462. doi: 10.1038/sj.emboj.7601773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang L, Li S, Dorf ME. 2012. NEMO binds ubiquitinated TANK-binding kinase 1 (TBK1) to regulate innate immune responses to RNA viruses. PLoS One 7:e43756. doi: 10.1371/journal.pone.0043756. [DOI] [PMC free article] [PubMed] [Google Scholar]