

Abstract
Fibroblast growth factor 21 (FGF21) has emerged as an important beneficial regulator of glucose and lipid homeostasis but its levels are also abnormally increased in insulin-resistant states in rodents and humans. The synthetic retinoid Fenretinide inhibits obesity and improves glucose homeostasis in mice and has pleotropic effects on cellular pathways. To identify Fenretinide target genes, we performed unbiased RNA-seq analysis in liver from mice fed high-fat diet ± Fenretinide. Strikingly, Fgf21 was the most downregulated hepatic gene. Fenretinide normalised elevated levels of FGF21 in both high-fat diet-induced obese mice and in genetically obese-diabetic Leprdbmice. Moreover, Fenretinide-mediated suppression of FGF21 was independent of body weight loss or improved hepatic insulin sensitivity and importantly does not induce unhealthy metabolic complications. In mice which have substantially decreased endogenous retinoic acid biosynthesis, Fgf21 expression was increased, whereas acute pharmacological retinoid treatment decreased FGF21 levels. The repression of FGF21 levels by Fenretinide occurs by reduced binding of RARα and Pol-II at the Fgf21 promoter. We therefore establish Fgf21 as a novel gene target of Fenretinide signalling via a retinoid-dependent mechanism. These results may be of nutritional and therapeutic importance for the treatment of obesity and type-2 diabetes.
Fibroblast growth factor (FGF) 21 is a predominantly liver derived hormone and is being investigated as an anti-obesity/anti-diabetes therapy1. FGF21 binds cognate receptors FGFR and β-klotho, which are expressed at high levels in adipose tissue, to mediate its metabolic effects including control of lipid metabolism, body weight and glucose homeostasis1,2. Hepatic Fgf21 expression is increased by stimuli such as fasting, protein/amino acid restriction and fructose ingestion via a range of transcription factors, including peroxisome proliferator-activated receptor-alpha (PPARα) and carbohydrate response-element binding protein (ChREBP) 13,4,5.
Circulating FGF21 levels are also elevated in states of metabolic stress in both rodents and humans and are positively correlated with body mass index and insulin resistance6. This has to led the hypothesis that a rise in serum FGF21 levels may be a predictor for metabolic syndrome and type-2 diabetes and that these may be states of relative “FGF21-resistance”7. Furthermore, diverse treatments to improve glycaemic control in humans can decrease elevated levels of FGF21 (e.g. with metformin, rosiglitazone, insulin, insulinotropic agents, bariatric surgery, lifestyle modification, fish oil supplements or exercise8,9,10,11,12,13,14). Thus, the biological role of FGF21 in obesity and insulin resistance is still not fully explained and further understanding the regulation of FGF21 with the aim of modulating physiological levels maybe key in maximizing its therapeutic potential.
The synthetic retinoid 4-hydroxy(phenyl)retinamide (Fenretinide, FEN) is widely studied as a cancer therapeutic due to its favourable toxicological profile and is currently undergoing phase II clinical trials for treatment of insulin resistance in obese humans with hepatic steatosis15,16. We have previously shown that FEN prevents obesity and improves insulin sensitivity in both high fat diet (HFD)-fed male and female mice17,18,19. FEN acts via several different mechanisms including induction of retinoid signalling, increased hepatic lipid oxidation and inhibition of the final step of ceramide biosynthesis in multiple tissues to exert its metabolic effects20,21,22. FEN can also directly inhibit gene expression of the satiety hormone leptin and reduce serum levels in mice, but does not have measureable effects on food intake or energy expenditure17,18. The relationship between FEN and leptin signalling is not fully understood and it is not known whether FEN can modulate Fgf21 levels.
To further understand the signalling mechanisms of FEN, we performed RNA-seq analysis in liver from FEN-treated mice fed a HFD, which we had previously characterised18,22. Interestingly, Fgf21 was the gene most downregulated by FEN treatment. We therefore further tested the potential role of FEN in regulating hepatic Fgf21 in mouse models of diet and genetically-induced obesity and in wild-type animals.
Results
Hepatic Fgf21 is down regulated in HFD-fed mice receiving Fenretinide treatment
To identify targets of FEN signalling, RNA-seq analysis was performed in liver from C57BL/6 mice fed a HFD ± 0.04% FEN for 20 weeks. Representative mice (n = 4) from the whole cohort were selected based on the extensive phenotyping performed previously (data in Supplementary Table S1)18. Chronic FEN treatment resulted in significant upregulation of 377 genes and 487 were significantly downregulated (Fig. 1a).
Figure 1. Hepatic Fgf21 is down regulated in HFD-fed mice receiving Fenretinide treatment.
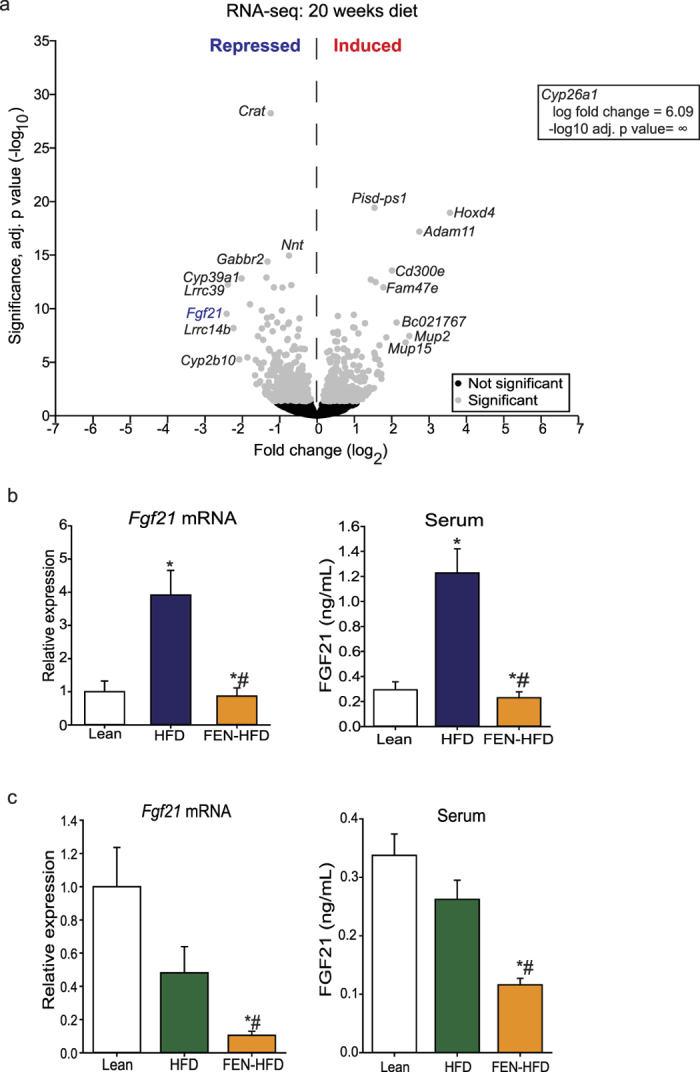
(a) Volcano plot of DESeq2 analysis of liver RNA-seq from C57BL/6 mice fed HFD ± FEN for 20 weeks (FEN-HFD; n = 4 mice per group). Grey circles denote significantly changed gene expression (−log10 adj. p value < 0.05); black circles denote gene expression unchanged. (b) Liver mRNA expression measured by qPCR and serum FGF21 levels in mice fed chow (lean, n = 5), HFD (n = 7) or FEN-HFD (n = 8) for 20 weeks, following 5-hour fast. (c) as in (b) except diets were fed for 7 days: chow (lean, n = 5), HFD (n = 7) or FEN-HFD (n = 8). Liver gene expression was normalized to Hprt. Significant differences were determined by one-way ANOVA followed by post-hoc tests. Differences are marked *p < 0.05 vs lean or #p < 0.05 vs HFD.
The most upregulated genes were classic retinoid targets (cytochrome P450-type enzyme Cyp26a1, transcription factor homeobox D4, Hoxd4 and metalloprotease Adam11) which were induced between 6.7 and 68-fold (Fig. 1a). The most significantly downregulated gene was carnitine acyltransferase (Crat) whilst, Fgf21 was the most downregulated gene, repressed 5.4-fold. Both of these genes are established PPARα targets involved in control of lipid metabolism3,23.
To determine whether FEN can oppose PPARα signalling, we also performed RNA-seq analysis in liver from mice fed a HFD ± FEN for 7 days, which had also been previously characterised (data in Supplementary Table S1)18. Acute FEN treatment resulted in significant upregulation of several classic retinoid target genes and Fgf21 trended to be downregulated. However, FEN treatment did not change the expression of PPARα target genes or target genes of other transcription factors identified as having a role in inducing FGF21, e.g. ChREBP1 (Table 1)3,4,24,25.
Table 1. Hepatic expression of genes regulated by transcription factors that also regulate Fgf21.
| Gene | Log2 fold change | adj. p value | q value |
|---|---|---|---|
| Transcription factors | |||
| Mlxipl (ChREBP) | +0.123 | 0.301 | 1.000 |
| Nr1h3 (LXRα) | −0.095 | 0.431 | 1.000 |
| Nr1c1 (PPARα) | +0.040 | 0.786 | 1.000 |
| Nr1c3 (PPARγ) | +0.419 | 1.000 | 1.000 |
| Nr1d1 (Rev-erbα) | −0.261 | 0.254 | 1.000 |
| ChREBP targets | |||
| Gck | −0.378 | 0.046 | 0.894 |
| Pck1 | +0.410 | 1.000 | 1.000 |
| Elovl6 | −0.288 | 1.000 | 1.000 |
| LXR targets | |||
| Apoe | −0.211 | 0.198 | 1.000 |
| Fas | −0.072 | 0.648 | 1.000 |
| Cyp7a1 | −0.196 | 1.000 | 1.000 |
| PPARα/PPARγ targets | |||
| Acot1 | +0.448 | 1.000 | 1.000 |
| Ehhadh | +0.211 | 0.786 | 0.163 |
| Vnn1 | +0.496 | 0.094 | 0.094 |
| Cidec | +0.159 | 1.000 | 1.000 |
| Cd36 | −0.211 | 0.150 | 1.000 |
| Mogat1 | +0.088 | 0.723 | 1.000 |
| Rev-erbα targets | |||
| Arntl (BMAL1) | +0.060 | 0.786 | 1.000 |
| Per1 | −0.021 | 0.585 | 1.000 |
| Cry1 | +0.104 | 1.000 | 1.000 |
DESeq2 analysis of liver RNA-seq of expression of transcription factors known to regulate Fgf21 and some of their key target genes in mice fed HFD (n = 4) vs HFD +0.04% FEN (FEN-HFD, n = 4) for 7 days.
Gene expression analysis in mice from the whole cohort (n = 7–8), including chow-fed lean animals, confirmed the decrease in Fgf21 expression in long-term treated mice and FEN also completely normalised elevated serum FGF21 levels (Fig. 1b). In mice receiving the acute diet intervention (HFD ± FEN for 7 days), HFD feeding did not increase Fgf21, however FEN decreased FGF21 expression and serum levels more than 50% below normal levels (Fig. 1c). Thus, FEN is able to inhibit Fgf21 expression both chronically in obese mice and more acutely in lean mice, without inducing parallel changes to known major regulators of hepatic Fgf21.
Fenretinide inhibits Fgf21 expression and improves glucose homeostasis without preventing weight gain in Lepr db genetically obese mice
To investigate the relationship between FEN and leptin signalling and determine effects on FGF21 levels in non-diet-induced obesity, we used the Leprdb genetic mouse model. Unlike in HFD mice, FEN did not inhibit rapid weight gain in Leprdb mice fed a 10% fat diet (Fig. 2a). FEN did decrease fasting hyperglycaemia and markedly attenuated glucose intolerance in Leprdb mice (Fig. 2b,c), in association with increased serum insulin (Fig. 2d) and increased pancreatic islet mass (Fig. 2e). Thus, FEN can improve glucose homeostasis independent of leptin signalling.
Figure 2. Fenretinide inhibits Fgf21 expression and improves glucose homeostasis without preventing weight gain in Leprdb genetically obese mice.

(a) Body weight of C57BL/6 lean mice on chow (lean, n = 8; error bars too small to be shown clearly) and C57BL/6 Leprdbmice on 10% fat (Leprdb, n = 7) or 10% fat diet +FEN (FEN-Leprdb, = 7). (b) Glucose (1 mg/g body weight) Tolerance Test (GTT) and (c) AUC at week 4. (d) serum insulin levels at the end of the study, following a 5 h fast. (e) Representative photos of haematoxylin and eosin staining of pancreas sections (10x magnification) and quantification of pancreatic islet size (n = 3 per group). (f) Hepatic gene expression and serum levels of FGF21. Gene expression was normalised to Nono. (g) PG-WAT gene expression, normalised to yWhaz. Significant differences were determined by one-way ANOVA followed by post-hoc tests or student’s t-test for PG-WAT Fgf21 FEN-Leprdb vs Leprdb and Leprdb vs lean groups and β-klotho FEN-Leprdb vs Leprdb and Leprdb vs lean groups. Differences are marked *p < 0.05 vs lean or #p < 0.05 vs Leprdb. Quantification of ceramide (h) and dihydroceramide (i) species in liver. (j) Total liver levels of ceramide, dihydroceramide and the dihydroceramide/ceramide ratio. Significant differences were determined by one-way ANOVA followed by post-hoc tests or student’s t-test for ceramides: C18:0 FEN-Leprdb vs Leprdb and FEN-Leprdb vs lean groups, C20:0 FEN-Leprdb vs Leprdb and FEN-Leprdb vs lean groups, C24:1 FEN-Leprdb vs Leprdb groups; dihydroceramides: C16:0 FEN-Leprdb vs Leprdb and Leprdb vs lean groups, C18:0 FEN-Leprdb vs Leprdb and Leprdb vs lean groups, C20:1 FEN-Leprdb vs Leprdb groups, C20:0 Leprdb vs lean groups, C24:0 FEN-Leprdb vs lean and Leprdbvs lean groups. Differences are marked *p < 0.05 vs lean or #p < 0.05 vs Leprdb.
Hepatic expression and circulating levels of FGF21 were elevated in Leprdb mice but were completely normalised by FEN treatment (Fig. 2f). FEN also suppressed the elevation of Fgf21 in perigonadal white adipose tissue (PG-WAT) but had no effect on the downregulation of Fgfr1 or β-klotho (Fig. 2g). Several PG-WAT metabolic genes (e.g. Pparγ and Glut4) were markedly downregulated in Leprdb mice. FEN co-ordinately increased the expression of these genes to levels similar to those measured in lean mice and also normalised the elevated expression of PG-WAT Rbp4 (Table 2).
Table 2. Gene expression, serum factors and metabolites in Lepr db mice.
| Gene | Lean | Leprdb | FEN-Leprdb |
|---|---|---|---|
| Adipose: metabolic genes | |||
| C/ebpα | 1.00 ± 0.13 | 0.52 ± 0.05* | 0.89 ± 0.19 |
| Chrebp1 | 1.00 ± 0.14 | 0.40 ± 0.07* | 0.65 ± 0.10* |
| Fas | 1.00 ± 0.23 | 0.26 ± 0.03* | 0.58 ± 0.14# |
| Glut4 | 1.00 ± 0.13 | 0.36 ± 0.06* | 0.63 ± 0.14 |
| Insulin receptor | 1.00 ± 0.12 | 0.32 ± 0.04* | 0.41 ± 0.08* |
| Pparγ | 1.00 ± 0.14 | 0.52 ± 0.09* | 0.83 ± 0.19 |
| Scd1 | 1.00 ± 0.14 | 0.55 ± 0.06* | 0.69 ± 0.06* |
| Adipose: retinoid genes | |||
| Crbp1 | 1.00 ± 0.28 | 0.81 ± 0.17 | 1.07 ± 0.37 |
| Raldh1 | 1.00 ± 0.26 | 0.54 ± 0.13 | 0.65 ± 0.18 |
| Rarγ | 1.00 ± 0.16 | 0.37 ± 0.09* | 0.56 ± 0.11* |
| Rbp4 | 1.00 ± 0.38 | 3.61 ± 0.99* | 0.88 ± 0.46# |
| Adipose: adipokine genes | |||
| Adiponectin | 1.00 ± 0.18 | 0.22 ± 0.06* | 0.21 ± 0.03* |
| Leptin | 1.00 ± 0.24 | 2.30 ± 0.83 | 1.49 ± 0.39 |
| Resistin | 1.00 ± 0.18 | 0.19 ± 0.06* | 0.11 ± 0.02* |
| Adipose: inflammation genes | |||
| Cd11b | 1.00 ± 0.15 | 3.34 ± 0.95* | 2.98 ± 0.54* |
| F4/80 | 1.00 ± 0.32 | 24.47 ± 5.66* | 22.91 ± 6.16* |
| Liver: retinoid genes | |||
| Crbp1 | 1.00 ± 0.15 | 1.75 ± 0.27* | 22.26 ± 10.98*,# |
| Cyp26a1 | 1.00 ± 0.11 | 0.31 ± 0.07* | 47.77 ± 3.13*,# |
| Lrat | 1.00 ± 0.09 | 2.70 ± 0.29* | 3.41 ± 0.36* |
| Rbp4 | 1.00 ± 0.09 | 1.98 ± 0.20* | 1.30 ± 0.13*,# |
| Rarβ | 1.00 ± 0.14 | 3.19 ± 0.47* | 8.79 ± 1.02*,# |
| Liver: inflammation genes | |||
| Cd11b | 1.00 ± 0.25 | 3.12 ± 1.32 | 3.32 ± 0.41* |
| F4/80 | 1.00 ± 0.16 | 4.94 ± 1.78* | 3.10 ± 1.05* |
| Hypothalamus genes | |||
| Foxo1a | 1.00 ± 0.18 | 0.43 ± 0.04* | 0.52 ± 0.13* |
| Melanocortin 4 receptor | 1.00 ± 0.26 | 0.49 ± 0.06 | 0.37 ± 0.03* |
| Npy | 1.00 ± 0.15 | 1.61 ± 0.32* | 1.85 ± 0.39 |
| Npy receptor | 1.00 ± 0.22 | 0.62 ± 0.07 | 0.44 ± 0.05* |
| Pomc | 1.00 ± 0.12 | 0.16 ± 0.06* | 0.22 ± 0.04* |
| Ptp1b | 1.00 ± 0.13 | 0.48 ± 0.06* | 0.49 ± 0.06* |
| Raldh2 | 1.00 ± 0.13 | 0.76 ± 0.08* | 0.96 ± 0.21 |
| Stat3 | 1.00 ± 0.20 | 0.49 ± 0.04* | 0.46 ± 0.08* |
| Tgfβ1 | 1.00 ± 0.26 | 0.54 ± 0.07 | 0.56 ± 0.09 |
| Serum leptin | 5.60 ± 1.63 | 131.48 ± 3.34* | 134.88 ± 6.09* |
| Liver triglycerides | 8.34 ± 0.89 | 53.75 ± 5.40* | 61.97 ± 9.73* |
Relative mRNA in PG-WAT, liver and hypothalamus and serum factors and metabolites in lean or Leprdb mice+/−FEN. Expression was normalised to Nono in liver or yWhaz in adipose and hypothalamus. Data are shown as mean ± SEM and significance determined by one-way ANOVA followed by post-hoc tests or student’s t-test for liver: Crbp1 Leprdb vs lean, Rbp4 FEN-Leprdb vs lean, Cd11b FEN-Leprdb vs lean and F4/80 FEN-Leprdb vs lean and Leprdb vs lean groups; PG-WAT: Scd1 Leprdb vs lean, Fas FEN-Leprdb vs Leprdb, Pparγ Leprdb vs lean, C/ebpα Leprdb vs lean, Fgf21 Leprdb vs lean, Rarγ FEN-Leprdb vs lean, Cd11b Leprdb vs lean, Fgf21 FEN-Leprdb vs Leprdb and Leprdb vs lean and β-klotho FEN-Leprdb vs lean and Leprdb vs lean groups; hypothalamus: Npy Leprdb vs lean, and Raldh2 Leprdb vs lean groups. Differences are marked *p < 0.05 vs lean; #p < 0.05 vs Leprdb.
FEN strongly increased hepatic expression of classic retinoid target genes, as expected (Table 2). FEN had no effect on many genes, circulating factors and metabolites known to have a key role in the control of appetite, adiposity, gluconeogenesis, macrophage infiltration or inflammation (Table 2).
Previous work has shown that FEN treatment results in alterations in ceramide and dihydroceramide levels in HFD obese mice, in association with improvements in glucose homeostasis and insulin sensitivity20,22. To determine whether FEN could have the same effect in genetically-obese mice, lipid analysis was performed in liver tissue from Leprdb mice. A number of ceramide species elevated in Leprdb mice were partially decreased by FEN treatment, including C18:0, C20:0 and C22:0 (Fig. 2h). With regards to the immediate precursor to ceramide in its biosynthetic pathway, FEN treatment also resulted in an increase in dihydroceramides, seven out of nine species measured (Fig. 2i), and increased the ratio of dihydroceramide to ceramide (Fig. 2j). Inhibiting de novo hepatic ceramide biosynthesis may be a mechanism by which FEN can improve insulin sensitivity and glycaemia in mice.
Thus, FEN improved glucose homeostasis without inhibiting obesity or decreasing tissue inflammation in Leprdbmice. FEN strongly modulated retinoid and metabolic genes in adipose and liver and decreased production of ceramide biosynthesis in liver. FEN also inhibited the elevation in hepatic and adipose Fgf21 induced by obesity and impaired glucose homeostasis in Leprdb mice, demonstrating that the effects of FEN on Fgf21 gene expression are common to both diet- and genetically-induced obesity.
Fenretinide treatment inhibits hepatic Fgf21 expression in Alb-cre-Ptp1b −/− mice, independent of effects on glucose homeostasis
To test whether directly improving hepatic insulin sensitivity and glucose homeostasis in the background of HFD-induced obesity could lead to a reduction in FGF21 levels, we examined mice with a hepatocyte-specific knock-out of PTP1B. Ptp1bfl/fl (fl/fl) and hepatocyte-specific Ptp1b−/− (Alb-cre-Ptp1b−/−) male mice were fed HFD ± FEN for 17 weeks. As previously reported26,27, Alb-cre-Ptp1b−/− mice showed no difference in body weight compared to fl/fl controls. FEN inhibited weight gain and adiposity (data not shown) in many Alb-cre-Ptp1b−/− mice but this did not reach statistical significance (Fig. 3a).
Figure 3. Fenretinide treatment inhibits hepatic Fgf21 expression in Alb-cre-Ptp1b−/− mice, independent of effects on glucose homeostasis.

(a) Body weight of fl/fl male mice on HFD (fl/fl-HFD, n = 9) or HFD +FEN (fl/fl-FEN-HFD, n = 9) or hepatocyte-specific-Ptp1b−/− male mice on HFD (Alb-cre-Ptp1b−/−-HFD, n = 5) or FEN-HFD (Alb-cre-Ptp1b−/−-FEN-HFD, n = 7). (b) GTT (1 mg/g glucose) and AUC (c) at week 15. (d) Serum insulin and (e) serum leptin levels at the end of the experiment, following a 5-hour fast. (f) Hepatic gene expression of retinoid and metabolic genes, normalised to Hprt. (g) Hepatic mRNA expression and serum levels of FGF21. Significant differences were determined by two-way ANOVA followed by post-hoc tests or student’s t-test for GTT AUC Alb-cre-Ptp1b−/−-FEN-HFD vs fl/fl-HFD; Rarβ FEN-HFD (both fl/fl and Alb-cre-Ptp1b−/−) vs fl/fl-HFD; Fgf21 FEN-HFD (both fl/fl and Alb-cre-Ptp1b−/−) vs fl/fl-HFD and FEN-HFD vs HFD in Alb-cre-Ptp1b−/−. Differences are marked *p < 0.05 vs fl/fl-HFD, #p < 0.05 vs Alb-cre-Ptp1b−/−-HFD or † vs fl/fl-FEN-HFD.
Alb-cre-Ptp1b knock-out resulted in improved glucose homeostasis in both HFD+/− FEN mice. Alb-cre-Ptp1b−/− mice fed HFD appeared to be the most glucose tolerant but this was not statistically significant (Fig. 3b,c). Alb-cre-Ptp1b knock-out alone did not alter serum insulin (Fig. 3d) or leptin (Fig. 3e) levels but FEN treatment decreased both factors in each genotype. FEN also increased retinoid gene expression in both fl/fl and Alb-cre-Ptp1b−/− mice, but there was no effect on hepatic Rbp4 expression (Fig. 3f). Importantly, Fgf21 expression and serum levels were similar in Alb-cre-Ptp1b−/− and fl/fl mice with HFD-induced obesity, but FEN strongly decreased FGF21 in both groups (Fig. 3g). Thus, hepatic Fgf21 levels are not decreased as a result of improved hepatic insulin signalling alone, thereby providing further evidence for a direct effect of FEN on hepatic Fgf21, unrelated to improved insulin sensitivity and glucose homeostasis. However, diverse treatments to improve glycaemic control in humans can decrease elevated levels of FGF21, so it is unclear if there is a direct relationship between glucose homeostasis and FGF21 levels.
Fenretinide inhibits hepatic Fgf21 and induces changes to gene expression in lean mice fed a normal-fat diet
Next, we tested whether FEN could decrease body weight, improve glucose homeostasis and decrease FGF21 levels in lean mice fed a normal-fat diet. FEN had no effect on body weight, food intake (Fig. 4a,b) or glucose tolerance (Fig. 4c) but altered retinoid gene expression in PG-WAT, with increased Rarβ and Cyp26a1 and decreased Rbp4 expression (Fig. 4d). FEN also increased hepatic Rarβ, Cyp26a1 and Crbp1 (Fig. 4e). Strikingly, FEN decreased hepatic Fgf21 and circulating protein levels in these mice (Fig. 4f). These results suggest that FEN can inhibit basal hepatic Fgf21 expression but cannot decrease body weight or improve glucose homeostasis under normal physiological conditions.
Figure 4. Fenretinide inhibits hepatic Fgf21 and induces changes to gene expression in lean mice fed a normal-fat diet.

(a) Body weight of C57BL/6 mice on 10% fat diet (n = 10) or 10% fat +FEN (FEN-10% fat, n = 10). (b) Weekly food intake of mice during weeks 2–7. (c) GTT (2 mg/g body weight) at week 18. Student’s t-test was used to determine significant differences (none). Expression of retinoid target genes was measured in PG-WAT (d) and liver (e). Gene expression was normalized to the geomean of 18s, NoNo and yWhaz (PG-WAT) or geomean of NoNo, yWhaz and β-actin (liver). (f) hepatic gene expression and serum levels of FGF21. Significant differences were determined by student’s t-test. Differences are marked *p < 0.05 vs control.
Mice with reduced RA synthesis have increased Fgf21 expression
To establish whether our findings were retinoid dependent, we used mice lacking the retinol metabolising enzyme retinaldehyde dehydrogenase 1 (Raldh1−/−; liver western blot Fig. 5a shows deletion), which have an approximately 70% reduction in normal RA levels28. Raldh1−/− mice showed decreased fat mass (Fig. 5b,c) and improved glucose tolerance (Fig. 5d) compared to controls. Raldh1−/− mice showed increased hepatic gene expression of Fgf21 but not circulating levels (Fig. 5e). These data suggest that Fgf21 may be directly suppressed by retinoid-dependent signalling mechanisms.
Figure 5. Mice with reduced RA synthesis have increased Fgf21 expression.

(a) Representative immunoblot of hepatic RALDH1 levels from C57BL/6 wild type (n = 7 for whole group) and C57BL/6-Raldh1−/− mice (n = 8 for whole group). (b) Body weight and fat mass (c) of all mice. (d) GTT (1 mg/g glucose) and AUC analysis at week 10. (e) Hepatic Fgf21 expression (normalized to Hprt) and FGF21 serum levels. Significant differences were determined by student’s t-test. Differences are marked *p < 0.05 vs wildtype.
Acute retinoid treatment results in inhibition of hepatic Fgf21 expression and RARα binding at the Fgf21 promoter is reduced with Fenretinide treatment
Since FEN is an activator of both retinoid- and non-retinoid signalling pathways, we tested whether Fgf21 could be directly inhibited by RA. RA and FEN both increased hepatic target genes Cyp26a1 and Rarβ in lean C57BL/6 mice as expected (Fig. 6a) and in parallel decreased hepatic gene expression and circulating levels of FGF21 at two hours post-injection (Fig. 6b,c).
Figure 6. Acute retinoid treatment results in inhibition of hepatic Fgf21 expression and RARα binding at the Fgf21 promoter is reduced with Fenretinide treatment.

C57BL/6 mice were on chow diet and ad libitum fed before a single injection of olive oil vehicle or 50 mg/kg RA or FEN for 2 hours, n = 8 for each group. (a) Hepatic gene expression of retinoid targets normalised to yWhaz. (b) Hepatic Fgf21 expression and (c) serum FGF21 levels. Significant differences were determined by one-way ANOVA followed by post-hoc tests or student’s t-test for Cyp26a1 FEN vs RA, Rarβ FEN vs vehicle, Fgf21 mRNA FEN vs vehicle and FGF21 serum FEN vs vehicle. Differences are marked *p < 0.05 vs vehicle or #p < 0.05 vs RA. (d) ChIP-qPCR of RARα binding at the hepatic Fgf21 RARE in mice injected with vehicle or 50 mg/kg FEN for 2 hours (n = 2 IP’s from 3 individual mice per group). IgG antibody was used to measure non-specific binding (IgG ab, n = 1). (e) ChIP-qPCR of RARα and polymerase (Pol) II binding at the hepatic Fgf21 and Cyp26a1 RAREs in mice fed HFD or HFD +FEN (FEN-HFD) for 20 weeks (n = 1 IP from 4 individual mice per group). IgG antibody was used to measure non-specific binding (IgG ab). Significant differences were determined by student’s t-test. Differences are marked *p < 0.05.
A putative retinoic acid response element (RARE) was previously reported within the Fgf21 promoter region29. We confirmed the presence of this RARE by ChIP-qPCR and observed that 2-hour FEN treatment resulted in decreased RARα binding at this site (Fig. 6d). Furthermore, both RARα and polymerase (Pol) II binding were decreased at the Fgf21 RARE in mice chronically fed FEN-HFD, compared to HFD-fed mice (Fig. 6e). Binding of both RARα and Pol-II were increased at RARE 1 of Cyp26a1 (Fig. 6e), which is induced by retinoid signaling. Together, these results suggest that retinoid signalling has a direct inhibitory effect upon Fgf21 gene expression.
To test whether acutely blocking RAR signalling could prevent the retinoid-induced suppression of Fgf21, we performed experiments in mouse primary hepatocytes. Treatment with GW7647, a potent PPARα agonist, led to the induction of Fgf21 and Cd36 (Supplemental Figure S1a). However, RA treatment did not lead to the induction of Rarβ or the suppression of Fgf21 in these cells (Supplemental Figure S1a). Strikingly, Rarα gene expression was found to be more than 10-fold lower in mouse primary hepatocytes than in mouse liver tissue (Supplemental Figure S1b). Thus, we suggest that down-regulation of RAR-signalling can prevent the retinoid-induced suppression of Fgf21.
Discussion
FGF21 is an important regulator of lipid and glucose metabolism and is undergoing trials in humans to evaluate its efficacy as a treatment for obesity and type-2 diabetes3. However, circulating levels of FGF21 are elevated in obesity and insulin-resistant states, therefore it is not clear if attempts to (in)directly increase levels further will be of therapeutic benefit in humans8,11,14. We have now identified FGF21 as a novel retinoid-dependent target of FEN and thus found a novel direct way to normalise elevated levels in both diet- and genetically-induced obese states whilst, at the same time, improving metabolic dysfunction.
Many others have reported that diverse treatments to improve glycaemic control in humans can decrease elevated levels of FGF21, e.g. with metformin, bariatric surgery or exercise8,9,12,14. Although the mechanism by which FGF21 is physiologically induced by fasting, protein restriction and sugar intake is relatively clear, the molecular pathway by which the paradoxical elevation of FGF21 occurs in metabolically dysregulated states is still unknown. We have shown that HFD-feeding itself does not directly induce hepatic FGF21, suggesting that chronic dysregulation of lipid and glucose homeostasis is responsible for the elevation in obese-insulin-resistant states. Moreover, improving hepatic insulin sensitivity and glucose homeostasis in the background of HFD-induced obesity did not decrease FGF21 levels in mice lacking hepatic PTP1B. Thus, more research is required to fully understand the nutritional and pharmacological regulation of FGF21 in physiological and patho-physiological states.
There have been some studies into the negative regulation of FGF21 e.g. PGC-1α potentiates the repressive activity of Rev-Erbα in concert with recruitment of the corepressors NCoR2 and HDAC3 on the FGF21 promoter in primary hepatocytes24. Our data, that RARα binding is reduced by FEN treatment at the Fgf21 promoter, suggests that Fgf21 is also repressed by RAR-mediated signaling in vivo. LXR is also reported to inhibit FGF21 via recruitment of HDAC325. It is therefore possible that RARα may function via a similar mechanism to inhibit Fgf21 expression. FEN treatment did not change the expression of Rev-Erbα or LXR target genes (Table 1).
Results from cultured cells have not provided clear evidence for a direct mechanism of retinoid-induced suppression of Fgf21. Fgf21 was shown to be induced by RARβ signaling in human HepG2 cells29. We found that down-regulation of Rarα in mouse primary hepatocytes prevented the retinoid-induced suppression of Fgf21. It is likely that these irreconcilable results are due to the molecular and physiological differences between cultured cells and in vivo systems. Directly activating/blocking RAR signalling in mice via the portal circulation could be a more suitable method to test these findings in the future.
We and others have recently identified FEN as a potential safe treatment for diet-induced obesity and type-2 diabetes in mice17,18,19,22. FEN can directly inhibit leptin gene transcription but does not have measureable effects on food intake or energy expenditure17,18. Here, we have established that FEN does not inhibit excess weight gain driven by hyperphagia and low energy expenditure due to disrupted leptin signalling in Leprdb mice. These findings suggest that FEN may interact with leptin signalling to inhibit adiposity, but it is unlikely that a direct link can be determined given the difference in potency of effects between FEN and leptin on these energy balance parameters.
In contrast to the lack of an anti-obesity effect in Leprdb mice, FEN decreased fasting hyperglycaemia and glucose intolerance and increased pancreatic islet mass and circulating insulin. However, this result contrasts with our studies in HFD-obese mice where FEN decreased serum insulin in parallel with beneficial effects on adiposity and glucose homeostasis18 and to the results shown here in Alb-cre Ptp1b−/− HFD-fed mice, where FEN treatment also resulted in decreased serum insulin. These results suggest that with the genetic loss of leptin-signalling, FEN may improve insulin sensitivity via a direct effect on the pancreas, perhaps mediated by RA signalling. Normally, leptin inhibits insulin secretion and biosynthesis to adapt glucose homeostasis to the amount of body fat. However, in obesity, a state of leptin resistance contributes to dysregulation of the adipo-insular axis and promotes the development of hyperinsulinaemia30. Moreover, RA can induce pancreatic development and affect the function of pancreatic β-cells, thus RA signalling may be involved in complex cross-talk with insulin and leptin signalling in normal physiology and this may be impaired in pathophysiological states31.
However, in the more physiological model of HFD-induced obesity, the mechanism of FEN action to improve glucose homeostasis is more likely due to an effect on adipose tissue to normalise impaired mitochondrial fatty acid beta-oxidation and tricarboxylic acid flux22. We and others have already shown that FEN can inhibit the final step in ceramide synthesis through blockade of the enzyme dihydroceramide desaturase 1 in adipose tissue20,22. Here, we have now shown that ceramide synthesis is also blocked by FEN in liver in a genetic model of severe obesity and insulin resistance. Excess levels of ceramides directly mediate impaired insulin signalling (via inhibition of Akt phosphorylation and dysregulation of mitochondrial fatty acid oxidation32,33). Thus, FEN may improve glucose homeostasis in mice via decreased ceramide accumulation and normalisation of metabolic flux in adipose, liver and muscle, and/or metabolic flux between these insulin-sensitive tissues, independent of effects on the regulation of FGF21. Further data analysis of our RNA-seq data may reveal the pathways that are altered in association with the beneficial effects of FEN-treatment.
Overall, the list of effects upon FEN treatment now includes suppressing the elevation of FGF21 in insulin resistant states. Since the pleotropic effects of FEN are all beneficial and not at all detrimental, our findings suggest targeting the multiple molecular defects that are manifest with metabolic disease may be an achievable goal.
Methods
Animals
Experiments were approved by the University of Aberdeen Ethics Board and performed following UK Home Office licenses PPL60/3951 and PPL70/7911 under the Animals (Scientific Procedures) Act 1986. Male C57BL/6 mice fed HFD ± 0.04% FEN were previously reported (Supplementary Table S1)18,22. HFD used in respective mouse cohorts through-out this study contained 45% lard-based fat. Male C57BL/6 Leprdb and lean wild-type/heterozygous littermates were from Charles River (Edinburgh, UK). Mice were group-housed (2–3 obese/8 lean mice per cage) with conditions as previously reported18. Male C57BL/6 Raldh1−/− and wild-type mice were bred in-house and described previously18. Ptp1bfl/fl and albumin-cre-Ptp1b−/− mice with hepatocyte-specific deletion of protein tyrosine phosphatase (PTP)-1B
(Alb-cre-Ptp1b−/−) were previously generated26,27. Alb-cre-Ptp1b−/− mice were fed HFD ± FEN as previously reported for C57BL/6 mice18,22. In FEN-diet studies, both Leprdb and C57BL/6 mice were fed 10% fat diet (D12450B, Research Diets, New Brunswick, NJ). Lean controls, C57BL/6 mice for injection studies and Raldh1−/− mice were fed chow diet18. Glucose tolerance test (GTT) and fat mass measurements by Echo MRI or DEXA scan were performed as previously described5,18. GTT area under the curve was calculated using time zero glucose level for each mouse. Mice were killed following a 5 h fast (except for injection studies), tissues rapidly dissected, flash frozen in liquid nitrogen and stored at −80 °C until further use.
Tissue and serum analysis
Tissue serum factors, liver triacylglycerols and gene expression were measured as previously described5. Most stable reference genes were selected by screening a panel of common reference genes with RefFinder34. Mouse primer sequences used for qPCR are listed in Supplementary Table S2. Pancreas tissues for IHC were rapidly dissected, fixed in formalin and embedded in paraffin wax using a Peloris II tissue processor (Leica Biosystems, Milton Keynes, UK). Embedded tissue was cut into 4 μM-thick sections and mounted onto glass slides. Tissue sections were dewaxed, stained with haematoxylin and eosin, and visualized using an Axioskop 40 microscope (Zeiss, Cambridge, UK). Islet area size was quantified using ImageJ software (NIH, Bethesda, MD, USA).
RNA sequencing
Total RNA was extracted from frozen liver using TriZOL reagent (Sigma Aldrich, Irvine, UK) to manufacturer’s instructions. RNA was purified using RNeasy miniprep kit (Qiagen, Manchester, UK) and quantified on a Bioanalyzer 2000 (Agilent Technologies, Edinburgh, UK). Ribosomal RNA was removed from samples using the Ribo-Zero rRNA removal kit (Illumina, San Diego, CA, USA). Sequencing libraries were prepared using the TruSeq RNA Library Preparation Kit v2 (Illumina) following the low sample protocol, using 1 μg RNA. Libraries were sequenced on the NextSeq-500 Desktop sequencer platform (Illumina) with a 2 × 75 paired-end read length giving 719 million raw reads (average depth 45 million reads per sample). Reads were filtered using the FASTQ Toolkit v2.0.0 in the Illumina BaseSpace cloud computing environment to remove adapter sequences and reads with phred score <20 or length <20 bp35. Filtered reads were checked using FastQC before aligning with STAR 2.0 to the Mus musculus UCSC mm10 genome and differential expression measured using DESeq2 with default parameters36,37,38,39,40.
Quantification of liver dihydroceramides and ceramides
Extraction of liver lipids was performed according to the method described by Folch et al.41. Ceramides and dihydroceramides were isolated by solid phase extraction chromatography using C17:0 ceramide and C12:0 dihydroceramide (Avanti Polar Lipids, Alabaster, Al, USA) as internal standards. Samples were analysed by liquidchromatography–mass spectrometry (LC–MS) using a Thermo TSQ Quantum Ultra mass spectrometer (Thermo Scientific, Hemel Hempstead, UK), equipped with a heated electrospray ionisation (HESI) probe and coupled to a Thermo Accela 1250 UHPLC system. Samples were analysed in positive ion mode over the mass to charge (m/z) range 200–2000. The samples were injected on to a Kinetex C8 column (2.1 mm_100 mm, 2.6 μm). Mobile phase A consisted of 90% (v/v) H2O2 and 10% (v/v) acetonitrile with 0.1% (v/v) formic acid. Mobile phase B consisted of 90:10 isopropanol/acetonitrile with 0.1% (v/v) formic acid. The initial conditions for analysis were 80% B and the percentage of mobile phase B was increased to 100% over 15 minutes and held for 1 min before re-equilibration with the starting conditions for 4 min. All solvents were LC–MS grade (Fisher Scientific, Loughborough, UK). The raw LC–MS data were processed with Thermo Xcalibur v2.1 (Thermo Scientific) and concentration of ceramide and dihydroceramide molecular species determined by comparison to calibration curves generated with C16:0 and C24:1 standards (Avanti). Total ceramide and dihydroceramide concentrations were calculated from the summed concentrations of all the monitored molecular species. All values were normalised to the wet weight of liver.
Immunoblotting and Chromatin Immunoprecipitation (ChIP)
Tissue lysates were prepared and immunoblotting performed as described previously42,43. ALDH1A1 (RALDH1; Abcam; ab52492) and β-actin (Pierce Scientific; PA1-21176) primary antibodies were at 1/1000 dilution; peroxidase-conjugated goat-anti-rabbit secondary antibody (Anaspec; 28177) at 1/5000 dilution. ChIP was performed using the SimpleChIP kit (9002, Cell Signalling, Danvers, MA) with antibodies: RARα (C15310155, Diagenode) and Pol II (sc-3001, Santa Cruz).
Statistical analysis
Sequencing data was analysed with DESeq2 and presented as p values normalised for a false discovery rate of 0.05. All other data was analysed using Sigmaplot 13.0 and presented as mean ± SEM and compared using two-way or one-way ANOVA with post-hoc tests or student’s t-test as indicated.
Additional Information
How to cite this article: Morrice, N. et al. Elevated Fibroblast growth factor 21 (FGF21) in obese, insulin resistant states is normalised by the synthetic retinoid Fenretinide in mice. Sci. Rep. 7, 43782; doi: 10.1038/srep43782 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
The authors would like to thank undergraduate student Aleksandra Kowalczuk (University of Aberdeen) for assisting in experiments and Dr. Emma K. Lees (School of Health Sciences, Liverpool Hope University, Liverpool, UK) for invaluable discussions concerning the regulation of FGF21. We thank Dr. Calum Sutherland and Dr. Amy Cameron (both Division of Molecular and Clinical Medicine, Ninewells Hospital and Medical School, University of Dundee, Scotland, UK) for technical support and expertise in performing hepatocyte studies. Fenretinide was a generous gift of T. Martin (Johnson & Johnson, New Brunswick, NJ) and U. Thumeer (Cilag AG, Schaffhausen, Switzerland), for use completely without restriction or obligation. Quantitative-PCR was carried out using the qPCR Core Facility (Institute of Medical Sciences, University of Aberdeen). RNA-sequencing was carried out at the University of Aberdeen Centre for Genome Enabled Biology and Medicine. Pancreas histology was performed by Dr Linda Davidson (Department of Histology, Aberdeen Royal Infirmary, NHS Grampian, Foresterhill Health Campus, Aberdeen, UK). This study was supported by the British Heart Foundation Intermediate Basic Research Fellowship FS/09/026 to N. Mody, RCUK fellowship to MD, EFSD/Lilly Programme Grant to MD and N. Mody, Tenovus Scotland grants G10/04 and G14/14 to N. Mody, University of Aberdeen Centre for Genome Enabled Biology and Medicine (CGEBM) PhD studentship to N. Morrice and Biotechnology and Biological Sciences Research Council studentship to GDM.
Footnotes
The authors declare no competing financial interests.
Author Contributions N. Morrice, G.D.M., M.D., P.D.W. and N. Mody designed experiments. N. Morrice, G.D.M., S.R.T., K.S., J.R., P.D.W. and N. Mody performed experiments. N. Morrice, G.D.M., K.S., M.K.D., M.D., P.W.D. and N. Mody analysed the data. N. Morrice made the figures. N. Morrice and N. Mody wrote the manuscript. N. Morrice, N. Mody and M.D. edited the manuscript.
References
- Markan K. R. et al. Circulating FGF21 is Liver Derived and Enhances Glucose Uptake During Refeeding and Overfeeding. Diabetes (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giralt M., Gavalda-Navarro A. & Villarroya F. Fibroblast growth factor-21, energy balance and obesity. Mol. Cell. Endocrinol. 418 Pt 1, 66–73 (2015). [DOI] [PubMed] [Google Scholar]
- Badman M. K. et al. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell. Metab. 5, 426–437 (2007). [DOI] [PubMed] [Google Scholar]
- Dushay J. R. et al. Fructose ingestion acutely stimulates circulating FGF21 levels in humans. Mol. Metab. 4, 51–57 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees E. K. et al. Methionine restriction restores a younger metabolic phenotype in adult mice with alterations in fibroblast growth factor 21. Aging Cell. 13, 817–827 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. et al. Serum FGF21 levels are increased in obesity and are independently associated with the metabolic syndrome in humans. Diabetes 57, 1246–1253 (2008). [DOI] [PubMed] [Google Scholar]
- Fisher F. M. et al. Obesity is a fibroblast growth factor 21 (FGF21)-resistant state. Diabetes 59, 2781–2789 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H. et al. Effect of Metformin on Fibroblast Growth Factor-21 Levels in Patients with Newly Diagnosed Type 2 Diabetes. Diabetes Technol. Ther. 18, 120–126 (2016). [DOI] [PubMed] [Google Scholar]
- Srinivasa S. et al. Effects of lifestyle modification and metformin on irisin and FGF21 among HIV-infected subjects with the metabolic syndrome. Clin. Endocrinol. (Oxf) 82, 678–685 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K. et al. Effects of rosiglitazone on fasting plasma fibroblast growth factor-21 levels in patients with type 2 diabetes mellitus. Eur. J. Endocrinol. 161, 391–395 (2009). [DOI] [PubMed] [Google Scholar]
- Yang M., Dong J., Liu H., Li L. & Yang G. Effects of short-term continuous subcutaneous insulin infusion on fasting plasma fibroblast growth factor-21 levels in patients with newly diagnosed type 2 diabetes mellitus. PLoS One 6, e26359 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haluzikova D. et al. Laparoscopic sleeve gastrectomy differentially affects serum concentrations of FGF-19 and FGF-21 in morbidly obese subjects. Obesity (Silver Spring) 21, 1335–1342 (2013). [DOI] [PubMed] [Google Scholar]
- Qin Y. et al. Fish Oil Supplements Lower Serum Lipids and Glucose in Correlation with a Reduction in Plasma Fibroblast Growth Factor 21 and Prostaglandin E2 in Nonalcoholic Fatty Liver Disease Associated with Hyperlipidemia: A Randomized Clinical Trial. PLoS One 10, e0133496 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi H., Tanisawa K., Sun X., Kubo T. & Higuchi M. Endurance Exercise Reduces Hepatic Fat Content and Serum Fibroblast Growth Factor 21 Levels in Elderly Men. J. Clin. Endocrinol. Metab. 101, 191–198 (2016). [DOI] [PubMed] [Google Scholar]
- Zanardi S. et al. Clinical trials with retinoids for breast cancer chemoprevention. Endocr. Relat. Cancer 13, 51–68 (2006). [DOI] [PubMed] [Google Scholar]
- Chojkier M. A Randomized, Double-blind Study of the Effects of Fenretinide Administered in Subjects with Obesity, https://clinicaltrials.gov/ct2/show/NCT00546455, (19/12/2016) (2013).
- Preitner F., Mody N., Graham T. E., Peroni O. D. & Kahn B. B. Long-term Fenretinide treatment prevents high-fat diet-induced obesity, insulin resistance, and hepatic steatosis. Am. J. Physiol. Endocrinol. Metab. 297, E1420–9 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcilroy G. D. et al. Fenretinide treatment prevents diet-induced obesity in association with major alterations in retinoid homeostatic gene expression in adipose, liver, and hypothalamus. Diabetes 62, 825–836 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearer K. D. et al. Fenretinide prevents obesity in aged female mice in association with increased retinoid and estrogen signaling. Obesity (Silver Spring) 23, 1655–1662 (2015). [DOI] [PubMed] [Google Scholar]
- Bikman B. T. et al. Fenretinide prevents lipid-induced insulin resistance by blocking ceramide biosynthesis. J. Biol. Chem. 287, 17426–17437 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mody N. & Mcilroy G. D. The mechanisms of Fenretinide-mediated anti-cancer activity and prevention of obesity and type-2 diabetes. Biochem. Pharmacol. (2014). [DOI] [PubMed] [Google Scholar]
- Mcilroy G. D. et al. Fenretinide mediated retinoic acid receptor signalling and inhibition of ceramide biosynthesis regulates adipogenesis, lipid accumulation, mitochondrial function and nutrient stress signalling in adipocytes and adipose tissue. Biochem. Pharmacol. 100, 86–97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakhshandehroo M. et al. Comprehensive analysis of PPARalpha-dependent regulation of hepatic lipid metabolism by expression profiling. PPAR Res. 2007, 26839 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estall J. L. et al. PGC-1alpha negatively regulates hepatic FGF21 expression by modulating the heme/Rev-Erb(alpha) axis. Proc. Natl. Acad. Sci. USA 106, 22510–22515 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer A. et al. Fasting-induced FGF21 is repressed by LXR activation via recruitment of an HDAC3 corepressor complex in mice. Mol. Endocrinol. 26, 1980–1990 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delibegovic M. et al. Liver-specific deletion of protein-tyrosine phosphatase 1B (PTP1B) improves metabolic syndrome and attenuates diet-induced endoplasmic reticulum stress. Diabetes 58, 590–599 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen C. et al. Inducible liver-specific knockdown of protein tyrosine phosphatase 1B improves glucose and lipid homeostasis in adult mice. Diabetologia 56, 2286–2296 (2013). [DOI] [PubMed] [Google Scholar]
- Molotkov A. & Duester G. Genetic evidence that retinaldehyde dehydrogenase Raldh1 (Aldh1a1) functions downstream of alcohol dehydrogenase Adh1 in metabolism of retinol to retinoic acid. J. Biol. Chem. 278, 36085–36090 (2003). [DOI] [PubMed] [Google Scholar]
- Li Y., Wong K., Walsh K., Gao B. & Zang M. Retinoic acid receptor beta stimulates hepatic induction of fibroblast growth factor 21 to promote fatty acid oxidation and control whole-body energy homeostasis in mice. J. Biol. Chem. 288, 10490–10504 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seufert J. Leptin effects on pancreatic beta-cell gene expression and function. Diabetes 53 Suppl 1, S152–8 (2004). [DOI] [PubMed] [Google Scholar]
- Rhee E. J. & Plutzky J. Retinoid metabolism and diabetes mellitus. Diabetes Metab. J. 36, 167–180 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratford S., Hoehn K. L., Liu F. & Summers S. A. Regulation of insulin action by ceramide: dual mechanisms linking ceramide accumulation to the inhibition of Akt/protein kinase B. J. Biol. Chem. 279, 36608–36615 (2004). [DOI] [PubMed] [Google Scholar]
- Keung W. et al. Inhibition of carnitine palmitoyltransferase-1 activity alleviates insulin resistance in diet-induced obese mice. Diabetes 62, 711–720 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie F., Xiao P., Chen D., Xu L. & Zhang B. miRDeepFinder: a miRNA analysis tool for deep sequencing of plant small RNAs. Plant Mol. Biol. (2012). [DOI] [PubMed] [Google Scholar]
- Illumina Inc., BaseSpace, https://basespace.illumina.com/home/index (19/12/2016) (2016).
- Andrews, S. FastQC, http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (19/12/2016) (2016).
- Dobin A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W. J. et al. The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom K. R. et al. The UCSC Genome Browser database: 2015 update. Nucleic Acids Res. 43, D670–81 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M. I., Huber W. & Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch J., Lees M. & Sloane Stanley G. H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 226, 497–509 (1957). [PubMed] [Google Scholar]
- Delibegovic M. et al. Improved glucose homeostasis in mice with muscle-specific deletion of protein-tyrosine phosphatase 1B. Mol. Cell. Biol. 27, 7727–7734 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees E. K. et al. Effects of hepatic protein tyrosine phosphatase 1B and methionine restriction on hepatic and whole-body glucose and lipid metabolism in mice. Metabolism 64, 305–314 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.