

Abstract
High-fat diets (HFD) are thought to contribute to the development of metabolism-related diseases. The long-term impact of HFD may be mediated by epigenetic mechanisms, and indeed, HFD has been reported to induce DNA methylation changes in white adipose tissue (WAT) near metabolism related genes. However, previous studies were limited to a single WAT depot, a single time-point and primarily examined the pre-pubertal period. To define dynamic DNA methylation patterns specific for WAT depots, we investigated DNA methylation of Pparg2 and Leptin in gonadal adipose tissue (GAT) and subcutaneous adipose tissue (SAT), at baseline and after 6, 12 and 24 weeks of HFD exposure in adult mice. HFD induced hypermethylation of both the Leptin promoter (max. 19.6% at week 24, P = 2.6·10−3) and the Pparg2 promoter in GAT (max. 10.5% at week 12, P = 0.001). The differential methylation was independent of immune cell infiltration upon HFD exposure. In contrast, no differential methylation in the Pparg2 and Leptin promoter was observed in SAT. Leptin and Pparg2 DNA methylation were correlated with gene expression in GAT. Our study shows that prolonged exposure to HFD in adulthood is associated with a gradually increasing DNA methylation level at the Leptin and Pparg2 promoters in a depot-specific manner.
Obesity induced by exposure to a high content of saturated fat diet (HFD) is characterized by hypertrophy and hyperplasia of adipocytes in white adipose tissue (WAT). It is followed by a chronic state of mild inflammation and changes in adipokine secretion, a phenomenon particularly apparent in metabolically active visceral WAT depots, including gonadal adipose tissue (GAT), rather than subcutaneous adipose tissue (SAT)1,2. Epigenetic changes, such as at the level of DNA methylation, are suggested to contribute to long-term changes in adipokine secretion2,3,4,5. In particular, HFD exposure has been consistently linked to differences in DNA methylation near the genes Leptin and Pparg2 (the adipocyte-specific isoform 2 of Pparg)6,7,8. The adipokine Leptin is a critical signalling component regulating food intake, energy homeostasis, and exhibits potent immunomodulatory functions9,10. The role of Leptin in satiety signaling is thought be related to an activation of PI3 kinase in the hypothalamus11. While in adipose tissue, leptin influences insulin responsiveness via suppressor of cytokine signalling 3 expression which inhibits auto-phosphorylation of the insulin receptor and down-regulates Leptin responsiveness12,13. In addition to its role in insulin signalling, Leptin has been shown to strongly inhibit lipid synthesis in epididymal adipocytes of normal lean Zucker diabetic fatty rats14. Pparg2 is a master regulator of adipogenesis and is involved in adipocyte differentiation and maturation as well as fat storage and glucose metabolism. Its activation it thought to have insulin sensitizing effects6,15,16. Many of the genes activated by Pparg2 stimulate lipid uptake by adipocytes and adipogenesis or are involved in glucose homeostasis via glucose transporter type 4 (Glut4) and c-Cbl–associated protein (CAP) in adipocytes15. Moreover, PPARg2 can control the expression of numerous adipokines such as leptin and tumor necrosis factor-α (TNF-α) in adipose tissue, which can reduce insulin sensitivity17.
Until now, studies investigating promoter DNA methylation of Leptin and Pparg2 have primarily focused on HFD exposure in utero or in early life18,19, while few studies examined the effect of HFD in adulthood, a period which may be particularly relevant to the development of obesity in humans (Table 1). Furthermore, insight into the dynamics of DNA methylation differences over prolonged HFD exposure remains scarce and studies did not address the possibility that DNA methylation differences could be confounded by infiltration of immune cells in WAT after HFD exposure. Importantly, the potential difference in response across WAT depots has rarely been explored, although striking differences in morphology and function between depots have been established20,21,22,23. In particular SAT and GAT have been implicated in HFD induced obesity6,20,21,23,24,25,26. GAT and other abdominal fat depots drive the development of obesity-associated metabolic disorders25,26, while SAT is considered to be a ‘safe’ storage depot for excess energy without these detrimental effects. Consistent with this view, transplantation of SAT in diet induced obese mice attenuates metabolic dysregulation while its removal exacerbates the condition27. Furthermore, GAT is much more prone to inflammation than SAT and its surgical removal attenuates the development of metabolic liver disease in HFD-treated male C57BL/6J mice26.
Table 1. Effects of adult HFD exposure on Leptin and Pparg2 DNA methylation and gene expression reported in literature.
| Gene | Study | Species/Strain/Sex | Group size | Tissue | HFD | Time | Start diet | Expression | DNA methylation |
|---|---|---|---|---|---|---|---|---|---|
| Leptin | 28 | Male C57BL/6 J | n = 12 chow/HF | Epididymal fat (GAT) | 34.9% fat by wt | 4/8/12/18 weeks | 4/5 weeks | mRNA Leptin↑ | Leptin↑ |
| 29 | n = 15 chow/HF | ||||||||
| Pparg2 | 7 | n = 3 chow/HF | Inguinal and Epididymal fat (GAT) | n.a. | 16 weeks | 4 weeks | mRNA Pparγ2↓ | Pparγ2↑ | |
| Leptin | 6 | Wistar rats | n = 5 chow/n = 6 HF | Retroperitoneal fat | 59.2% energy from fat | 11 weeks | post weaning | n.a. | Leptin↑ |
| 46 | Wistar rats | n = 12 chow/HF | Retroperitoneal fat | 45% fat by wt | 20 weeks | 3 weeks | n.a. | Leptin↑ |
Here, we report on dynamic changes in DNA methylation of the Pparg2 and Leptin promoters during prolonged HFD exposure (at baseline and 6, 12 and 24-week of HFD exposure) in adult male C57BL/6J mice in two different WAT depots, GAT and SAT. In addition, we analysed the impact of immune cell infiltration on DNA methylation and investigated the association between DNA methylation and expression of Leptin and Pparg2.
Results
HFD and body and fat depot weight
We investigated the effect of HFD on bodyweight and mass of GAT and SAT depots as compared with chow after 0 (baseline), 6, 12, or 24 weeks of exposure (n = 11/12 per group). Food intake in HFD and chow groups was isocaloric. Body weight increased with 34.3% after 24 weeks of exposure (P < 2.2·10−16; Supplementary Table S1). Concomitantly, GAT showed a steep increase in mass during the initial phase of weight gain, reaching its maximum at 12 weeks of exposure (t12: 88.8%, P = 4.4·10−12), while SAT mass increased more gradually up to 89% after 24 weeks of HFD exposure (P = 3.2·10−11). The chow control group showed minor and non-significant increases over time confirming that the changes observed were due to the HFD exposure and not an age-related effect (Supplementary Table S1).
Pparg2 and Leptin methylation during HFD
At baseline, DNA methylation levels of the promoters of Pparg2 and Leptin (Fig. 1a,b) were similar in GAT (P = 0.68, Fig. 1c) and SAT (P = 0.93, Fig. 1d). Over time differences in DNA methylation between the two fat depots were observed in mice exposed to HFD. HFD exposure was associated with increased Leptin DNA methylation in GAT (PHFD = 2.69·10−25) over time (Pdynamic = 8.46·10−3). Analysis of the individual CpG sites (Supplementary Fig. S1) revealed a maximum increase in DNA methylation of 20% compared to control mice fed a chow diet at the CpG site 373 base pairs downstream of the transcription start site after 24 weeks of HFD exposure (CpG-373, P = 7.76·10−10, Fig. 2a). Hence, whereas the weight of the GAT reached its maximum at 12 weeks, Leptin DNA methylation did at week 24. In SAT, no difference in DNA methylation of the Leptin promoter in response to HFD was found (P = 0.82, Fig. 2c and Supplementary Fig. S2).
Figure 1. Average DNA methylation of the promoter region of Leptin and Pparg2 before HFD exposure.

(a,b) Schematic overview of the measured Leptin (a) and Pparg2 (b) location using reference genome (GRCm38/mm10) and amplicon size; CpGs displayed in red could not be analysed (c,d) Average DNA methylation of the Leptin and Pparg2 promoter (y-axis) in each depot for each CpG site relative to the transcription start site (x-axis). Columns represent the mean ± SEM.
Figure 2. Average DNA methylation after HFD exposure over time.

Leptin (a,c) and Pparg2 (b,d) promoter DNA methylation pattern over time observed in GAT (a,b) and SAT (c,d). Line represent the mean ± SEM.
DNA methylation of the Pparg2 promoter region also increased after HFD exposure (P = 8.91·104). In-depth analyses of individual CpG sites in GAT revealed a hypermethylated state of 2 out of 3 measured CpG sites (Supplementary Fig. S3), where CpG + 96 showed the greatest increase in DNA methylation after 12 weeks of exposure compared to the control group (10%, P = 6.56·10−4, Fig. 2b). Again, no differential DNA methylation was observed in SAT (P = 0.46; Fig. 2d, Supplementary Fig. S4). A formal statistical interaction test confirmed the depot-specific DNA methylation due to HFD exposure (Leptin: P = 3.65·10−68, Pparg2: P = 7.04·10−4).
DNA methylation changes are not driven by the infiltration of immune cells
The increasing storage of lipids in adipose tissue is associated with an increased number of crown-like structures, which are thought to constitute of dead adipocytes that are surrounded by macrophages21. This change in cell type composition in the adipose tissue may confound the relationship between HFD and DNA methylation, as the macrophages may have differential DNA methylation compared to adipose tissue. To exclude this confounding effect, we quantified the number of CLS in GAT for a subset of the mice (n = 24). A steep increase in CLS formation was observed after 12 weeks of HFD exposure, with a maximum of 39 CLS per 1000 cells (3.9%) after 24 weeks of exposure to the diet. Next, we compared the number of CLS to the average DNA methylation over time. It was noted that the association between CLS formation and DNA methylation varied profoundly for distinct methylation states in both the Pparg2 and Leptin promoter region (Fig. 3a,b). Subsequently, the effect of CLS formation on DNA methylation was investigated by correcting for CLS formation in a linear mixed model (see statistical analysis). This analysis revealed that CLS formation did not have a significant effect on the DNA methylation (Leptin; P = 0.2, Pparg2; P = 0.4). Moreover, the effect of HFD exposure on DNA methylation remained significant after correction for CLS in both genes (Leptin; P = 0.004, Pparg2; P = 0.02). Together, these results indicate that CLS formation did not drive the changes in DNA methylation associated with exposure to a HFD.
Figure 3. CLS formation over time and relationship with average DNA methylation.

(a,b) Average DNA methylation correlated with CLS formation per 1000 cells. Individual colours depict different time points.
Correlation in Leptin and Pparg2 promoter DNA methylation and gene expression
To study the functional consequences of the observed DNA methylation differences of Pparg2 and Leptin, we compared the observed DNA methylation to gene expression of Pparg2 and Leptin in GAT. Leptin promoter DNA methylation was positively correlated with gene expression of Leptin (Fig. 4a). The correlation with gene expression varied per individual CpG site, with the strongest correlation observed at CpG-51 (r = 0.47, P = 2.0·10−10, Fig. 4b). The positive correlation obtained from microarray data was confirmed by real-time RT-qPCR for all CpG sites (Supplementary Fig. S5). Furthermore, plasma leptin levels increased over time indicating that the observed effects on DNA methylation and Leptin gene expression resulted in an effect on protein level (Fig. 5a).
Figure 4. Comparison between Leptin and Pparγ2 gene expression patterns and DNA methylation.

(a,c) Pearson correlation coefficient between the log expression fold change and individual CpG DNA methylation in GAT and SAT. Columns represent the mean ± SEM (b,d) Correlation analysis of DNA methylation of an individual CpG site of Leptin and Pparg2 against the log fold change gene expression.
Figure 5. Functional changes for Leptin and Pparg2.

(a) Leptin plasma levels over time; columns represent the mean ± SEM (b) Gene expression changes of Pparg2 target genes over time.
Analysis of Pparg2 promoter revealed a negative correlation between DNA methylation and gene expression (Fig. 4c). Again, it was observed that the correlation varied for individual CpG sites, with the strongest correlation observed in CpG + 96 (r = −0.53 P = 1.1·10−6, Fig. 4d). Although the lack of a Pparg2-specific assay precluded direct validation of this result using qPCR, the expression of Pparg2 target-genes were significantly reduced at 12 and 24 weeks of HFD feeding in GAT in line with a lower Pparg2 expression (Fig. 5b).
Discussion
We studied the longitudinal effects of long term HFD exposure on DNA methylation of the Leptin and Pparg2 promoters in two fat depots, GAT and SAT, in adult mice during the development of obesity. We found consistently accumulating DNA methylation changes in the promoter regions of the Leptin and Pparg2 genes in GAT over a period of prolonged HFD exposure, whereas DNA methylation remained unaffected in WAT.
In GAT, an increase of 6% was found in Leptin promoter DNA methylation starting after 6 weeks of HFD exposure, which reached a maximum increase of 20% after 24 weeks of HFD exposure. Hypermethylation in the Leptin promoter after long-term HFD exposure observed is consistent with previous studies in rats6 and mice30,31 after long-term high caloric diet treatment (about 15% increase in DNA methylation). However, discrepancies in the dynamics were observed as some mice studies reported an initial decrease in Leptin promoter DNA methylation, after which the DNA methylation increased gradually during high caloric diet exposure28,29. Different dynamics may be due to the different ages at the start of high caloric diet treatment (e.g. 4–5 weeks of age31 versus 12 weeks in this study) and differences in energy density and composition of the diets used (e.g. cafeteria diet in17, a high fat diet with a supra-physiological fat content of 60% kcal31 versus a more physiological diet we used with a fat content of 45% kcal as is comparable to human diets in Finland and Crete30).
At baseline, DNA methylation was similar between GAT and SAT. After HFD exposure, however, an increase in promoter methylation for both the Pparg2 and the Leptin promoter was found in GAT, but not SAT. The difference in DNA methylation between controls and HFD exposed animals increased over time, but remained relatively stable from 12 weeks onwards. Our results show that HFD induces increased Pparg2 and the Leptin promoter DNA methylation in a depot specific manner. DNA methylation changes were observed in metabolically active GAT but not in SAT.
The HFD-induced changes in DNA methylation specifically in GAT indicate differential regulation of the Leptin and Pparg2 genes across depots. The underlying mechanism and physiological consequences of this phenomenon remain unclear, but may be related to reprogramming of the adipokine secretion profile in a depot specific manner. Adipokines are known to reduce the adverse metabolic effects of saturated adipose tissue25.
Both Leptin and Pparg2 genes are known to play a role in negative feedback control upon the reduction of body fat. Adipocytes have the ability to regulate Leptin expression, which in turn can suppress food intake and permit energy expenditure31. Yet, with increasing obesity, tissues can become insensitive to Leptin which is thought to contribute to the progression of obesity32. It is thought that the increase in promoter DNA methylation may counteract the steep increase in Leptin gene expression28. Although it is known that DNA methylation and gene expression can also be positively correlated33, an alternative explanation is that Leptin promoter DNA methylation is not sufficient to decrease the Leptin expression to normal levels30. Analysis of plasma leptin showed that the increased DNA methylation and associated gene expression translate in an parallel increase in circulating leptin, indicating an effect on protein level. Hypermethylation of the Pparg2 promoter, which has been found to be negatively associated with Pparg2 expression7, is hypothesized to reflect an adaptation to further prevent hyperplasia and hypertrophy in already saturated (maximally expanded) adipose tissue by decreasing Pparg2 expression7,32. A decrease in Pparg2 expression results in the decrease in adipose differentiation, insulin responsiveness, lipid uptake and storage through the induction of target genes16. However, it is important to note that the observed changes in DNA methylation are not sufficient to make claims regarding protein functionality or biological effects. An analysis of the changes in expression of Pparg2 target genes indicate that the transcriptional activity of Pparg2 was reduced upon HFD. Future (in vitro) studies should be performed that focus on the more acute effects of dietary constituents on DNA methylation and how these effects translate into changed expression of target genes and the functional effects (e.g. effect on protein translation or indirect measures such as insulin sensitivity or adipose differentiation).
The inter-depot differences in DNA methylation we report, indicate differential regulation of the Pparg2 and Leptin gene expression in GAT and SAT. This corresponds with previous literature showing that primarily GAT is responsible for the development of metabolic imbalance23,25,34. The depot specific response is thought to arise due to differences in specific adipocyte characteristics8. Differences in SAT and GAT in the adult stages have been extensively reported in literature, showing that visceral fat contains a greater percentage of larger adipocytes, is metabolically more active and contains more metabolism-related receptors35,36. Furthermore, it has been observed that adipogenic progenitors are more abundant in SAT than WAT leading to increased proliferation in SAT but not WAT in response to a high fat diet37. The latter could explain the higher sensitivity, early saturation, metabolic dysfunction and DNA methylation changes found in GAT and not SAT after HFD exposure. The presence of inter-depot differences is supported by recent lineage tracing studies showing that both WAT depots originate from different lineages and therefore develop into functionally specific depots24,38,39,40. Furthermore, both depots are thought to develop during two different developmental stages. GAT develops postnatally whereas SAT develops prenatally during embryonic day 14 and 1824.
It is well-known that saturated GAT accumulates CLS20,25, a phenomenon also observed in our study. Because adipose tissue and macrophages are known to have a differential DNA methylation profiles, the infiltration of macrophages could have contributed to the observed differential DNA methylation in GAT41,42. This potential confounding effect has been ignored in previous studies. Importantly, we could exclude that CLS formation drove our findings, implicating that the differential DNA methylation reflects changes in regulation of the Leptin and Pparg2 genes in adipocytes.
In conclusion, we show that prolonged HFD exposure during adulthood dynamically changes DNA methylation and expression of Leptin and Pparg2 genes in GAT. Our study demonstrates that DNA methylation changes induced by prolonged HFD exposure are fat depot specific and may primarily occur in metabolically active depots that become saturated such as GAT, while DNA methylation in SAT remains unaffected. The depot-specific changes in epigenetic regulation may be a starting-point in unravelling the putatively adverse adipokine profiles produced by saturated fat depots that contribute to obesity-induced metabolic imbalance.
Materials and Methods
Animals and experimental model
Tissues and plasma were obtained from a subset of mice of a large time-resolved cohort study in C57BL/6J mice26. Briefly, eighty 12-week old C57BL/6J mice (Charles River Laboratories, France) were treated with a high fat diet (HFD) with a high content of saturated fat or a control chow diet as previously reported26. All animals were housed in a temperature- and humidity-controlled room with ad libitum access to food. The animals were sacrificed by CO2 asphyxiation after 0, 6, 12 and 24 (n = 12) weeks of exposure to the diet. After termination, GAT and SAT tissue were collected and partly fixed in formalin for histological analysis as reported26 or snap-frozen in liquid nitrogen and stored at −80 °C until further use, e.g. for DNA methylation and mRNA expression analyses performed herein. The histological characterization of the tissues is reported in ref. 26. All animal experiments were performed in compliance with the European Union Council Directive 2010/63/EU and approved by an independent Animal Care and Use Committee of the Netherlands Organization of Applied Scientific Research (Zeist, The Netherlands).
DNA methylation analysis
Regions in the promoters of the Leptin and Pparg2 genes to target with region-specific DNA methylation assay were taken from previous experimental studies on dietary exposures in rodents7,43 (for more detailed information regarding promotor location see Supplementary Methods Leptin, Pparg2). Locus-specific DNA methylation was measured using the mass-spectrometry method Epityper (Agena Biosciences®, Germany). Primers were designed, assays run and data processed as described previously44. In brief, all primers were designed with a T7-promoter tag using® EpiDesigner BETA software (http://www.epidesigner.com/) based on the sequences obtained from UCSC genome browser mm10 (http://genome.ucsc.edu/). In silico mass spectrometry using R package RSeqMeth was performed to determine the CpG coverage and base mass of the expected PCR products. Details of the primers are shown in Supplementary Methods PCR. DNA was isolated using a phenol/chloroform extraction protocol. Sodium bisulfite conversion was performed on 500 ng genomic DNA using the EZ-96 DNA methylation kit (Zymo Research®, USA). To account for possible batch effects the plate was designed with a similar distribution in HFD and chow exposed samples. The bisulfite treated samples were amplified by performing touchdown PCRs using selected PCR primers containing a T7-promotor tag and a 10-mer tag on the reverse and forward primer, respectively (specific PCR conditions are summarized in Supplementary Methods PCR). In vitro transcription and T-cleavage reaction were performed using the hMC assay.
DNA methylation ratios were determined by the ratio of C and G spectral peaks using the mass spectrometry–based method Epityper MassARRAY compact MALDITOF (Agena Biosciences®, Germany) and analysed by EpiTYPER software 1.2. All measurements were performed in triplicate and individual CpG measurement with ≤2 success rate or with a standard deviation ≥0.1 were discarded. For statistical analysis the average of the triplicate measurements was used.
Microarray and RT-PCR analysis
Total RNA was extracted from adipose tissues using glass beads and RNAbee (Tel-Test Inc, Friendswood, USA). RNA integrity was examined using the RNA 6000 Nano Lab-on-a-Chip kit and a Bioanalyzer 2100 (Agilent Technologies®, Amstelveen, The Netherlands). The Illumina® TotalPrep™ RNA Amplification Kit (Ambion, art.No.AM-IL1791) was used to synthesize biotin labeled cRNA starting with 500 ng total RNA. The biotinylated cRNA was then hybridized onto the MouseRef-8 Expression BeadChip was 750 ng. Lastly, the default setting of Illumina’s Genomestudio v1.1.1 was used for Gene Expression analysis. All the quality control data of this BeadChip were within specifications of the microarray service provider (Service XS, Leiden, the Netherlands). The microarray gene expression data were validated using quantitative real-time PCR for Leptin using established protocols and primer/probe sets45. Ingenuity Pathway Analysis (IPA) was used to analyse microarray data and Pparg2 target genes. The upstream regulator analysis tool of IPA was used to determine the transcriptional activity of Pparg2 transcription factor essentially as reported45. A negative Z-score <−2 indicated a reduced transcriptional activity based on the direction of gene expression changes of target genes.
Biochemical analyses
Plasma leptin levels were determined by a Quantikine ELISA for murine leptin (R&D Systems®, UK).
Statistical analyses
First, the effect of HFD exposure on the developed fat mass and bodyweight was analysed using a one-way analysis of variance that accounted for the effect of individual differences in time, HFD exposure and the interaction between time and exposure.
Secondly, exposure and/or time specific differences in DNA methylation after HFD exposure in the whole measured region were analysed. To analyse this effect, linear mixed models were used. The analyses accounted for individual differences between CpG sites (CpG ID ∗ b1), age (Time ∗ b3) and individual variation of the mice (Mouse ID ∗ b2). To test for time specificity, the interaction between time and exposure was added to the model (Equation 1):
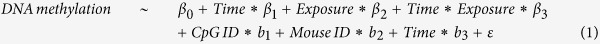 |
Furthermore, the effect of HFD between tissues was evaluated using a linear mixed model similar to the abovementioned model, however depot DNA methylation was added as a fixed effect and the interaction between Exposure * Depot DNA methylation was investigated instead of the interaction between Exposure*Time.
To calculate the effect in individual CpG sites over time, pairwise t-tests were applied to determine the effects of HFD exposure for every time point. P-values were corrected for multiple comparisons following the Bonferroni procedure and considered significant at Padj ≤ 0.05.
The effect of HFD exposure on expression levels of Pparg2 and Leptin was investigated using a linear mixed model corrected for age (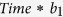 ) (Equation 2):
) (Equation 2):
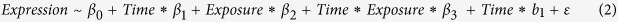 |
The relationship between gene expression and DNA methylation was calculated based on the Pearson correlation coefficients between gene expression and DNA methylation per CpG and corrected for multiple testing using the Bonferroni procedure.
Finally, to test for the confounding effect of CLS formation on DNA methylation a linear mixed model was applied on the data. The variable CLS (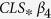 ) was added to the model as fixed effect and the interaction between time and exposure was removed (Equation 3):
) was added to the model as fixed effect and the interaction between time and exposure was removed (Equation 3):
 |
All analyses were performed using R statistics version 3.1.2 (http://www.r-project.org/) with the packages: ggplot2 and lme4.
Additional Information
How to cite this article: Zwamborn, R. A. J. et al. Prolonged high-fat diet induces gradual and fat depot-specific DNA methylation changes in adult mice. Sci. Rep. 7, 43261; doi: 10.1038/srep43261 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
This work was funded by TNO research programs ‘Predictive Health Technologies’ and ‘Enabling Technology Systems Biology’ and financially supported by the European Union’s Seventh Framework Program (FP7/2007–2011) under grant agreement No. 259679 (IDEAL).
Footnotes
The authors declare no competing financial interests.
Author Contributions R.A.J.Z., R.C.S., I.Z., P.C.A.M., H.E.D.S., S.D., R.K., T.K., B.T.H. conceived and performed the experiments. R.A.J.Z., R.C.S., B.T.H. wrote the draft manuscript. P.C.A.M., R.K., T.K., L.V., K.H.T., P.E.S. critically revised the manuscript. All authors read and approved the final manuscript.
References
- Trayhurn P. & Wood I. S. Signalling role of adipose tissue: adipokines and inflammation in obesity. Biochemical Society Transactions 33, 1078–1081 (2005). [DOI] [PubMed] [Google Scholar]
- Ding Y. et al. DNA hypomethylation of inflammation-associated genes in adipose tissue of female mice after multigenerational high fat diet feeding. International Journal of Obesity 38, 198–204 (2014). [DOI] [PubMed] [Google Scholar]
- Slater-Jefferies J. L. et al. Feeding a protein-restricted diet during pregnancy induces altered epigenetic regulation of peroxisomal proliferator-activated receptor-gamma in the heart of the offspring. Journal of developmental origins of health and disease 2, 250–255 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attig L. et al. Dietary alleviation of maternal obesity and diabetes: increased resistance to diet-induced obesity transcriptional and epigenetic signatures. PLoS One 8, e66816 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterland R. A. et al. Maternal methyl supplements increase offspring DNA methylation at Axin Fused. Genesis 44, 401–406 (2006). [DOI] [PubMed] [Google Scholar]
- Milagro F. I. et al. High fat diet-induced obesity modifies the methylation pattern of leptin promoter in rats. Journal of physiology and biochemistry 65, 1–9 (2009). [DOI] [PubMed] [Google Scholar]
- Fujiki K., Kano F., Shiota K. & Murata M. Expression of the peroxisome proliferator activated receptor gamma gene is repressed by DNA methylation in visceral adipose tissue of mouse models of diabetes. BMC biology 7, 38 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinnick K. E. & Karpe F. DNA methylation of genes in adipose tissue. Proceedings of the Nutrition Society 70, 57–63 (2011). [DOI] [PubMed] [Google Scholar]
- Jung U. J. & Choi M. S. Obesity and its metabolic complications: the role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int J Mol Sci 15, 6184–6223 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero P. et al. Leptin and TNF-alpha promoter methylation levels measured by MSP could predict the response to a low-calorie diet. Journal of physiology and biochemistry 67, 463–470 (2011). [DOI] [PubMed] [Google Scholar]
- Yadav A., Kataria M. A., Saini V. & Yadav A. Role of leptin and adiponectin in insulin resistance. Clin Chim Acta 417, 80–84 (2013). [DOI] [PubMed] [Google Scholar]
- Senn J. J. et al. Suppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytes. J Biol Chem 278, 13740–13746 (2003). [DOI] [PubMed] [Google Scholar]
- Bjorbaek C., El-Haschimi K., Frantz J. D. & Flier J. S. The role of SOCS-3 in leptin signaling and leptin resistance. J Biol Chem 274, 30059–30065 (1999). [DOI] [PubMed] [Google Scholar]
- Wang M. Y., Lee Y. & Unger R. H. Novel form of lipolysis induced by leptin. J Biol Chem 274, 17541–17544 (1999). [DOI] [PubMed] [Google Scholar]
- Ahmadian M. et al. PPAR [gamma] signaling and metabolism: the good, the bad and the future. Nature medicine 99, 557–566 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tontonoz P. & Spiegelman B. M. Fat and beyond: the diverse biology of PPARgamma. Annu. Rev. Biochem 77, 289–312 (2008). [DOI] [PubMed] [Google Scholar]
- Hollenberg A. N. et al. Functional antagonism between CCAAT/Enhancer binding protein-alpha and peroxisome proliferator-activated receptor-gamma on the leptin promoter. J Biol Chem 272, 5283–5290 (1997). [DOI] [PubMed] [Google Scholar]
- Palou M. et al. Protective effects of leptin during the suckling period against later obesity may be associated with changes in promoter methylation of the hypothalamic pro-opiomelanocortin gene. British journal of nutrition 106, 769–778 (2011). [DOI] [PubMed] [Google Scholar]
- Muhlhausler B. & Smith S. R. Early-life origins of metabolic dysfunction: role of the adipocyte. Trends Endocrinol. Metab 20, 51–57 (2009). [DOI] [PubMed] [Google Scholar]
- Altintas M. M. et al. Mast cells, macrophages, and crown-like structures distinguish subcutaneous from visceral fat in mice. Journal of lipid research 52, 480–488 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murano I. et al. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. Journal of lipid research 49, 1562–1568 (2008). [DOI] [PubMed] [Google Scholar]
- Tchkonia T. et al. Identification of depot-specific human fat cell progenitors through distinct expression profiles and developmental gene patterns. American journal of physiology. Endocrinology and metabolism 292, E298–307 (2007). [DOI] [PubMed] [Google Scholar]
- Wajchenberg B. L., Giannella-Neto D., Da Silva M. E. & Santos R. F. Depot-specific hormonal characteristics of subcutaneous and visceral adipose tissue and their relation to the metabolic syndrome. Hormone and metabolic research 34, 616–621 (2001). [DOI] [PubMed] [Google Scholar]
- Wang Q. A., Tao C., Gupta R. K. & Scherer P. E. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nature medicine 19, 1338–1344 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Beek L. et al. The limited storage capacity of gonadal adipose tissue directs the development of metabolic disorders in male C57Bl/6J mice. Diabetologia 58, 1601–1609 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder P. et al. Surgical removal of inflamed epididymal white adipose tissue attenuates the development of non-alcoholic steatohepatitis in obesity. Int J Obes 40, 675–684 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster M. T. et al. Subcutaneous adipose tissue transplantation in diet-induced obese mice attenuates metabolic dysregulation while removal exacerbates it. Physiological reports 1, e00015 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W. et al. Epigenetic modification of the leptin promoter in diet-induced obese mice and the effects of n-3 polyunsaturated fatty acids. Scientific reports 4 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia L. et al. Time-specific changes in DNA methyltransferases associated with the leptin promoter during the development of obesity. Nutr Hosp 30, 1248–1255 (2014). [DOI] [PubMed] [Google Scholar]
- Hu F. B., Manson J. E. & Willett W. C. Types of dietary fat and risk of coronary heart disease: a critical review. J Am Coll Nutr 20, 5–19 (2001). [DOI] [PubMed] [Google Scholar]
- Myers M. G., Cowley M. A. & Munzberg H. Mechanisms of leptin action and leptin resistance. Annu. Rev. Physiol 70, 537–556 (2008). [DOI] [PubMed] [Google Scholar]
- Guilherme A., Virbasius J. V., Puri V. & Czech M. P. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell Biol 9, 367–377 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonder M. J. et al. Disease variants alter transcription factor levels and methylation of their binding sites. Nature Genetics 49, 131–138 (2015). [DOI] [PubMed] [Google Scholar]
- Bjørndal B., Burri L., Staalesen V., Skorve J. & Berge R. K. Different adipose depots: their role in the development of metabolic syndrome and mitochondrial response to hypolipidemic agents. Journal of obesity 2011 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinou K. et al. Structural and functional properties of deep abdominal subcutaneous adipose tissue explain its association with insulin resistance and cardiovascular risk in men. Diabetes Care 37, 821–829 (2014). [DOI] [PubMed] [Google Scholar]
- Smith S. R. & Zachwieja J. J. Visceral adipose tissue: a critical review of intervention strategies. Int. J. Obes. Relat Metab Disord 23, 329–335 (1999). [DOI] [PubMed] [Google Scholar]
- Joe A. W., Yi L., Even Y., Vogl A. W. & Rossi F. M. Depot-specific differences in adipogenic progenitor abundance and proliferative response to high-fat diet. Stem Cells 27, 2563–2570 (2009). [DOI] [PubMed] [Google Scholar]
- Berry D. C., Stenesen D., Zeve D. & Graff J. M. The developmental origins of adipose tissue. Development 140, 3939–3949 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry R., Rodeheffer M. S., Rosen C. J. & Horowitz M. C. Adipose Tissue-Residing Progenitors (Adipocyte Lineage Progenitors and Adipose-Derived Stem Cells (ADSC)). Current Molecular Biology Reports 1, 101–109 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau Y. Y. et al. Visceral and subcutaneous fat have different origins and evidence supports a mesothelial source. Nature cell biology 16, 367–375 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefterova M. I. et al. Cell-specific determinants of peroxisome proliferator-activated receptor gamma function in adipocytes and macrophages. Mol Cell Biol 30, 2078–2089 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugii S. & Evans R. M. Epigenetic codes of PPARgamma in metabolic disease. FEBS Lett 585, 2121–2128 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan C., Liu X., Shen W., Deckelbaum R. J. & Qi K. The regulation of leptin, leptin receptor and pro-opiomelanocortin expression by N-3 PUFAs in diet-induced obese mice is not related to the methylation of their promoters. Nutr Metab (Lond) 8, 31–31 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchiman H. E. et al. Design, measurement and processing of region-specific DNA methylation assays: the mass spectrometry-based method EpiTYPER. Frontiers in genetics 6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang W. et al. Coordinated and interactive expression of genes of lipid metabolism and inflammation in adipose tissue and liver during metabolic overload. PLoS One 8, e75290 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uriarte G., Paternain L., Milagro F. I., Martinez J. A. & Campion J. Shifting to a control diet after a high-fat, high-sucrose diet intake induces epigenetic changes in retroperitoneal adipocytes of Wistar rats. J Physiol Biochem 69, 601–611 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.