

Abstract
An E. coli expression system offers a mean for rapid, high yield and economical production of Hepatitis B Virus core (HBc) particles. However, high-level production of HBc particles in bacteria is demanding and optimisation of HBc particle yield from E. coli is required to improve laboratory-scale productivity for further drug delivery applications. Production steps involve bacterial culture, protein isolation, denaturation, purification and finally protein assembly. In this study, we describe a modified E. coli based method for purifying HBc particles and compare the results with those obtained using a conventional purification method. HBc particle morphology was confirmed by Atomic Force Microscopy (AFM). Protein specificity and secondary structure were confirmed by Western Blot and Circular Dichroism (CD), respectively. The modified method produced ~3-fold higher yield and greater purity of wild type HBc particles than the conventional method. Our results demonstrated that the modified method produce a better yield and purity of HBc particles in an E. coli-expression system, which are fully characterised and suitable to be used for drug delivery applications.
The past quarter century has brought biomedical and health sciences researchers to focus on nanotechnologies. Cross-disciplinary research bringing together engineering, biology, physics and chemistry resulted in the production of a vast and diverse number of nanovectors, which have been explored in drug delivery and imaging1. In nanotechnology, 1–100 nm diameter particles are referred to as nanoparticles. Cancer-related nano-technology studies have shown substantial development in producing nanoparticles with different sizes, chemical nature and structure, all of which influence their applications including administration for targeted therapeutic and imaging moieties2,3. Nanoparticles can be developed from different materials: organic, inorganic or biological (macro) molecules. The latter, referred to as bio-nanoparticles can be formed by assembly of protein subunits4. One popular example is ferritin, a naturally occurring human protein, which is able to self-assemble into spherical capsids or cages5. However, the most promising protein cages are considered to be virus-derived nanoparticles or virus-like particles (VLPs). VLPs are self-assembling, non-replicative and non-pathogenic particles, which structurally mimic the viral capsid lacking the viral genome6. They can either be isolated directly after the coat protein expression in eukaryotic cells, or assembled in vitro from coat protein subunits purified from any recombinant host; ranging from mammalian cells to bacteria7,8.
As the first VLP candidate and the first icosahedral VLP carriers, HBc particles remain the most flexible and promising model for knowledge-based display of foreign peptide sequences9,10. HBc particles are hollow nanoparticles ranging from 30 to 34 nm in diameter with 7 nm thick envelopes11. These core particles are icosahedral nucleocapsids with primarily triangulation number T = 3 or T = 4 symmetry, each containing 180–240 units of 21 kDa core monomers12. Their self-assembly property and ability to assemble and dis-assemble freely through altering the urea concentration, modifying salt concentration and reducing disulfide bonds13,14,15, have been utilised to allow for encapsulation of various drugs16 and other biomolecules including nuclease17, oligonucleotides18 and siRNA19.
HBc particles can be expressed in homologous and heterologous expression systems. Escherichia coli offers a means for rapid, high yield and economical production of recombinant proteins20,21. However, high-level production of recombinant bio-nanoparticles in bacteria on a laboratory-scale is challenging. Challenges include: (1) biomass production of specific-gene selection cells22, (2) degradation of protein by host cell proteases23, (3) protein isolation from inclusion bodies24 and (4) successful protein folding25. Therefore, a yield optimisation study of HBc particles expression in E. coli is required to improve the productivity of the laboratory-scale for further drug delivery applications.
This study focused on developing an improved method (I) for producing wild type HBc (WT-HBc) particles in high yields, suitable for drug delivery applications. For comparison, WT-HBc particles were also prepared by the “conventional” method (C) reported previously by others26, Production steps involve bacterial culture, protein isolation, denaturation, purification and finally VLP assembly. The protein yield and purity were determined by Nanodrop™. HBc particle morphology was confirmed by Atomic Force Microscopy (AFM). Protein specificity and secondary structure were confirmed by Western Blot and Circular Dichroism (CD), respectively. We concluded that the modified method produced ~3 fold higher yield and greater purity of HBc particles than the conventional method.
Results
Optimisation of the WT-HBc particles expression and assembly method
In this study, we aimed at developing an improved method (I) for producing wild type HBc (WT-HBc) particles in high yields, suitable for drug delivery applications. For comparison, WT-HBc particles were also prepared by the “conventional” method (C) reported previously by others26. The four steps involved including bacterial culture, protein isolation, denaturing and assembly are summarised in Fig. 1. The major differences between the two methods and the rationale for these modifications will be highlighted here.
Figure 1. A scheme summarising WT-HBc particles production in E. coli expression system.

WT-HBc particles were prepared using one of the two methods; the “conventional” or “improved” method.
E. coli was chosen as the host organism to produce HBc particles on a laboratory scale, due to the advantages of relatively simple fermentation design and high final cell densities20. Auto-Induction Media Terrific Broth (AIM-TB) was used in the new method to replace Luria-Bertani Broth (LB) used previously. AIM-TB supports E. coli growth to a cell density 10-fold or higher in a shake flask26,27.
A protease inhibitor and RNase were included in the lysis buffer of the new method (Fig. 1), specifically to protect HBc proteins from cleavage by endogenous proteases and to remove contaminating RNA28,29.
Urea is used as a denaturing agent to solubilise the inclusion bodies containing WT-HBc particles. In the “conventional” method, a moderate urea concentration (4 M) in dissociation buffer was used compared to 8 M used in the new method. The higher urea concentration was expected to denature HBc particles to monomers more efficiently13. The His-tag, located at the C-terminus of HBc protein, resides inside the fully assembled particles30,31, so efficient denaturing of the HBc particles makes the His-tag more available for His-tag affinity chromatography, used in a later step of purification.
Purified WT-HBc protein, in the monomer form, was then re-folded via the assembly step to restore the core particles structure. In the “conventional” method, after affinity chromatography urea was removed by dialysis against Tris/EDTA/NaCl assembly buffer, where pronounced precipitation was associated with the sudden drop in urea concentration. In the new method; the purified WT-HBc monomers were re-folded by a stepwise reduction in urea concentration first to 2 M during column elution then to 0.5 M in dialysis against assembly buffer (DTT/EDTA/Tris/NaCl/CaCl2) then dialysis against buffer containing no urea. This altered assembly strategy was designed to improve the efficiency of protein re-assembly by reducing random protein folding, induced by disulfide bonds formation32,33, thereby increasing the yield of appropriately assembled WT-HBc particles.
Protein yield, particle morphology and size
Significantly higher protein yields (p < 0.001) from 1 l growth medium, were obtained for WT-HBc prepared using the new method (3.21 ± 0.21 mg) compared with the conventional method (1.21 ± 0.30 mg) (Table 1). Height AFM images of HBc prepared by both methods revealed relatively monodisperse and spherical structures, observed only in case of the assembled particles. The “halo” observed suggested the presence of core-shell type structures. HBc mean diameters measured by AFM were 36.61 ± 8.51 nm and 33.77 ± 4.58 nm (p = 0.45 (>0.05)), respectively (Fig. 2). The OD260/OD280 values, used to determine the purity of the protein from nucleic acids contamination, are shown in Table 1. WT-HBc (I) showed a significantly lower value of OD260/OD280 ratio (0.58 ± 0.01) than WT-HBc (C) (1.02 ± 0.04) suggesting the higher nucleic acid contamination in the latter. A OD260/OD280 ratio greater than 0.6 indicates nucleic acid contamination in the protein samples34. In addition, as shown in Fig. 3 (cropped gels) and Supplementary Information Figure S1 (full length gels), some unknown protein bands were observed on SDS-PAGE at molecular weight lower than 20 kDa, in case of WT-HBc (C) (Fig. 3A, Supplementary Figure S1a), but not for WT-HBc (I) (Fig. 3B, Supplementary Figure S1b). This result confirmed the impurities observed in the WT-HBc (C) samples. Overall, these results indicated that the new method provides HBc particles of an improved yield and purity.
Table 1. Yield and nucleic acid contamination characterisation of HBc particles prepared using “Conventional” or “Improved” methods.
| Method | HBc code | Protein yield (mg)[1],[2] | OD260:OD280[1],[2] |
|---|---|---|---|
| Conventional | WT-HBc (C) | 1.21 ± 0.3 | 1.02 ± 0.04 |
| Improved | WT-HBc (I) | 3.21 ± 0.21*** | 0.58 ± 0.01*** |
[1] Values were obtained with Nanodrop from 1 litre bacteria containing media normalised to LB media.
[2] Results are expressed an average ± SD (n = 3). ***p = 0.00024 (<0.001) (One-way ANOVA).
Figure 2. Morphological analysis of purified HBc core particles with Atomic Force Microscopy (AFM).

(A) AFM images using tapping mode AFM (TM-AFM) and (B) Histogram analysis of assembled HBc particles. HBc particles were deposited on the mica substrates and measurements were carried out in air at 25 °C, using a Bruker Dimension ICON with Scan Assist. Dis-assembled HBc were achieved by dilution in distilled water at 40 °C for 10 min. Core shell structure was observed for the assembled particles for both formulations. Histograms were obtained for n = 50, analysed using WSxM v5.0 Developed 6.2 and Origin 7.5 software. Scan size is 300 nm × 300 nm. Scale bars are 60 nm.
Figure 3. Elution profile of WT-HBc monomers from Ni2+-chelate affinity chromatography column (cropped gels).
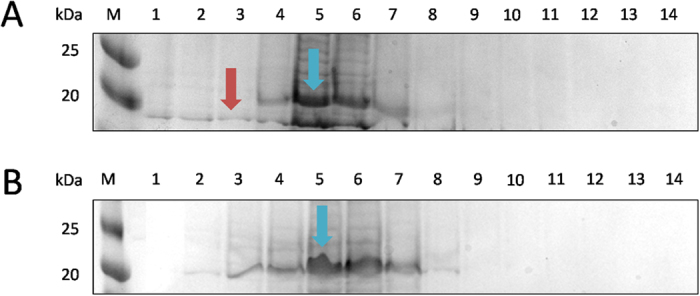
A column with 6 ml of cOmplete™ His-Tag Purification Resin was equilibrated with 3-times bed-volume (18 ml) of the dissociation buffer. The column was loaded with the protein probe and washed with 18 ml dissociation buffer. Bound proteins were eluted with 14 ml of elution buffer and collected in 1 ml fractions. The aliquots of each fractions were subjected to SDS-PAGE gel electrophoresis and stained with Coomassie Brilliant Blue. M, marker in kDa; Numbers 1–14 represent aliquots of the respective elution fractions for (A) WT-HBc (C) or (B) WT-HBc (I) monomers. Blue arrows represent WT-HBc monomers (21 kDa). Higher number of fractions (7 fractions, lane 2–8) were collected in case of WT-HBc (I) compared to WT-HBc (C) (5 fractions, lane 4–8). Some unknown protein bands were observed at molecular weight lower than 20 kDa (red arrow).
Protein specificity and secondary structure analysis of the purified WT-HBc particles
Western blot analysis was performed to confirm the protein specificity of WT-HBc particles. The presence of HBc antigen and His-tag, as biochemical markers, was confirmed. Specific protein bands, positive for anti-6-His and anti-HBc antibodies, were observed at 21 kDa26 (Fig. 4A (cropped blot), Supplementary Information Figure S2 (full length blots)) confirming the successful expression of WT-HBc particles by both methods.
Figure 4. Protein specificity and secondary structure analysis of HBc particles.
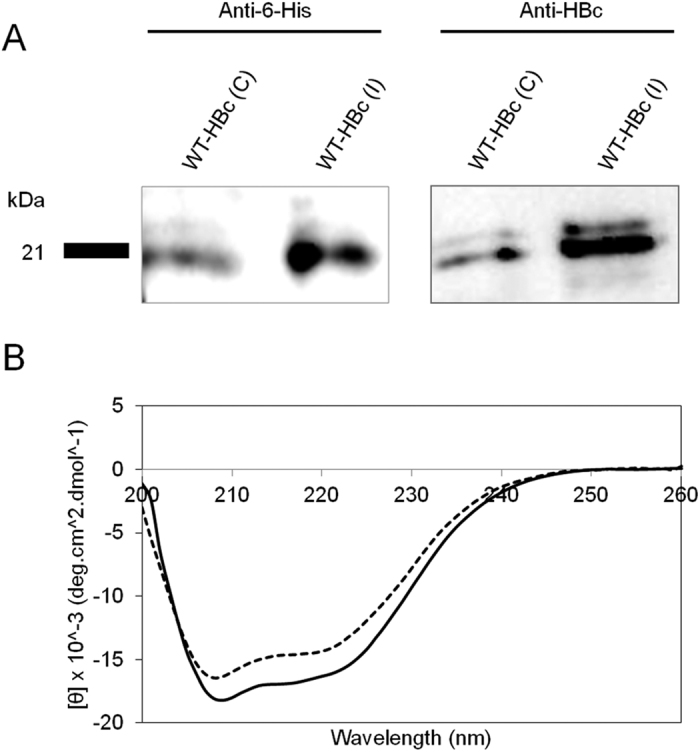
(A) Western blotting (cropped blot) and (B) Circular Dichroism (CD) analysis of WT-HBc particles. Denatured HBc samples were subjected to SDS-PAGE followed by immuno-blotting using anti-6-His and anti-HBc antibodies. Results confirmed the presence of specific protein bands at 21 kDa. CD graph shows the overall conformation of far-UV CD analysis of WT-HBc core particles. WT-HBc (C) (solid line) and WT-HBc (I) (dashed line) exhibited spectra typical of α-helix-containing proteins with minima at 220 and 208 nm. The CD spectra are typical of WT-HBc core monomer secondary structure.
The typical secondary structure of the WT-HBc35 monomer was confirmed by far-UV circular dichroism (CD). WT-HBc particles from both preparations displayed a predominantly α-helix conformation with well-defined characteristic negative bands at 208 and 222 nm35 (Fig. 4B).
Discussion
HBc particles have been expressed extensively in several types of hosts including E. coli36. However, it has been a challenge to produce a high yield of purified HBc particles on a laboratory-scale. In the conventional method of HBc particles production in E. coli, LB media is used as a culture medium37. LB media can support E. coli growth at an OD600 0.7–0.8 under normal shaking incubation conditions38. However due to its mid-exponential growth phase, the pH of the cultures drops to pH 4 to 5 upon saturation, causing the antibiotics such as ampicillin to be degraded, resulting in a partial or no selection of cells. Under these conditions, 80% of the total cell population may not contain the intended plasmid, preventing these cells from being induced to express the target protein, resulting in low protein yields23. One approach to overcome this problem is to use an auto-induction media such as the AIM-TB media39. Auto-induction is based on the function of lac operon regulatory elements using a mixture of glucose, glycerol and lactose under diauxic growth conditions. During the initial growth period, glucose is preferentially used as a carbon source as a first sugar source. As the glucose is depleted, usually in mid to late log phase, the second source of sugar, lactose is taken up and converted by β-galactosidase to the inducer allolactose. Allolactose causes the release of lac repressor from its specific binding sites in the DNA, which induces the expressions of T7 RNA polymerase from the lacUV5 promoter, which thereby unblocks T7lac promoters, allowing expression of the target protein by T7 RNA polymerase. This transition from the un-induced to induced state under metabolic control of the expression host is one of the factors contributed to the higher amount of protein expressed in the bacteria. Furthermore, the increased richness of the AIM-TB media endorse to the higher amount of lysate harvested23.
Inclusions of protease inhibitors and RNase in a lysis buffer might be the important parameters for HBc isolation from the E. coli lysate. Bacterial cells contain many different types of proteases. As soon as cells are disrupted, proteases are released and can quickly degrade any protein, causing a drastic reduction in the protein yield during isolation and purification. Therefore, inclusion of protease inhibitors in the new method prevents their proteolytic activity which can cause cleavage of HBc protein fractions28. HBc particles can be contaminated with the host RNA when expressed in an E. coli expression system40. Addition of RNase in the early stage of purification removes contaminating RNA, hence preventing any remnant non sequitur activity for further studies29.
In this study, we isolated WT-HBc particles from protein inclusion bodies, which are insoluble protein aggregates, that can be easily separated by centrifugation from the rest of the soluble bacterial cytoplasmic proteins41. In order to solubilise the inclusion bodies, the extraction of protein is generally performed in the presence of denaturing agents. Here, a strategy of urea usage has been chosen for its compatibility with protein folding and most chromatographic procedures42,43. In the “conventional” method, a moderate urea concentration (4 M) in dissociation buffer was only able to solubilise the protein inclusion bodies partially with undissolved material observed after overnight incubation. However, in the new method, with 8 M urea in the dissociation buffer, the protein inclusion bodies were completely solubilised and dissociated WT-HBc proteins. A high concentration of urea acts to enhance the aqueous solubility of protein by weakening the hydrophobic interactions and stabilising the solvation of the unfolded protein. This allows a greater number of non-polar side chains to be exposed to water molecules, thus resulting in a completely solubilised protein in solution44,45.
The abrupt removal of urea to re-fold HBc monomers by dialysis against Tris/EDTA/NaCl assembly buffer led to precipitation, most probably due to the random proteins re-folding, which may have resulted in a low yield of the purified assembled WT-HBc particles. Here, a strategy of adding a reducing agent, DTT and a divalent salt, CaCl2 was used in the new method, helping to reduce the protein aggregation problem. DTT, a thiol reagent was used to reduce the disulfide bonds of proteins, which helps to prevent intra-molecular and intermolecular disulfide bonds from forming between cysteine residues of the protein. Ca2+ ion can also promote HBc particles assembly. It is suggested that the ionic strength from the calcium ions increases the electrostatic attractions between the proteins monomers thus act as an electrostatic switch for the protein self-assembly46. So when a highly concentrated WT-HBc monomers solution was dialysed under these conditions, the proteins re-folded at the correct geometry bonding, stabilising the protein structure, resulting in high re-folding yield of HBc particles and fewer aggregation problems16,47,48.
Another study by Zlotnick et al. demonstrated that the usage of assembly effectors could enhance HBc particles assembly. The usage of HAP1 [methyl 4-(2-chloro-4-fluorophenyl)-6-methyl-2-(pyridin-2-yl)- 1,4-dihydropyrimidine-5-carboxylate], a type of heteroaryldihydropyrimidine molecules, together with 150 mM NaCl was found to accelerate HBc particles assembly kinetics, resulting in 65% particles assembly, compared with particles assembled in 150 mM NaCl only (25% assembly)49,50. The addition of assembly effector in the future might be a good approach to improve protein assembly yield of HBc particles.
The organisation of HBc particles is known to be largely α-helical and quite different from previously known viral capsid proteins with β-sheet jellyroll packings51,52. Association of two amphipathic α-helical hairpins results in the formation of a dimer with a four-helix bundle as the major central feature. The four-helix bundles protrude, forming spikes, presents as a central part of major immudominant region of the HBc particles53. Here, both WT-HBc particles exhibited typical spectra of α-helix-containing proteins structure.
Conclusion
The experimental work has focused on the initial development of functional nano-assemblies of virus-like particles for therapeutic applications in addition to the development of “proposed” novel biocompatible and therapeutically efficient nanocarriers for cancer. We demonstrated an improved method to produce a better yield and purity of HBc particles in an E. coli-expression system. The HBc particles produced are fully characterised and are suitable to be used to encapsulate drugs/biomolecules or to be bioengineered with targeting molecules.
Materials
Luria-Bertani-Broth Miller, tryptone, yeast extract powder, MOPS, ampicillin sodium, IPTG dioxan free, AIM-terrific broth base including trace elements, Tris base Ultra-Pure, EDTA disodium and dithiothreitol (DTT) were obtained from ForMedium™ (UK). Urea (carbamide) were obtained from Melford (UK). Imidazole, Glycerol, 2-mercapoethanol, N, N, N’, N’-tetramethylethylenediamine, RNase A (from bovine pancreas) and bromophenol blue were obtained from Sigma Life Science (UK). Complete™ ULTRA Tablets, glass vials Protease Inhibitor Cocktail and cOmplete™ His-Tag Purification Resin were from Roche (Germany). Acetic acid ≥99.0% (T) and skimmed milk powder were obtained from Fluka Analytical (Switzerland). Triton® X-100, sodium carbonate and ammonium persulfate were obtained from Sigma-Aldrich (Germany). HEPES and Brilliant Blue R were obtained from Sigma (UK). Sodium dodecyl sulphate was obtained from BDH Laboratory (UK). 30% Acrylamide/Bis Solution, 37.5:1, Precision Plus Protein™ Dual Xtra Standards, Precision Protein StrepTactin-HRP Conjugate and Clarity Western enhanced chemiluminescence (ECL) Substrates were obtained from Bio-Rad Laboratories (USA). Mouse Anti-Hepatitis B Virus Antibody, core antigen (ayw), clone 10E11 was from Merck Millipore (USA). HCL was obtained from Aeros Organic (Germany). Anti 6-His affinity purified antibody was obtained from Bethyl Laboratories Inc. (USA). SnakeSkin™ Dialysis Tubing, 10 K MWCO was from Thermo Scientific (USA). Escherichia Coli, BL21 (DE3) was obtained from Dr. Mitla Garcia Maya, Randall Division of Cell and Molecular Biophysics, King’s College London, UK. Plasmid pET-22b(+)-WT-HBc-His6 encoding C-terminally His tagged HBc protein (residues 1–183) Accession No. CAA59538 was obtained from Associate Professor Chiaki Ogino, Chemical and Science Engineering Department, Kobe University, Japan.
Methods
Expression, purification and assembly of WT-HBc particles
For gene expression of WT-HBc protein, the plasmid for expression of WT-HBc (pET-22b(+)-WT-HBc-His6); was transfected into E.coli BL21 (DE3) strain. E. coli transfected WT-HBc was expressed, purified and assembled via the following methods:
Conventional method
E. coli transfected WT-HBc (15 μl) was cultured in 10 ml of LB media in the presence of 100 μg/ml ampicillin and grown at 37 °C for 16 h using an incubator shaker. The culture was then diluted with 500 ml of fresh LB media in the presence of 100 μg/ml ampicillin and grown to OD600 0.7–0.8 at 37 °C. The culture was induced by adding isopropyl-β-thiogalactopyranoside (IPTG) to a final concentration of 0.1 mM at 25 °C overnight. Cells were harvested at 4500 g, 4 °C for 15 min. Pelleted cells were re-suspended in the 30 ml of lysis buffer (50 mM Tris, 100 mM NaCl, 5 mM EDTA, 0.2% v/v Triton X-100, pH 8.0). The cells were lysed on ice by three cycles of probe sonication for 1 min each with 1 min intervals to avoid heating the material. The supernatant was removed by centrifugation at 18,500 g, 4 °C for 30 min. The core particles in the cell pellet were washed in 30 ml of lysis buffer and collected by centrifugation at 18,500 g, 4 °C for 30 min. The cell pellet containing WT-HBc particles was denatured in 40 ml of dissociation buffer (4 M urea, 200 mM NaCl, 50 mM sodium carbonate, 10 mM 2-mercaptoethanol, pH 9.5) by overnight incubation at 4 °C. Then, the pellet was discarded by centrifugation at 18,500 g, 4 °C for 30 min.
Soluble fraction containing contaminating proteins was separated from HBc particle proteins using Ni2+-chelate affinity chromatography. A column with 6 ml of cOmplete™ His-Tag Purification Resin was equilibrated with 3-times bed-volume (18 ml) of dissociation buffer. The column was loaded with the protein probe and washed with 18 ml of dissociation buffer. Bound HBc particle proteins were eluted with 14 ml of elution buffer (4 M urea, 200 mM NaCl, 50 mM sodium carbonate, 10 mM 2-mercaptoethanol, 1 M imidazole, pH 9.5). The eluted material was collected in 1 ml fractions. The aliquots of each fraction were subjected to sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and stained with Coomassie Brilliant Blue (CBB) to analyse their purity.
Fractions containing the HBc protein were re-assembled to particles by the removal of the urea in assembly buffer (500 mM NaCl, 50 mM Tris, 0.5 mM EDTA, pH 7.0). Specifically, protein fractions were dialysed against 5 l of dialysis buffer using SnakeSkin™ Dialysis Tubing, 10 K MWCO at 4 °C for overnight. Assembled HBc particles were filtered using 0.44 μm filter to remove any aggregates.
Improved method
E. coli transfected WT-HBc (15 μl) was cultured in 10 ml of AIM-TB media (10 g/l tryptone, 5 g/l yeast extract, 3.3 g/l (NH4)2SO4, 6.8 g/l KH2PO4, 7.1 g/l Na2HPO4, 0.5 g/l glucose, 2.0 g/l α-lactose, 0.15 g/l MgSO4) in the presence of 100 μg/ml ampicillin and grown at 37 °C for 16 h using an incubator shaker. The culture was then diluted with 500 ml of fresh AIM-TB media in the presence of 100 μg/ml ampicillin and grown at 25 °C for 72 h. Cells were harvested at 4500 g, 4 °C for 15 min. Pelleted cells were re-suspended in the 30 ml of lysis buffer (50 mM Tris, 100 mM NaCl, 5 mM EDTA, 0.2% v/v Triton X-100, 1x cOmplete™ protease inhibitor pH 8.0). The cells were treated with RNase A at a final concentration of 5 μg/ml at 4 °C overnight. The lysate was sonicated using a probe sonicator on ice by three cycles for 1 min each with 1 min intervals to avoid heating the material. The supernatant was removed by centrifugation at 18,500 g, 4 °C for 30 min. The core particles in the cell pellet were washed in 30 ml of lysis buffer and collected by centrifugation at 18,500 g, 4 °C for 30 min. The cell pellet containing WT-HBc particles was denatured in 40 ml of dissociation buffer (8 M urea, 200 mM NaCl, 50 mM sodium carbonate, 10 mM 2-mercaptoethanol, pH 9.5) by overnight incubation at 4 °C. Then, the pellet was discarded by centrifugation at 18,500 g, 4 °C for 30 min.
The soluble fraction containing-contaminating proteins were separated from HBc particle proteins using Ni2+-chelate affinity chromatography. A column with 6 ml of cOmplete™ His-Tag Purification Resin was equilibrated with 3-times bed-volume (18 ml) of dissociation buffer. The column was loaded with the protein probe and washed with 18 ml of dissociation buffer. Bound HBc particle proteins were eluted with 14 ml of elution buffer (2 M urea, 200 mM NaCl, 50 mM sodium carbonate, 10 mM 2-mercaptoethanol, 1 M imidazole, pH 9.5). The eluted material was collected in 1 ml fractions. The aliquots of each fraction were subjected to SDS-PAGE and stained with CBB to analyse their purity.
Fractions containing the HBc protein were re-assembled to particles by the removal of urea. Specifically, protein fractions were dialysed against 2 l of dialysis buffer 1 (0.5 M urea, 100 mM Tris, 150 mM NaCl, 2 mM DTT, 1 mM EDTA, 10 mM CaCl2, pH 8.0) using SnakeSkin™ Dialysis Tubing, 10 K MWCO at 4 °C for 4 h, allowing HBc protein to start assembled. Then, the solution was dialysed against dialysis buffer 2 (100 mM Tris, 150 mM NaCl, 1 mM EDTA, 10 mM CaCl2, pH 8.0) to completely removed urea and DTT, hence assembled HBc particles. HBc particles were filtered using 0.44 μm filter to remove any aggregates.
All protein concentrations were measured using NanoDrop™ ND-1000 UV-Vis Spectrophotometer.
Tapping mode Atomic Force Microscopy (TM-AFM) analysis of purified WT-HBc particles
A 10 μg/ml sample of purified WT-HBc particles (100 μl) was deposited on mica surfaces for 5 min and then flushed with air. TM-AFM on the mica substrates were carried out in air at 25 °C using a Bruker Dimension ICON with Scan Assist. The surfaces were imaged with a general purpose-tapping tip made by MikroMasch in Estonia (NSC15/no Al, tip radius <10 nm; tip height = 20–25 μm; cone angle <40°; cantilever thickness = 3.5–4.5 μm; cantilever width = 32–28 μm; cantilever length = 120–130 μm; frequency f0 = 265–400 kHz; force constant k = 20–75 N m−1, VEECO, USA). The statistical analysis of the AFM images was carried out using WSxM v5.0 Developed 6.2 software (Spain).
SDS-PAGE and western blot analysis of WT-HBc particles
The expression of each WT-HBc monomer was confirmed by western blotting. The purified WT-HBc particles were analysed by SDS-PAGE and electro-transferred onto a nitrocellulose membrane. For the detection of the His6-tag, rabbit anti-6-His antibody was used as a primary antibody at 1:1000 dilution followed by HRP-linked anti-rabbit at 1:1000 dilution. For the detection of the HBc, mouse anti-HBc antibody was used as a primary antibody at 1:1000 dilution, followed by HRP-linked anti-mouse at 1:1000 dilution. For staining of the ladder, Precision Protein StrepTactin-HRP Conjugate was used at 1:10,000 dilution. The specific bands were detected with enhanced chemiluminescence (ECL) detection system. The membrane was imaged using the ChemiDocTMMP and analysed with Image Lab 4.1 software.
Circular Dichroism (CD) for protein secondary structure analysis
CD thermal scan measurements were performed on a Chirascan spectrometer (Applied Photophysics, Leatherhead, UK) supplied with a thermoelectric temperature control system. Temperature-dependent conformational changes were measured for WT-HBc particles (0.1 mg/ml) in Tris buffer (5 mM, pH 8.0). CD spectra of the samples were recorded from 260 to 180 nm using a 0.5 mm cuvette at 6 °C before starting the thermal scan. The temperature sensitivity was then tested by increasing the temperature from 6 to 94 °C at 1 °C/min heating rate and 2 °C/step. At the end of the thermal scan, the sample was equilibrated at 94 °C and the CD spectrum was recorded. Once the thermal scan was completed, the samples were cooled to 6 °C and equilibrated for 15 min before recording the CD spectra. Data analysis was performed using Applied Photophysics Chirascan software, and the transition temperatures were determined with Global 3 analysis software for dynamic multimode spectroscopy.
Statistics
For all experiments, data were presented as mean ± SD, where n denotes the number of repeats. Independent variable Student t tests were performed using one-way ANOVA. The t-value, degrees of freedom and two-tailed significance (p-value) were determined. *p < 0.05, **p < 0.01 and ***p < 0.001.
Additional Information
How to cite this article: Bin Mohamed Suffian, I. F. et al. Yield Optimisation of Hepatitis B Virus Core Particles in E. coli Expression System for Drug Delivery Applications. Sci. Rep. 7, 43160; doi: 10.1038/srep43160 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
We would like to acknowledge Mr. William Luckhurst for the assistance in the AFM usage. IFBMS would like to thank Public Service Department, Government of Malaysia for the Excellence Student Programme studentship. We acknowledge funding from Biotechnology and Biological Sciences Research Council (BBSRC; (BB/J008656/1)).
Footnotes
The authors declare no competing financial interests.
Author Contributions Study design: I.F.B.M.S., M.G.M., P.B., T.B., Y.N., C.O., A.K., K.T.A.J. Data analysis and interpretation: I.F.B.M.S., M.G.M., P.B., T.B., K.T.A.J. Drafting manuscript: I.F.B.M.S., P.B., Y.N., A.R.B.M.J.P., K.T.A.J. All the authors critically reviewed the manuscript. All authors read and approved the final manuscript.
References
- Ferrari M. Cancer nanotechnology: opportunities and challenges. Nature reviews. Cancer 5, 161–171, doi: 10.1038/nrc1566 (2005). [DOI] [PubMed] [Google Scholar]
- Hoption Cann S. A., van Netten J. P. & van Netten C. Dr William Coley and tumour regression: a place in history or in the future. Postgraduate medical journal 79, 672–680 (2003). [PMC free article] [PubMed] [Google Scholar]
- Anderson R. J. & Schneider J. Plasmid DNA and viral vector-based vaccines for the treatment of cancer. Vaccine 25 Suppl 2, B24–34, doi: 10.1016/j.vaccine.2007.05.030 (2007). [DOI] [PubMed] [Google Scholar]
- Li L. et al. Evaluation of specific delivery of chimeric phi29 pRNA/siRNA nanoparticles to multiple tumor cells. Molecular bioSystems 5, 1361–1368, doi: 10.1039/b903428e (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita I., Iwahori K. & Kumagai S. Ferritin in the field of nanodevices. Biochimica et biophysica acta 1800, 846–857, doi: 10.1016/j.bbagen.2010.03.005 (2010). [DOI] [PubMed] [Google Scholar]
- Garcea R. L. & Gissmann L. Virus-like particles as vaccines and vessels for the delivery of small molecules. Current opinion in biotechnology 15, 513–517, doi: 10.1016/j.copbio.2004.10.002 (2004). [DOI] [PubMed] [Google Scholar]
- Ramqvist T., Andreasson K. & Dalianis T. Vaccination, immune and gene therapy based on virus-like particles against viral infections and cancer. Expert opinion on biological therapy 7, 997–1007, doi: 10.1517/14712598.7.7.997 (2007). [DOI] [PubMed] [Google Scholar]
- Szecsi J. et al. Induction of neutralising antibodies by virus-like particles harbouring surface proteins from highly pathogenic H5N1 and H7N1 influenza viruses. Virology journal 3, 70, doi: 10.1186/1743-422x-3-70 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke B. E. et al. Improved immunogenicity of a peptide epitope after fusion to hepatitis B core protein. Nature 330, 381–384, doi: 10.1038/330381a0 (1987). [DOI] [PubMed] [Google Scholar]
- Ulrich R. et al. Immunogenicity of recombinant core particles of hepatitis B virus containing epitopes of human immunodeficiency virus 1 core antigen. Archives of Virology 126, 321–328, doi: 10.1007/BF01309705 (1992). [DOI] [PubMed] [Google Scholar]
- Lewellyn E. B. & Loeb D. D. The arginine clusters of the carboxy-terminal domain of the core protein of hepatitis B virus make pleiotropic contributions to genome replication. J Virol 85, 1298–1309, doi: 10.1128/JVI.01957-10 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyvne S. A., R. A. C. A. G. W. L. The crystal structure of the human hepatitis B virus capsid. Molecular Cell 3, 771–780 (1999). [DOI] [PubMed] [Google Scholar]
- Ceres P. & Zlotnick A. Weak protein-protein interactions are sufficient to drive assembly of hepatitis B virus capsids. Biochemistry 41, 11525–11531 (2002). [DOI] [PubMed] [Google Scholar]
- Zlotnick A., Ceres P., Singh S. & Johnson J. M. A small molecule inhibits and misdirects assembly of hepatitis B virus capsids. J Virol 76, 4848–4854, doi: 10.1128/jvi.76.10.4848-4854.2002 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S. & Zlotnick A. Observed hysteresis of virus capsid disassembly is implicit in kinetic models of assembly. The Journal of biological chemistry 278, 18249–18255, doi: 10.1074/jbc.M211408200 (2003). [DOI] [PubMed] [Google Scholar]
- Lee K. W. & Tan W. S. Recombinant hepatitis B virus core particles: association, dissociation and encapsidation of green fluorescent protein. Journal of virological methods 151, 172–180, doi: 10.1016/j.jviromet.2008.05.025 (2008). [DOI] [PubMed] [Google Scholar]
- Beterams G., Böttcher B. & Nassal M. Packaging of up to 240 subunits of a 17 kDa nuclease into the interior of recombinant hepatitis B virus capsids. Febs Lett 481, 169–176 (2000). [DOI] [PubMed] [Google Scholar]
- Cooper A. & Shaul Y. Recombinant viral capsids as an efficient vehicle of oligonucleotide delivery into cells. Biochemical and biophysical research communications 327, 1094–1099, doi: 10.1016/j.bbrc.2004.12.118 (2005). [DOI] [PubMed] [Google Scholar]
- Choi K. M., Choi S. H., Jeon H., Kim I. S. & Ahn H. J. Chimeric capsid protein as a nanocarrier for siRNA delivery: stability and cellular uptake of encapsulated siRNA. ACS nano 5, 8690–8699, doi: 10.1021/nn202597c (2011). [DOI] [PubMed] [Google Scholar]
- Rosano G. L. & Ceccarelli E. A. Recombinant protein expression in Escherichia coli: advances and challenges. Front Microbiol 5, 172, doi: 10.3389/fmicb.2014.00172 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana S. & Deb J. K. Strategies for efficient production of heterologous proteins in Escherichia coli. Applied microbiology and biotechnology 67, 289–298, doi: 10.1007/s00253-004-1814-0 (2005). [DOI] [PubMed] [Google Scholar]
- Makrides S. C. Strategies for achieving high-level expression of genes in Escherichia coli. Microbiological Reviews 60, 512–538 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier F. W. Protein production by auto-induction in high density shaking cultures. Protein expression and purification 41, 207–234 (2005). [DOI] [PubMed] [Google Scholar]
- Palmer I. & Wingfield P. T. Preparation and extraction of insoluble (inclusion-body) proteins from Escherichia coli. Curr Protoc Protein Sci Chapter 6, Unit 6.3, doi: 10.1002/0471140864.ps0603s38 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graslund S. et al. Protein production and purification. Nature methods 5, 135–146, doi: 10.1038/nmeth.f.202 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura Y. et al. Granting specificity for breast cancer cells using a hepatitis B core particle with a HER2-targeted affibody molecule. Journal of biochemistry 153, 251–256, doi: 10.1093/jb/mvs140 (2013). [DOI] [PubMed] [Google Scholar]
- Tartof K. D. & Hobbs C. A. Improved media for growing plasmid and cosmid clones (1987).
- Kahne D. & Still W. C. Hydrolysis of a peptide bond in neutral water. Journal of the American Chemical Society 110, 7529–7534, doi: 10.1021/ja00230a041 (1988). [DOI] [Google Scholar]
- Kang J., Lee M. S. & Gorenstein D. G. Application of RNase in the purification of RNA-binding proteins. Analytical biochemistry 365, 147–148, doi: 10.1016/j.ab.2007.03.003 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlotnick A. et al. Dimorphism of hepatitis B virus capsids is strongly influenced by the C-terminus of the capsid protein. Biochemistry 35, 7412–7421, doi: 10.1021/bi9604800 (1996). [DOI] [PubMed] [Google Scholar]
- Zlotnick A. et al. Localization of the C terminus of the assembly domain of hepatitis B virus capsid protein: implications for morphogenesis and organization of encapsidated RNA. Proc Natl Acad Sci USA 94, 9556–9561 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeng K. S., Hu C. P. & Chang C. M. Differential formation of disulfide linkages in the core antigen of extracellular and intracellular hepatitis B virus core particles. J. Virol. 65, 3924–3927 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pumpens P. & Grens E. Hepatitis B core particles as a universal display model: a structure-function basis for development. Febs Lett 442, 1–6, doi: 10.1016/S0014-5793(98)01599-3 (1999). [DOI] [PubMed] [Google Scholar]
- Glasel J. A. Validity of nucleic acid purities monitored by 260nm/280nm absorbance ratios. BioTechniques 18, 62–63 (1995). [PubMed] [Google Scholar]
- Greenfield N. J. Using circular dichroism spectra to estimate protein secondary structure. Nature protocols 1, 2876–2890, doi: 10.1038/nprot.2006.202 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen H. P. & Mortensen K. K. Advanced genetic strategies for recombinant protein expression in Escherichia coli. Journal of biotechnology 115, 113–128, doi: 10.1016/j.jbiotec.2004.08.004 (2005). [DOI] [PubMed] [Google Scholar]
- Rosano G. L. & Ceccarelli E. A. Recombinant protein expression in Escherichia coli: advances and challenges. Frontiers in Microbiology 5, 172, doi: 10.3389/fmicb.2014.00172 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivashanmugam A. et al. Practical protocols for production of very high yields of recombinant proteins using Escherichia coli. Protein science : a publication of the Protein Society 18, 936–948, doi: 10.1002/pro.102 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blommel P. G., Becker K. J., Duvnjak P. & Fox B. G. Enhanced bacterial protein expression during auto-induction obtained by alteration of lac repressor dosage and medium composition. Biotechnology progress 23, 585–598, doi: 10.1021/bp070011x (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broos K. et al. Expression, purification and characterization of full-length RNA-free hepatitis B core particles. Protein expression and purification 54, 30–37, doi: 10.1016/j.pep.2007.02.006 (2007). [DOI] [PubMed] [Google Scholar]
- Cabrita L. D. & Bottomley S. P. In Biotechnology Annual Review Vol. Volume 10 31–50 (Elsevier, 2004). [DOI] [PubMed] [Google Scholar]
- Rudolph R. & Lilie H. In vitro folding of inclusion body proteins. The FASEB Journal 10, 49–56 (1996). [PubMed] [Google Scholar]
- Patra A. K. et al. Optimization of Inclusion Body Solubilization and Renaturation of Recombinant Human Growth Hormone from Escherichia coli. Protein expression and purification 18, 182–192, doi: http://dx.doi.org/10.1006/prep.1999.1179 (2000). [DOI] [PubMed] [Google Scholar]
- Watlaufer D. B., Malik S. K., Stoller L. & Coffin R. L. Nonpolar Group Participation in the Denaturation of Proteins by Urea and Guanidinium Salts. Model Compound Studies. Journal of the American Chemical Society 86, 508–514, doi: 10.1021/ja01057a045 (1964). [DOI] [Google Scholar]
- Rossky P. J. Protein denaturation by urea: slash and bond. Proc Natl Acad Sci USA 105, 16825–16826, doi: 10.1073/pnas.0809224105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y., Gyoo Park S., Yoo J. H. & Jung G. Calcium ions affect the hepatitis B virus core assembly. Virology 332, 454–463, doi: 10.1016/j.virol.2004.11.019 (2005). [DOI] [PubMed] [Google Scholar]
- Birnbaum F. & Nassal M. Hepatitis B virus nucleocapsid assembly: primary structure requirements in the core protein. J Virol 64, 3319–3330 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochmann D., Jauk E. & Zimmer A. Drug delivery of oligonucleotides by peptides. European journal of pharmaceutics and biopharmaceutics: official journal of Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik e.V 58, 237–251, doi: 10.1016/j.ejpb.2004.03.031 (2004). [DOI] [PubMed] [Google Scholar]
- Zlotnick A. et al. In vitro screening for molecules that affect virus capsid assembly (and other protein association reactions). Nature protocols 2, 490–498, doi: 10.1038/nprot.2007.60 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne C. R., Finn M. G. & Zlotnick A. Global Structural Changes in Hepatitis B Virus Capsids Induced by the Assembly Effector HAP1. Journal of Virology 80, 11055–11061, doi: 10.1128/jvi.00933-06 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowther R. A. et al. Three-dimensional structure of hepatitis B virus core particles determined by electron cryomicroscopy. Cell 77, 943–950 (1994). [DOI] [PubMed] [Google Scholar]
- Wynne S. A., Crowther R. A. & Leslie A. G. The crystal structure of the human hepatitis B virus capsid. Mol Cell 3, 771–780 (1999). [DOI] [PubMed] [Google Scholar]
- Paul Pumpens E. G. HBV Core Particles as a Carrier for B Cell/T Cell Epitopes. Intervirology 44, 98–114 (2001). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.