

Abstract
Mucopolysaccharidosis type I (MPS IH) is a lysosomal storage disease (LSD) caused by inactivating mutations to the alpha-L-iduronidase (IDUA) gene. Treatment focuses on IDUA enzyme replacement and currently employed methods can be non-uniform in their efficacy particularly for the cardiac and craniofacial pathology. Therefore, we undertook efforts to better define the pathological cascade accounting for treatment refractory manifestations and demonstrate a role for the renin angiotensin system (RAS) using the IDUA−/− mouse model. Perturbation of the RAS in the aorta was more profound in male animals suggesting a causative role in the observed gender dimorphism and angiotensin receptor blockade (ARB) resulted in improved cardiac function. Further, we show the ability of losartan to prevent shortening of the snout, a common craniofacial anomaly in IDUA−/− mice. These data show a key role for the RAS in MPS associated pathology and support the inclusion of losartan as an augmentation to current therapies.
Introduction
The renin angiotensin system (RAS) controls systemic pressure and fluid balance and there is growing evidence that supports the existence of multiple, autonomously acting, intra-organ RASs that regulate the local levels of angiotensin II (ang II) (Lubel et al 2008; van Esch et al 2008; De Mello and Frohlich 2011). Ang II signals primarily through the angiotensin type 1 (AT1R) or type 2 (AT2R) receptors. AT1R signaling has been shown to activate multiple pathways with an associated increase in cytokine and matrix metalloproteinase (MMP) gene expression while AT2R signaling countermands these effects (Berk 2003).
A pathogenic signature consisting of MMP upregulation, tissue architecture erosion, and transcriptional dysregulation is observed in the aorta of human and animal models of the lysosomal storage disease (LSD) mucopolysaccharidosis type IH (MPS IH/Hurler syndrome). We hypothesized that RAS dysregulation is a contributing pathological feature of MPS IH and would therefore benefit from angiotensin receptor blockade (ARB) with losartan.
Lysosomal enzymes mediate the degradation of glycosaminoglycans (GAG) and a lack of IDUA leads to the accumulation of GAG metabolites causing systemic pathology (Neufeld and Meunzer 2001). Treatment with recombinant enzyme replacement therapy and/or hematopoietic cell transplantation can resolve some pathogenic features; however, these do not uniformly extend to the cardiac, and skeletal systems representing a significant therapeutic gap (Neufeld and Meunzer 2001; Herati et al 2008; Gabrielli et al 2010).
Therefore we undertook studies in the IDUA−/− animal model that closely mimics human MPS IH (Clarke et al 1997) and observed a molecular profile in aortic tissue consistent with overactive RAS signaling that was more pronounced in male mice. We initiated ARB with losartan in adult and juvenile male animals and performed serial echocardiographic (echo) analyses and observed improved cardiac functionality when treatment was initiated in juvenile but not adult mice. Finally, the skulls of ARB treated and untreated animals were analyzed and demonstrated improvement in the craniofacial skeletal manifestations in response to losartan. Collectively, our data show the benefit of losartan and indicate that future therapies for LSDs could include ARB.
Materials and methods
Animals
Wild-type (WT) C57BL6 and IDUA−/− (C57BL6-MPS) (Clarke et al 1997) mice were obtained from Jackson Laboratories (Bar Harbor, ME). Four groups of mice were employed (Fig. 1a). Group 1 consisted of five adult IDUA −/− mice and four age-matched WT male mice that underwent a single ultrasound examination between 7–8 months of age. In group 2, aortic arch tissue (n = 3 each male/female WT and 4 each male or female IDUA−/−) was harvested and analyzed by quantitative reverse transcription PCR (qRT-PCR). Group 3 was comprised of eight adult IDUA−/− mice that were either untreated (n = 4) or treated (n = 4) with angiotensin receptor blockade. Cardiac ultrasounds were performed on these mice prior to initiation of ARB and approximately every 6 weeks thereafter. One untreated IDUA−/− mouse expired at 234 days and the remainder of the IDUA mice underwent elective sacrifice at mean ages of 341.8 or 292.3 days for treated or untreated groups, respectively. Group 4 consisted of juvenile mice in an ARB treatment trial that were segregated thusly: IDUA−/− and WT males receiving ARB (N = 6 MPS mice; N = 6 WT mice); IDUA−/− and WT without treatment (N = 6 MPS mice; N = 5 WT mice). Cardiac ultrasounds were performed on all mice prior to initiation of protocol and approximately every 6 weeks until elective sacrifice at 262 ± 9 days of age for IDUA−/− +/− ARB mice and 208 ± 4 days of age for WT mice, respectively. One spontaneous death occurred (IDUA−/− no ARB at age 138 days).
Fig. 1.

Treatment groups, aortic enlargement, and gene expression analysis. a Treatment groups. Group 1 was comprised of five adult IDUA−/− mice and four age-matched WT mice of 7 and 8 months of age that were analyzed by echocardiography immediately prior to elective sacrifice. Group 2 was n = 3 each male/female WT and 4 each IDUA−/− (age 6–8 months) whose aortic root/arch tissue was harvested for gene quantitative expression analysis. Group 3 was n = 4 each IDUA−/− male animals that were left untreated or were treated with losartan. Group 4 were juvenile IDUA−/− or WT male mice whose numbers were split into six each in the no treatment or ARB group, respectively. X = untreated group, q weekly refers to the weekly replacement of losartan in the animals water. Ascending aorta echo analysis. Seven-month-old male mice were analyzed by high-resolution echo and the diameter of their aorta measured. A representative image is shown for b WT and c IDUA−/− mice and mean measurements (determined at orange line) were: WT = 1.38 mm ± 0.87 mm and IDUA−/− = 2.87 mm ± 0.88 mm and, p = 0.012). d Targeted gene expression analysis in MPS male and female mice. Aortic root/arch tissue was analyzed by qRT-PCR and was internally normalized to the GAPDH housekeeping gene with further standardization to reference sex and age matched C57/Bl6 mice. Relative quantitation using the 2-ddCT methodology is shown with standard deviation. * = p < 0.01 by Student’s t-test. ACE = angiotensin converting enzyme or ACE2 variant. AT = angiotensin receptor 1 or 2, CTS = cathepsin D or S, MMP = matrix metalloproteinase, TGF = transforming growth factor, OPN = osteopontin, ELN = elastin. WT mice (n = 3), IDUA−/− each for age matched male or female mice (n = 4)
Angiotensin receptor blockade
Losartan (AK Scientific, Union City, CA) was administered weekly in the drinking water at a dose of 0.6 g/L.
High resolution cardiac ultrasound
Two-dimensional, pulse-wave and color Doppler cardiac ultrasound was performed on anesthetized (2 % isoflurane) mice by a blinded single experienced user using either a Visual Sonics 770 Imaging System with 30-mHz transducer (group 1) or a Visual Sonics 2100 Imaging System with 18–38 mHz transducer (groups 3 and 4). Two-dimensional and M-mode images were obtained in parasternal long- and short-axis for determination of left ventricular chamber dimensions in diastole (LVID) and systole (LVIS) for calculation of shortening fraction (SF). M-mode measurements were obtained after discontinuation of isoflurane as the mice were emerging from anesthesia heart rates of ~500 bpm. Two-dimensional and pulse-wave and color Doppler images were obtained from modified high right parasternal view for measurement of ascending aorta measured at the level of the right pulmonary artery during peak expansion of the vessel, and for determination of the presence of aortic regurgitation. Pulse-wave and color Doppler interrogation of the descending aorta for the presence of flow-reversal (‘run-off’) was made from a modified high right lateral long-axis view. Measurement of the aortic sinus and sinotubular ridge were made at maximal valve opening from a modified left parasternal long-axis view. All measurements were made in triplicate and values averaged. Data are summarized with the mean +/− standard deviation and differences in outcomes between groups at the final measurement were evaluated adjusting for baseline values with robust variance estimation for confidence intervals and p-values (Frison and Pocock 1992; Senn 2006). Analyses were conducted using R v3.1.1 software (https://www.r-project.org/). Premature spontaneous death in both adult and juvenile mice occurred only in the untreated mice.
Gene expression
Aortic arch tissue was harvested and total RNA was isolated using the RNeasy Plus Micro Kit (Qiagen, Valencia, CA) followed by reverse transcription using the SuperScript® VILO™ cDNA Synthesis Kit (ThermoFisher, Waltham, MA). Target gene enrichment was accomplished using the TaqMan® PreAmp Master Mix Kit (ThermoFisher, Waltham, MA) and TaqMan® quantitative RT-PCR using the following probes (all from ThermoFisher):
| Probe: | Gene: |
|---|---|
| Mm00802048_m1 | (ACE) |
| Mm01159003_m1 | (ACE2) |
| Mm01957722_s1 | (AT1R) |
| Mm01341373_m1 | (AT2R) |
| Mm00515586_m1 | (CTSD) |
| Mm01255859_m1 | (CTSS) |
| Mm00439498_m1 | (MMP-2) |
| Mm00442991_m1 | (MMP-9) |
| Mm00500554_m1 | (MMP-12) |
| Mm01178820_m1 | (TGF-β1) |
| Mm00436767_m1 | (SPP1/OPN) |
| Mm00514670_m1 | (ELN) |
| Mm99999915_g1 | (GAPDH) |
The expression data was acquired in duplicate using the StepOnePlus™ Real-Time PCR System and analyzed using the 2−ΔΔCt method.
Geometric morphometric analysis
Skulls were scanned using a Scanco VivaCT at a resolution of 35 μm. 3D reconstructions from these scans were then landmarked using a standard set of 54 landmarks (Hallgrimsson et al 2009, 2015; Percival et al 2016). The sample analyzed for craniofacial shape variation consists of IDUA−/− mice (age range of 2–8 months), 49 C57BL/6 J mice along with five untreated and six losartan treated male IDUA−/− juvenile animals from group 4. Craniofacial shape was assessed by geometric morphometric methodology using the Procrustes superimposition as previously described (Mitteroecker and Gunz 2009). We obtained age-standardized residuals for each individual and then used principal components analysis to visualize variation within the entire dataset. To compare groups, we used the Procrustes distance permutation test and the Mahalanobis distance permutation test in Morpho J (Klingenberg 2011). Canonical variates analysis was used to visualize shape variation between groups but not to determine the existence of those differences.
Glycosaminoglycan quantification
Tissues were homogenized in RLB (Promega, Madison, WI) and analyzed using the dimethylmethylene blue methodology as described previously (Osborn et al 2011).
Results
Adult mouse echocardiography and gene expression
Similar to Marfan syndrome (MFS) we observed dilatation of the aorta in IDUA−/− mice (Merk et al 2012) (Figs. 1b and c). However, in contrast to the dysregulation of TGF-β seen in MFS, we did not observe significant changes to the TGF-β expression levels (Fig. 1d) (Ma et al 2008; Metcalf et al 2010). Rather, the expression of MMP genes was elevated, a finding that is in agreement with MFS and previous IDUA−/− animal studies (Braunlin et al 2006; Ikonomidis et al 2006; Tolar et al 2009). Cathepsin D (CTSD), cathepsin S (CTSS), and osteopontin (OPN) gene upregulation was noted which is significant in that cathepsin S can potentiate Ang II signaling (Qin et al 2012), cathepsin D is capable of local conversion of Ang I to Ang II (De Mello and Danser 2000), and Ang II results in OPN gene upregulation with subsequent vascular remodeling (deBlois et al 1996).
Analysis of the receptors and enzymes directly associated with the RAS revealed a gender dimorphism (Fig. 1d). Male IDUA−/− mice had higher AT1R levels than females and WT and females had higher angiotensin-converting enzyme 2 variant (ACE2) compared to male IDUA−/− mice. These findings are highly significant given the fact that estrogens are associated with the upregulation of AT2R (Armando et al 2002) and ACE2 (Shenoy et al 2009; Hilliard et al 2013) gene expression suggesting the observed gender disparity (Tolar et al 2009) may be linked to the RAS. These findings are of additional importance as they provided impetus for ARB in IDUA−/− mice as it has been documented in MFS that blockade of AT1R with losartan shunts Ang II to the AT2R for its mediation of cardiac benefit (Habashi et al 2011).
ARB treatment for MPS
Studies were structured to employ adult (~6 month; group 3) or juvenile (~2 month; group 4) IDUA−/− animals (Fig. 1a). As a control we administered losartan to WT animals and performed echo analyses at baseline and >200 days later. These animals showed a decrement in shortening fraction (SF) compared to untreated with no further statistically significant changes (Supplementary Table 1).
ARB in adult IDUA−/− animals (group 3) showed no significant differences over any baseline parameter after 3.5 months of losartan treatment (Supplementary Table 2). These data showed a lack of improvement in IDUA−/− mice when ARB was initiated at an advanced age due to established disease pathology that was unable to be reversed. Subsequently, weanling IDUA−/− mice all underwent baseline cardiac ultrasound at ~2 months (62 ± 7 days) of age (Fig. 1a, group 4). We noted that the aortic sinus and ascending aorta were larger than WT mice by 2 months of age and to insure uniformity and accuracy of assessment we stratified the treatment groups by size-matching the ascending aorta within the IDUA−/− groups. As such, the animals in the losartan treatment group were older (69.7 vs 55.8 days, p = 0.0016).
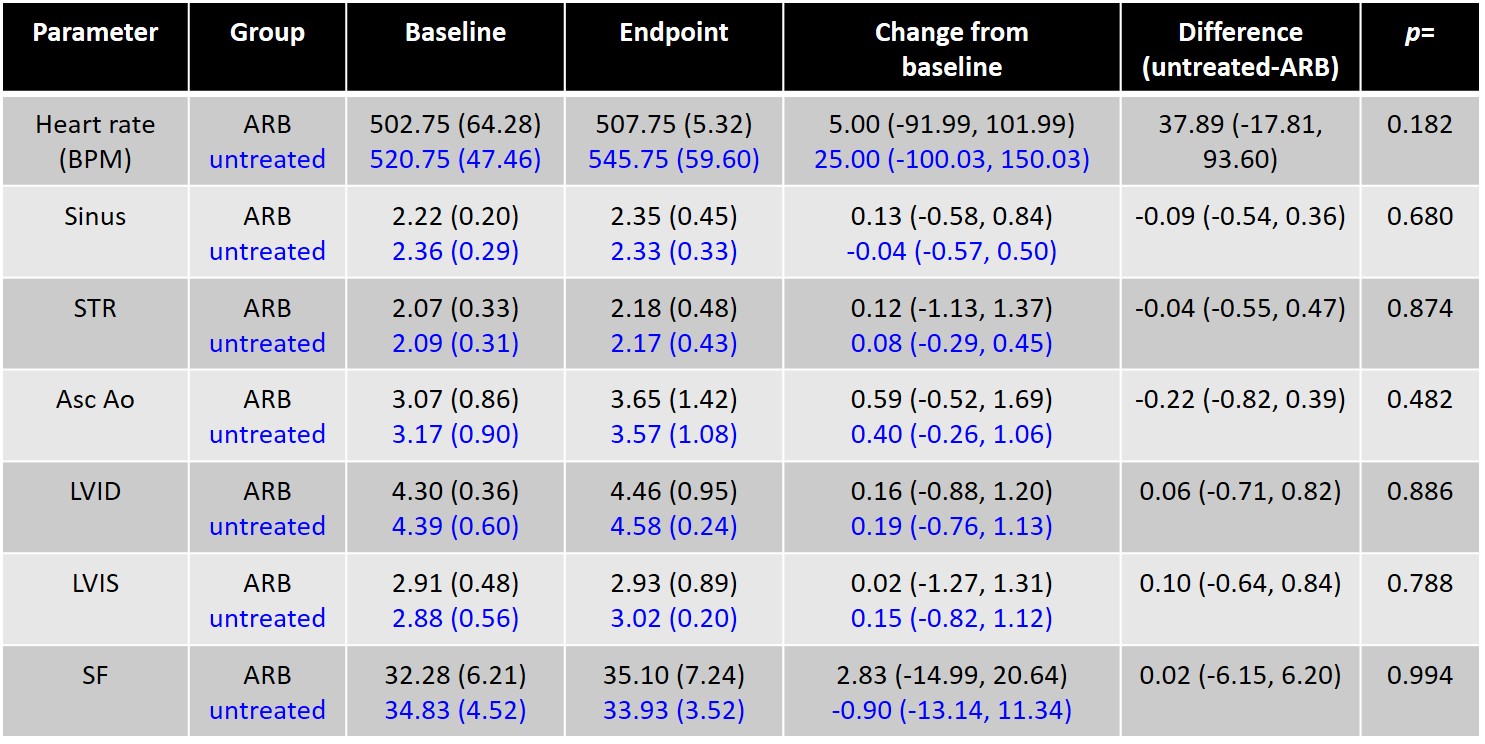
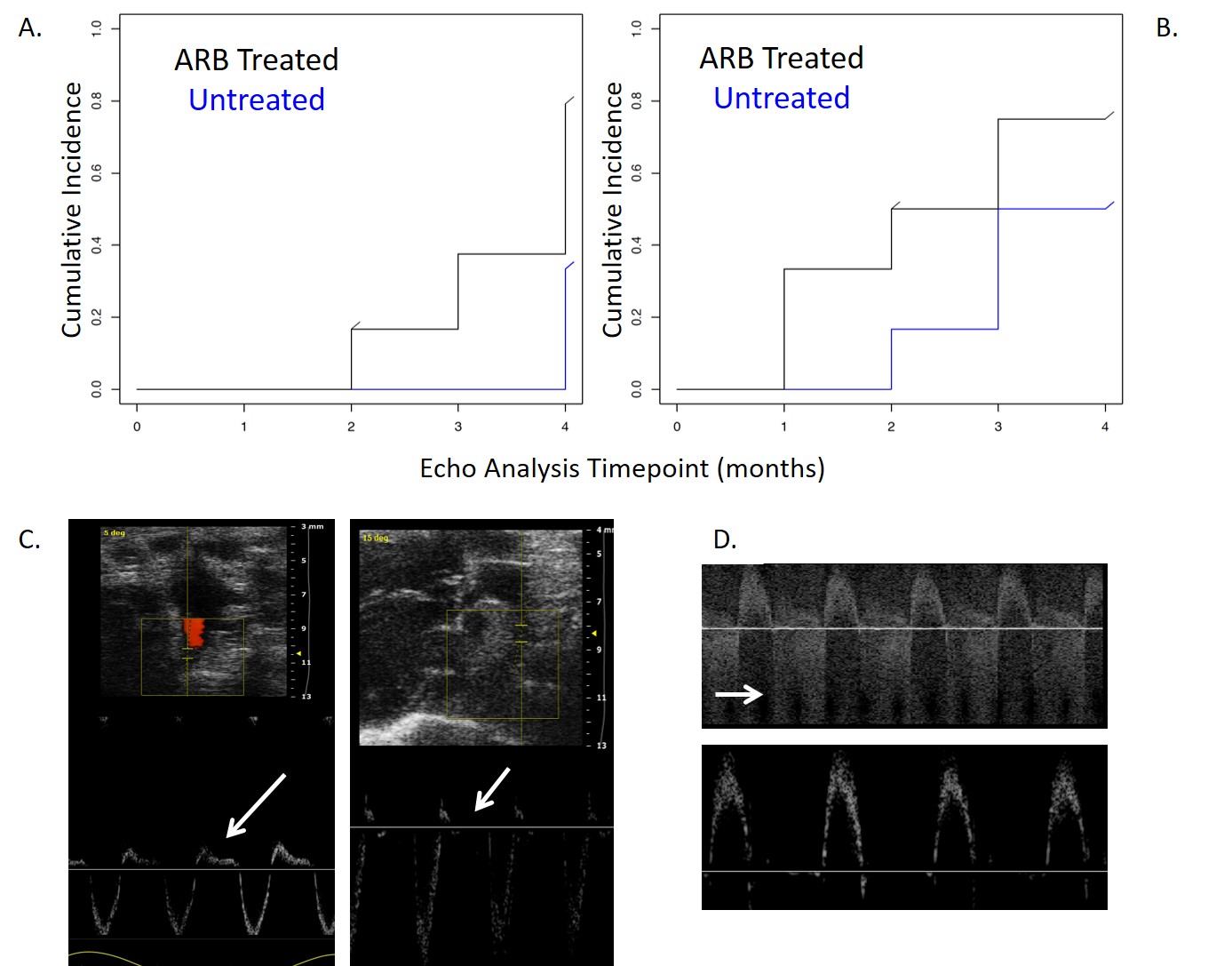
The echo data is shown in Table 1 and untreated IDUA−/− mice showed enlargement as measured by increased left ventricular internal diameter at diastole (LVID) and systole (LVIS). Further, the SF, a measure of left ventricular function, was decreased; all hallmarks of LSD cardiopathology (Table 1). ARB prevented these manifestations as evidenced by a stabilized LVID and LVIS at the endpoint compared to the baseline with a significantly increased SF (Table 1). Additionally, the time to development of aortic insufficiency/regurgitation was earlier for untreated IDUA−/− mice, despite their younger age, when compared to losartan treated mice (Supplementary Fig. 1). Further, none of the IDUA−/− plus ARB mice developed descending aortic flow reversal while 80 % of untreated IDUA−/− mice did (Supplementary Fig. 1). To our knowledge this is the first demonstration of the benefit of losartan in improving the functional capacity of the heart in the IDUA−/− model if initiated in younger, but not older, animals.
Table 1.
Juvenile IDUA−/− animal echo data
| Parameter | Group | Baseline | Endpoint | Change from baseline | Difference (untreated-ARB) | p= |
|---|---|---|---|---|---|---|
|
| ||||||
| Heart rate | ARB | 499.17 (37.95) | 522.67 (45.91) | 53.50 (6.78, 100.22) | −10.15 (−51.93, 31.62) | 0.634 |
| untreated | 506.80 (130.32) | 542.80 (32.52) | 36.00 (−133.33, 205.33) | |||
|
| ||||||
| Sinus | ARB | 1.87 (0.10) | 2.19 (0.26) | 0.32 (0.05, 0.59) | 0.18 (−0.05, 0.41) | 0.130 |
| untreated | 1.85 (0.07) | 2.35 (0.24) | 0.50 (0.27, 0.73) | |||
|
| ||||||
| STR | ARB | 1.34 (0.12) | 1.84 (0.30) | 0.50 (0.20, 0.79) | 0.09 (−0.19, 0.37) | 0.536 |
| untreated | 1.47 (0.11) | 2.12 (0.34) | 0.65 (0.34, 0.96) | |||
|
| ||||||
| Asc Ao | ARB | 1.57 (0.13) | 2.93 (1.02) | 1.36 (0.37, 2.35) | −0.03 (−0.65, 0.60) | 0.934 |
| untreated | 1.62 (0.10) | 3.16 (0.51) | 1.53 (1.00, 2.07) | |||
|
| ||||||
| LVID | ARB | 3.97 (0.21) | 4.01 (0.64) | 0.05 (−0.63, 0.72) | 0.78 (0.15, 1.40) | 0.015 |
| untreated | 3.95 (0.38) | 4.76 (1.07) | 0.81 (−0.09, 1.71) | |||
|
| ||||||
| LVIS | ARB | 2.59 (0.44) | 2.32 (0.58) | −0.27 (−1.15, 0.61) | 1.16 (0.32, 2.00) | 0.007 |
| untreated | 2.40 (0.44) | 3.36 (0.98) | 0.96 (0.20, 1.72) | |||
|
| ||||||
| SF | ARB | 34.82 (8.64) | 42.72 (6.12) | 7.90 (−5.38, 21.18) | −12.34 (−18.56, −6.11) | <0.001 |
| untreated | 39.54 (5.55) | 30.00 (4.17) | −9.54 (−13.08, −6.00) | |||
n = 6 IDUA−/− animals were treated with losartan ARB and n = 6 were left untreated. Mean (SD) ECHO measurements at baseline and final time points (performed at postnatal 250 days for treated and 275d untreated) and change from baseline (95 % CI) are shown along with differences between groups adjusting for baseline values (95 % CI). Losartan treated animals are in black lettering and untreated are in blue. Statistically significant values/parameters are in bold
Gene expression in losartan treated IDUA−/− mice
The MMP-12 and ACE2 mRNA levels in the aorta whose gene expression levels were significantly altered by ARB (p = <0.05) are shown in Fig. 2a and b. The other analyzed parameters (ACE, AT1, AT2, CTSD, CTSS, MMP-2, MMP-9, TGF, OPN, and ELN) did not show statistically significant changes (data not shown). To rule out any effect of ARB on GAG accumulation levels, we assessed GAG concentration in the liver and kidney and found no statistical difference between the treatment and control groups (Fig. 2c). These data are consistent with the levels of losartan induced ACE2 upregulation and partial MMP-12 decrement observed in other therapeutic models employing ARB (Podowski et al 2012; Zhu et al 2015) and complement the functional improvement observed by echocardiography.
Fig. 2.

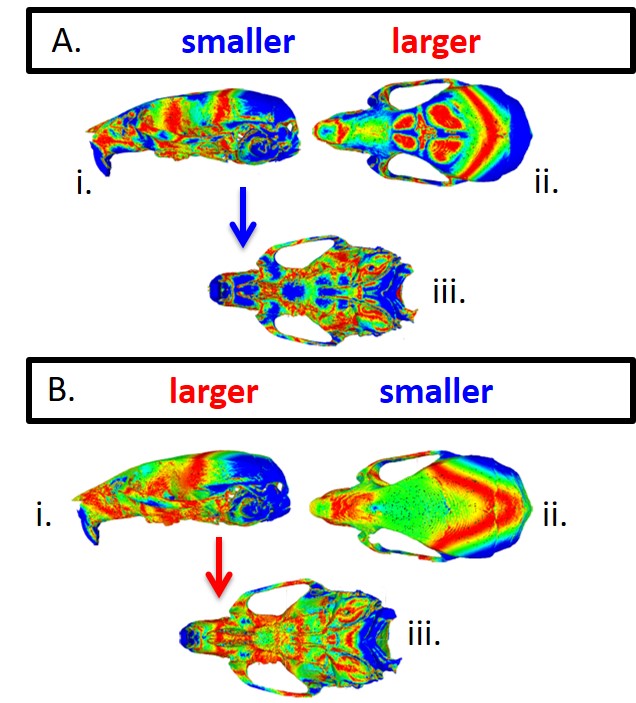
Quantitative gene expression and glycosaminoglycan storage analysis. Group 4 (juvenile) IDUA−/− mice (treated, n = 6 or untreated, n = 5 with losartan) were analyzed by qRT PCR for a MMP-12 and b ACE2 with normalization to GAPDH with untreated IDUA−/− animals in the cohort serving as the reference. p = 0.023 for MMP-12 and p = 0.010 for ACE2 (Student’s t-test). c GAG measurement. The liver and kidney of each animal was harvested for GAG measurement and is shown as micrograms of GAG per milligram of tissue. Standard error of the mean is shown and ns = not statistically significant. p = 0.277 for kidney and 0.725 for liver (Student’s t-test). Geometric morphometric analysis of three-dimensional craniofacial shape. d Principal components (PC) plot showing variation within and among the untreated IDUA−/− group controls (red dots), C57Bl/6 WT (purple dots), age and sex matched losartan treated (green dots) or untreated IDUA−/− (light blue dots) groups analyzed. For craniofacial shape e 3D morphs showing the shape variation along principal component 1. From left to right are representative images of untreated or losartan treated IDUA−/− or WT mice. Blue bracket indicates the region of the skull that was normalized by losartan
Craniofacial evaluation
A hallmark of IDUA−/− mouse skeletal disease is a prominent shortening of the snout (Clarke et al 1997). High-resolution micro-CT with geometric morphometric analysis was employed and, as determined by the Procrustes distance permutation test, untreated IDUA−/− mice differ significantly from C57BL/6 J mice in craniofacial shape (p < 0.0001) with IDUA−/− mice having shorter premaxillae, higher and more rounded neurocranial vaults, and smaller midfacial regions (Figs. 2d and e). Bone thickness heatmapping of IDUA−/− mice showed a significantly shorter snout that was improved by losartan treatment (Supplementary Fig. 2).
Discussion
Here we establish a role for the RAS in MPS pathology and show that ARB can ameliorate both the cardiac and craniofacial manifestations. Moreover, the differential expression of RAS responsive genes observed in male and female IDUA−/− mice provides a mechanistic explanation for the gender dimorphism observed in the IDUA−/− murine model (Tolar et al 2009). While human MPS patients do not appear to exhibit gender differences, the highly similar murine and human cardiac symptomology supports the investigation of ARB in MPS patients. An important consideration will be how well pediatric MPS patients tolerate losartan. Importantly, losartan has been evaluated for use in hypertensive pediatric patients with low incidence of side effects (Shahinfar et al 2005; Webb et al 2010). While hypertension was not evident in our model, the application of losatan in pediatric cardiac disease supports investigation in the context of MPS.
Targeting the entire pathogenic spectrum with combinatorial agents that treat the primary and secondary LSD-related pathologies has shown significant effect in a rodent model of globoid cell leukodystrophy (Hawkins-Salsbury et al 2015). To date such ‘mechanism-based’ strategies have been lacking in the most severe LSD, MPS IH, due to a poor understanding of the GAG-triggered secondary pathological cascade. Toward better defining the molecular drivers of MPS cardiac pathology Ma and colleagues assessed TGF-β gene expression in IDUA−/− mice (Ma et al 2008) due to its role in MFS aortic pathology that at the gross level shares striking similarity to MPS. Both we and Ma et al observed only a modest increase in TGF-β gene expression (Fig. 1d) with no TGF-β secondary messenger activation (Ma et al 2008). This indicated that the mediators of MFS and MPS pathology may be partially, but not equally, overlapping. Instead, Ma observed active STAT 1/3 and MAPK signaling and because these pathways are responsive to Ang II signaling via AT1R (McWhinney et al 1997; Venema et al 1998; Ma et al 2008) we assessed whether RAS signaling was operative in the aorta of IDUA−/− mice. We observed an increase in the levels of direct constituents of the RAS: ACE, AT1R, AT2R, as well as the Ang II responsive genes cathepsins S and D, osteopontin, MMP-2, and MMP-12 (Fig. 1d). Cathepsins are lysosomal proteases and cathepsin D is able to generate Ang II (Hu et al 2003), and cathepsin S has been implicated in cardiac disease and can be inhibited by ARB with losartan (Cheng et al 2012). Intriguingly, primary IDUA loss with GAG accumulation leads to secondary elevation of other lysosomal mRNAs (e.g., β-hexosaminidase) (Liu et al 2005) and this appears to extend to the cathepsin family as shown by our new data. These findings supported our hypothesis for a role of the RAS in the IDUA−/− mouse model of MPS.
We also extended our analyses to include female IDUA mice to determine whether their lower incidence of cardiac pathology was related to the RAS (Brosnihan et al 2008; Shenoy et al 2009; Tolar et al 2009). Our data show similar levels of AT2R mRNA in males and females but a gender disparity in AT1R, with males expressing elevated levels (Fig. 1d). The cardioprotective ACE2, whose expression is impacted by both estrogens and Ang II signaling through AT2R, was also increased in IDUA−/− females (Fig. 1d). Collectively these data provide an explanation for the lower incidence of cardiac disease in females. Previous studies show that Ang II associated signaling through the AT1 receptor is primarily responsible for pathogenic effects that can be counterbalanced by AT2R signaling (Habashi et al 2011; Mendoza-Torres et al 2015). Further, in MFS, RAS with losartan blockade but not with the ACE inhibitor enalapril resulted in decreased MMPs in the aorta and improved cardiac function (Habashi et al 2011). The mechanism for this is believed to be due to losartan AT1R blockade driving AngII to the AT2R receptor, resulting in cardiac benefit (Habashi et al 2011). Given that the IDUA−/− male mice have elevated AT1R and AT2R we administered losartan to adult or juvenile aged IDUA male mice to determine whether a beneficial effect could be achieved.
Similar to the losartan-mediated effects in murine models of MFS (Cook et al 2014) and muscular dystrophy (Spurney et al 2011), juvenile IDUA−/− mice exhibited increased SF in response to ARB (Table 1). Moreover, the chamber dimensions (LVID and LVIS) of losartan-treated mice were not significantly increased when compared to baseline values (LVID 3.97 mm at baseline vs 4.01 at final measurement and LVIS 2.59 mm beginning and 2.32 mm at endpoint), while untreated IDUA−/− animals showed a statistically significant decline of SF and pathological enlargement of the chambers (LVID 3.95 mm at baseline vs 4.76 at final measurement and LVIS 2.40 mm beginning and 3.36 mm at endpoint) (Table 1).
Molecular analysis of the aorta in ARB treated juvenile IDUA−/− mice showed a statistically significant decrement of MMP-12 and an elevation in ACE2 at levels consistent with previous reports (Figs. 2a and b) (Gallagher et al 2008; Zhang et al 2014; Zhu et al 2015). Thus our data show the benefit of ARBs in the treatment of MPS cardiac symptomology and supports our model for cardiac disease and therapeutic effect shown in Fig. 3: GAG accumulation results in secondary elevation of lysosomal enzymes (e.g., cathepsins) involved in local Ang II production. Additionally, a potentiating GAG:ang II cycle may manifest as it has been shown that ang II can stimulate GAG production(Shimizu-Hirota et al 2001) and pathogenic pathways related to GAGs and angII intersect to cause cardiac disease. In female animals that exhibit higher basal levels of ACE2, presumably driven by estrogen, the Ang II effects are attenuated accounting for the lower frequency of severe cardiac disease in MPS females (Hilliard et al 2013). In males however, local Ang II and elevated levels of AT1R drives a pathogenic cascade leading to depressed cardiac function and aortopathy characterized by upregulation of metalloproteinase genes, degradation of elastic fibers and subsequent vessel and chamber enlargement. Blockade of AT1R via losartan results in Ang II agonism of AT2R and improved cardiac symptomology.
Fig. 3.

Local RAS-driven model for MPS cardiac disease. a IDUA gene architecture and X indicates a gene mutation resulting in loss of IDUA enzymatic activity. b In the aorta, GAG accumulation results in upregulation of multiple genes including cathepsins that are capable of converting Ang I to Ang II. Ang II exerts its pathological effects through the type I angiotensin receptor (AT1R) that results in upregulation of MMPs and diminished ACE2. ARB shunts the Ang II to the AT2R through which beneficial effects (increased ACE2 and decreased MMP gene expression) are mediated. The structural benefits are an aorta with a smaller size, normalized shortening fraction, and less thickening of the chambers (observed by decreased LVID and LVIS)
Given the role of Ang II in bone development we also analyzed the skulls of IDUA−/− animals due to the fact that a manifestation of the disease is a shortened snout in animals and coarse facial features in humans (Clarke et al 1997; Neufeld and Meunzer 2001). Because any cardiac benefit related to ARB was observed only in animals treated at ~2 months of age (Table 1) we analyzed that cohort alone and observed a modest improvement in the shortening of the snout (Figs. 2d and e). These data are important as a dramatically blunted snout is prevalent at a relatively early age in IDUA−/− mice (12 weeks) (Clarke et al 1997) as well as are in agreement with another murine model with losartan that showed beneficial effects to the skeleton (Chen et al 2015).
At present the use of IDUA was not advocated and our study was intentionally done in the context of no therapeutic provision of IDUA enzyme. Our goal was to assess the efficacy of ARB, and our rationale was that in order to do so the experimental animals would need to be treated with ARB alone. Future studies will synergize the two therapeutic entities for treating the skeletal and cardiac manifestations concurrently as part of a multifaceted strategy to include defining the earliest stage treatment options that confer maximal therapeutic effect.
Conclusions
We demonstrate a role for the RAS in the pathological manifestations seen in the IDUA−/− mouse model. We observed differential levels of key effectors of the RAS in the aorta of male and female IDUA−/− mice suggesting the RAS as the cause of the observed gender disparity in cardiac disease penetrance. By treating male mice with losartan we observed improved cardiac function by echocardiography providing rationale for including losartan as an augmentation to current therapies.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors thank the Lindahl family for their vision and generosity in supporting this work. We are grateful to Nancy Griggs Morgan and Kelsey Vigoren for invaluable assistance in manuscript preparation and editing. We are also thankful for the generosity of the Children’s Cancer Research Fund, & the Corrigan Family. MJO is supported by 8UL1TR000114-02. JT is supported in part by R01 AR063070 and P01 CA065493. BRW is supported by NIH T32-HL007062. The present work was partly supported by research funds from the National Research Foundation of Korea (NRF-2015K1A4A3046807). Research reported in this publication was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health Award Number UL1TR000114 (MJO). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Compliance with ethical standards
Conflict of interest None.
Communicated by: Eva Morava
Electronic supplementary material The online version of this article (doi:10.1007/s10545-016-9988-z) contains supplementary material, which is available to authorized users.
References
- Armando I, Jezova M, Juorio AV, et al. Estrogen upregulates renal angiotensin II AT(2) receptors. Am J Physiol Renal Physiol. 2002;283:F934–F943. doi: 10.1152/ajprenal.00145.2002. [DOI] [PubMed] [Google Scholar]
- Berk BC. Angiotensin type 2 receptor (AT2R): a challenging twin. Scie STKE: Sig Trans Knowl Environ. 2003;2003:PE16. doi: 10.1126/stke.2003.181.pe16. [DOI] [PubMed] [Google Scholar]
- Braunlin E, Mackey-Bojack S, Panoskaltsis-Mortari A, et al. Cardiac functional and histopathologic findings in humans and mice with mucopolysaccharidosis type I: implications for assessment of therapeutic interventions in hurler syndrome. Pediatr Res. 2006;59:27–32. doi: 10.1203/01.pdr.0000190579.24054.39. [DOI] [PubMed] [Google Scholar]
- Brosnihan KB, Hodgin JB, Smithies O, Maeda N, Gallagher P. Tissue-specific regulation of ACE/ACE2 and AT1/AT2 receptor gene expression by oestrogen in apolipoprotein E/oestrogen receptor-alpha knock-out mice. Exp Physiol. 2008;93:658–664. doi: 10.1113/expphysiol.2007.041806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Grover M, Sibai T, et al. Losartan increases bone mass and accelerates chondrocyte hypertrophy in developing skeleton. Mol Genet Metab. 2015;115:53–60. doi: 10.1016/j.ymgme.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng XW, Shi GP, Kuzuya M, Sasaki T, Okumura K, Murohara T. Role for cysteine protease cathepsins in heart disease: focus on biology and mechanisms with clinical implication. Circulation. 2012;125:1551–1562. doi: 10.1161/CIRCULATIONAHA.111.066712. [DOI] [PubMed] [Google Scholar]
- Clarke LA, Russell CS, Pownall S, et al. Murine mucopolysaccharidosis type I: targeted disruption of the murine alpha-L-iduronidase gene. Hum Mol Genet. 1997;6:503–511. doi: 10.1093/hmg/6.4.503. [DOI] [PubMed] [Google Scholar]
- Cook JR, Carta L, Benard L, et al. Abnormal muscle mechanosignaling triggers cardiomyopathy in mice with Marfan syndrome. J Clin Invest. 2014;124:1329–1339. doi: 10.1172/JCI71059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Mello WC, Danser AH. Angiotensin II and the heart: on the intracrine renin-angiotensin system. Hypertension. 2000;35:1183–1188. doi: 10.1161/01.hyp.35.6.1183. [DOI] [PubMed] [Google Scholar]
- De Mello WC, Frohlich ED. On the local cardiac renin angiotensin system. Basic and clinical implications. Peptides. 2011;32:1774–1779. doi: 10.1016/j.peptides.2011.06.018. [DOI] [PubMed] [Google Scholar]
- deBlois D, Lombardi DM, Su EJ, Clowes AW, Schwartz SM, Giachelli CM. Angiotensin II induction of osteopontin expression and DNA replication in rat arteries. Hypertension. 1996;28:1055–1063. doi: 10.1161/01.hyp.28.6.1055. [DOI] [PubMed] [Google Scholar]
- Frison L, Pocock SJ. Repeated measures in clinical trials: analysis using mean summary statistics and its implications for design. Stat Med. 1992;11:1685–1704. doi: 10.1002/sim.4780111304. [DOI] [PubMed] [Google Scholar]
- Gabrielli O, Clarke LA, Bruni S, Coppa GV. Enzyme-replacement therapy in a 5-month-old boy with attenuated presymptomatic MPS I: 5-year follow-up. Pediatrics. 2010;125:e183–e187. doi: 10.1542/peds.2009-1728. [DOI] [PubMed] [Google Scholar]
- Gallagher PE, Ferrario CM, Tallant EA. Regulation of ACE2 in cardiac myocytes and fibroblasts. Am J Physiol Heart Circ Physiol. 2008;295:H2373–H2379. doi: 10.1152/ajpheart.00426.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habashi JP, Doyle JJ, Holm TM, et al. Angiotensin II type 2 receptor signaling attenuates aortic aneurysm in mice through ERK antagonism. Science. 2011;332:361–365. doi: 10.1126/science.1192152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallgrimsson B, Jamniczky H, Young NM, et al. Deciphering the palimpsest: studying the relationship between morphological integration and phenotypic covariation. Evol Biol. 2009;36:355–376. doi: 10.1007/s11692-009-9076-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallgrimsson B, Percival CJ, Green R, Young NM, Mio W, Marcucio R. Chapter twenty—morphometrics, 3D imaging, and craniofacial development. In: Yang C, editor. Current topics in developmental biology. Academic; New York: 2015. pp. 561–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins-Salsbury JA, Shea L, Jiang X, et al. Mechanism-based combination treatment dramatically increases therapeutic efficacy in murine globoid cell leukodystrophy. J Neurosci: Off J Soc Neurosci. 2015;35:6495–6505. doi: 10.1523/JNEUROSCI.4199-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herati RS, Ma X, Tittiger M, Ohlemiller KK, Kovacs A, Ponder KP. Improved retroviral vector design results in sustained expression after adult gene therapy in mucopolysaccharidosis I mice. J Gene Med. 2008;10:972–982. doi: 10.1002/jgm.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilliard LM, Sampson AK, Brown RD, Denton KM. The “his and hers” of the renin-angiotensin system. Curr Hypertens Rep. 2013;15:71–79. doi: 10.1007/s11906-012-0319-y. [DOI] [PubMed] [Google Scholar]
- Hu WY, Fukuda N, Ikeda Y, et al. Human-derived vascular smooth muscle cells produce angiotensin II by changing to the synthetic phenotype. J Cell Physiol. 2003;196:284–292. doi: 10.1002/jcp.10299. [DOI] [PubMed] [Google Scholar]
- Ikonomidis JS, Jones JA, Barbour JR, et al. Expression of matrix metalloproteinases and endogenous inhibitors within ascending aortic aneurysms of patients with Marfan syndrome. Circulation. 2006;114:I365–I370. doi: 10.1161/CIRCULATIONAHA.105.000810. [DOI] [PubMed] [Google Scholar]
- Klingenberg CP. Morpho J: an integrated software package for geometric morphometrics. Mol Ecol Resour. 2011;11:353–357. doi: 10.1111/j.1755-0998.2010.02924.x. [DOI] [PubMed] [Google Scholar]
- Liu Y, Xu L, Hennig AK, et al. Liver-directed neonatal gene therapy prevents cardiac, bone, ear, and eye disease in mucopolysaccharidosis I mice. Mol Ther. 2005;11:35–47. doi: 10.1016/j.ymthe.2004.08.027. [DOI] [PubMed] [Google Scholar]
- Lubel JS, Herath CB, Burrell LM, Angus PW. Liver disease and the renin-angiotensin system: recent discoveries and clinical implications. J Gastroenterol Hepatol. 2008;23:1327–1338. doi: 10.1111/j.1440-1746.2008.05461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Tittiger M, Knutsen RH, et al. Upregulation of elastase proteins results in aortic dilatation in mucopolysaccharidosis I mice. Mol Genet Metab. 2008;94:298–304. doi: 10.1016/j.ymgme.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhinney CD, Hunt RA, Conrad KM, Dostal DE, Baker KM. The type I angiotensin II receptor couples to Stat1 and Stat3 activation through Jak2 kinase in neonatal rat cardiac myocytes. J Mol Cell Cardiol. 1997;29:2513–2524. doi: 10.1006/jmcc.1997.0489. [DOI] [PubMed] [Google Scholar]
- Mendoza-Torres E, Oyarzun A, Mondaca-Ruff D, et al. ACE2 and vasoactive peptides: novel players in cardiovascular/renal remodeling and hypertension. Ther Adv Cardiovasc Dis. 2015;9:217–237. doi: 10.1177/1753944715597623. [DOI] [PubMed] [Google Scholar]
- Merk DR, Chin JT, Dake BA, et al. miR-29b participates in early aneurysm development in Marfan syndrome. Circ Res. 2012;110:312–324. doi: 10.1161/CIRCRESAHA.111.253740. [DOI] [PubMed] [Google Scholar]
- Metcalf JA, Ma X, Linders B, et al. A self-inactivating gamma-retroviral vector reduces manifestations of mucopolysaccharidosis I in mice. Mol Ther. 2010;18:334–342. doi: 10.1038/mt.2009.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitteroecker P, Gunz P. Advances in geometric morphometrics. Evol Biol. 2009;36:235–247. [Google Scholar]
- Neufeld EF, Meunzer J. The metabolic and molecular bases of inherited disease. McGraw-Hill; New York: 2001. The mucopolysaccharidoses. [Google Scholar]
- Osborn MJ, McElmurry RT, Lees CJ, et al. Minicircle DNA-based gene therapy coupled with immune modulation permits long-term expression of alpha-L-iduronidase in mice with mucopolysaccharidosis type I. Mol Ther. 2011;19:450–460. doi: 10.1038/mt.2010.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percival CJ, Liberton DK, Pardo-Manuel de Villena F, Spritz R, Marcucio R, Hallgrimsson B. Genetics of murine craniofacial morphology: diallel analysis of the eight founders of the Collaborative Cross. J Anat. 2016;228:96–112. doi: 10.1111/joa.12382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podowski M, Calvi C, Metzger S, et al. Angiotensin receptor blockade attenuates cigarette smoke-induced lung injury and rescues lung architecture in mice. J Clin Invest. 2012;122:229–240. doi: 10.1172/JCI46215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y, Cao X, Guo J, et al. Deficiency of cathepsin S attenuates angiotensin II-induced abdominal aortic aneurysm formation in apolipoprotein E-deficient mice. Cardiovasc Res. 2012;96:401–410. doi: 10.1093/cvr/cvs263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senn S. Change from baseline and analysis of covariance revisited. Stat Med. 2006;25:4334–4344. doi: 10.1002/sim.2682. [DOI] [PubMed] [Google Scholar]
- Shahinfar S, Cano F, Soffer BA, et al. A double-blind, dose–response study of losartan in hypertensive children. Am J Hypertens. 2005;18:183–190. doi: 10.1016/j.amjhyper.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Shenoy V, Grobe JL, Qi Y, et al. 17beta-Estradiol modulates local cardiac renin-angiotensin system to prevent cardiac remodeling in the DOCA-salt model of hypertension in rats. Peptides. 2009;30:2309–2315. doi: 10.1016/j.peptides.2009.09.005. [DOI] [PubMed] [Google Scholar]
- Shimizu-Hirota R, Sasamura H, Mifune M, et al. Regulation of vascular proteoglycan synthesis by angiotensin II type 1 and type 2 receptors. J Am Soc Nephrol. 2001;12:2609–2615. doi: 10.1681/ASN.V12122609. [DOI] [PubMed] [Google Scholar]
- Spurney CF, Sali A, Guerron AD, et al. Losartan decreases cardiac muscle fibrosis and improves cardiac function in dystrophin-deficient mdx mice. J Cardiovasc Pharmacol Ther. 2011;16:87–95. doi: 10.1177/1074248410381757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolar J, Braunlin E, Riddle M, et al. Gender-related dimorphism in aortic insufficiency in murine mucopolysaccharidosis type I. J Heart Valve Dis. 2009;18:524–529. [PMC free article] [PubMed] [Google Scholar]
- van Esch JH, van Gool JM, de Bruin RJ, et al. Different contributions of the angiotensin-converting enzyme C-domain and N-domain in subjects with the angiotensin-converting enzyme II and DD genotype. J Hypertens. 2008;26:706–713. doi: 10.1097/HJH.0b013e3282f465d2. [DOI] [PubMed] [Google Scholar]
- Venema RC, Venema VJ, Eaton DC, Marrero MB. Angiotensin II-induced tyrosine phosphorylation of signal transducers and activators of transcription 1 is regulated by Janus-activated kinase 2 and Fyn kinases and mitogen-activated protein kinase phosphatase 1. J Biol Chem. 1998;273:30795–30800. doi: 10.1074/jbc.273.46.30795. [DOI] [PubMed] [Google Scholar]
- Webb NJ, Lam C, Loeys T, et al. Randomized, double-blind, controlled study of losartan in children with proteinuria. Clin J Am Soc Nephrol. 2010;5:417–424. doi: 10.2215/CJN.06620909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Li B, Wang B, Zhang J, Wu J, Morgan T. Alteration of cardiac ACE2/Mas expression and cardiac remodelling in rats with aortic constriction. Chin J Physiol. 2014;57:335–342. doi: 10.4077/CJP.2014.BAD268. [DOI] [PubMed] [Google Scholar]
- Zhu L, Carretero OA, Xu J, et al. Activation of angiotensin II type 2 receptor suppresses TNF-alpha-induced ICAM-1 via NF-small ka, CyrillicB: possible role of ACE2. Am J Physiol Heart Circ Physiol. 2015;309:H827–H834. doi: 10.1152/ajpheart.00814.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.